")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 14
Long-term safety and efficacy of formoterol fumarate inhalation solution in patients with moderate-to-severe COPD
Authors Hanania NA, Sethi S , Koltun A , Ward JK, Spanton J, Ng D
Received 16 August 2018
Accepted for publication 28 November 2018
Published 27 December 2018 Volume 2019:14 Pages 117—127
DOI https://doi.org/10.2147/COPD.S173595
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Russell
Nicola A Hanania,1 Sanjay Sethi,2 Arkady Koltun,3 Jonathan K Ward,4 Jacqui Spanton,4 Dik Ng4
1Asthma Clinical Research Center, Section of Pulmonary and Critical Care Medicine, Baylor College of Medicine, Houston, TX, USA; 2Pulmonary, Critical Care, and Sleep Medicine, University at Buffalo Jacobs School of Medicine and Biomedical Sciences, Buffalo, NY, USA; 3Global Medical Affairs, Mylan Inc., Canonsburg, PA, USA; 4Mylan Global Respiratory Group, Mylan Pharma UK Ltd., Sandwich, Kent, UK
Background: Formoterol fumarate inhalation solution (FFIS; Perforomist®) is a long-acting β2-agonist (LABA) marketed in the US as a nebulized COPD maintenance treatment. Because long-term LABA use was associated with a potential increased risk of exacerbation or death in asthma patients, the US Food and Drug Administration (FDA) requested a postmarketing commitment study to evaluate long-term safety in COPD patients.
Methods: This was a multicenter, randomized, double-blind, placebo-controlled, noninferiority study. Patients (N=1,071; mean age, 62.6 years; 48.5% male; 89.7% white) with moderate-to-severe COPD on stable COPD therapy received FFIS (20 µg; n=541) or placebo (n=530) twice daily. The primary end point was the combined incidence of respiratory death, first COPD-related ER visit, or first COPD exacerbation-related hospitalization during 1 year post randomization. Noninferiority to placebo was concluded if the two-sided 90% CI of the HR of FFIS to placebo was <1.5. Secondary end points included spirometry.
Results: The planned 1-year treatment period was completed by 520 patients; 551 discontinued prematurely (FFIS: 45.7%; placebo: 57.4%). The median treatment duration was approximately 10 and 7 months for FFIS and placebo, respectively. Among 1,071 randomized patients, 121 had ≥1 primary event (FFIS: 11.8%; placebo: 10.8%). The estimated HR of a primary event with FFIS vs placebo was 0.965 (90% CI: 0.711, 1.308), demonstrating that FFIS was noninferior to placebo. No respiratory deaths were observed in the FFIS group. Adverse events were similar for FFIS vs placebo (patients with ≥1 treatment-emergent adverse events: 374 [69.1%] vs 369 [69.6%], respectively). Compared with placebo, FFIS demonstrated statistically greater improvements from baseline in trough FEV1, FVC, percent predicted FEV1, and patient-reported outcomes (Transition Dyspnea Index).
Conclusions: Nebulized FFIS was noninferior to placebo with respect to safety in patients with moderate-to-severe COPD. Additionally, fewer treatment withdrawals and larger lung function improvements were observed with FFIS compared with placebo when added to other maintenance COPD therapies.
Keywords: COPD, safety, bronchodilators, long-acting beta2-agonists, nebulization
Introduction
COPD is a highly prevalent condition1 with a significant morbidity and mortality2 and commonly coexists with cardiovascular disease.3 Long-acting inhaled bronchodilators, including long-acting β2-agonists (LABAs) and anticholinergic drugs (LAMAs), are a cornerstone of pharmacotherapy, especially for those with moderate-to-severe symptoms or those at a higher risk for COPD exacerbations.4,5
Cardiovascular events are leading causes of hospitalization and mortality in COPD, particularly in patients with mild-to-moderate disease.6,7 Sin and Man6 reported that for every 10% decrease in FEV1, all-cause mortality increased by 14%, cardiovascular mortality increased by 28%, and the risk for a nonfatal coronary event increased by almost 20%. Current evidence from clinical studies supports the use of LABA in COPD, with a review of 20 studies (8,774 LABA-treated patients) finding no evidence of an association between LABA treatment and increased exacerbations, COPD-related adverse events (AEs), or deaths.8 Some other studies, however, have suggested that long-term use of LABAs may increase the risk of cardiovascular complications in patients with obstructive airway disease, defined as asthma or COPD.9–11 The potential association of long-term LABA use with an increased risk of cardiovascular complications and mortality in asthma has prompted the US Food and Drug Administration (FDA) to request postmarketing commitment studies evaluating long-term safety and tolerability of LABAs in COPD patients.12
Formoterol fumarate inhalation solution (FFIS; Perforomist®) is a LABA marketed in the US as a nebulized maintenance treatment for bronchoconstriction in COPD.13–15 This Phase IV clinical trial evaluated the long-term safety of this drug in patients with moderate-to-severe COPD (ClinicalTrials.gov, NCT01488019). A preliminary abstract of this study was previously published: Hanania N, Sethi S, Koltun A, Ward J, Spanton J, Ng D. Evaluation of the Safety of Long-Term Use of Perforomist® (Formoterol Fumarate) Inhalation Solution in Patients with Moderate to Severe Chronic Obstructive Pulmonary Disease (COPD). Am J Respir Crit Care Med. 2017;195:A5473 [Abstract].
Methods
Study design and conduct
The study was a multicenter, randomized, double-blind, placebo-controlled, parallel-group, noninferiority study to evaluate the long-term safety of FFIS in patients with COPD. The study was conducted between March 27, 2012, and January 27, 2016, at 106 US sites and was carried out in accordance with Good Clinical Practice guidelines contained within the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH-E6),16 the principles of the Declaration of Helsinki, and the US Code of Federal Regulations. Three ethical review boards were used for the trial. All investigator sites used a Central Institutional Review Board (New England Institutional Review Board [NEIRB]) except for three sites, which used their local institutional review board (IRB; additional information can be found in the “Supplementary materials” section, Table S1 and Table S2). The local IRBs, ethics committees, and health authorities at each of the 106 study centers approved the protocol and all amendments. All patients provided written informed consent.
