")
Back to Journals » Cancer Management and Research » Volume 11
Loss of ARID1A promotes proliferation, migration and invasion via the Akt signaling pathway in NPC
Authors Yang Y, Wang X, Yang J, Duan J, Wu Z, Yang F, Zhang X , Xiao S
Received 1 March 2019
Accepted for publication 10 May 2019
Published 29 May 2019 Volume 2019:11 Pages 4931—4946
DOI https://doi.org/10.2147/CMAR.S207329
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Alexandra R. Fernandes
Yang Yang,1,* Xiaoyu Wang,1,* Junjun Yang,2 Jingling Duan,1 Zhen Wu,3 Fan Yang,1 Xiaoling Zhang,4 Shengjun Xiao1
1Department of Pathology, the Second Affiliated Hospital, Guilin Medical University, Guilin 541199, People’s Republic of China; 2Department of Stomatology, The Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430000, People’s Republic of China; 3Xiangya Medical College of South Central University, Changsha 413000, People’s Republic of China; 4Department of Physiology, Faculty of Basic Medical Science, Guilin Medical University, Guilin 541199, People’s Republic of China
*These authors contributed equally to this work
Background: AT-rich interactive domain-containing protein 1A (ARID1A) is a member of the switch/sucrose nonfermentable chromatin remodeling complex, which has been observed to be mutated in various tumors. The loss of ARID1A is reported to be frequently associated with PI3K/Akt pathway activation.
Objective: The roles of ARID1A in nasopharyngeal carcinoma (NPC) have not been reported until now. The aim of this research was to explore the clinical significance and potential mechanism of ARID1A in NPC development and progression.
Methods: ARID1A expression levels were investigated in human NPC tissues and cell lines. The effects of ARID1A knockdown on nasopharyngeal cancer cell proliferation, migration and invasion were evaluated in vitro using CCK8, wound healing, transwell and flow cytometry assays. The expression of relevant proteins was evaluated by Western blot assays.
Results: In this study, ARID1A was significantly downregulated in NPC tissues and cells. Furthermore, low ARID1A expression was significantly associated with aggressive clinicopathological characteristics and poor survival in NPC patients. Depletion of endogenous ARID1A by siRNA promoted proliferation, migration and invasion in CNE1 and HNE1 cells. Additionally, ARID1A knockdown increased the phosphorylation of Akt in NPC cells. High levels of p-Akt were also observed in NPC biopsies and correlated with ARID1A downregulation. These results imply that the loss of ARID1A could activate Akt signaling. In addition, MK-2206 (a highly selective inhibitor of Akt) partially suppressed NPC cell proliferation, migration and invasion, which were induced by ARID1A knockdown.
Conclusion: Our findings indicate that ARID1A plays an essential role in modulating the Akt pathway, functions as a tumor suppressor in NPC and may be a potential target for NPC treatment.
Keywords: nasopharyngeal carcinoma, SWI/SNF, ARID1A, PI3K/Akt pathway, Akt inhibitor
Introduction
Nasopharyngeal carcinoma (NPC) is a common malignant head and neck tumor in southern China, North Africa and Southeast Asia,1–3 and the incidence rate of NPC is up to 0.2%.4,5 The etiology of NPC development and progression may be closely related to geographic areas, genetic factors, environmental factors and Epstein–Barr virus infection.6,7 Although the treatment of NPC has improved greatly in recent years, the rate of distant metastasis is as high as 14.1%.8 Thus, it will be of great clinical value to explore the underlying molecular mechanisms of NPC progression.
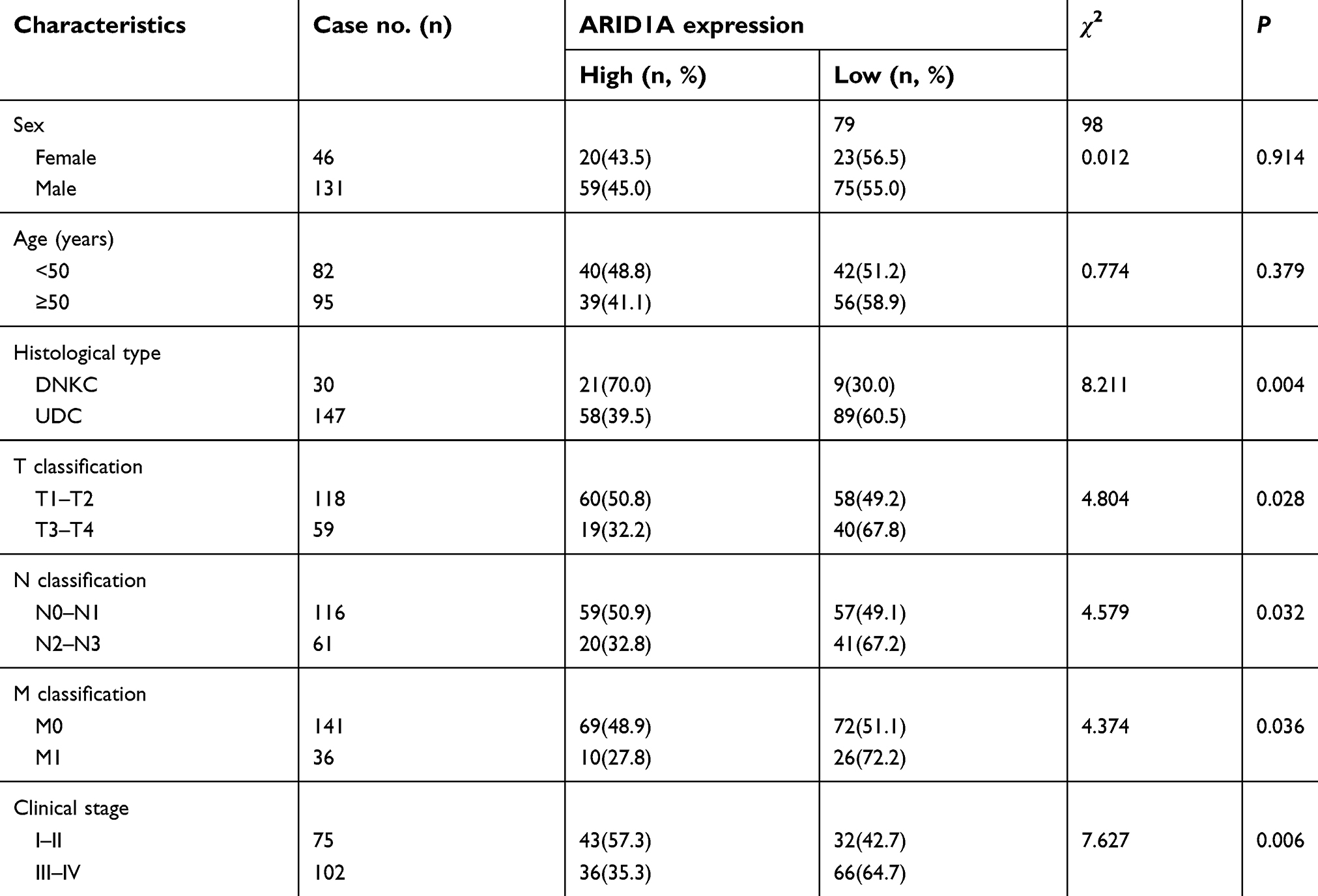 | Table 1 Correlations between ARID1A expression and the clinicopathological features of 177 NPC patients |
