Back to Journals » International Journal of Nanomedicine » Volume 12
Low-density lipoprotein-coupled micelles with reduction and pH dual sensitivity for intelligent co-delivery of paclitaxel and siRNA to breast tumor
Authors Zhu WJ, Yang SD, Qu CX, Zhu QL, Chen WL ![]() , Li F, Yuan ZQ, Liu Y, You BG, Zhang XN
, Li F, Yuan ZQ, Liu Y, You BG, Zhang XN ![]()
Received 1 November 2016
Accepted for publication 3 March 2017
Published 26 April 2017 Volume 2017:12 Pages 3375—3393
DOI https://doi.org/10.2147/IJN.S126310
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Wen-jing Zhu,1,* Shu-di Yang,1,* Chen-xi Qu,1 Qiao-ling Zhu,1,2 Wei-liang Chen,1 Fang Li,1 Zhi-qiang Yuan,1 Yang Liu,1 Ben-gang You,1 Xue-nong Zhang1
1Department of Pharmaceutics, College of Pharmaceutical Sciences, Soochow University, Suzhou, 2Department of Clinical Medicine, Nanjing Gulou Hospital, Nanjing, People’s Republic of China
*These authors contributed equally to this work
Abstract: Multidrug resistance (MDR) is a major obstacle for the clinical therapy of malignant human cancers. The discovery of RNA interference provides efficient gene silencing within tumor cells for reversing MDR. In this study, a new “binary polymer” low-density lipoprotein–N-succinyl chitosan–cystamine–urocanic acid (LDL–NSC–SS–UA) with dual pH/redox sensitivity and targeting effect was synthesized for the co-delivery of breast cancer resistance protein small interfering RNA (siRNA) and paclitaxel (PTX). In vivo, the co-delivering micelles can accumulate in tumor tissue via the enhanced permeability and retention effect and the specific recognition and combination of LDL and LDL receptor, which is overexpressed on the surface of tumor cell membranes. The siRNA–PTX-loaded micelles inhibited gene and drug release under physiological conditions while promoting fast release in an acid microenvironment or in the presence of glutathione. The micelles escaped from the lysosome through the proton sponge effect. Additionally, the micelles exhibited superior antitumor activity and downregulated the protein and mRNA expression levels of breast cancer resistance protein in MCF-7/Taxol cells. The biodistribution and antitumor studies proved that the siRNA–PTX-loaded micelles possessed prolonged circulation time with a remarkable tumor-targeting effect and effectively inhibited tumor growth. Therefore, the novel dual pH/redox-sensitive polymers co-delivering siRNA and PTX with excellent biocompatibility and effective reversal of MDR demonstrate a considerable potential in cancer therapy.
Keywords: multidrug resistance, micelle, co-delivery, environmentally sensitive, tumor targeting
Introduction
Chemotherapy is a major treatment approach for cancer, but this strategy often fails because of multidrug resistance (MDR).1,2 MDR is caused by the overexpression of multiple efflux transport proteins, including ATP-binding cassette (ABC) transporters,3 which operate as exporters in cells and hydrolyze ATP to yield the energy necessary for transport. Polyspecific ABC transporters, including P-glycoprotein (ABCB1), MRP1 (ABCC1), and breast cancer resistance protein (BCRP; ABCG2), are associated with MDR.4,5 ABCG2 was discovered in the multidrug-resistant breast cancer cell line MCF-7/AdrVp.6,7 This protein is expressed in various organs with barrier functions and numerous types of cancer.8 ABCG2 induces MDR via the transportation of diverse molecules with amphipathic characteristics, and the diversity of its substrates ranges from anticancer drugs to fluorescence dyes.9,10 Thus, the exploration of potent, selective, and safe methods to inhibit the expression of efflux proteins and restore the intracellular drug concentration has opened a possibility to overcome MDR.
The discovery of RNA interference (RNAi) has helped in pioneering the application of gene silencing as a new method in the therapy of multiple diseases.11 Recent studies have demonstrated that small interfering RNA (siRNA), which is an important component participating in RNAi, is used for efficient and specific gene silencing within tumor cells12,13 and is a potential therapeutic agent in numerous diseases.14–18 RNAi mediated by siRNA can be incorporated into the RNA-induced silencing complex to degrade the target messenger RNA (mRNA) and inhibit the expression of the target protein.19,20 However, siRNA-based therapies are limited by the poor intracellular penetration of naked siRNA because of its strong negative charge and the serum instability of RNA molecules.21 Thus, various delivery vectors have been extensively explored to enhance the delivery and internalization of siRNA.
Viral vectors are efficient for siRNA delivery; however, the clinical applications of viral vectors are limited by their tumorigenicity, cytotoxicity, and immunogenicity.22 Thus, nonviral siRNA delivery systems based on polymers, lipids, and proteins have been developed.23–25 Low-density lipoprotein (LDL) isolated from human plasma contains a hydrophobic core surrounded by a phospholipid monolayer with apolipoprotein B-100, which can be recognized by the LDL receptor (LDLr) that is abundantly expressed in tumor membranes via active lysine residues.26–29 LDL is an effective vehicle of lipophilic drugs because of its endogenous degradability, safety and compatibility with cell membranes, and capability to escape from the reticuloendothelial system.30,31 In the present study, LDL was used as a ligand for targeting tumor cells and incorporated with cholesterol-conjugated BCRP siRNA as a gene carrier.
Various polymeric micelles demonstrate considerable potential in antitumor drug delivery.32,33 However, polymeric micelles still face tremendous challenges, including the burst release of encapsulated drugs. Therefore, delivery systems with predictable and controlled drug-release properties should be explored. Acid-triggered drug release can be achieved inside tumor tissue (pH 6.5) or lysosomal compartments (pH 5.3) of cancer cells by using micelle-bearing pH-sensitive blocks, such as histidine or imidazole groups.34,35 Polymers responding to an acid microenvironment can promote the drug release in tumor cells and escape from lysosomes through the proton sponge effect. In addition, the glutathione (GSH) concentration in intracellular compartments is 100–1,000 times higher than that in extracellular fluids.36,37 Thus, reduction-sensitive drug delivery systems have attracted considerable attention in cancer therapy because of their capacity for “active” tumor-specific drug release.
In this study, a novel dually pH/redox-sensitive polymer N-succinyl chitosan–cystamine–urocanic acid (NSC–SS–UA) was synthesized. NSC, a derivative of chitosan (CS) with improved water solubility and security,38,39 served as the hydrophilic shell. SS and UA exhibiting reduction and pH sensitivity, respectively, were used as the hydrophobic core for paclitaxel (PTX) loading. Then, LDL was grafted by an amide bond to obtain a new “binary polymer” LDL–NSC–SS–UA, which contained siRNA-loaded LDL nanoparticles and PTX-loaded micelles with dual pH/redox sensitivity and targeting effect. “Binary polymer” micelles that co-deliver siRNA and PTX were designed to downregulate BCRP expression, overcome MDR, and exert a synergistic antitumor effect (Figure 1). The properties and co-delivering ability of the micelles were investigated. The cellular uptake, tumor-targeting, and antitumor activity of siRNA–PTX/LDL–NSC–SS–UA in vitro and in vivo were further confirmed in this study.
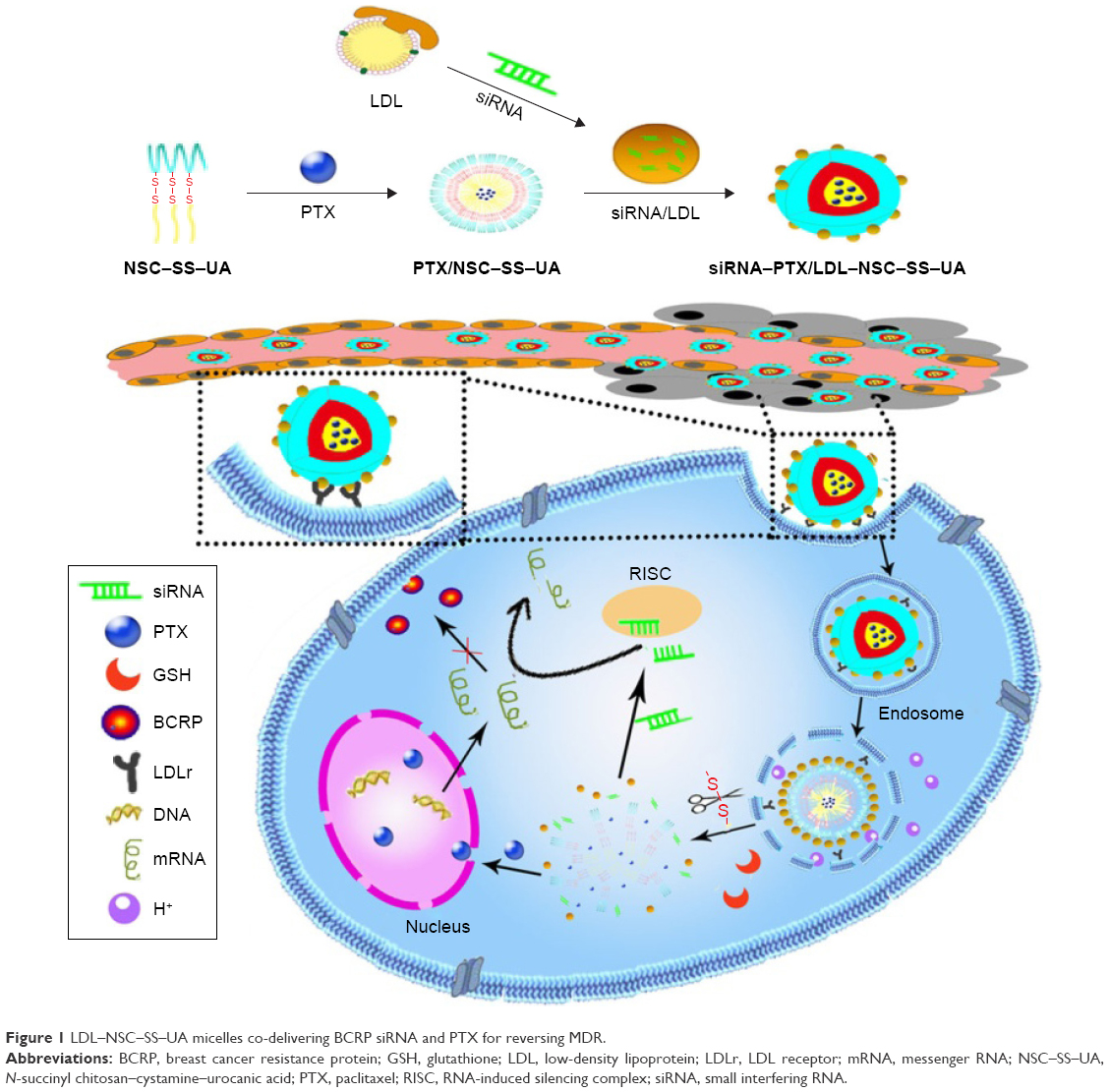 | Figure 1 LDL–NSC–SS–UA micelles co-delivering BCRP siRNA and PTX for reversing MDR. |
Materials and methods
Materials
Virus-inactivated human plasma was supplied by the First Affiliated Hospital of Soochow University, Suzhou, People’s Republic of China. Cholesterol-conjugated siRNA (chol-siRNA) with respective sense and antisense patterns of 5′-GGCCUUGGGAUACUUUGAATT-(cholesterol)-3′ and 5′-UUCAAAGUAUCCCAAGGCCTT-3′ was obtained from Ribobio Co., Ltd. (Guangzhou, People’s Republic of China). Targeting BCRP mRNA was chemically synthesized at Sangon Biotech Co., Ltd. (Shanghai, People’s Republic of China). CS with a molecular weight (MW) of 8–10 kDa was purchased from Xingcheng Biochemical Co., Ltd. (Nantong, People’s Republic of China). Cystamine dihydrochloride and UA were both purchased from Beijing Acros Technology Co., Ltd. (Beijing, People’s Republic of China). GSH, 1-(3-dimethylaminopropyl)-3-ethyl carbon carbodiimide hydrochloride (EDC·HCl), and N-hydroxysuccinimide (NHS) were obtained from Aladdin-Reagent Co., Ltd. (Shanghai, People’s Republic of China). PTX was acquired from Meilun Biological Technology Co., Ltd. (Dalian, People’s Republic of China). Fetal bovine serum (FBS), Roswell Park Memorial Institute (RPMI)-1640, and Dulbecco’s Modified Eagle’s Medium (DMEM) medium were purchased from Hyclone (Thermo-Fisher Biochemical Products Co., Ltd., Beijing, People’s Republic of China). All other chemicals were of analytical grade and used without further purification.