Patients and treatments
Patients were men or women of nonchildbearing potential (age ≥40 years) with a medical diagnosis of COPD17 who had experienced ≥1 COPD exacerbations within the past year and had a current or former smoking history of >10 pack-years. At screening, patients were required to demonstrate a post-bronchodilator (albuterol) FEV1/FVC ratio of <0.7 and an FEV1 of 30%–70% of the predicted normal value after withholding LABA-containing medications for ≥48 hours. Concomitant therapy was managed during the study as follows: LABA, LABA-containing combination products, combination products containing short-acting β2-agonists and anticholinergics, nebulized and oral β2-agonists, orally inhaled nedocromil or cromolyn sodium, and theophylline background medications were all prohibited; inhaled short- or long-acting anticholinergics, inhaled corticosteroids (ICSs; monotherapy only), leukotriene modifiers, and oral phosphodiesterase-4 inhibitors were allowed. Albuterol administered by a metered dose inhaler was allowed as necessary but was requested to be withheld for 6 hours before spirometry assessment was performed at each study visit. The following were also permitted, providing the appropriate washout periods were observed prior to spirometry: inhaled short-acting anticholinergics (8 hours), inhaled long-acting anticholinergics (48 hours), leukotriene modifiers (24 hours), and phosphodiesterase-4 inhibitors (24 hours).
Assessments
Following the screening period (≤28 days prior to randomization), eligible patients were randomized 1:1 to receive FFIS (20 μg) or matching placebo administered twice daily (morning and evening) for 52 weeks using a Trek® S (PARI, Midlothian, VA, USA) portable nebulizer. All study personnel, as well as the sponsor, site monitors, and patients, were blinded to treatment allocation throughout the study. Predose FEV1, FVC, and inspiratory capacity (IC) were measured via spirometry according to techniques consistent with the American Thoracic Society guidelines for spirometry.18 Assessments were made at baseline, prior to the first dose, and at specified timepoints during the treatment period (3, 6, 9, and 12 months). The Saint George’s Respiratory Questionnaire (SGRQ), a validated instrument that evaluates health-related quality of life,19 and the Baseline Dyspnea Index (BDI)/Transition Dyspnea Index (TDI) questionnaire, an instrument to assess the effect of treatment on dyspnea,20 were administered at baseline, prior to the first dose, and at specified timepoints during the treatment period (3, 6, 9, and 12 months). A diary was used for recording dosing times, rescue medication use, concomitant medications, and AEs throughout the 1-year treatment period. Subjects were questioned at each scheduled visit (at baseline, 3, 6, 9, and 12 months) if they had any AEs since the previous visit. They were also questioned if they had any hospitalizations, emergency room visits, or other unscheduled health care provider visits since the previous visit, and if so, relevant details of the events and whether they were related to COPD.
Study end points
The primary end point was the combined incidence of respiratory death, first COPD-related ER visit, or first COPD exacerbation-related hospitalization (whichever occurred first from the time of randomization to the end of the study). Safety and tolerability were assessed based on reported AEs and SAEs, clinical laboratory findings, vital signs, physical examinations, and/or electrocardiogram readings obtained before, during, and after the treatment period. The relationship of a treatment-emergent adverse event (TEAE) to the study medication was determined by the investigator and characterized as probably related, possibly related, or not related.
An independent mortality adjudication board (MAB), which assigned causality of death and relationship to COPD, determined respiratory death. The MAB consisted of three physicians who each had clinical and research expertise in internal medicine and pulmonary and critical care medicine.
Sample size and statistical analyses
The statistical design was based on demonstrating noninferiority, defined as a ≤50% higher risk (HR 1.5) of a primary event (ie, combined incidence of respiratory death, first COPD-related ER visit, or first COPD exacerbation-related hospitalization) with FFIS vs placebo. Noninferiority was to be declared if the upper limit of the two-sided 90% CI of the HR of FFIS to placebo was wholly less than 1.5. The study was adequately powered for an observed event rate of 15% in the placebo group and assumed that the event rate for FFIS was lower than placebo under the one-sided alternative hypothesis (HR ≤0.8). The HR (90% CI) for the primary analysis was estimated from a Cox proportional hazards model with treatment group, center(s), and baseline bronchodilator reversibility as covariates. Sparse centers were pooled on a geographical basis for analysis purposes. Patients without an observed primary event were censored at the time of withdrawal of study treatment or on completion of study. A sensitivity analysis was conducted to assess the effects of events recorded during the follow-up period after early discontinuation from study treatment and included all events through 1 year after randomization.
For the secondary efficacy end points (spirometry end points, total SGRQ, and focal TDI scores), changes from baseline were analyzed using a repeated measures model with terms for the baseline score, treatment, geographical region, visit, treatment by visit, and baseline score by visit as the independent variables. Subjects were included as a random effect. The statistical model assumed that missing data were missing at random. Rescue medication usage was descriptively summarized.
Results
All safety and efficacy data were summarized and analyzed in all randomized patients who took at least one dose of study medication. For the safety analyses, subjects were analyzed according to the actual treatment they received for the majority of the study (>50% of the double-blind treatment period). For the efficacy analyses, patients were analyzed according to their assigned treatment at randomization.
Subject disposition
Of the 1,071 patients randomized (FFIS, n=541; placebo, n=530), 520 patients completed the 1-year treatment period as planned and 551 patients discontinued prematurely (FFIS, n=247 [45.7%]; placebo, n=304 [57.4%]; Figure 1). All subjects received the treatment they were randomized to receive for the majority of the study. One subject did not receive the assigned treatment for 24.1% of their treatment period. The primary reasons for discontinuation were similar between the treatment groups and included withdrawal of consent (19.8%), AEs (15.7%), and patients lost to follow-up (8.4%). The time to withdrawal in the two treatment arms is illustrated by the Kaplan–Meier plot in Figure 2.