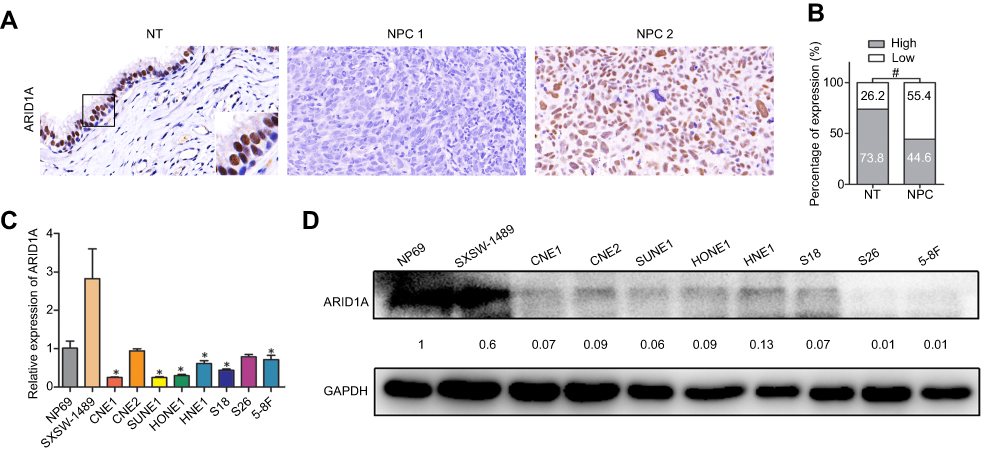 | Figure 1 ARID1A was downregulated in NPC tissues and cell lines. (A) ARID1A expression in noncancerous nasopharyngeal biopsies and NPC tissues based on IHC. a: High expression of ARID1A in noncancerous nasopharyngeal epithelia; b: Low expression of ARID1A in NPC biopsies; c: High expression of ARID1A in NPC biopsies. The brown staining indicates ARID1A immunoreactivity. (B) ARID1A expression was significantly lower in the NPC biopsies than in the noncancerous nasopharyngeal biopsies. (C–D) Expression levels of ARID1A in the indicated nasopharyngeal epithelial and NPC cell lines were examined by qRT-PCR (C) and Western blotting (D).Abbreviations: NPC, nasopharyngeal carcinoma; ARID1A, AT-rich interacting domain-containing protein 1A; IHC, immunohistochemical staining. |
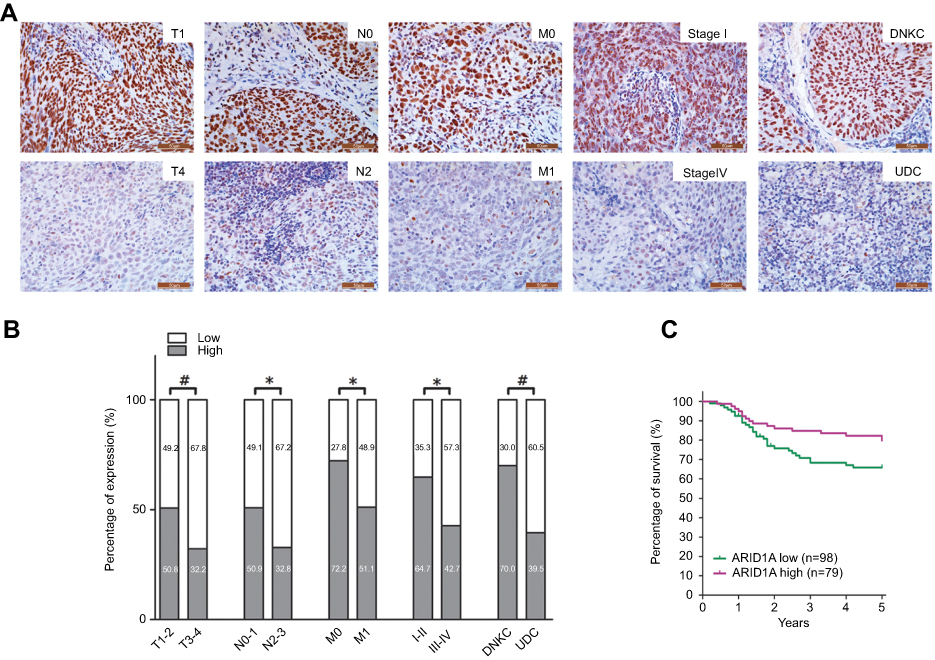 | Figure 2 Low ARID1A expression was associated with advanced TNM stage and poor prognosis in NPC patients. (A) Representative images of ARID1A expression in NPC biopsies with different TNM stages. High expression of ARID1A was observed for the T1, N0, M0, and I stage and DNKC subtype NPC biopsies, while low expression of ARID1A was detected for the T4, N3, M1, and IV stage and UDC subtype tumors. (B) Numbers and percentages of cases with high or low expression of ARID1A according to different clinicopathological features. (C) Low ARID1A expression was associated with a poor prognosis in NPC patients. *P<0.05, **P<0.01, #P<0.001.Abbreviations: NPC, nasopharyngeal carcinoma; ARID1A, AT-rich interacting domain-containing protein 1A; DNKC, differentiated nonkeratinized squamous carcinoma; UDC, undifferentiated nonkeratinized carcinoma. |
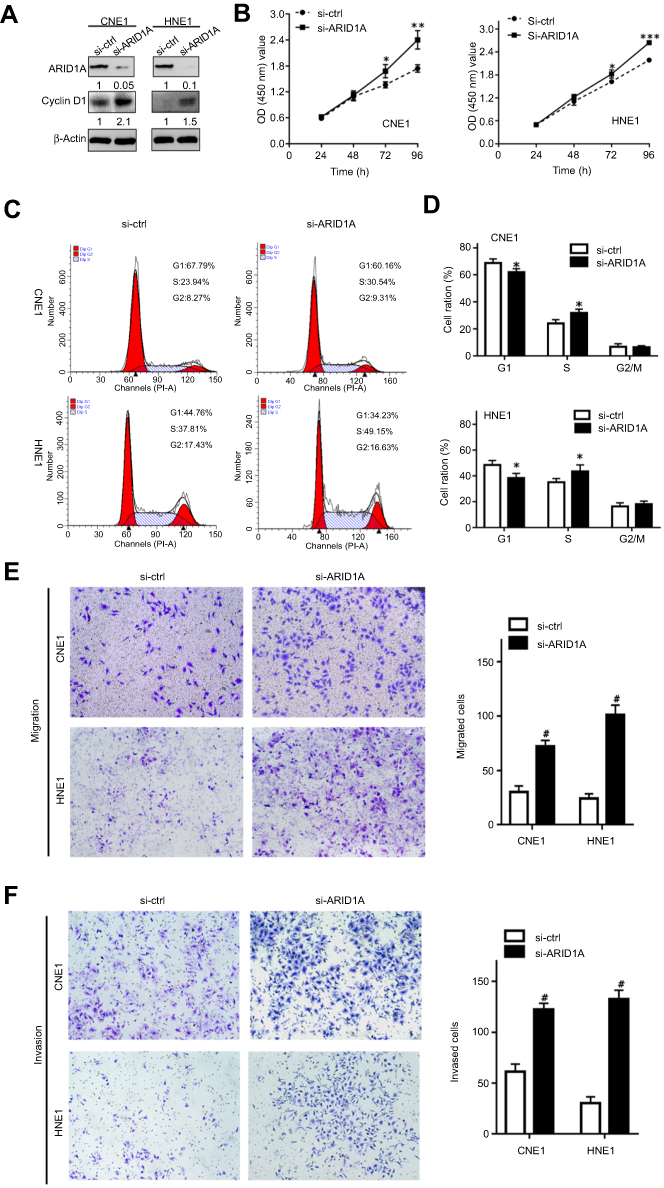 | Figure 3 Silencing ARID1A promotes NPC cell proliferation, migration and invasion. (A) According to Western blot assay results, ARID1A was successfully knocked down by siRNA in CNE1 and HNE1 cells, which also resulted in Cyclin D1 upregulation. (B) The effects of ARID1A silencing on cell proliferation were measured by CCK8 assay. (C) Cell cycle distribution was analyzed by flow cytometry at 48 hrs post-culture. (D) Relative cell distribution percentage of ARID1A-silenced (si-ARID1A) and control (si-ctrl) NPC cells in each cell cycle phase. (E and F) Motile and invasive activities of the si-ARID1A and si-ctrl groups of NPC cells based on transwell assays. The average number of cells per field was determined from three repeated independent experiments. *P<0.05, **P<0.01, *** or #P<0.001.Abbreviations: NPC, nasopharyngeal carcinoma; ARID1A, AT-rich interacting domain-containing protein 1A. |
Switch/sucrose nonfermentable (SWI/SNF) is a conserved chromatin remodeling complex that plays an essential role in various cellular processes, such as development, differentiation, proliferation and DNA repair.9 This complex has helicase and ATPase activities and is thought to regulate the transcription of certain genes by altering the chromatin structure around those genes.10 SWI/SNF is composed of a core subunit, which is either BRM or BRG1, and a few noncatalytic subunits. The noncatalytic subunits are also called BRG1- or BRM-associated factors (BAFs). Genes encoding subunits of SWI/SNF (BAF) chromatin remodeling complexes are collectively mutated in 10–20% of all human cancers. Among these genes, AT-rich interacting domain-containing protein 1A (ARID1A) is the most frequently mutated.11
ARID1A is located in the chromosome 1p36 region and is also known as BAF250a, p270 or SMARCF1. ARID1A has been found to be mutated in various cancers, including endometrioid carcinoma,12 ovarian clear cell carcinoma,13 breast cancer,14 liver cancer,15 gastric cancer,16 urothelial carcinoma17 and pancreatic cancer.18 These findings show that ARID1A plays a key role in carcinogenesis and is a potential tumor suppressor.