Human breast cancer cell MCF-7 and human PTX-resistant breast cancer cell MCF-7/Taxol from Jiangsu Province Key Laboratory of Biotechnology and Immunology (Suzhou, People’s Republic of China) were cultured in RPMI 1640 supplemented with 10% FBS under 5% CO2 humidified conditions at 37°C.
Female nude mice (BALB/C, nu/nu) (16±2 g) were purchased from the Shanghai SLAC Laboratory Animals Co. Ltd (Shanghai, People’s Republic of China) and raised in specific pathogen-free conditions. All animal protocols were approved by the Institutional Animal Care and Use Committee of Soochow University and were in compliance with the Guidelines for the Care and Use of Laboratory Animals (Chinese-National-Research-Council, 2006) and Animal Research: Reporting of In Vivo Experiments guidelines.
Synthesis and characterization of NSC–SS–UA conjugates
NSC was synthesized based on CS as previously reported.40 Then, NSC–SS was synthesized by reacting the carboxyl groups of NSC and the amine groups of SS. In brief, 2.0 g of NSC was dissolved in 40 mL of distilled water, added with EDC·HCl (2.92 g) and NHS (1.76 g), and then stirred for 0.5 h at 40°C to activate the carboxyl groups of NSC. Then, the aqueous solution of SS (0.84 M, 40 mL) was added drop wise, and the reaction was carried out at 50°C for 12 h. The product was dialyzed (MW =3,500) against distilled water for 3 days and then lyophilized to obtain NSC–SS.
NSC–SS–UA was synthesized by NSC–SS and UA under the catalysis of EDC·HCl and NHS. In brief, UA (0.86 g), EDC·HCl (2.38 g), and NHS (1.42 g) were dissolved in 50 mL of N, N-dimethylformamide by stirring at 50°C. Then, the reaction solution was added drop wise to 250 mL of aqueous solution of NSC–SS (96.3 mM). After being stirred for another 30 h, the reaction product was dialyzed (MW =3,500) against distilled water for 3 days and then lyophilized to obtain NSC–SS–UA.
The chemical structures of NSC–SS and NSC–SS–UA were characterized by Fourier-transform infrared (FT-IR) spectrometry (Varian, Palo Alto, CA, USA) and 1H nuclear magnetic resonance (1HNMR) spectroscopy (400 MHz, Varian, Palo Alto, CA, USA). FT-IR spectra were studied using the potassium bromide method. For 1HNMR measurements, the copolymers were dissolved in D2O, and the degree of substitution (DS) was studied by 1HNMR spectroscopy.
Preparation of siRNA/LDL complex
LDL was isolated from virus-inactivated human plasma by sequential density gradient ultracentrifugation41 and then stored at 4°C within 2 weeks. Protein concentration was determined using the bicinchoninic acid (BCA) method, and the particle distribution of LDL was measured by dynamic light scattering (DLS) (Zeta-Sizer; Malvern Nano-ZS90, Malvern, UK).
Lipophilic chol-siRNA could insert itself into the lipid monolayer of LDL at room temperature and form chol-siRNA/LDL complex. Chol-siRNA (2 optical density) was dissolved in 125 μL of diethyl pyrocarbonate-treated water and then stored at −20°C. The siRNA solution and LDL (mole ratio of 30:1)41,42 were mixed at room temperature and then incubated for 0.5 h to obtain siRNA/LDL complex.
RNase protection assay and serum stability
The protection of siRNA encapsulated in LDL from RNase A was evaluated by 12% polyacrylamide gel electrophoresis. Chol-siRNA and siRNA/LDL complex containing 0.06 nmol siRNA were separately incubated with 4 mL of RNase A (20 μg·mL−1) at 37°C for different hours (1, 3, and 6 h). Ethylenediaminetetraacetic acid (0.25 M, pH 8.0) was added to deactivate RNase A, and the remaining siRNA was tested by 12% polyacrylamide gel electrophoresis stained with 0.01% GelRed. The untreated sample was used as a control.
To study serum stability, the siRNA/LDL complex and equivalent LDL were diluted with phosphate-buffered saline (PBS, 200 nM) and then mixed with 50% FBS. Subsequently, 5% glucose (50 μL) was mixed with the same FBS volume in 96-well cell plates. The mixtures were incubated at 37°C for various durations (0.5, 1, 3, 6, 12, 24, 36, and 72 h), and the turbidity of the mixtures was measured by the absorbance at 415 nm to study the serum stability of the complex.
Preparation of siRNA–PTX/LDL–NSC–SS–UA micelles
Drug-loaded PTX/NSC–SS–UA micelles were prepared via film dispersion. In brief, PTX (2 mg) and NSC–SS–UA (10 mg) were uniformly dispersed in 10 mL of methanol. The solvent was removed by rotary evaporation, and the drug and carrier were re-dissolved in 10 mL of distilled water. Later, the mixed solution was filtered twice with a 0.22 μm Millipore filter to yield the final PTX-loaded micelles.
EDC·HCl (5 mg), NHS (1 mg), and siRNA/LDL (200 μL) were added to 5 mL of PTX/NSC–SS–UA solution (1 mg·mL−1) by stirring for 5 min at 40°C.43 The pH of the solution was regulated to 8–9 by adding 5% NaOH solution. After being stirred for another 2 h, the resulting solution was filtered through a 0.22 μm Millipore filter to obtain siRNA–PTX/LDL–NSC–SS–UA micelles.
Evaluation of siRNA–PTX/LDL–NSC–SS–UA micelles
FT-IR spectroscopy was used to determine the structure of LDL–NSC–SS–UA. The particle distribution and micellar morphology of siRNA–PTX/LDL–NSC–SS–UA were investigated by DLS (Nicomp-380 ZLS particle sizing system; PSS Co., Port Richey, FL, USA) and transmission electron microscopy (JEOL Ltd., Tokyo, Japan), respectively.
The critical micelle concentration (CMC) was measured using the pyrene fluorescent probe method as previously reported.44 About 0.5 mL volume of the acetone solution of pyrene (1 μM) was collected in a test tube, and the solvent was volatilized in nitrogen atmosphere. Then, 5 mL of the block copolymer solution at different concentrations was added. A FLS920 fluorescence spectrometer was used to monitor the fluorescence spectra at an excitation wavelength of 336 nm. CMC was reckoned as the cross-point when extrapolating the intensity ratio of I372/I383 at different concentration regions.
pH/redox responding test of LDL–NSC–SS–UA micelles
LDL–NSC–SS–UA micelles (2 mg·mL−1) were prepared through probe sonication. The size and electric potential changes of the micelles dissolved in different PBS solutions, including pH 7.4, pH 5.3, pH 7.4 with GSH (100 mM), and pH 5.3 with GSH (100 mM), were detected using DLS to investigate the pH and redox responsive behaviors of the micelles.
Loading and release of PTX
The drug loading content (LC%) and encapsulation efficiency (EE%) of the LDL–NSC–SS–UA micelles were determined via ultrafiltration centrifugation, and the concentration of PTX was measured by reversed-phase high-performance liquid chromatography (Agilent1100, Agilent, Technologies Santa Clara, CA, USA) with a reversed-phase column (Hypersil ODS-C18, 250×4.6 mm, 5 μm; Thermo Fisher Scientific, Waltham, MA, USA). The mobile phase was acetonitrile–water (70:30, v/v), flow rate was 1.0 mL·min−1 at 27°C, and the detection wavelength was 227 nm.
The release of PTX from the PTX/NSC–SS–UA micelles in vitro was studied by using the dialysis bag method45 at 37°C under different conditions. In brief, 5 mL of PTX/NSC–SS–UA was added into a dialysis bag (molecular weight cut off =3,500) against 100 mL of PBS with different pH and GSH levels (pH 7.4; pH 7.4+ GSH [10 mM]; pH 5.3 and pH 5.3+ GSH [10 mM]) and then stirred at 37°C. The corresponding medium (1 mL) was replaced with equal volumes of fresh medium at each predesigned time, and the free Taxol® group was set as a control.
Cytotoxicity assay
The in vitro cytotoxicity of polymers and PTX in different formulations to MCF-7 and MCF-7/Taxol cells was studied by MTT assay. MCF-7 and MCF-7/Taxol cells were seeded in 96-well plates at a density of 2,000–5,000/well and then incubated at 37°C. Blank micelles NSC–SS–UA, LDL–NSC–SS–UA, drug-loaded micelles PTX/NSC–SS–UA, PTX/LDL–NSC–SS–UA, and free Taxol in 100 μL of medium were added to 96-well plates. After incubation for 48 h, 20 μL of MTT in PBS (5 mg·mL−1) was added to each well. After incubation for another 4 h, the medium was replaced with dimethyl sulfoxide (150 μL/well) to dissolve the blue–purple formazan crystal. The absorbance was determined with a microplate reader (ELx808, BioTek; Winooski, VT, USA) at 570 nm wavelength, and the cell viability (%) of the treated cells was calculated as follows:
|
|
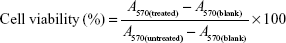
where A570(treated) and A570(untreated) are the absorbance of the test and control samples at 570 nm wavelength, respectively, and A570(blank) is the absorbance of blank wells.
The effect of siRNA on drug cytotoxicity in vitro was analyzed by MTT assay. SiRNA–PTX/LDL–NSC–SS–UA micelles with different siRNA concentrations (PTX concentration was 2 μg·mL−1) were incubated with MCF-7 and MCF-7/Taxol cells at 37°C for different times as previously described.
Disruption of lysosomal membranes
To evaluate the disruption effect of lysosomal membranes of the micelles, acridine orange (AO) was used as an indicator of acidic organelle integrity in MCF-7 cells, which can be charged in acidic compartments. When excited with blue light, AO in acidic organelles can emit red fluorescence, whereas AO in cytosol and nuclei emit green fluorescence.46 If the lysosomal membranes are disrupted, the red fluorescence from AO becomes weak, and the green fluorescence becomes strong. In the present study, MCF-7 cells were seeded on glass slides for 24 h at 37°C and then treated with PBS (pH 7.4) and siRNA–PTX/LDL–NSC–SS–UA micelles for different times (0.5, 1, and 2 h). Then, the cells were incubated with 1 mL of AO (10 μg·mL−1) for 15 min, followed by fluorescent microscopy observation (IX 51; Olympus Corp., Tokyo, Japan) (Excitation wavelength [Ex] =488 nm, emission wavelength [Em] =617 nm for red; Ex =488 nm, Em =528 nm for green).