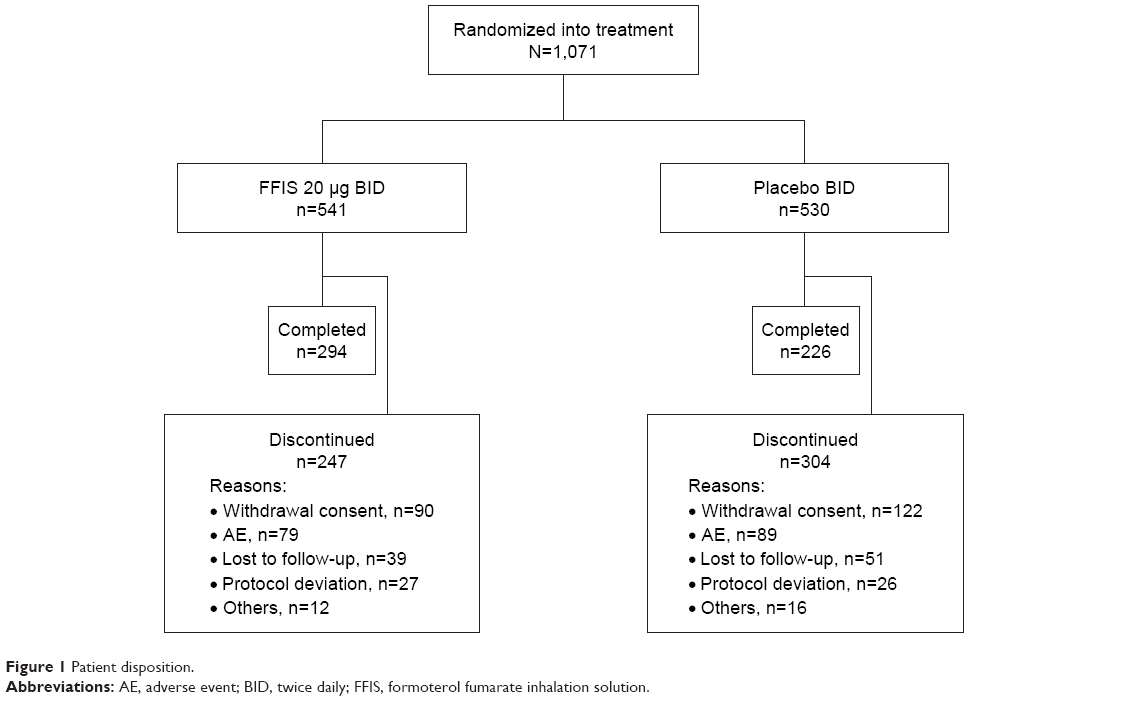 | Figure 1 Patient disposition. |
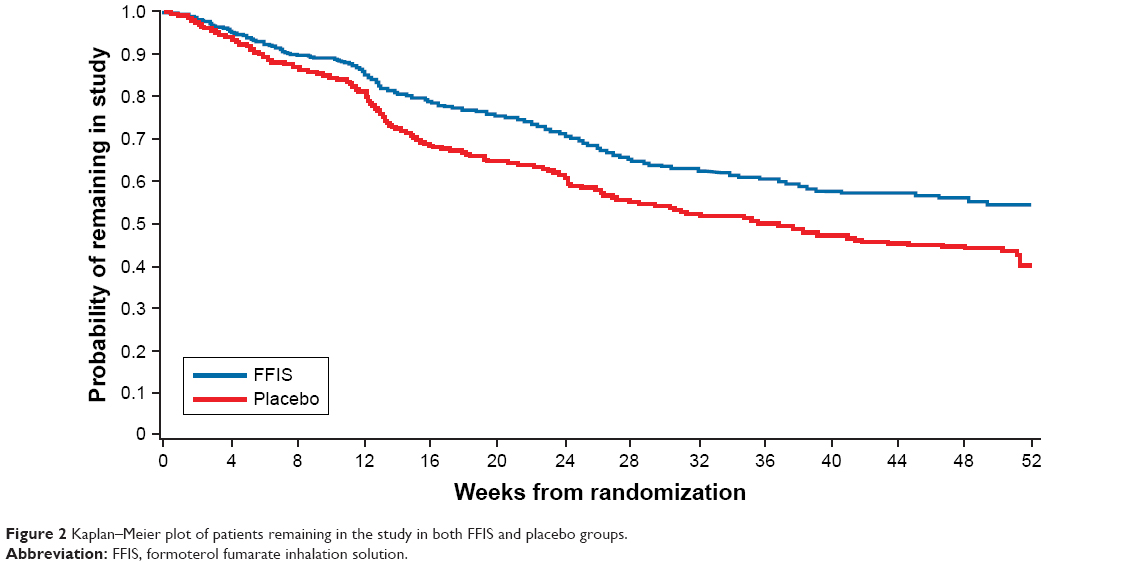 | Figure 2 Kaplan–Meier plot of patients remaining in the study in both FFIS and placebo groups. |
Baseline demographic and clinical characteristics were generally well-matched across treatment groups (Table 1). The median pack-year history of smoking was 43 pack-years, and more than half (52.3%) of all enrolled patients were current smokers. The majority of patients had moderate (51.1%) or severe (47.7%) COPD at screening using GOLD spirometry criteria.17 Prior ICS usage, either as monotherapy or as a fixed-dose combination, was comparable between the FFIS (53.0%) and placebo (51.1%) patients, and most patients (97.5%) took at least one concomitant medication (other than rescue medication) during the study. Baseline spirometry results (FEV1, FVC, FEV1 percent predicted, and IC), SGRQ, and BDI scores were comparable between the treatment groups (Table 1). The use of concomitant medications was similar between the treatment groups. The most frequently used concomitant medication drug classes were glucocorticoids (58.9%), anticholinergics (41.8%), and 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors (39.1%).