However, the expression and function of ARID1A in NPC have not been reported until now. In the present study, we demonstrated that ARID1A expression was downregulated and associated with Akt signaling pathway activation in NPC tissues and cells. Furthermore, ARID1A knockdown by siRNA promoted NPC cell proliferation, migration and invasion, and MK-2206 (a highly selective inhibitor of Akt) partially rescued these biological changes. Thus, these findings indicated that ARID1A functions as a tumor suppressor in NPC and may be a potential target for NPC treatment.
Materials and methods
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the Institutional Review Board (IRB) of the Second Affiliated Hospital of Guilin Medical College (Guilin, China) and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. The cells used for research were approved by the IRB of the Second Affiliated Hospital of Guilin Medical College.
Patients and samples
A total of 177 paraffin-embedded NPC biopsies and 61 noncancerous nasopharyngeal epithelial biopsies (ie, chronic nasopharyngitis tissues for immunohistochemistry assays) were obtained from the Department of Pathology, the Second Affiliated Hospital of Guilin Medical College, China, between 2005 and 2009. None of the 177 NPC patients received preoperative radiotherapy or chemotherapy. The patients whose tissues were used provided written informed consent.
RNA isolation and quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from NPC cells using TRIzol Reagent (TaKaRa, Dalian, China) according to the manufacturer’s instructions. Then, mRNA was reverse transcribed into cDNA using a PrimeScript RT Reagent Kit (TaKaRa). We evaluated mRNA levels using SYBR Green qRT-PCR master mix with a StratageneMx3005P qRT-PCR System (Agilent, Santa Clara, CA, USA). The relative mRNA expression levels were normalized to those of GAPDH. The primers used in the qRT-PCR assay are listed in Table S1.
Immunohistochemical staining (IHC) and microscopic analysis
After deparaffinization, rehydration and antigen retrieval, tissue slides (4 μm thick) were blocked with 3% H2O2 for 10 mins and incubated with anti-ARID1A and anti-p-Akt antibodies (Table S2) at 4°C overnight. The slides were then stained with a secondary antibody at 37°C for 20 mins. Subsequently, the sections were stained with diaminobenzidine, counterstained with hematoxylin and washed with water. Histopathology and immunohistochemistry analyses were performed independently by two pathologists without knowledge of the clinicopathological results of the patients. As ARID1A was expressed in almost all tumor cells, only staining intensity was evaluated for the IHC scores. Staining intensity was classified as 0 (negative); 1 (weak); 2 (moderate); and 3 (strong). The scores 0 and 1 were defined as low, and the scores 2 and 3 were defined as high. Slides with conflicting evaluations were reassessed, and a consensus was reached, as we have fully described previously.
Cell culture
Human NPC cell lines (namely, CNE1, CNE2, SUNE1, HONE1, HNE1, S18, S26, and 5-8F) and NP69 and SXSW-1489 cells were kindly provided by Prof. Dong Xiao (Institute of Cancer Research, Southern Medical University, Guangzhou, China). NPC cells were cultured in RPMI 1640 medium (Gibco, Carlsbad, CA, USA) supplemented with 10% FBS(Gibco), while NP69 and SXSW-1489 cells were maintained in keratinocyte/serum-free medium (Invitrogen). All cells were grown at 37°C with 5% CO2.
Transient siRNA transfection
ARID1A depletion was performed by transfecting CNE1 and HNE1 cells with ARID1A siRNA (sc-43628, Santa Cruz Biotech, CA, USA). The control cells were transfected with scrambled siRNA (sc-36869, Santa Cruz Biotech). Cells were incubated with the transfection mixture for 6 hrs, and the medium was then changed. After transfection for 48 hrs, ARID1A expression levels were checked by Western blot assays. Finally, we chose a dose of 0.552 μg for transfection.
CCK8 assay
CNE1 cells (4,000 cells/plate) and HNE1 cells (3,000 cells/plate) were grown in 96-well plates (Costar, CA, USA). After the cells were cultured overnight, they were transfected with siRNA and/or treated with MK2006 (Selleckchem, TX, USA). Then, the cells were cultured for 1–4 days. Ten microliters of Cell Counting Kit-8 assay reagent (CCK-8, Dojindo, Kumamoto, Japan) were added to each well, followed by another 2 hrs incubation. The absorbance of each well was measured at 450 nm at different time points with a microplate reader.
Wound healing assay
After the cells were transfected with siRNA and/or treated with MK-2206, their migratory ability was evaluated by wound repair assays. The monolayer cells were scratched equally with the narrow edge of a 1,000 μL pipette tip and cultured with serum-free media. Then, the wound healing distance was documented at 24 and 48 hrs.
Transwell migration and invasion assays
To evaluate cell migration capability by transwell assay, 6.5 mm transwell chambers with 8 µm micropores were used (CorningCostar, Manassas, VA, USA). For the invasion assay, the transwell inserts were coated with Matrigel (BD Biosciences, San Jose, CA, USA). A total of 2×104 cells/well (migration assay) or 4×104 cells/well (invasion assay) in 200 μL serum-free 1,640 were seeded on top of the filter, and 1,640 medium containing 10% FBS (500 μL) was added to the bottom wells for incubation at 37°C. After 24 hrs (migration assay) or 48 hrs (invasion assay), the cells had migrated or invaded to the undersurface of the transwell membrane. The filters were washed with PBS thrice and then fixed with 4% paraformaldehyde for 15 mins. Next, the cells were stained with a 0.1% crystal violet solution for 30 mins. The numbers of migrated or invaded cells in five different fields were counted using a microscope.
Flow cytometry
After treatment, cells were washed with PBS thrice and fixed with 70% cold ethanol at 4°C overnight. Then, the cells were washed, centrifuged, suspended and stained with 50 μg/mL propidium iodide and 1 mg/mL RNase in PBS. The cell cycle was measured using a FACScan flow cytometer (BD Biosciences) and analyzed by ModFit software (BD Biosciences).