Cellular uptake and distribution of drug-loaded micelles in vitro
Subcellular distribution of micelles
The intracellular release and distribution of drugs in different formulations were examined by confocal laser scanning microscopy (CLSM), where coumarin-6 (C6) was used to replace PTX. MCF-7 and MCF-7/Taxol cells in the exponential phase were cultured in six-well plates at 37°C for 24 h and then transfected with C6/NSC–SS–UA, C6/LDL–NSC–SS–UA (200 nM), or siRNA-C6/LDL–NSC–SS–UA (200 nM) for different times (0.5, 1, and 2 h). The lysosome of MCF-7 cells was stained with Lyso Tracker™ Green (100 nM for 2 h), and the nuclei of MCF-7 and MCF-7/Taxol cells were stained with Hoechst 33258 (5 mg·mL−1 for 10 min). The intracellular distribution of drug-loaded micelles was examined under a Zeiss LSM 510 META confocal laser scanning microscope (Carl Zeiss, Jena, Germany).
Quantitative uptake of micelles
PTX was replaced with C6 to examine the quantitative uptake of drug-loading micelles in vitro. MCF-7 and MCF-7/Taxol cells in the exponential phase were cultured in six-well plates for 24 h at 37°C and then transfected with C6/NSC–SS–UA, C6/LDL–NSC–SS–UA (200 nM), or siRNA–C6/LDL–NSC–SS–UA (200 nM) for different times (0.5, 1, and 2 h). Then, the cells were washed three times with PBS (pH 7.4), harvested, and then resuspended with 500 μL of PBS (pH 7.4). The fluorescence intensity of C6 in cells was quantified through flow cytometry.
Dynamic phagocytosis of micelles
Dynamic phagocytosis was observed by a live cell station (Cell’ R, Olympus Corp.) where PTX was replaced with C6. In brief, MCF-7 and MCF-7/Taxol cells in the exponential phase were seeded in a glass bottom dish and then cultured for 24 h at 37°C. The nuclei of the cells were stained with Hoechst 33258 (5 mg·mL−1 for 10 min) and then injected with C6/NSC–SS–UA, C6/LDL–NSC–SS–UA (5 μg·mL−1 C6), or siRNA–C6/LDL–NSC–SS–UA (100 nM siRNA). Differential interference contrast (DIC), fluorescein isothiocyanate (FITC), and 4′,6-diamidino-2-phenylindole (DAPI) staining images were captured every 10 s for 1 h.
Uptake kinetics of micelles in MCF-7/Taxol cells
The uptake kinetics of the micelles was computed on the basis of the results of the live cell station. In brief, we hypothesized that the micelles entered the cells and were eliminated from the cells at speeds of K0 and K, respectively:
|
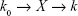
As the incubation time was prolonged, K was proportional to the amount of micelles within the cells (X), which is the algebraic sum of the uptake rate (k0) and the elimination speed (kX), and k0 and k can be obtained by nonlinear fitting:
|
|
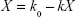
|
|
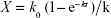
Reverse transcription-polymerase chain reaction (RT-PCR) for the detection of intracellular mRNA level
MCF-7/Taxol cells were cultured in six-well plates until 70% confluence was reached and then incubated with chol-siRNA, PTX/LDL–NSC–SS–UA, or siRNA–PTX/LDL–NSC–SS–UA (2 μg·mL−1 PTX) for 24 or 48 h. Total mRNA was extracted with Trizol and then quantified using QuawellQ5000 (Quawell Technology Int., Sunnyvale, CA, USA). The complementary DNA (cDNA) was synthesized based on the isolated mRNA using first-strand cDNA Synthesis Kit (Beyotime, Haimen, People’s Republic of China) and then used for RT-PCR by PCR kit with Taq (Beyotime). The RT-PCR was performed at 95°C for 3 min, 40 cycles at 94°C for 0.5 min, 60°C for 0.5 min, 72°C for 1 min, and then 72°C for 10 min to terminate the reaction. The products were detected with 1.5% agarose gels and stained by GelRed.
Western blot for the detection of intracellular BCRP protein content
MCF-7/Taxol cells were incubated with chol-siRNA, PTX/LDL–NSC–SS–UA, or siRNA–PTX/LDL–NSC–SS–UA (2 μg·mL−1 PTX) and then incubated with radio-immunoprecipitation assay cell lysis buffer (Cell Signaling Technology, Danvers, MA, USA) for 15 min on ice. Then, protein concentration was determined with the BCA Protein Assay Kit (Beyotime). Total proteins were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis gel and then transferred to Immobilon®-P polyvinylidene difluoride membranes. After blocking for 1 h, the blots were probed by anti-BCRP/ABCG2 monoclonal antibody (1:500; Proteintech Group, Rosemont, IL, USA) or anti-ß-actin monoclonal antibody (1:500; San Ying Biotechnology, Wuhan, People’s Republic of China) as a control. The fluorescence of the blots was detected by ChemiScope 3300 Mini (Clinx Science Instruments Co., Ltd., Shanghai, People’s Republic of China).
Near-infrared fluorescence (NIRF) imaging for studying the targeting properties in vivo
The targeting properties in vivo of the two types of micelles with 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine (Dir) as the probe were investigated using a NIRF imaging system. Tumor-bearing nude mice were established by the subcutaneous inoculation of MCF-7 cells into female Balb/c nude mice until the tumor volume reached 100–200 mm3. Dir-labeled micelles Dir/NSC–SS–UA and Dir/LDL–NSC–SS–UA (50 μg·mL−1 Dir) at 0.2 mL/10 g were intravenously injected into the mice through the tail vein. At designated time intervals (12, 24, and 48 h), the fluorescence distribution in the mice was detected using the NIRF imaging system. The tumors and major organs were also imaged (Dir: λex =748 nm; λem =780 nm).
Antitumor effect in vivo
Thirty tumor-bearing nude mice were divided into six groups randomly (five in each group), and then injected with normal saline, Taxol, LDL–NSC–SS–UA, PTX/LDL–NSC–SS–UA, siRNA/LDL–NSC–SS–UA, or siRNA–PTX/LDL–NSC–SS–UA (PTX: 0.5 mg·kg−1, siRNA: 0.1 mg·kg−1) every 3 days. The tumor size and mouse weight were measured every 2 days, and the tumor volume was calculated as follows:
|
|
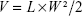
where V is the tumor volume, and L and W are the longer diameter and smaller diameter, respectively.
Statistical analysis
All quantitative data were expressed as the mean ± standard deviation of three experiments. Data were analyzed by analysis of variance. Statistical significance is presented as *P<0.05, **P<0.01, and ***P<0.001.
Discussion
Synthesis and characterization of NSC–SS–UA
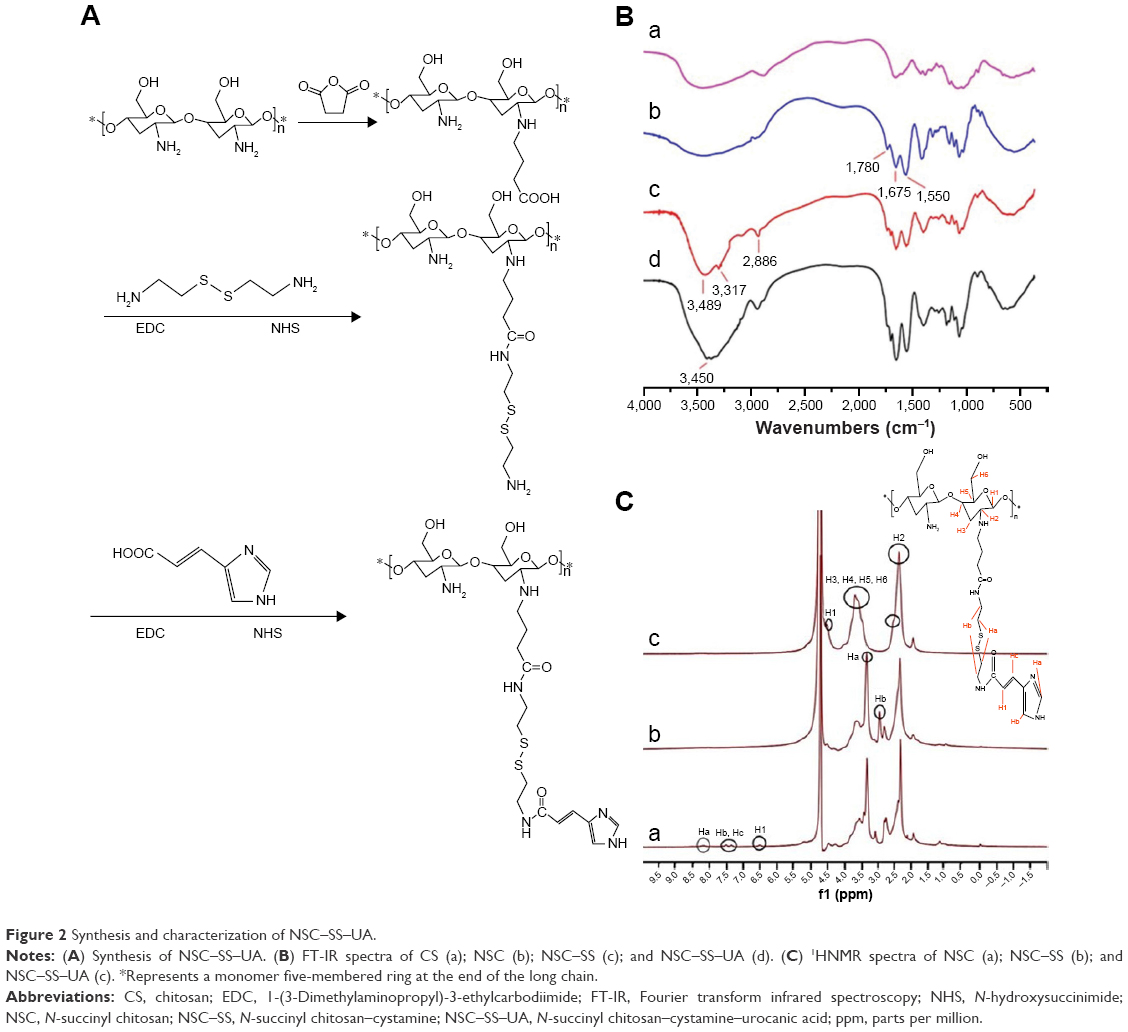
As shown in Figure 2A, NSC–SS–UA was synthesized under the catalysis of EDC and NHS, and cystamine was used as a connecting bridge to conjugate NSC and UA through its two terminal amine groups.
 | Figure 2 Synthesis and characterization of NSC–SS–UA. |
The FT-IR spectra of CS, NSC, NSC–SS, and NSC–SS–UA are illustrated in Figure 2B. In the spectrum of NSC–SS, the stretching vibration of –C=O and the in-plane bending vibration of –N–H– were observed at 1,675 and 1,550 cm−1, respectively, suggesting the formation of NH-CO, and the absorption peak of free carboxyl groups at 1,780 cm−1 was weakened compared with that of CS. This result suggests that the succinyl groups were grafted onto the carboxyl groups of CS successfully. In the spectrum of NSC–SS, the symmetric and asymmetric stretching vibrations of –NH2 groups were at 3,317 and 3,489 cm−1, respectively. The absorption peak of 3,450 cm−1 corresponded to the –C–H– stretching vibration of SS, indicating that SS was successfully grafted to NSC. In addition, the strong absorption peak at 1,657 cm−1 corresponded to the –N–H– stretching vibration in the aromatic ring, indicating the synthesis of NSC–SS–UA.