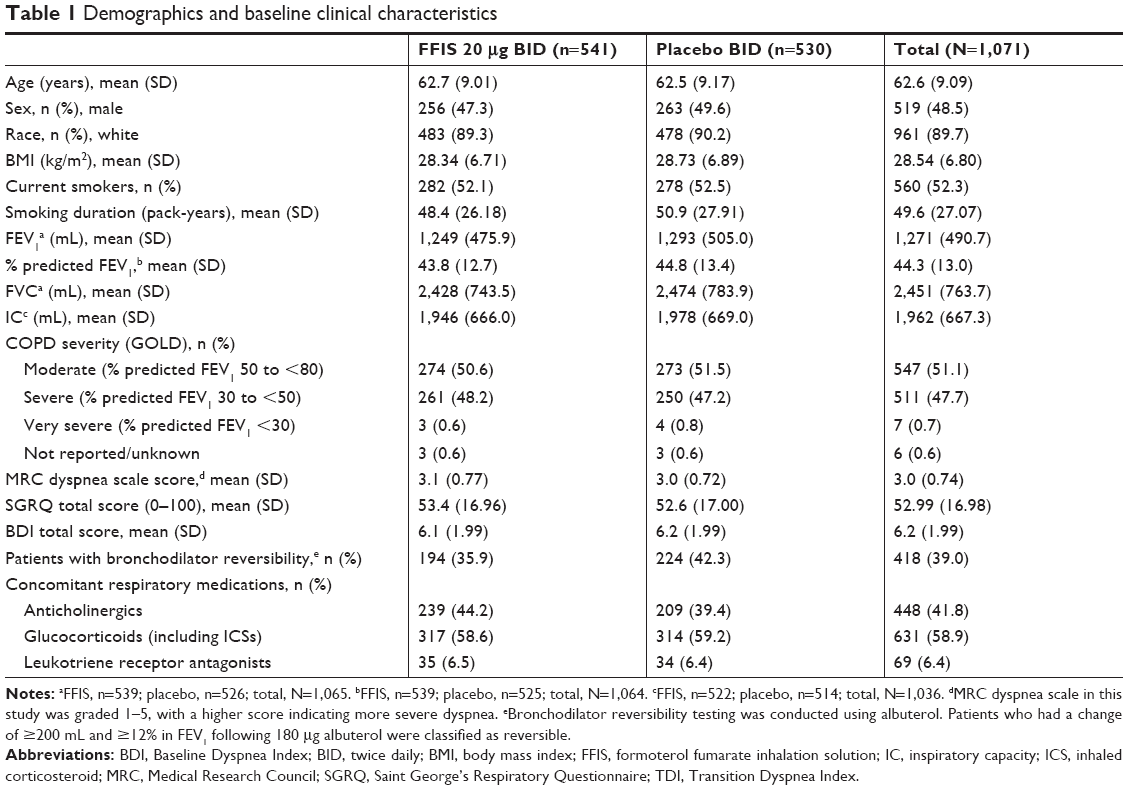 | Table 1 Demographics and baseline clinical characteristics |
Treatment compliance, defined as taking 80%–120% of the prescribed doses of study medication, was 84.5% for FFIS-treated patients and 82.5% for placebo-treated patients. The number of patients exposed to study medication was 5%–10% higher in the FFIS group than that in the placebo group throughout the 1-year treatment period. The extent of exposure was greater for FFIS compared with placebo; the median duration of exposure was 316 days (~10 months) for FFIS and 216 days (~7 months) for placebo.
Safety outcomes
Primary end point
The primary analysis assessed the combined incidence of respiratory death, first COPD-related ER visit, or first COPD exacerbation-related hospitalization. Among 1,071 randomized patients, 121 had ≥1 primary end point events (FFIS, 11.8% [64 patients]; placebo, 10.8% [57 patients]; Table 2 and Figure 3). The Kaplan–Meier estimate of the cumulative probability of a primary end point event at week 52 was 15.5% in the FFIS group and 14.9% in the placebo group, with an estimated HR of FFIS to placebo of 0.965 (90% CI: 0.711, 1.308), demonstrating that FFIS was noninferior to placebo. A sensitivity analysis was conducted that also included primary events reported through 1 year after randomization for those patients who discontinued study drug early (irrespective of any alternative treatment received after withdrawal of study medication). The results were consistent with the primary analysis with an estimated HR of FFIS to placebo of 1.079 (90% CI: 0.801, 1.453).