Western blot analysis
Cells were collected after treatment for 48 hrs, and protein was extracted using RIPA buffer (Solarbio, Beijing, China). Protein concentrations were measured with a BCA Protein Assay Kit (Beyotime, Beyotime Biotechnology, Shanghai, China). Total protein was separated on an 8% SDS-PAGE gel and transferred onto polyvinylidene difluoride (PVDF) membranes (Amersham Pharmacia Biotech, Piscataway, NJ, USA), which were blocked with 5% fat-free milk in TBST for 2 hrs at room temperature. The membranes were then incubated at 4°C overnight with primary antibodies (Table S2). The membranes were washed with TBST three times and incubated with an HRP-conjugated secondary antibody (1:5,000) at room temperature for 1 hr. Protein detection was performed using enhanced chemiluminescence (ECL).
Results
ARID1A is downregulated in NPC tissues and cells
To determine ARID1A expression levels in NPC tissues, 177 NPC tissues and 61 noncancerous epithelial tissues were detected by IHC. The IHC staining results revealed significantly decreased ARID1A expression in NPC specimens (Figure 1A and B & Table S3). We next quantitatively evaluated ARID1A expression in NPC cell lines and immortalized nasopharyngeal epithelial cells using qRT-PCR and Western blotting. Compared to that in immortalized nasopharyngeal epithelial cells, ARID1A expression was significantly downregulated in NPC cell lines (Figure 1C and D).
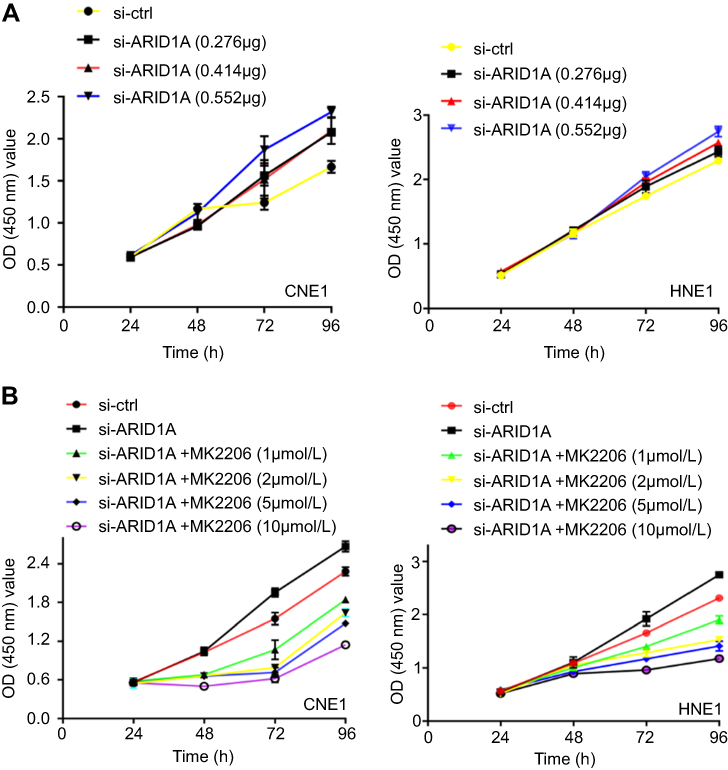 | Figure S1 The effects of different concentration of siRNA and MK2206 on cell growth of NPC cells. |
Low ARID1A expression was associated with advanced TNM stage and poor prognosis in NPC patients
The relationships between ARID1A expression and the clinicopathologic characteristics of NPC patients are summarized in Table 1. No significant association was identified between ARID1A expression and the age (P=0.914) or sex (P=0.379) of 177 NPC patients (Table 1). ARID1A expression was, however, significantly associated with the histological type (P=0.004), tumor size (T classification) (P=0.028), lymph node metastasis (N classification) (P=0.032), distant metastasis (M classification) (P=0.036) and clinical stage (P=0.006) (Table 1). Low ARID1A expression was more frequently observed in undifferentiated nonkeratinized carcinoma (UDC), T3-T4, N2-N3, M1, and stage III-IV tumors than in differentiated nonkeratinized squamous carcinoma (DNKC), T1-T2, N0-N1, M0, and stage I-II tumors (Figure 2A and B and Table 1). In addition, low ARID1A expression was associated with a poor prognosis in NPC patients (Figure 2C). These data indicate that ARID1A loss is a key molecular event in advanced cases of NPC, and ARID1A may be involved in the progression of NPC.
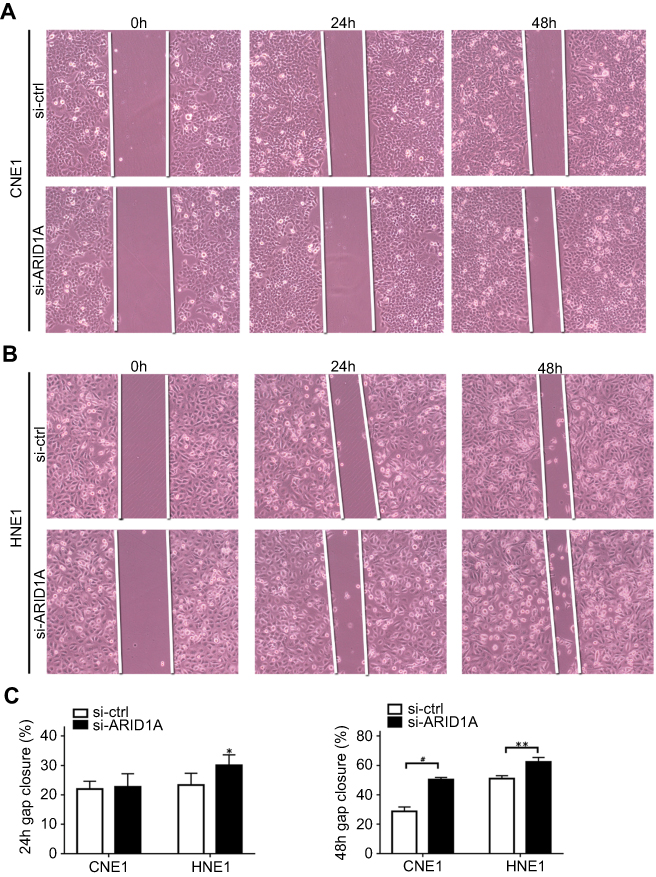 | Figure S2 ARID1A knockdown promoted cell migration in NPC cells by wound scratch assays. *P<0.01, **P<0.05, #P<0.001. |
Silencing ARID1A increases NPC cell proliferation, migration, invasion and an EMT-like molecular phenotype
To examine the functional significance of ARID1A in NPC cells, CNE1 and HNE1 cells were transiently transfected with siRNA. According to Western blot assay results, ARID1A was successfully knocked down by siRNA in both cell lines (Figure 3A). Flow cytometry showed that compared to scrambled siRNA, ARID1A siRNA significantly decreased the G0/G1 phase cell population and increased the S phase cell population for both cell lines (Figure 3C and D). The CCK8 assay demonstrated that ARID1A silencing obviously enhanced the proliferation of both cell lines (Figure 3B). Cyclin D1 was upregulated in both cell lines after ARID1A silencing. In addition, wound healing assays and transwell assays were performed to evaluate the migration and invasion of CNE1 and HNE1 cells. Depleting endogenous ARID1A by siRNA increased the invasion and migration of both cell lines (Figure 3E and F and Figure S2). In addition, E-cadherin was downregulated, and Vimentin was upregulated (Figure 7). These results suggested that ARID1A silencing increased the migration, invasion and epithelial–mesenchymal transition (EMT)-like molecular phenotype of NPC cells. *P<0.05, **P<0.01, #P<0.001.