The 1H-NMR spectrum signals of NSC were at δ=4.5 (H1), δ=2.61 (H2), δ=3.35–3.83 (H3 to H6), and δ=2.35 (–CH2–) (Figure 2C) because the electron cloud density of H2 was changed by the–NH2 succinylation of CS. Compared with the NSC spectrum, the characteristic peaks of SS were found at δ=3.5 (Ha) and δ=3.0 (Hb). The spectrum signals of UA were at δ=8.23 (Ha), δ=7.61 (Hb), δ=7.5 (Hc), and δ=6.51 (H1), suggesting that UA was successfully introduced into NSC–SS. The DS values of SS and UA conjugated to NSC were 38.4% and 18.0%, respectively, according to the integral areas of 1HNMR spectrum peaks.
Evaluation of the siRNA/LDL complex
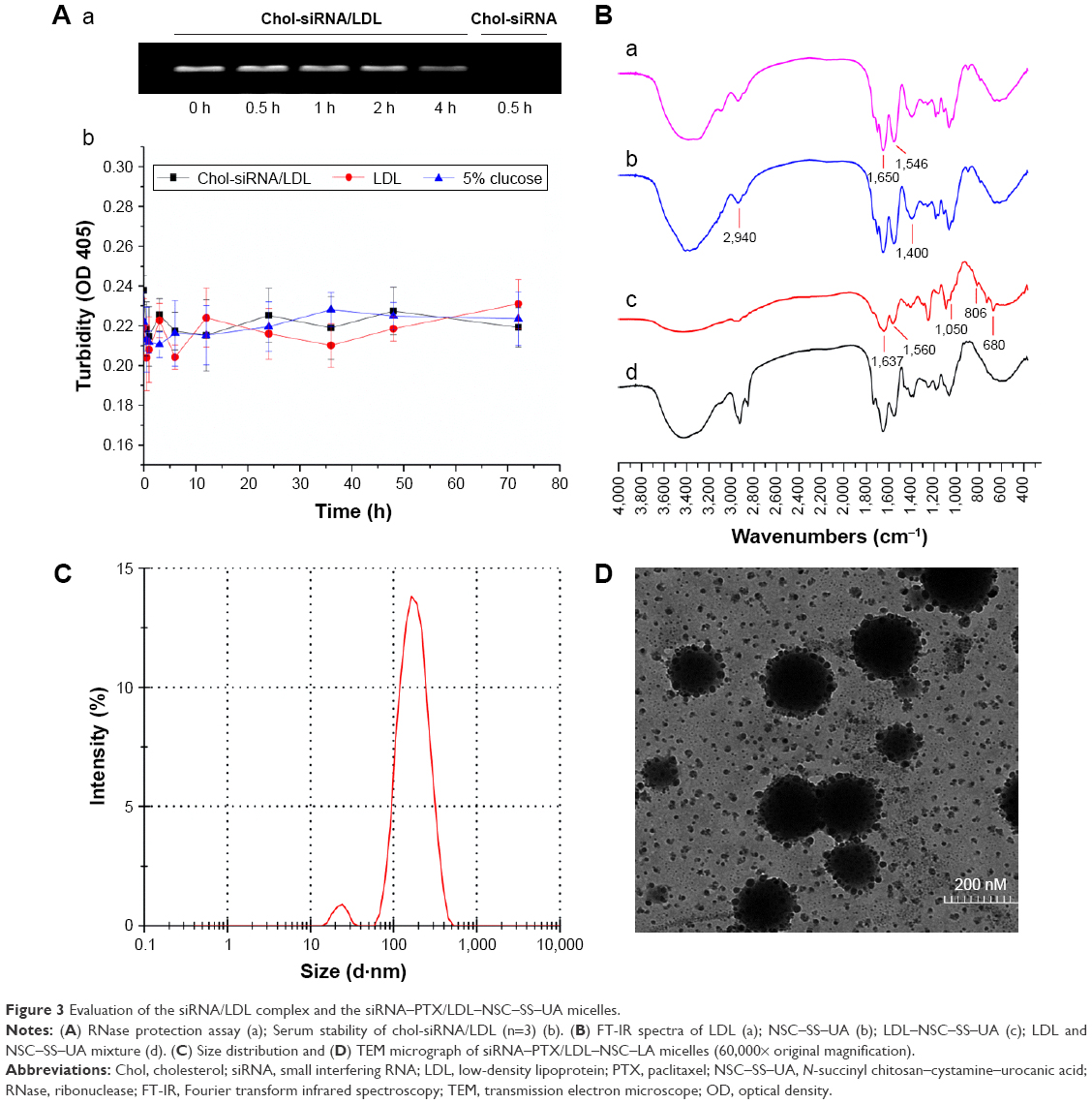
The density of LDL isolated from virus-inactivated human plasma was 0.796±0.08 g·mL−1. The mean particle size and polydispersity index (PDI) of LDL were 24.36±0.62 nm and 0.173, respectively (Figure S1), and the zeta potential was −1.34±0.11 mV, which is consistent with the literature.41
Free siRNA was rapidly degraded by the nucleases in blood or body fluids; thus, siRNA should be protected from nucleases. Figure 3A shows that after incubation with RNase A (20 μg·mL−1) for 0.5 h, free chol-siRNA was almost completely degraded, whereas the chol-siRNA encapsulated in LDL was protected from degradation for up to 2 h. The serum stability of the siRNA/LDL complex was evaluated through turbidity changes during incubation with 50% FBS. Aggregation was not observed for LDL and siRNA/LDL complex with 5% glucose within 72 h, suggesting that LDL could protect siRNA from the aggregation caused by nonspecific interactions with serum proteins.
 | Figure 3 Evaluation of the siRNA/LDL complex and the siRNA–PTX/LDL–NSC–SS–UA micelles. |
Evaluation of siRNA–PTX/LDL–NSC–SS–UA micelles
The structure of LDL–NSC–SS–UA was evaluated by FT-IR spectroscopy. As shown in Figure 3B, the absorption peaks of amides I and II from the peptide bond of LDL were at 1,650 and 1,546 cm−1, respectively; however, the two peaks shifted to1,637 and 1,546 cm−1 after NSC–SS–UA coupling, respectively. The stretching and bending vibration peaks of –OH from NSC–SS–UA were at 2,940 and 1,400 cm−1, respectively, which weakened or disappeared in the spectrum of LDL–NSC–SS–UA. The absorption peaks at 1,050, 806 and 680 cm−1 also indicate the introduction of the succinyl group. The FT-IR spectra imply that the reaction that occurred between LDL and NSC–SS–UA was chemical coupling instead of physical mixture.
SiRNA–PTX/LDL–NSC–SS–UA possessed a mean particle size of 167.90±1.46 nm (PDI =0.196) (Figure 3C) and a zeta potential of −21.91±1.25 mV, which was suitable for the enhanced permeability and retention (EPR) effect in the tumor tissues. The transmission electron micrograph (×60,000) of siRNA–PTX/LDL–NSC–SS–UA in Figure 3D reveals the uniform spherical morphology of micelles with a particle size of 148.1±1.12 nm. The surface of NSC–SS–UA was coupled with LDL, which can specifically bind with the LDLr of the cell surface. LDLr is reportedly ingested by MCF-7 cells even though the concentration of LDL is relatively saturated.2 Thus, the micelles could show effective tumor-targeting capability despite the existence of free LDL molecules.
CMC was measured within the LDL–NSC–SS–UA concentration range of 1 μg·mL−1 to 1 mg·mL−1 (Figure 4A), and the CMC of the micelles was calculated to be 5.62×10−3 mg·mL−1 according to the plots of I1/I3 and log concentration (Figure 4B). The results imply that the LDL–NSC–SS–UA polymeric micelles were relatively stable during dilution and exhibited considerable potential as a suitable drug carrier.
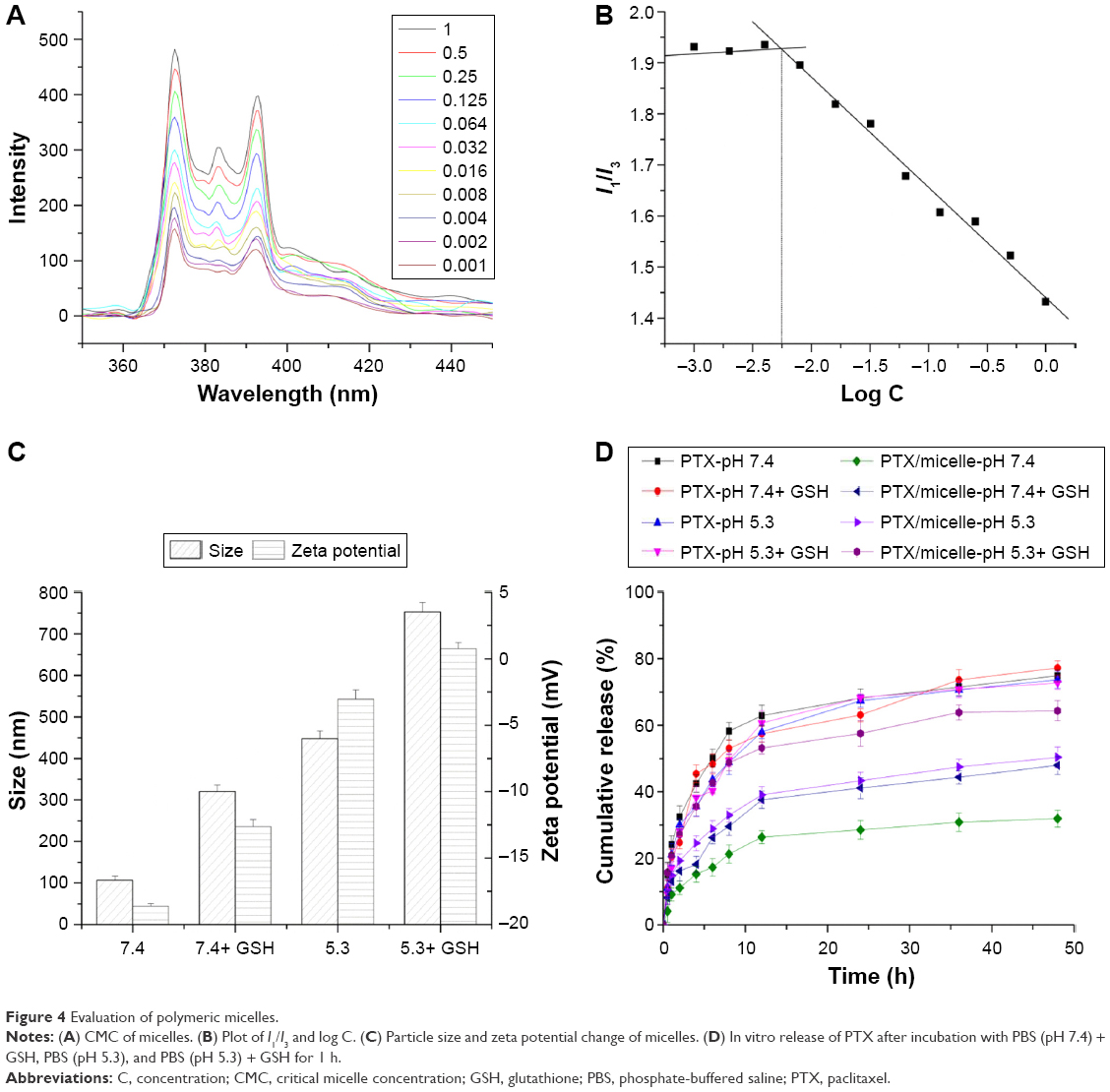 | Figure 4 Evaluation of polymeric micelles. |
The pH/redox-responsive property of LDL–NSC–SS–UA was investigated via the changes in the size and zeta potential of the micelles after incubation with weak acidic PBS and PBS with GSH (10 mM). As shown in Figure 4C, the LDL–NSC–SS–UA micelles swelled from 105.7 to 295.3, 396.1 and 712.4 nm after incubation with PBS (pH 7.4) + GSH, PBS (pH 5.3), and PBS (pH 5.3) + GSH, respectively, for 1 h. In addition, the zeta potential gradually increased with the decrease in pH or increase in GSH concentration. This result suggests that the stability of the micelles was destroyed. Changes in the particle size and zeta potential were due to the protonation of imidazole groups in acidic solution and the fracture of disulfide bonds in the presence of GSH, which resulted in the rapid breakdown of hydrophilic/hydrophobic balance and the rapid intracellular release of drugs and genes.