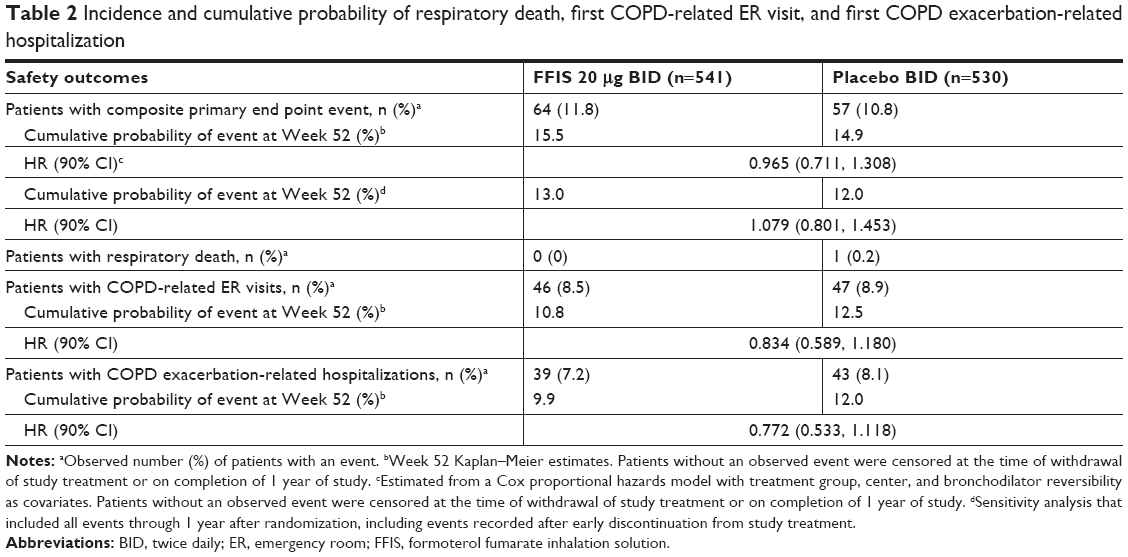 | Table 2 Incidence and cumulative probability of respiratory death, first COPD-related ER visit, and first COPD exacerbation-related hospitalization |
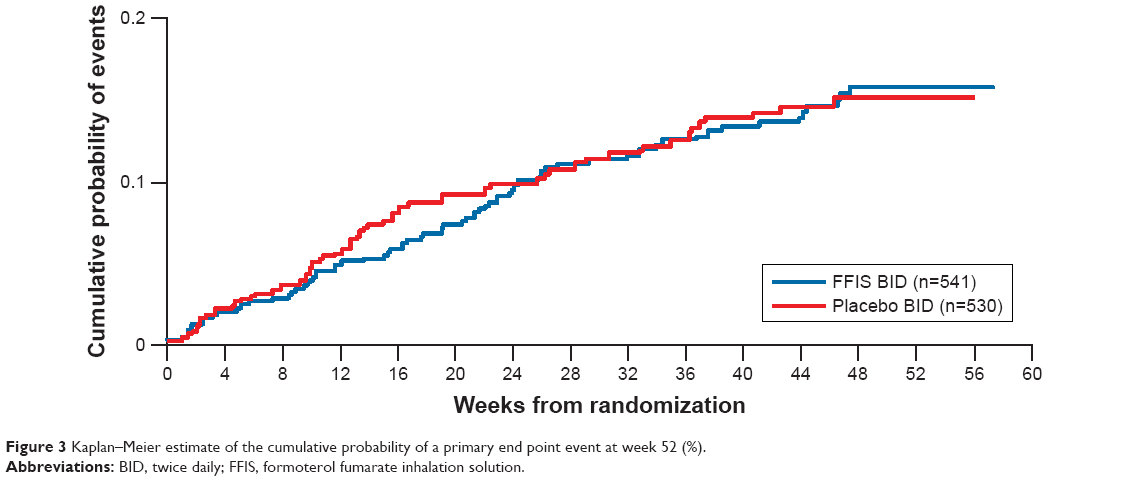 | Figure 3 Kaplan–Meier estimate of the cumulative probability of a primary end point event at week 52 (%). |
Secondary end points
The individual components of the primary end point were assessed. Patients could have been included in more than one event category for the individual components of the primary end point since the primary end point only selected the first incidence of any one of the three events.
No respiratory-related deaths occurred in the FFIS-treated group, and one respiratory-related death (COPD) occurred in the placebo group. The estimated cumulative probability of a COPD-related ER visit at Week 52 was 10.8% in the FFIS group compared with 12.5% in the placebo group, with an HR of FFIS to placebo of 0.834 (90% CI: 0.589, 1.180; Table 2). The estimated cumulative probability of a COPD exacerbation-related hospitalization at Week 52 was 9.9% for FFIS compared with 12.0% for placebo, with an HR of FFIS to placebo of 0.772 (90% CI: 0.533, 1.118).
A total of 148 (27.4%) subjects in the FFIS group and 138 (26.0%) subjects in the placebo group had at least one protocol-defined COPD exacerbation recorded. The cumulative probabilities of an event at Week 52 were similar (34.7% for the FFIS group vs 34.0% for the placebo group). While the percentage of patients with COPD exacerbations was comparable between the FFIS and placebo groups, the time to the first exacerbation was longer for FFIS compared with that for placebo, with the time at which at least 30% of patients had an event estimated as 43.3 and 36.9 weeks, respectively.
AEs
The reported TEAE profile was consistent with the established safety profile of FFIS,13 and no new safety concerns were reported. The number of patients who experienced any TEAEs was comparable between the FFIS-treated patients (n=374 [69.1%]; Table 3) and those taking placebo (n=369 [69.6%]). The most common TEAEs by system organ class were respiratory, thoracic, and mediastinal disorders (FFIS: 43.3%; placebo: 40.4%) and infections and infestations (FFIS: 29.8%; placebo: 29.6%). The incidence of study medication-related TEAEs was comparable between the FFIS (18.3%) and placebo (20.9%) groups. The most common medication-related TEAEs by preferred term were COPD (worsening and exacerbations; FFIS: 30.3%; placebo: 27.4%), dyspnea (FFIS: 6.5%; placebo: 7.5%), and cough (FFIS: 7.2%; placebo: 4.5%).