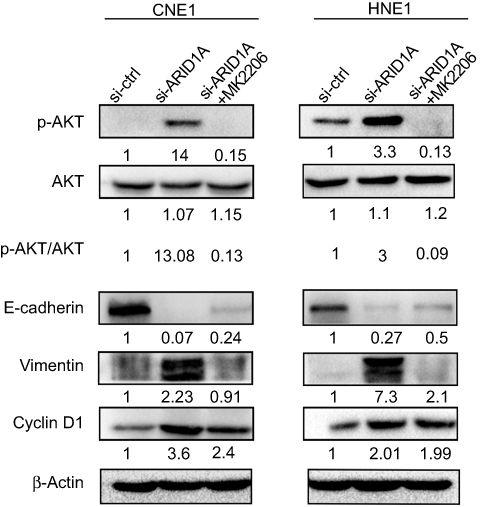 | Figure 7 MK-2206 rescued the changes in Cyclin D1 and EMT-associated markers in ARID1A-silenced NPC cells. Western blot analysis of the expression of ARID1A, p-Akt, Akt, Cyclin D1, E-cadherin and Vimentin. MK-2206 rescued the changes in Cyclin D1, E-cadherin and Vimentin expression in ARID1A-silenced NPC cells.Abbreviations: NPC, nasopharyngeal carcinoma; ARID1A, AT-rich interacting domain-containing protein 1A. |
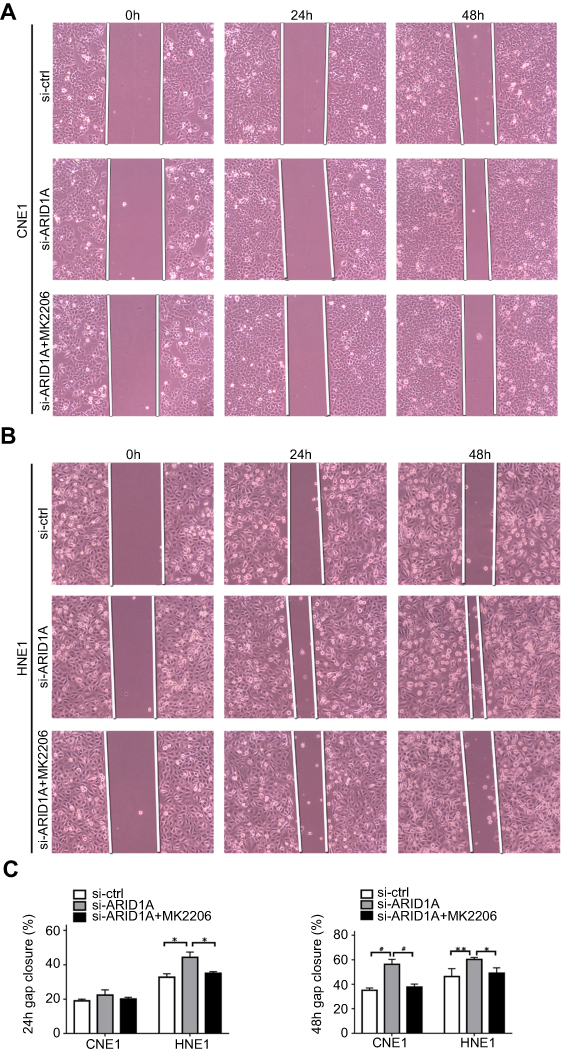 | Figure S3 MK2006 partially rescued the enhanced migration ability induced by ARID1A silencing in NPC cells by wound scratch assays. *P<0.05, **P<0.01, #P<0.001. |
ARID1A downregulation was correlated with Akt pathway activation in NPC cells and clinical NPC biopsies
The Western blot results showed that depletion of endogenous ARID1A by siRNA increased Akt phosphorylation in CNE1 and HNE1 cells (Figure 4A). The IHC results from clinical NPC biopsies revealed that low ARID1A expression levels were significantly associated with high p-Akt protein levels (Figure 4B and C and Table S4). These results suggested that the loss of ARID1A was associated with Akt signaling pathway activation in NPC cells and NPC biopsies.
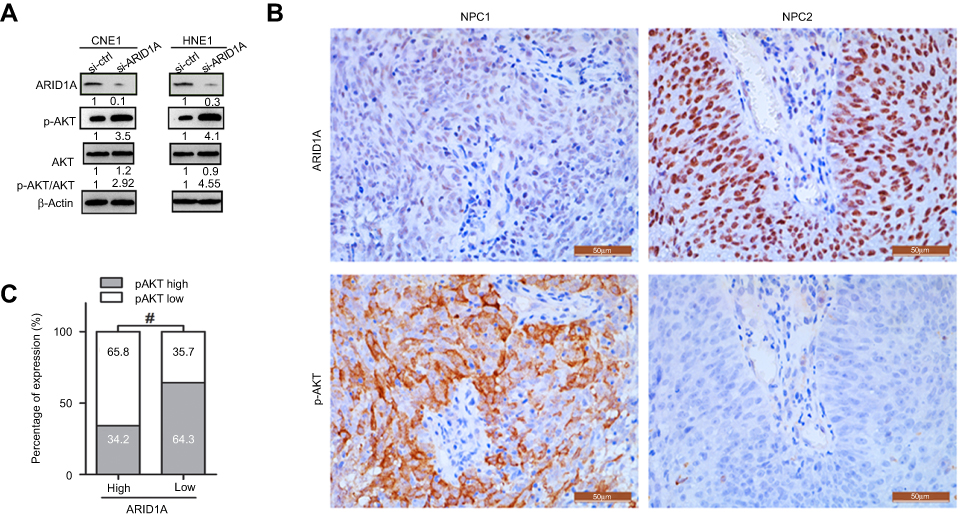 | Figure 4 Silencing ARID1A was correlated with Akt pathway activation in NPC cells and clinical NPC biopsies. (A) Depletion of endogenous ARID1A by siRNA increased the phosphorylation of Akt in CNE1 and HNE1 cells. (B and C) Representative images revealed that low ARID1A expression levels were significantly associated with high p-Akt levels in NPC biopsies. #P<0.001.Abbreviations: NPC, nasopharyngeal carcinoma; ARID1A, AT-rich interacting domain-containing protein 1A. |
MK-2206 partially rescued the changes in proliferation, migration, and invasion and in the EMT-like molecular phenotype induced by ARID1A silencing in NPC cells
Since silencing ARID1A led to Akt signaling activation in NPC cells, we hypothesized that inhibiting Akt signaling could reverse the biological effects induced by ARID1A silencing. MK-2206, a highly selective Akt inhibitor, was used to block Akt signaling. A CCK8 assay confirmed that MK-2206 significantly inhibited proliferation in ARID1A-silenced NPC cells (Figure 5A). Flow cytometry showed that MK-2206 arrests the cell cycle at the G1/S checkpoint in ARID1A-silenced NPC cells (Figure 5B and C). Wound healing assays and transwell assays demonstrated that MK-2206 obviously suppressed the invasive and migratory abilities of ARID1A-silenced NPC cells (Figure 6A–C and Figure S3). Moreover, E-cadherin downregulated Vimentin and Cyclin D1 in ARID1A-silenced NPC cells (Figure 7). These results suggest that Akt signaling plays an important role in ARID1A silencing-induced proliferation, migration, and invasion and EMT-like molecular phenotypes in NPC cells.