The EE% and LC% of the PTX/LDL–NSC–SS–UA micelles were 94.81%±1.37% and 17.86%±1.13%, respectively, which were determined via ultrafiltration centrifugation. The in vitro release of PTX from the micelles is shown in Figure 4D. The release of PTX from PTX/LDL–NSC–SS–UA at pH 7.4 PBS was significantly inhibited by the self-assembly of micelles, with an accumulated release of 31.91%±2.83% within 48 h. However, the micelles showed a rapid drug release in pH 5.3 PBS or PBS with GSH (10 mM). In particular, the accumulated release ratio of PTX from PTX/LDL–NSC–SS–UA was as high as 64.97%±3.51% in pH 5.3 PBS with GSH (10 mM), which is almost double the release in pH 7.4 PBS. The protonation of imidazole groups in an acidic environment contributed to the rapid release, which led to the hydrophilicity of the hydrophobic core of the micelles. In addition, this condition resulted in the fracture of disulfide bonds under the reductive condition, which caused the rapid breakdown of the hydrophilic/hydrophobic balance. The result indicates an extracellular stable and intracellular release in tumor, which benefits preferred cancer therapy.
Cytotoxicity assay
The safety of polymers NSC–SS–UA and LDL–NSC–SS–UA was investigated with MTT assay, as shown in Figure 5A. Up to 83.54% and 81.35% of the cells survived after treatment with NSC–SS–UA and LDL–NSC–SS–UA, respectively, for 48 h at the concentration of 128 μg·mL−1, which demonstrated the preferable safety of the two blank micelles.
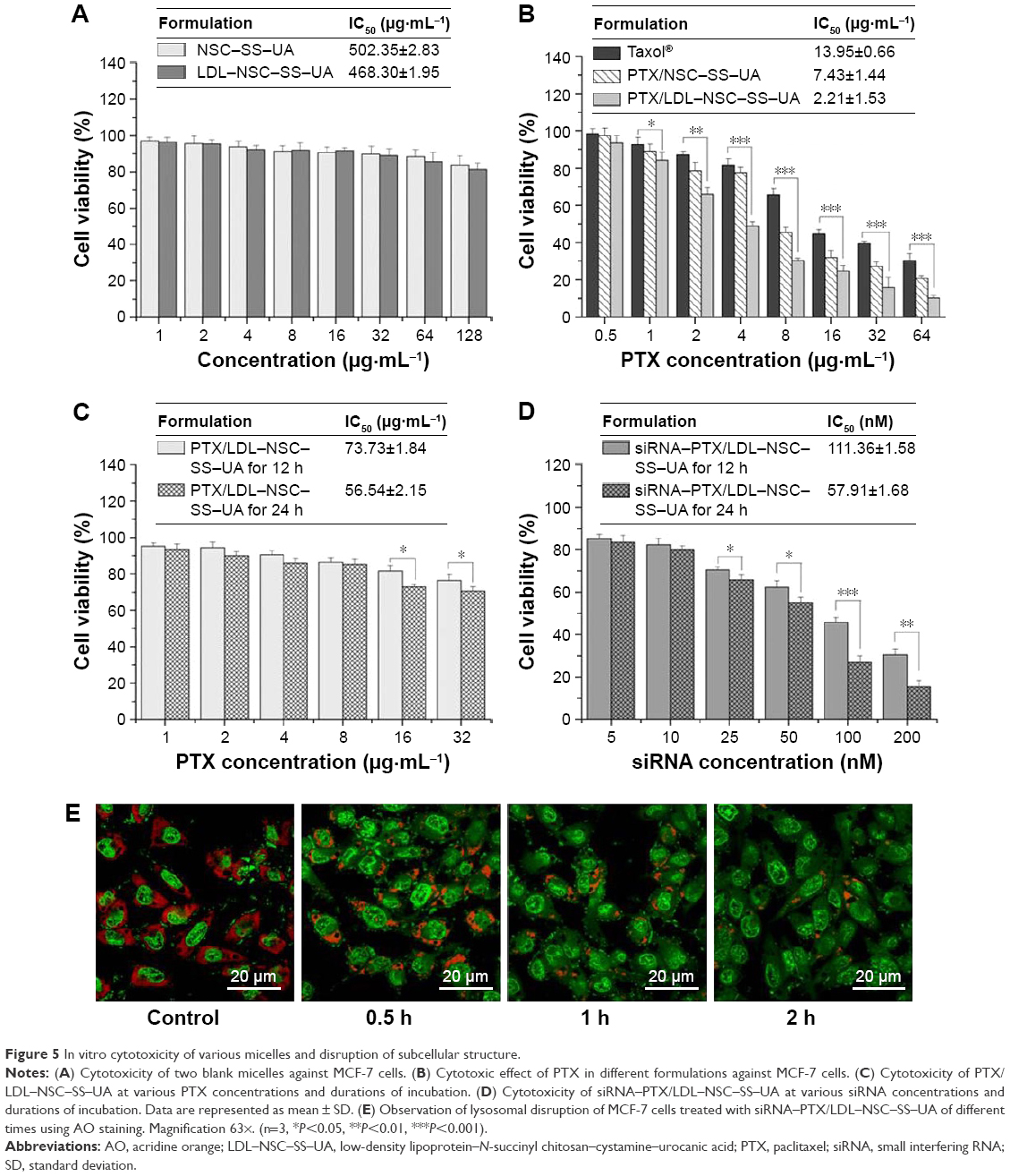 | Figure 5 In vitro cytotoxicity of various micelles and disruption of subcellular structure. |
The different formulations of PTX inhibited cellular growth in a dose-dependent manner, as shown in Figure 5B. PTX/LDL–NSC–SS–UA displayed the strongest inhibition to MCF-7 cells, followed by PTX/NSC–SS–UA and Taxol, as well as good blood compatibility (Figure S2). This result can be attributed to the higher uptake and the rapid intracellular release of the drug from the PTX/LDL–NSC–SS–UA micelles.
MTT assay was performed on MCF-7/Taxol cells to compare the toxicity of PTX/LDL–NSC–SS–UA and siRNA–PTX/LDL–NSC–SS–UA. As shown in Figure 5C, the cytotoxicity of the two types of micelles increased in a dose-dependent manner. However, the cytotoxicity of PTX/LDL–NSC–SS–UA against MCF-7/Taxol cells was not evident; 76.48% and 70.59% of the cells survived at the PTX concentration of 32 μg·mL−1 for 12 and 24 h, respectively. Meanwhile, only 30.47% and 15.59% of the cells survived under the same conditions, respectively, when the gene concentration of siRNA–PTX/LDL–NSC–SS–UA was 200 nM (2 μg·mL−1 PTX) (Figure 5D). This phenomenon could be attributed to the increase in efflux function of the drugs from MCF-7/Taxol cells without siRNA, and the intracellular drug concentration was too low to kill cells, suggesting MDR. However, siRNA could reduce the expression of efflux proteins and maintain a high intracellular drug concentration to enhance cytotoxicity.
Disruption of subcellular structure
To demonstrate the influence of the siRNA–PTX/LDL–NSC–SS–UA micelles on the subcellular structure, we monitored the disruption of lysosomal membranes by using AO as an indicator (Figure 5E). Fluorescence microscopy showed that the acidic lysosomes of MCF-7 cells treated with PBS (pH 7.4) exhibited red fluorescence, and almost no green fluorescence was observed in the cytosol. However, the red fluorescence from AO markedly decreased after treatment with siRNA–PTX/LDL–NSC–SS–UA for 0.5, 1, and 2 h. This result indicates that the micelles can effectively disrupt the lysosomal membranes by the protonation of imidazole groups in an acidic environment, escape from lysosomes into the cytosol to avoid the degradation of drugs and genes in the lysosomes, and potentially contribute to the accessibility of drugs to target organelles, ultimately eliciting an antitumor effect.
Cellular uptake and distribution of drug-loading micelles in vitro
To demonstrate whether the micelles can be internalized by cancer cells, we evaluated their cellular uptake on MCF-7 cells. As shown in Figure 6A, the fluorescence of C6 in different formulations gradually increased with time, which initially accumulated in the endo/lysosomes and gradually diffused into the whole cytoplasm. Co-location with endo/lysosomes could provide micelles with a weakly acidic microenvironment to release PTX rapidly. However, the fluorescence of C6 in LDL–NSC–SS–UA was stronger than that in NSC–SS–UA. This finding indicates that the LDL–NSC–SS–UA micelles could be taken up by MCF-7 cells more rapidly, which is consistent with the result of the live cell station (Figure 6B). The divergence in uptake could be attributed to the interaction between ApoB100 and LDLr, as well as to the protection by the LDL layer during transport.