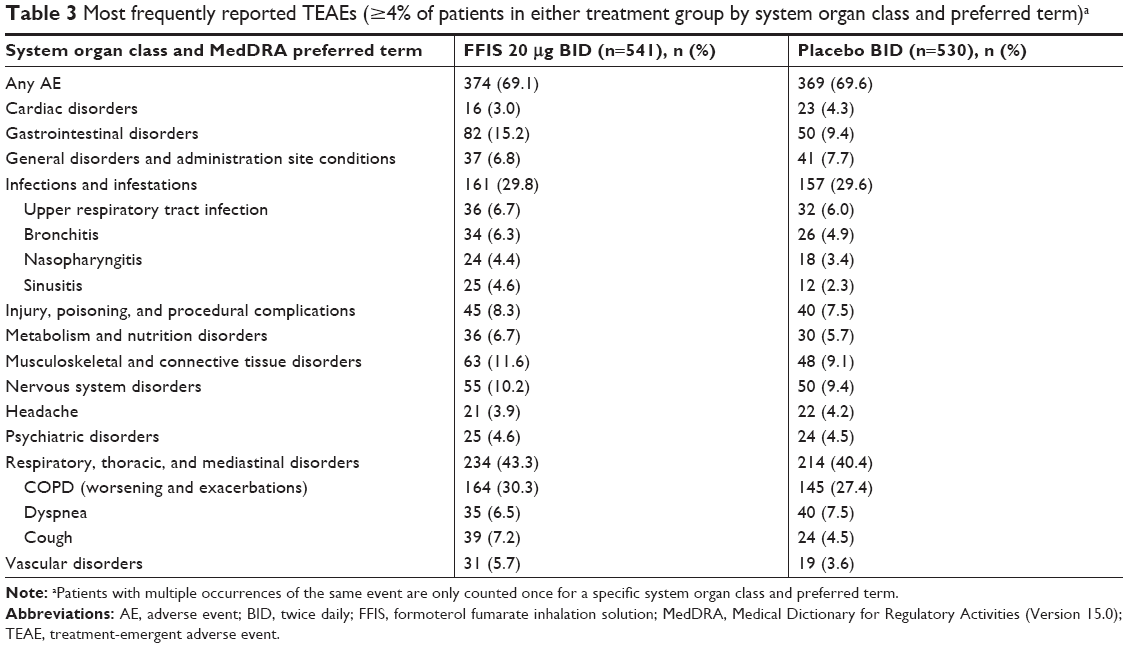 | Table 3 Most frequently reported TEAEs (≥4% of patients in either treatment group by system organ class and preferred term)a |
The percentage of patients who experienced treatment-emergent serious AEs was comparable between the FFIS (15.9%) and placebo (16.0%) groups (Table 4). The most common treatment-emergent serious AE by preferred term was COPD (worsening and exacerbations; FFIS: 7.2%; placebo: 7.5%). A total of 13 patients died during the study, of which three (0.6%) were in the FFIS group and 10 (1.9%) were in the placebo group. No fatalities were considered to be related to study treatment.
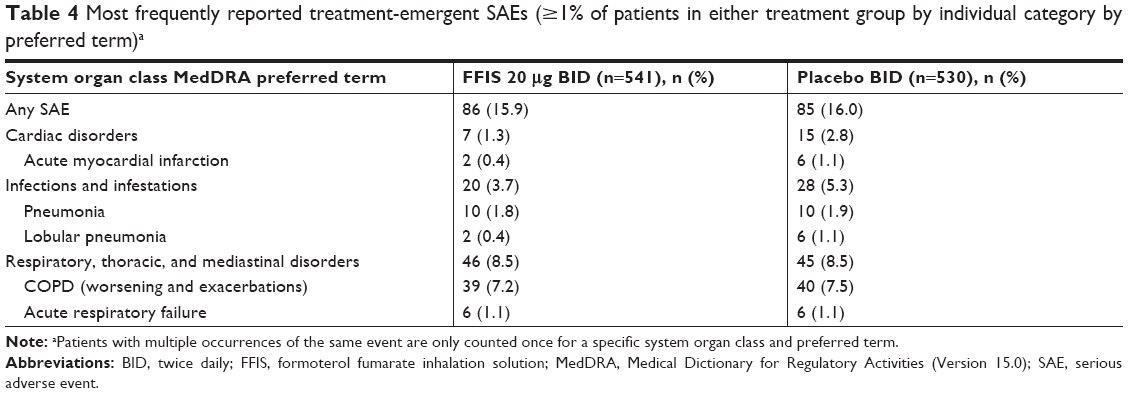 | Table 4 Most frequently reported treatment-emergent SAEs (≥1% of patients in either treatment group by individual category by preferred term)a |
Efficacy outcomes
Improvements from baseline in spirometry end points were all numerically greater in the FFIS group compared with the placebo group at all visits during the study (Figure 4). Estimated differences (FFIS–placebo) in the improvements were statistically significant for FEV1 (3- and 6-month visits; P<0.05), FVC (all visits; P<0.005), and % predicted FEV1 (3-, 6-, and 9-month visits; P<0.05). None of the differences were statistically significant for IC.