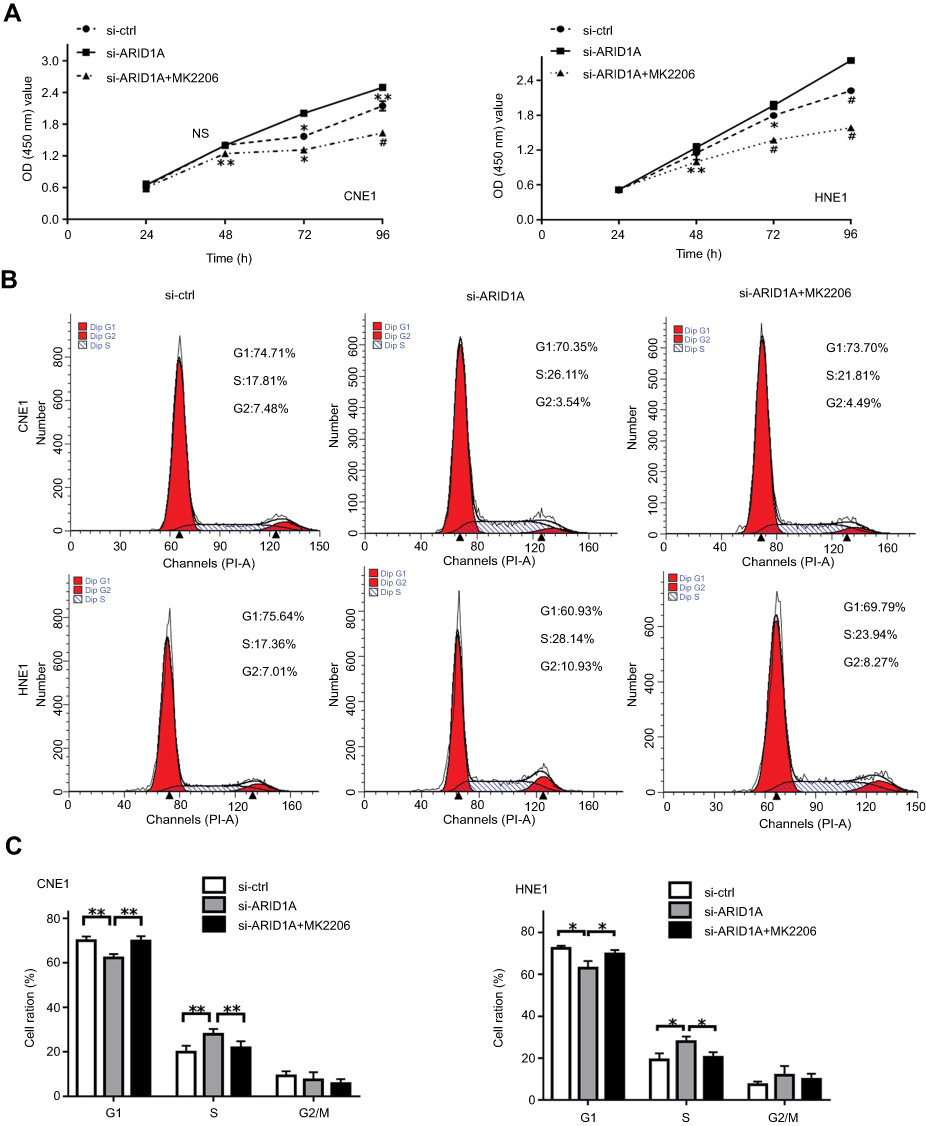 | Figure 5 MK-2206 partially rescued the increased proliferation induced by ARID1A silencing in NPC cells. (A) MK-2206 inhibited proliferation in ARID1A-silenced NPC cells according to a CCK8 assay. (B and C) MK-2206 inhibited G1/S transition in ARID1A-silenced NPC cells according to flow cytometry. *P<0.05, **P<0.01, #P<0.001.Abbreviations: NPC, nasopharyngeal carcinoma; ARID1A, AT-rich interacting domain-containing protein 1A. |
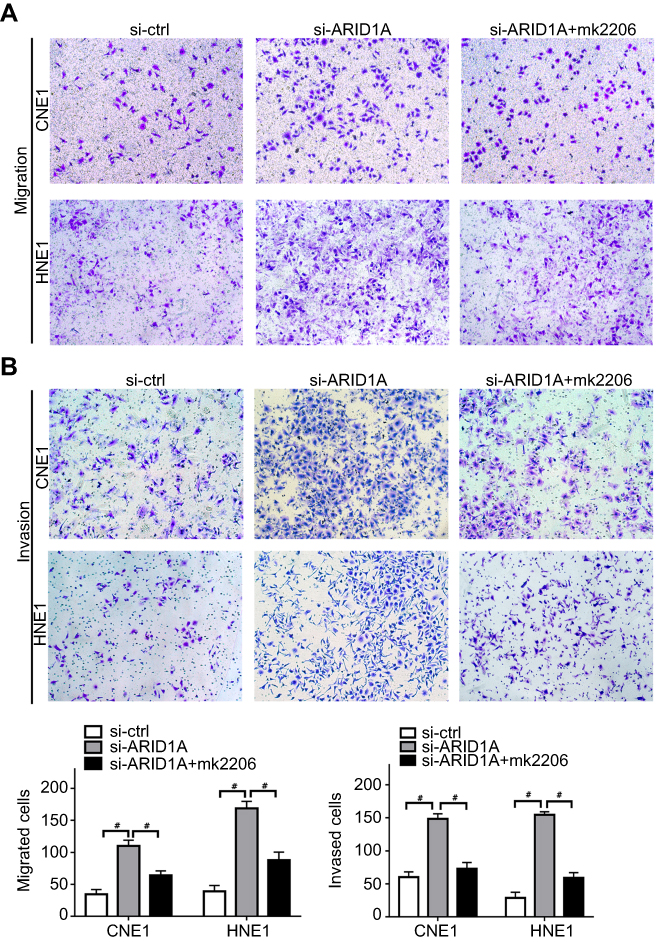 | Figure 6 MK-2206 partially rescued the enhanced migration and invasion ability induced by ARID1A silencing in NPC cells. (A–C). MK-2206 inhibited the motility (A) and invasion (B) of ARID1A-silenced NPC cells. # P<0.001.Abbreviations: NPC, nasopharyngeal carcinoma; ARID1A, AT-rich interacting domain-containing protein 1A. |
Discussion
Since next-generation sequencing methods have enabled genome-wide analyses, ARID1A mutations in ovarian endometrioid carcinoma and ovarian clear cell carcinoma were first reported in 2010.10 Subsequently, ARID1A mutations have been observed in many other human malignancies, including breast cancer,19 stomach cancer,20,21 pancreatic cancer22 and lung adenocarcinoma.23 These data imply that ARID1A is a potential tumor suppressor in carcinogenesis. Despite these findings, to the best of our knowledge, no further reports regarding the roles of ARID1A in NPC have been published.
Here, we reveal that ARID1A is downregulated in NPC tissues and cells and that low ARID1A expression is significantly associated with aggressive clinicopathological characteristics, such as larger tumor size, lymph node metastasis, distant metastasis, advanced clinical stage, poor differentiation, and poor prognosis in NPC patients. Collectively, the results regarding ARID1A loss in the advanced-stage cases imply that ARID1A functions as a tumor suppressor in the progression of NPC.
Several reports have confirmed that ARID1A mutations are involved in cancer cell proliferation and tumor growth.24,25 Enhanced cell proliferation was found in breast cancer cells and ovarian cancer cells following ARID1A knockdown, whereas the restoration of wild-type ARID1A expression in ARID1A-mutated cells significantly inhibited cell proliferation.25,26 In line with these findings, our data also showed that silencing ARID1A obviously enhanced the proliferation of CNE1 and HNE1 cells. Additionally, upregulated cyclin D1 and accelerated G1/S transition were observed in ARID1A-silenced cells. The T classification data from the human NPC biopsies clearly show that ARID1A was significantly downregulated in large-sized tumors. These results indicate that ARID1A negatively modulates NPC proliferation.