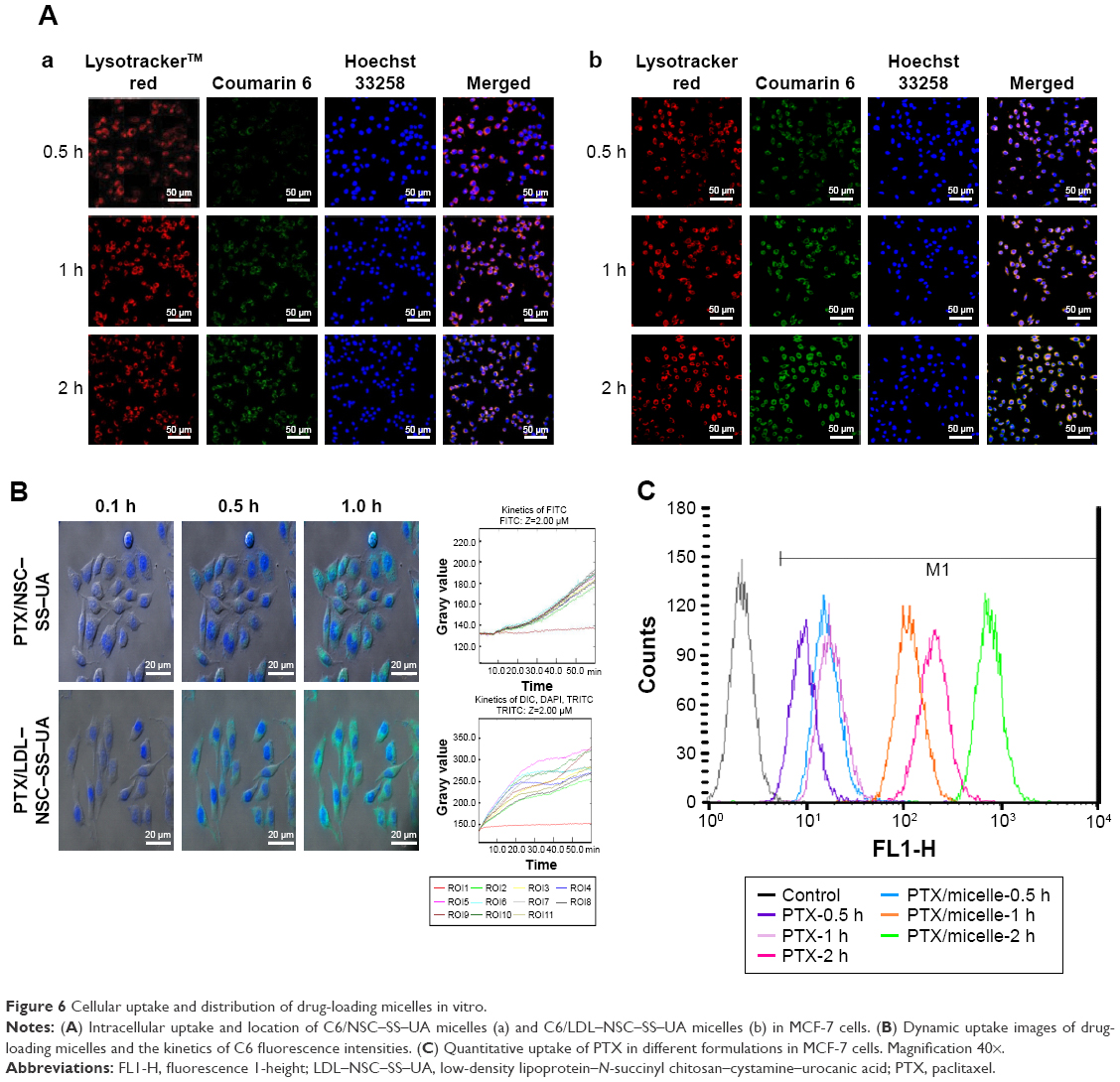 | Figure 6 Cellular uptake and distribution of drug-loading micelles in vitro. |
The uptake of Taxol and the PTX/NSC–SS–UA micelles by using fluorescence-activated cell sorter is displayed in Figure 6C. The micelles were more easily taken up than free Taxol. This result can be attributed to the fact that the cellular uptake of the micelles mainly occurred through endocytosis (shown as Figure S3), whereas that of Taxol occurred through diffusion. Furthermore, the intracellular acidic environment with reducing condition could promote drug release.
Cellular uptake and distribution of the siRNA–PTX/LDL–NSC–SS–UA micelles in vitro
The cellular uptake and intracellular distribution of the drug- and gene-co-transported micelles in MCF-7/Taxol cells were confirmed by CLSM (Figure 7A). The co-localization of C6 (green fluorescence) and siRNA (red fluorescence) in the cytoplasm indicates the simultaneous delivery of drugs and genes in MCF-7/Taxol cells after incubation with siRNA-C6/micelle. Preponderant cellular uptake and intracellular release were further confirmed by the live cell station (Figure 7B), where C6 and siRNA were delivered by the micelles in the cells and then released into the cytoplasm almost simultaneously. Two-color flow cytometry was performed to detect the quantitative uptake (Figure 7C), where both red- and green-positive cells were detected. The fluorescence distribution table indicates that the siRNA-C6/micelle could simultaneously deliver drugs and genes into cells, which agrees with the aforementioned observations.
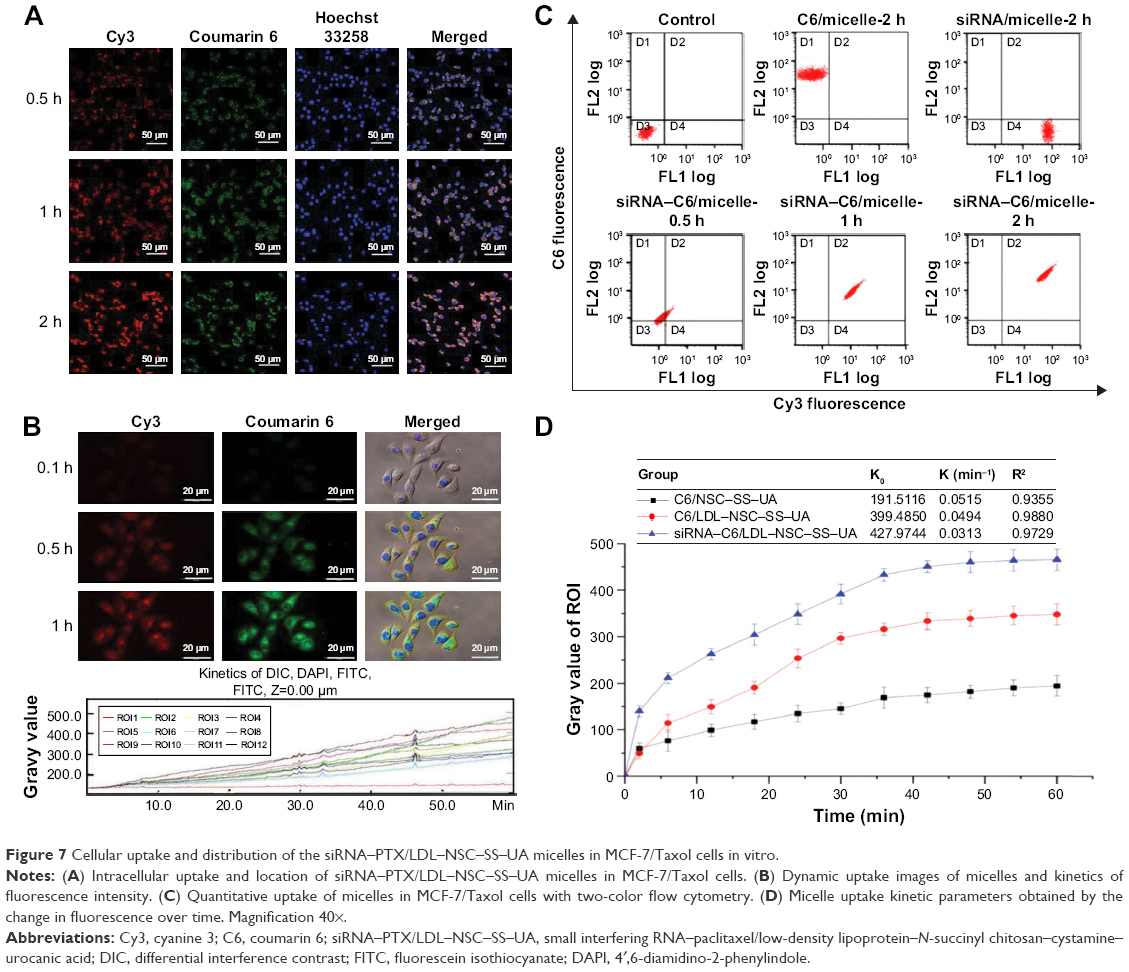 | Figure 7 Cellular uptake and distribution of the siRNA–PTX/LDL–NSC–SS–UA micelles in MCF-7/Taxol cells in vitro. |
The dynamic changes in intracellular fluorescence from C6 and siRNA were measured after treatment with different micelles and were fitted to non-linear cellular uptake kinetics (Figure 7D). SiRNA–C6/LDL–NSC–SS–UA showed the highest uptake rate and the lowest elimination speed, followed by C6/LDL–NSC–SS–UA and C6/NSC–SS–UA. Divergence was attributed to the gene silencing effect by siRNA, which could reduce BCRP protein expression and increase the intracellular concentration of the drug. Furthermore, micelles with LDL were taken up by LDLr, which is highly expressed on the cell surface, and could circulate between the cytoplasm and cell membrane to accelerate the uptake efficiency of drugs and genes.
Assessment of gene-silencing effect in vitro
The intracellular mRNA and protein levels were measured through RT-PCR and Western blot, respectively, to detect the transfection efficiency of siRNA/micelles. The siRNA/micelle knocked down the mRNA and protein levels of BCRP in the cells compared with the micelles without siRNA and the untreated groups (Figure 8). In addition, a more significant knockdown efficacy occurred after incubation with increased siRNA concentration or prolonged incubation time. The inhibition rate of naked chol-siRNA at 24 h was stronger than that of the micelles, and the difference was attributed to its lipophilicity in interacting with the cell membrane. The PCR and Western blot results showed that the LDL–NSC–SS–UA micelles could deliver siRNA into the cells efficiently to knockdown the protein expression of BCRP and achieve a gene-silencing effect.
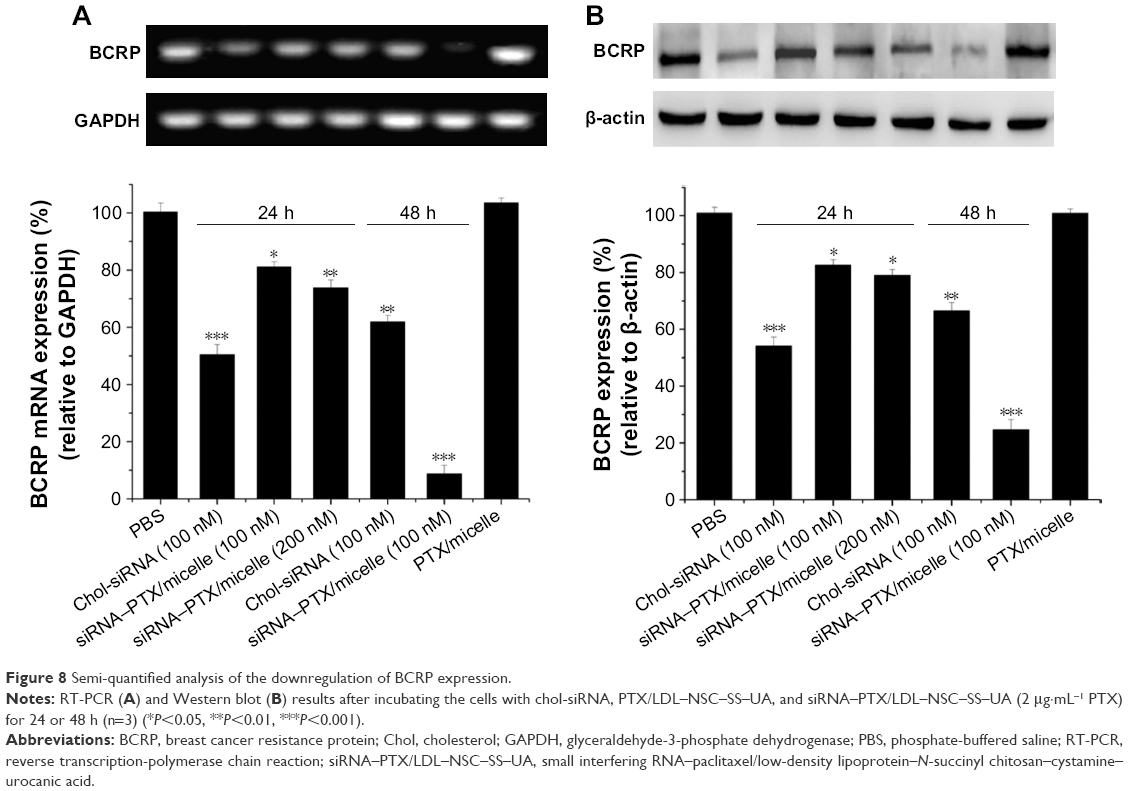 | Figure 8 Semi-quantified analysis of the downregulation of BCRP expression. |
Characterization of targeting properties in vivo
The targeting efficacies of NSC–SS–UA and LDL–NSC–SS–UA were evaluated using a NIRF imaging system that used Dir as a probe. As shown in Figure 9A, after treatment with the two types of micelles, most of the fluorescent signals accumulated in the liver within 12 h because of the rapid swallowing by mononuclear phagocytes, which were abundantly found in the liver. At 24 h post-injection, the fluorescence in the tumor was stronger than that in the organs for both types of micelles indicating that the micelles for co-delivery could circulate in the blood and ultimately accumulate in the tumor via the EPR effect. In addition, compared with the tumor-bearing nude mice treated with Dir/NSC–SS–UA, those treated with Dir/LDL–NSC–SS–UA showed a more significant accumulation of fluorescence in the tumor within 48 h after injection. This result indicates that the LDL-bearing micelles exerted a better target effect on the tumor for the active targeting effect of LDL. The fluorescence of tumors and major organs (heart, liver, spleen, lung, and kidney) also confirmed the effectiveness of the micelles in tumor-targeted delivery.