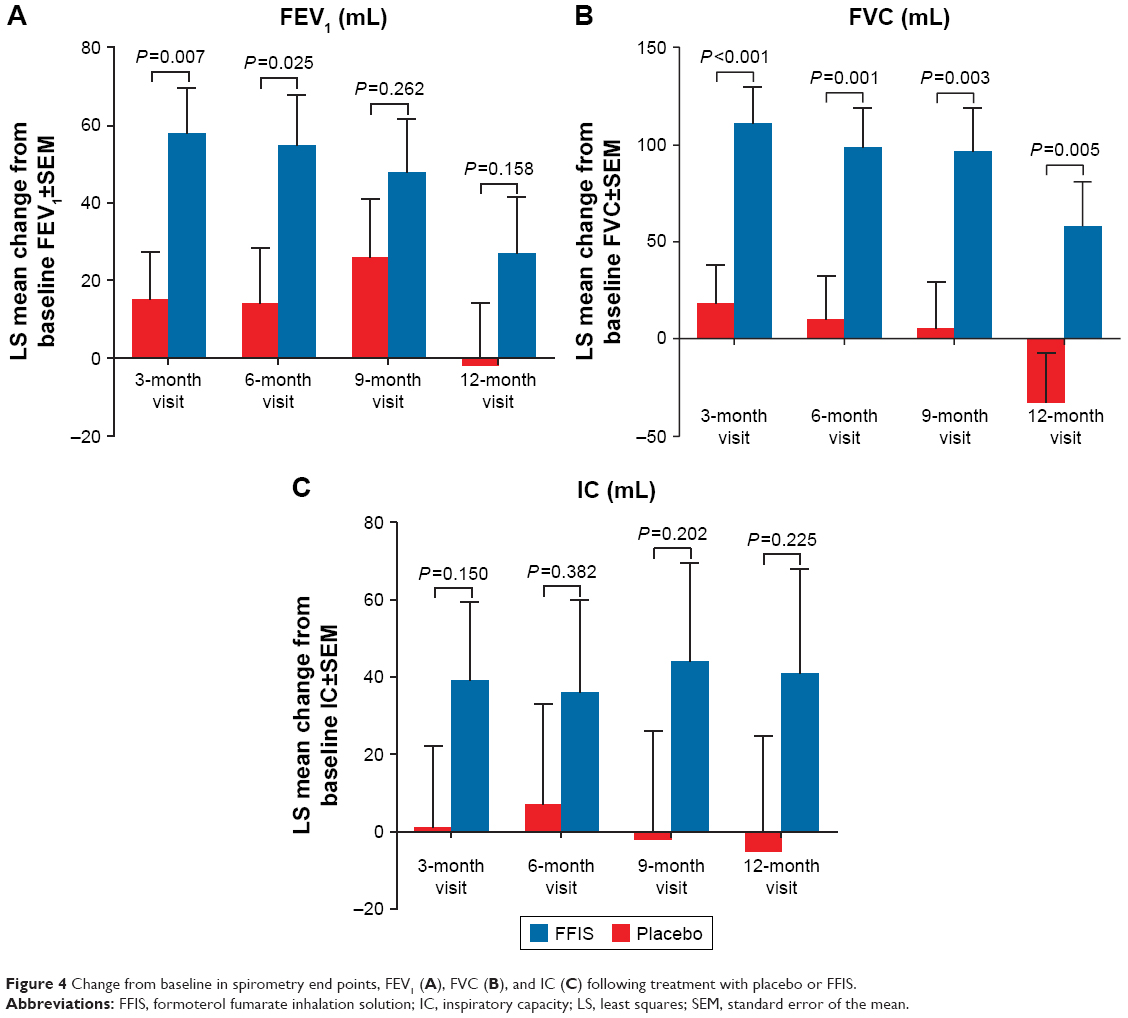 | Figure 4 Change from baseline in spirometry end points, FEV1 (A), FVC (B), and IC (C) following treatment with placebo or FFIS. |
The average daily rescue medication usage during the treatment period was numerically lower in the FFIS group (1.92 puffs/day) compared with that in the placebo group (2.35 puffs/day), although inferential analysis was not performed. The estimated mean (standard error [SE]) decrease from baseline (improvement) in SGRQ total score at month 3 was −3.25 (0.55) for the FFIS group compared with −2.03 (0.57) for the placebo group, although the treatment difference (−1.226 [95% CI: −2.674, 0.223]) was not statistically significant (P=0.097). The estimated mean (SE) increase from baseline (improvement) in focal TDI scores at month 3 was 0.7 (0.11) for the FFIS group compared with 0.0 (0.12) for the placebo group, and this difference (0.7 [95% CI: 0.4, 1.0]) was statistically significant (P<0.001).
Discussion
Our study demonstrates that treatment for 1 year with FFIS delivered by twice-daily nebulization in patients with moderate-to-severe COPD is not associated with an increase in the combined incidence of respiratory death, COPD-related ER visits, or COPD exacerbation-related hospitalizations compared with that with placebo. In general, safety outcomes were similar between FFIS and placebo groups, and drug-related life-threatening respiratory or cardiac events were not observed. Secondary end points of our study demonstrate a numerically lower probability of COPD-related ER visits or COPD-related hospitalizations with FFIS vs placebo. Although the study was not designed to evaluate efficacy, greater improvement in lung function, quality of life, dyspnea, and lower rescue medication use were observed with FFIS compared to placebo, despite patients being permitted to remain on other background treatments for COPD. The results were therefore consistent with previous studies demonstrating that FFIS is an effective and well-tolerated COPD maintenance therapy.13,14,21
Other clinical trials have also demonstrated the safety of long-term use of LABAs in patients with COPD. In a 3-year trial of salmeterol/fluticasone propionate (LABA/ICS combination), there was no excess of cardiac disorders, supporting the long-term safety of LABA and ICS agents.22 Additionally, a 1-year safety and efficacy study of another nebulized LABA (arformoterol)23 demonstrated no increased risk of respiratory death or COPD exacerbation-related hospitalizations vs placebo during 1 year of treatment. More recently, long-term safety of the LABA vilanterol, alone or in combination with the ICS fluticasone furoate, was confirmed in a large randomized, double-blind, placebo-controlled trial in patients with moderate COPD who had or were at high risk of cardiovascular disease.23
Strengths and limitations
Our study has several strengths and limitations. Similar to the Phase IV safety study of arformoterol,24 study strengths included a large patient cohort, a 1-year noninferiority study design that was powered to investigate severe respiratory-related safety outcomes as a primary end point, the use of an independent MAB, and evaluation of efficacy on top of other maintenance COPD therapies. Additionally, for patients who dropped out, efforts were made to follow them up for primary end point at 1 year. Study limitations included lower than expected event rates and the high percentage of patient withdrawals, which was at the higher end of that observed in some of the other long-term COPD studies (27%–50%).22–26 However, a sensitivity analysis that included those patients who discontinued study drug confirmed that FFIS was noninferior to placebo.
Conclusion
FFIS-nebulized solution administered twice daily did not increase the combined incidence of respiratory death, COPD-related ER visits, or COPD exacerbation-related hospitalizations compared with placebo over 1 year of treatment. In addition, fewer treatment withdrawals and larger lung function improvements were observed with the drug compared with placebo when added to other maintenance COPD therapies. These results add reassurance and are consistent with previous studies demonstrating the safety of long-term use of LABAs in patients with COPD.