Metastasis, which converts local tumors into systemic diseases, is a hallmark of cancer and is the leading cause of death in cancer patients.27 In the initiation process of metastasis, cancer cells detach from the primary site, migrate and invade the surrounding tissues. Cancer cells lose their cell polarity and cell–cell adhesion and gain migratory and invasive properties, a process named EMT.28 Loss of ARID1A is reported to promote metastasis in several human malignancies.29–31 ARID1A loss is also significantly associated with local lymph node metastasis and distant metastasis in hepatocellular carcinoma (HCC)29 and gastric cancer.31 ARID1A knockdown promotes cell migration and invasion, whereas ARID1A overexpression inhibits migration and invasion in HCC,29 gastric cancer31 and neuroblastoma cells. Moreover, ARID1A silencing decreases the expression of E-cadherin31 and increases the expression of N-cadherin.30 Our data implied that the loss of ARID1A promoted the mobility and EMT of NPC cells. Our data also demonstrated that ARID1A knockdown strongly enhanced the migratory and invasive abilities of CNE1 and HNE1 cells. Additionally, Vimentin upregulation and E-cadherin downregulation were observed. Consistent with the in vitro results, the loss of ARID1A was significantly associated with lymph node metastasis and distant metastasis in human NPC tissues. These results indicate that the loss of ARID1A promotes invasion and metastasis and induces an EMT-like molecular phenotype in NPC cells. Hence, ARID1A loss is a hallmark of NPC progression.
Accumulating evidence suggests that frameshift or nonsense ARID1A mutations are involved in carcinogenesis via various mechanisms, including the phosphatidylinositol-3-kinase (PI3K)/Akt signaling pathway reported in several malignancies, such as ovarian clear cell adenocarcinoma,32,33 pancreatic cancer,34 and gastric cancer.35 Activated Akt signaling results in enhanced cell survival, cell cycle progression, tumor cell invasion and EMT.36 As shown in Figure 4, ARID1A loss also increased Akt phosphorylation in NPC cells and NPC biopsies. These results indicate that the loss of ARID1A expression leads to Akt pathway activation in NPC cells.
Akt inhibitors radiosensitize gastric cancer cells37 and ARID1A-deficient pancreatic cancer cells.34 The loss of ARID1A sensitizes endometrial cancer cell lines and breast cancer cells to Akt inhibitors, which leads to increased apoptosis.38 To demonstrate the roles of activated Akt signaling in ARID1A silencing-induced changes in biological characteristics in NPC cells, MK-2206 was used to treat ARID1A-silenced NPC cells. MK-2206 partially rescued the effects on proliferation, migration and invasion in the ARID1A-silenced NPC cells. MK-2206 also increased the expression of Vimentin and Cyclin D1 and decreased the expression of E-cadherin in ARID1A-silenced NPC cells. These data imply that Akt signaling activation plays a crucial role in the ARID1A silencing-induced proliferation, migration, invasion and EMT-like molecular phenotype of NPC cells. However, the detailed mechanisms of ARID1A loss and Akt signaling activation still need to be investigated.
Conclusion
In conclusion, ARID1A functions as a tumor suppressor that inhibits EMT, prevents tumor progression and negatively modulates the Akt pathway in NPC cells. ARID1A disruption in NPC may be a potential diagnostic and prognostic predictor as well as a therapeutic target for NPC.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant Nos. 81560441 and 81760491 to S.J. Xiao) and the Natural Science Foundation of Guangxi Province of China (Grant No. 2015GXNSFAA139131 to S.J. Xiao).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Wei YC, Yang SF, Chang SL, et al. Periostin overexpression is associated with worse prognosis in nasopharyngeal carcinoma from endemic area: a cohort study. Onco Targets Ther. 2018;11(3205–3213). doi:10.2147/OTT.S163626
2. Sun X, Tong LP, Wang YT, et al. Can global variation of nasopharynx cancer be retrieved from the combined analyses of IARC cancer information (CIN) databases? PLoS One. 2011;6(7):e22039. doi:10.1371/journal.pone.0022039
3. Cao SM, Simons MJ, Qian CN. The prevalence and prevention of nasopharyngeal carcinoma in China. Chin J Cancer. 2011;30(2):114. doi:10.5732/cjc.010.10377
4. Tham IW, Lu JJ. Controversies and challenges in the current management of nasopharyngeal cancer. Expert Rev Anticancer Ther. 2010;10(9):1439. doi:10.1586/era.10.97
5. Zong J, Huang Q, Guo Q, et al. Evolution of the Chinese staging system for nasopharyngeal carcinoma. Chin Clin Oncol. 2016;5(2):19. doi:10.21037/cco.2016.03.04
6. Yee D, Hanson J, Lau H, et al. Treatment of nasopharyngeal carcinoma in the modern era: analysis of outcomes and toxicity from a single center in a nonendemic area. Cancer J. 2006;12(2):147.
7. Fang W, Xin L, Jiang Q, et al. Transcriptional patterns, biomarkers and pathways characterizing nasopharyngeal carcinoma of Southern China. J Transl Med. 2008;6(1):32. doi:10.1186/1479-5876-6-32
8. Lai SZ, Li WF, Chen L, et al. How does intensity-modulated radiotherapy versus conventional two-dimensional radiotherapy influence the treatment results in nasopharyngeal carcinoma patients? Int J Radiat Oncol Biol Phys. 2011;80(3):661–668. doi:10.1016/j.ijrobp.2010.03.024
9. Reisman D, Glaros S, Thompson EA. The SWI/SNF complex and cancer. Oncogene. 2009;28(14):1653–1668. doi:10.1038/onc.2009.4