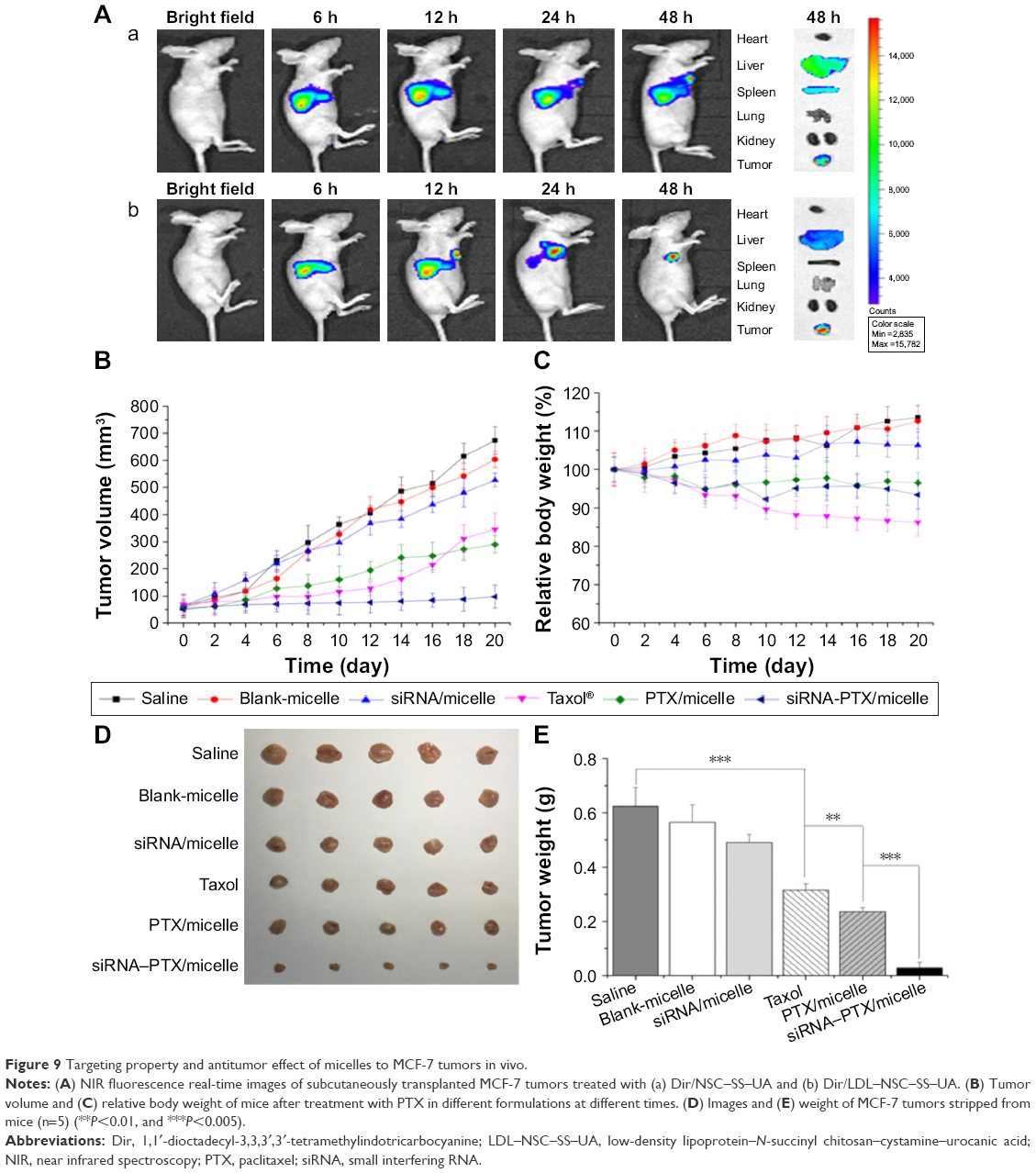 | Figure 9 Targeting property and antitumor effect of micelles to MCF-7 tumors in vivo. |
Antitumor effect in vivo
To investigate the synergistic antitumor activity of the siRNA–PTX/micelles in vivo, tumor-bearing nude mice were injected with different micelles to monitor the systemic toxicity and antitumor effect (Figure 9B–E). Compared with Taxol, the PTX-loaded micelles exhibited stronger antitumor activity. The siRNA–PTX/micelles exhibited the best antitumor effect among all formulations, possibly because of the synergistic antitumor activity of siRNA and PTX, as demonstrated by the comparison between the PTX/micelles and siRNA/micelles groups. The Taxol group did not exhibit a more significant tumor suppression effect but significantly decreased the weight of the mice compared with the PTX/micelle group. In addition, the tumor volume in the Taxol group increased rapidly after 12 days of treatment possibly because of the drug resistance of the tumor. Thus, we can conclude that the combination of genes and drugs could exert a strong tumor-suppressive effect in vivo.
Conclusions
We have developed and characterized siRNA–PTX/LDL–NSC–SS–UA micelles successfully for superior synergistic therapy and precise tumor localization. The binary polymer consisting of siRNA-loaded nanoparticles and PTX-loaded micelles was sensitive to pH and reduction surroundings, thereby enhancing the intracellular release of drugs and genes. The LDL nanoparticles demonstrated efficient cellular uptake, gene-silencing function, tumor-targeting effect, and siRNA protection from macrophage phagocytosis and RNase. Moreover, the siRNA–PTX/micelles could improve the tumor therapeutic efficacy by downregulating BCRP protein expression, decreasing drug efflux and increasing the intracellular drug concentration and reverse MDR. The siRNA–PTX/LDL–NSC–SS–UA micelles possessed enhanced stability, tumor accumulation and cell internalization. The results of this study prove the substantial potential of these materials for the clinical treatment of cancers.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos 81571788 & 81273463), Jiangsu Science and Technology Support Plan (BE2011670), and Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Disclosure
The authors report no conflicts of interest in this work.
References
Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13(10):714–726. | ||
Higgins CF. Multiple molecular mechanisms for multidrug resistance transporters. Nature. 2007;446(7137):749–757. | ||
Sun X, Bao J, Shao Y. Mathematical modeling of therapy-induced cancer drug resistance: connecting cancer mechanisms to population survival rates. Sci Rep. 2016;6:22498. | ||
Moitra K. Overcoming multidrug resistance in cancer stem cells. Biomed Res Int. 2015;2015:635745. | ||
Brzozowska N, Li KM, Wang XS, et al. ABC transporters P-gp and Bcrp do not limit the brain uptake of the novel antipsychotic and anticonvulsant drug cannabidiol in mice. PeerJ. 2016;4:e2081. | ||
Nakanishi T, Ross DD. Breast cancer resistance protein (BCRP/ABCG2): its role in multidrug resistance and regulation of its gene expression. Chin J Cancer. 2012;31(2):73–99. | ||
Lukyanova NY. Characteristics of homocysteine-induced multidrug resistance of human MCF-7 breast cancer cells and human A2780 ovarian cancer cells. Exp Oncol. 2010;32(1):10–14. | ||
Mao Q, Unadkat JD. Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport–an update. AAPS J. 2015;17(1):65–82. | ||
Litman T, Brangi M, Hudson E, et al. The multidrug-resistant phenotype associated with overexpression of the new ABC half-transporter, MXR (ABCG2). J Cell Sci. 2000;113(Pt 11):2011–2021. | ||
Jonker JW, Buitelaar M, Wagenaar E, et al. The breast cancer resistance protein protects against a major chlorophyll-derived dietary phototoxin and protoporphyria. Proc Natl Acad Sci U S A. 2002;99(24):15649–15654. | ||
Kwekkeboom RF, Lei Z, Doevendans PA, Musters RJ, Sluijter JP. Targeted delivery of miRNA therapeutics for cardiovascular diseases: opportunities and challenges. Clin Sci (Lond). 2014;127(6):351–365. | ||
Shi B, Zhang H, Bi J, Dai S. Endosomal pH responsive polymers for efficient cancer targeted gene therapy. Colloids Surf B Biointerfaces. 2014;119:55–65. | ||
Xue HY, Liu S, Wong HL. Nanotoxicity: a key obstacle to clinical translation of siRNA-based nanomedicine. Nanomedicine. 2014;9(2):295–312. | ||
Thibault M, Lavertu M, Astolfi M, Buschmann MD. Structure dependence of lysosomal transit of chitosan-based polyplexes for gene delivery. Mol Biotechnol. 2016;58(10):648–656. | ||
Gu J, Chen X, Xin H, Fang X, Sha X. Serum-resistant complex nanoparticles functionalized with imidazole-rich polypeptide for gene delivery to pulmonary metastatic melanoma. Int J Pharm. 2014;461(1–2):559–569. | ||
Leucuta SE. Subcellular drug targeting, pharmacokinetics and bioavailability. J Drug Target. 2014;22(2):95–115. | ||
Yoshida T, Lai TC, Kwon GS, Sako K. pH- and ion-sensitive polymers for drug delivery. Expert Opin Drug Deliv. 2013;10(11):1497–1513. | ||
Raemdonck K, Martens TF, Braeckmans K, Demeester J, De Smedt SC. Polysaccharide-based nucleic acid nanoformulations. Adv Drug Deliv Rev. 2013;65(9):1123–1147. | ||
Khan IN, Al-Karim S, Bora RS, Chaudhary AG, Saini KS. Cancer stem cells: a challenging paradigm for designing targeted drug therapies. Drug Discov Today. 2015;20(10):1205–1216. | ||
Messaoudi K, Clavreul A, Lagarce F. Toward an effective strategy in glioblastoma treatment. Part II: RNA interference as a promising way to sensitize glioblastomas to temozolomide. Drug Discov Today. 2015;20(6):772–779. | ||
Mehrotra N, Tripathi RM. Short interfering RNA therapeutics: nanocarriers, prospects and limitations. IET Nanobiotechnol. 2015;9(6):386–395. | ||
Vicentini FT, Borgheti-Cardoso LN, Depieri LV, et al. Delivery systems and local administration routes for therapeutic siRNA. Pharm Res. 2013;30(4):915–931. | ||
Heyes J, Palmer L, Bremner K, MacLachlan I. Cationic lipid saturation influences intracellular delivery of encapsulated nucleic acids. J Control Release. 2005;107(2):276–287. | ||
Aliabadi HM, Landry B, Sun C, Tang T, Uludag H. Supramolecular assemblies in functional siRNA delivery: where do we stand? Biomaterials. 2012;33(8):2546–2569. | ||
Shukla RS, Tai W, Mahato R, Jin W, Cheng K. Development of streptavidin-based nanocomplex for siRNA delivery. Mol Pharm. 2013;10(12):4534–4545. | ||
Brown MS, Herz J, Goldstein JL. LDL-receptor structure. Calcium cages, acid baths and recycling receptors. Nature. 1997;388(6643):629–630. | ||
Vyas SP, Sihorkar V. Endogenous carriers and ligands in non-immunogenic site-specific drug delivery. Adv Drug Deliv Rev. 2000;43(2–3):101–164. | ||
Huntosova V, Buzova D, Petrovajova D, et al. Development of a new LDL-based transport system for hydrophobic/amphiphilic drugdelivery to cancer cells. Int J Pharm. 2012;436(1–2):463–471. | ||
Zheng G, Chen J, Li H, Glickson JD. Rerouting lipoprotein nanoparticles to selected alternate receptors for the targeted delivery of cancer diagnostic and therapeutic agents. Proc Natl Acad Sci U S A. 2005;102(49):17757–17762. | ||
Kader A, Pater A. Loading anticancer drugs into HDL as well as LDL has little affect on properties of complexes and enhances cytotoxicity to human carcinoma cells. J Control Release. 2002;80(1–3):29–44. | ||
Nikanjam M, Gibbs AR, Hunt CA, Budinger TF, Forte TM. Synthetic nano-LDL with paclitaxel oleate as a targeted drug delivery vehicle for glioblastoma multiforme. J Control Release. 2007;124(3):163–171. | ||
Bei YY, Yuan ZQ, Zhang L, et al. Novel self-assembled micelles based on palmitoyl-trimethyl-chitosan for efficient delivery of harmine to liver cancer. Expert Opin Drug Deliv. 2014;11(6):843–854. | ||
Yuan ZQ, Li JZ, Liu Y, et al. Systemic delivery of micelles loading with paclitaxel using N-succinyl-palmitoyl-chitosan decorated with cRGDyK peptide to inhibit non-small-cell lung cancer. Int J Pharm. 2015;492(1–2):141–151. | ||
Lungu II, Radulescu M, Mogosanu GD, Grumezescu AM. pH sensitive core-shell magnetic nanoparticles for targeted drug delivery in cancer therapy. Rom J Morphol Embryol. 2016;57(1):23–32. | ||
Chen WL, Yang SD, Li F, et al. Tumor microenvironment-responsive micelles for pinpointed intracellular release of doxorubicin and enhanced anti-cancer efficiency. Int J Pharm. 2016;511(2):728–740. | ||
Li J, Huo M, Wang J, et al. Redox-sensitive micelles self-assembled from amphiphilic hyaluronic acid-deoxycholic acid conjugates for targeted intracellular delivery of paclitaxel. Biomaterials. 2012;33(7):2310–2320. | ||
Sun Y, Huang Y, Bian S, Liang J, Fan Y, Zhang X. Reduction-degradable PEG-b-PAA-b-PEG triblock copolymer micelles incorporated with MTX for cancer chemotherapy. Colloids Surf B Biointerfaces. 2013;112:197–203. | ||
Zhang CG, Zhu QL, Zhou Y, et al. N-Succinyl-chitosan nanoparticles coupled with low-density lipoprotein for targeted osthole-loaded delivery to low-density lipoprotein receptor-rich tumors. Int J Nanomed. 2014;9:2919–2932. | ||
Zhang CG, Zhu WJ, Liu Y, et al. Novel polymer micelle mediated co-delivery of doxorubicin and P-glycoprotein siRNA for reversal of multidrug resistance and synergistic tumor therapy. Sci Rep. 2016;6:23859. | ||
Zhang CG, Yang SD, Zhu WJ, et al. Distinctive polymer micelle designed for siRNA delivery and reversal of MDR1 gene-dependent multidrug resistance. J Biomed Mater Res B Appl Biomater. Epub 2016 Jul 13. | ||
Zhu QL, Zhou Y, Guan M, et al. Low-density lipoprotein-coupled N-succinyl chitosan nanoparticles co-delivering siRNA and doxorubicin for hepatocyte-targeted therapy. Biomaterials. 2014;35(22):5965–5976. | ||
Yang SD, Zhu WJ, Zhu QL, et al. Binary-copolymer system base on low-density lipoprotein-coupled N-succinyl chitosan lipoic acid micelles for co-delivery MDR1 siRNA and paclitaxel, enhances antitumor effects via reducing drug. J Biomed Mater Res B Appl Biomater. Epub 2016 Mar 23. | ||
Cao N, Cheng D, Zou S, Ai H, Gao J, Shuai X. The synergistic effect of hierarchical assemblies of siRNA and chemotherapeutic drugs co-delivered into hepatic cancer cells. Biomaterials. 2011;32(8):2222–2232. | ||
Sun H, Meng F, Cheng R, Deng C, Zhong Z. Reduction-responsive polymeric micelles and vesicles for triggered intracellular drug release. Antioxid Redox Signal. 2014;21(5):755–767. | ||
Kettiger H, Schipanski A, Wick P, Huwyler J. Engineered nanomaterial uptake and tissue distribution: from cell to organism. Int J Nanomed. 2013;8:3255–3269. | ||
Guo M, Mao H, Li Y, et al. Dual imaging-guided photothermal/photodynamic therapy using micelles. Biomaterials. 2014;35(16):4656–4666. |
Supplementary materials
Synthesis of the N-succinyl chitosan (NSC)
The synthesis of the NSC is described in Figure 2. Briefly, 5.0 g chitosan was dissolved in 100 mL of dimethyl sulfoxide, followed by adding 10.0 g succinic anhydride, stirring for 6 h at 60°C and kept overnight. Then the solidoid was soaked in ethanol for 1.5 h. After suction filtration, the deposit was dissolved in 50 mL of distilled water and 5% NaOH solution was used to regulate the pH to 10. Then the solution was filtered and the solidoid was precipitated with excessive acetone. After centrifuging, the sediment was successively washed three times with 70% ethanol and acetone, and then dried at 50°C for 48 h.
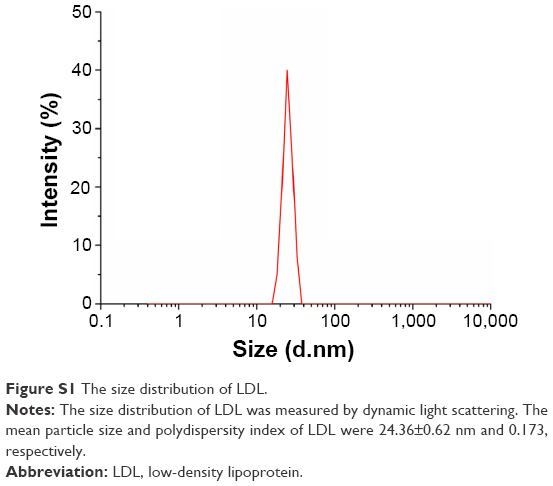 | Figure S1 The size distribution of LDL. |
Evaluation of LDL
Hemolysis activity of Taxol® and PTX-micelles in vitro
As shown in Figure S2, the hemolysis of PTX-micelles groups was far below that of Taxol group at the same PTX concentration, indicating that the polymer micelles have good blood compatibility and safety.
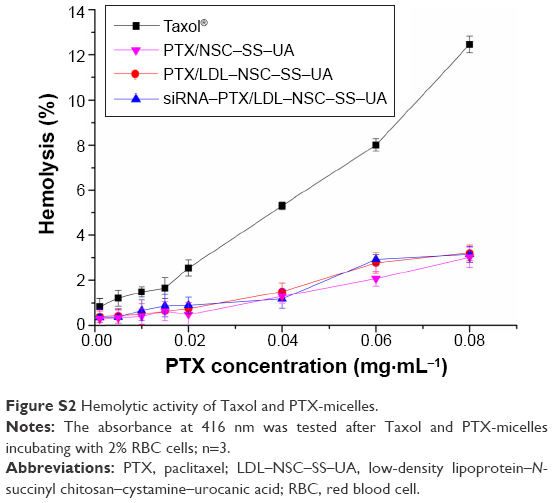 | Figure S2 Hemolytic activity of Taxol and PTX-micelles. |
Endocytosis mechanism study of blank micelles
The endocytic pathway of N-succinyl chitosan–cystamine–urocanic acid (NSC–SS–UA) and low-density lipoprotein–N-succinyl chitosan–cystamine–urocanic acid (LDL–NSC–SS–UA) micelles was elucidated via flow cytometry. As shown in Figure S3, chlorpromazine decreased 63.7% and 67.5% cellular uptake of NSC–SS–UA and LDL–NSC–SS–UA, respectively, indicating that both the micelles were mainly internalized by a clathrin-mediated endocytotic pathway into cancer cells. Methyl-β-cyclodextrin decreased 82.3% cellular uptake of LDL–NSC–SS–UA, but almost had no effect on the internalization of NSC–SS–UA suggesting that the uptake of micelles with LDL dependent on lipid rafts. In addition, tyrosine kinase and SR-BI acceptor also participated in the internalization of micelles. Low temperature also led to a significant decrease in cellular uptake indicating that the internalization of micelles was also energy-dependent.
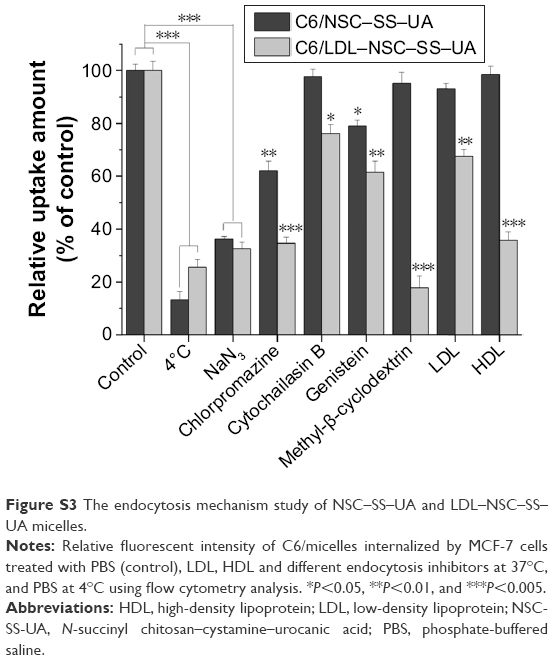 | Figure S3 The endocytosis mechanism study of NSC–SS–UA and LDL–NSC–SS–UA micelles. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.