10. Takeda T, Banno K, Okawa R, et al. ARID1A gene mutation in ovarian and endometrial cancers (Review). Oncol Rep. 2016;35(2):607–613. doi:10.3892/or.2015.4421
11. Wu JN, Roberts CWM. ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Discov. 2013;3(1):35–43. doi:10.1158/2159-8290.CD-12-0361
12. Levine DA. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497(7447):67. doi:10.1038/nature12113
13. Nishikimi K, Kiyokawa T, Tate S, et al. ARID1A expression in ovarian clear cell carcinoma with an adenofibromatous component. Histopathology. 2016;67(6):866–871. doi:10.1111/his.12721
14. Stephens PJ, Tarpey PS, Davies H, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486(7403):400–404. doi:10.1038/nature11017
15. Fujimoto A, Totoki Y, Abe T, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012;44(7):760–764. doi:10.1038/ng.2291
16. Wang K, Kan J, Yuen ST, et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet. 2011;43(12):1219–1223. doi:10.1038/ng.982
17. Network TCGA. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507(7492):315–322. doi:10.1038/nature12965
18. Shain AH, Giacomini CP, Matsukuma K, et al. Convergent structural alterations define SWItch/Sucrose NonFermentable (SWI/SNF) chromatin remodeler as a central tumor suppressive complex in pancreatic cancer. Proc Natl Acad Sci USA. 2012;109(5):1370–1371. doi:10.1073/pnas.1114817109
19. Cho HD, Lee JE, Jung HY, et al. Loss of tumor suppressor ARID1A protein expression correlates with poor prognosis in patients with primary breast cancer. J Breast Cancer. 2015;18(4):339–346. doi:10.4048/jbc.2015.18.4.339
20. Zang ZJ, Cutcutache I, Song LP, et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat Genet. 2012;44(5):570–574. doi:10.1038/ng.2246
21. Jones S, Li M, Parsons DW, et al. Somatic mutations in the chromatin remodeling gene ARID1A occur in several tumor types. Hum Mutat. 2015;33(1):100–103. doi:10.1002/humu.21633
22. Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491(7424):399. doi:10.1038/nature11547
23. Imielinski M, Berger A, Hammerman P, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150(6):1107–1120. doi:10.1016/j.cell.2012.08.029
24. Zhang Y, Xu X, Zhang M, et al. ARID1A is downregulated in non-small cell lung cancer and regulates cell proliferation and apoptosis. Tumour Biol. 2014;35(6):5701–5707. doi:10.1007/s13277-014-1755-x
25. Guan B, Wang TL, Iem S. ARID1A, a factor that promotes formation of SWI/SNF-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer Res. 2011;71(21):6718–6727. doi:10.1158/0008-5472.CAN-11-1562
26. Mamo A, Cavallone L, Tuzmen S, et al. An integrated genomic approach identifies ARID1A as a candidate tumor-suppressor gene in breast cancer. Oncogene. 2012;31(16):2090–2100. doi:10.1038/onc.2011.386
27. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013
28. Li L, Li W. Epithelial-mesenchymal transition in human cancer: comprehensive reprogramming of metabolism, epigenetics, and differentiation. Pharmacol Ther. 2015;150(33–46). doi:10.1016/j.pharmthera.2015.01.004
29. He F, Li J, Xu JF, et al. Decreased expression of ARID1A associates with poor prognosis and promotes metastases of hepatocellular carcinoma. J Exp Clin Cancer Res. 2015;34(1):47. doi:10.1186/s13046-015-0164-3
30. Li C, Xu ZL, Zhao Z, et al. ARID1A gene knockdown promotes neuroblastoma migration and invasion. Neoplasma. 2017;64(3):367. doi:10.4149/neo_2017_307
31. Yan HB, Wang XF, Zhang Q, et al. Reduced expression of the chromatin remodeling gene ARID1A enhances gastric cancer cell migration and invasion via downregulation of E-cadherin transcription. Carcinogenesis. 2014;35(4):867–876. doi:10.1093/carcin/bgt398
32. Yamamoto S, Tsuda H, Takano M, et al. Loss of ARID1A protein expression occurs as an early event in ovarian clear-cell carcinoma development and frequently coexists with PIK3CA mutations. Mod Pathol. 2012;25(4):615. doi:10.1038/modpathol.2011.189
33. Yamamoto S, Tsuda H, Takano M, et al. PIK3CA mutation is an early event in the development of endometriosis‐associated ovarian clear cell adenocarcinoma. J Pathol. 2011;225(2):189–194. doi:10.1002/path.2940
34. Yang L, Yang G, Ding Y, et al. Inhibition of PI3K/AKT signaling pathway radiosensitizes pancreatic cancer cells with ARID1A deficiency in vitro. J Cancer. 2018;9(5):890–900. doi:10.7150/jca.21306
35. Kim YB, Ham IH, Hur H, et al. Various ARID1A expression patterns and their clinical significance in gastric cancers. Hum Pathol. 2016;49(61–70). doi:10.1016/j.humpath.2015.10.008
36. Testa JR, Bellacosa A. AKT plays a central role in tumorigenesis. Proc Natl Acad Sci U S A. 2001;98(20):10983–10985. doi:10.1073/pnas.211430998
37. Lee D, Yu EJ, Ham IH, et al. AKT inhibition is an effective treatment strategy in ARID1A-deficient gastric cancer cells. Onco Targets Ther. 2017;10(4153–4159). doi:10.2147/OTT.S139664
38. Samartzis EP, Gutsche K, Dedes KJ, et al. Loss of ARID1A expression sensitizes cancer cells to PI3K- and AKT-inhibition. Oncotarget. 2014;5(14):5295–5303. doi:10.18632/oncotarget.2092
Supplementary material
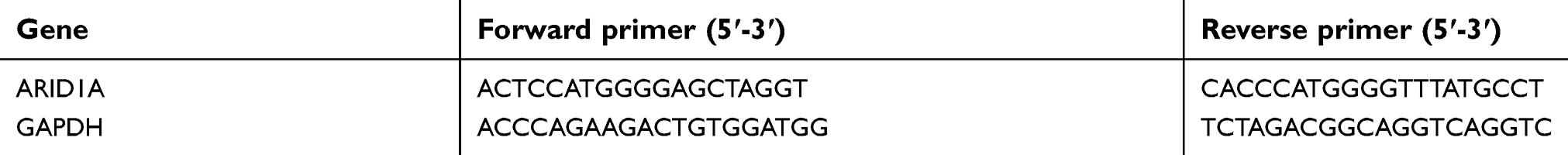 | Table S1 Primers used in quantitative real-time PCR (qRT-PCR) assay |
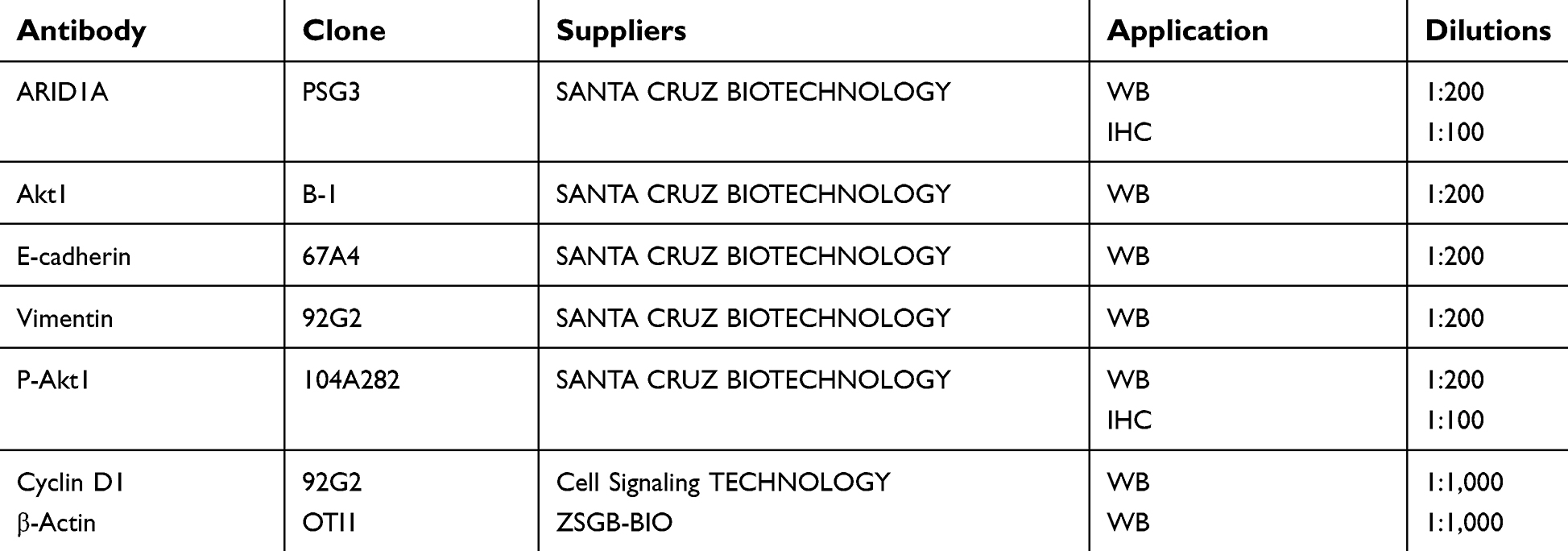 | Table S2 List of antibodies and suppliers used for immunohistochemistry or immunoblotting |
 | Table S3 Expression of ARID1A in 61 non-cancerous epithelial tissues and 177 NPC tissues |
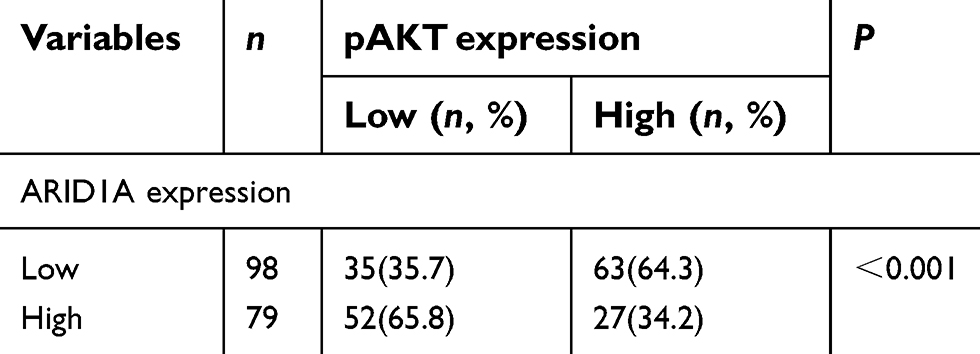 | Table S4 Association of ARID1A expression and pAKT in 177 NPC tissues |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.