Acknowledgments
Mylan Inc. (Canonsburg, PA, USA) funded both the study and medical writing support. The authors acknowledge Roger Hill, PhD, for medical writing and Paula Stuckart for editorial assistance in the preparation of the manuscript (Ashfield Healthcare Communications, Middletown, CT, USA). The abstract of this paper was presented at the 2017 CHEST Annual Meeting and the 2017 American Thoracic Society conference as an oral and poster presentation, respectively, with interim findings. The poster’s abstract was published as “Poster Abstracts” in CHEST, 2017;152(4):Suppl A769, https://doi.org/10.1016/j.chest.2017.08.800 and Am J Respir Crit Care Med. 2017;195:A5473, https://www.atsjournals.org/doi/abs/10.1164/ajrccm-conference.2017.195.1_MeetingAbstracts.A5473.
Disclosure
NAH received fees as a consultant or serving on advisory board from Roche/Genentech, Novartis, Boehringer Ingelheim, Mylan, and AstraZeneca and grant support from GlaxoSmithKline, Mylan, Boehringer Ingelheim, and Roche/Genentech. SS received research support from AstraZeneca and Mylan; he was on advisory committees, a consultant, and/or a speaker for Aradigm, AstraZeneca, Bayer Schering Pharma, Boehringer Ingelheim, Cempra, CSL, Behring, GlaxoSmithKline, Merck, Invacare, Pulmonx, Sunovion, and Theravance Biopharma. AK is an employee of Mylan Inc., USA. JKW, JS, and DN are employees of Mylan Pharma UK Ltd. The authors report no other conflicts of interest in this work.
References
Lung.org. [homepage on the Internet]. American lung association. COPD fact sheet [updated 2011]. Available from: http://www.lung.org/lung-disease/copd/resources/facts-figures/COPD-Fact-Sheet.html. Accessed July 20, 2017. | ||
Landis SH, Muellerova H, Mannino DM, et al. Continuing to Confront COPD International Patient Survey: methods, COPD prevalence, and disease burden in 2012–2013. Int J Chron Obstruct Pulmon Dis. 2014;9:597–611. | ||
Chen W, Thomas J, Sadatsafavi M, Fitzgerald JM. Risk of cardiovascular comorbidity in patients with chronic obstructive pulmonary disease: a systematic review and meta-analysis. Lancet Respir Med. 2015;3(8):631–639. | ||
Global initiative for chronic obstructive lung disease (GOLD) [homepage on the Internet]; 2016. Available from: http://goo.gl/ItQL3a. Accessed May 4, 2016. | ||
Barjaktarevic IZ, Arredondo AF, Cooper CB. Positioning new pharmacotherapies for COPD. Int J Chron Obstruct Pulmon Dis. 2015;10:1427–1442. | ||
Sin DD, Man SF. Chronic obstructive pulmonary disease as a risk factor for cardiovascular morbidity and mortality. Proc Am Thorac Soc. 2005;2(1):8–11. | ||
van den Berg ME, Stricker BH, Brusselle GG, Lahousse L. Chronic obstructive pulmonary disease and sudden cardiac death: A systematic review. Trends Cardiovasc Med. 2016;26(7):606–613. | ||
Decramer ML, Hanania NA, Lötvall JO, Yawn BP. The safety of long-acting β2-agonists in the treatment of stable chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2013;8:53–64. | ||
Chowdhury BA, dal Pan G. The FDA and safe use of long-acting beta-agonists in the treatment of asthma. N Engl J Med. 2010;362(13):1169–1171. | ||
Chowdhury BA, Seymour SM, Levenson MS. Assessing the safety of adding LABAs to inhaled corticosteroids for treating asthma. N Engl J Med. 2011;364(26):2473–2475. | ||
Wilchesky M, Ernst P, Brophy JM, Platt RW, Suissa S. Bronchodilator use and the risk of arrhythmia in COPD: part 2: reassessment in the larger Quebec cohort. Chest. 2012;142(2):305–311. | ||
FDA.gov. [homepage on the Internet]. FDA drug safety communication: New safety requirements for long-acting inhaled asthma medications called long-acting beta-agonists (LABAs); 2010. Available from: http://bit.ly/2ryDZit. Accessed June 16, 2017. | ||
Gross NJ, Donohue JF. Nebulized formoterol: a review of clinical efficacy and safety in COPD. Int J Chron Obstruct Pulmon Dis. 2010;5:223–232. | ||
Gross NJ, Nelson HS, Lapidus RJ, et al. Efficacy and safety of formoterol fumarate delivered by nebulization to COPD patients. Respir Med. 2008;102(2):189–197. | ||
Tashkin DP. A review of nebulized drug delivery in COPD. Int J Chron Obstruct Pulmon Dis. 2016;11:2585–2596. | ||
ICH. Integrated addendum to ICH harmonised guideline: Guideline for good clinical practice E6 (R2); 2015. Available from: https://goo.gl/CFOmR3. Accessed July 20, 2017. | ||
Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report. GOLD executive summary. Am J Respir Crit Care Med. 2017;195(5):557–582. | ||
Miller MR, Hankinson J, Brusasco V, et al. Standardisation of spirometry. Eur Respir J. 2005;26(2):319–338. | ||
Jones PW, Quirk FH, Baveystock CM, Littlejohns P. A self-complete measure of health status for chronic airflow limitation. The St George’s Respiratory Questionnaire. Am Rev Respir Dis. 1992;145(6):1321–1327. | ||
Mahler DA, Weinberg DH, Wells CK, Feinstein AR. The measurement of dyspnea. Contents, interobserver agreement, and physiologic correlates of two new clinical indexes. Chest. 1984;85(6):751–758. | ||
Baumgartner RA, Hanania NA, Calhoun WJ, Sahn SA, Sciarappa K, Hanrahan JP. Nebulized arformoterol in patients with COPD: a 12-week, multicenter, randomized, double-blind, double-dummy, placebo- and active-controlled trial. Clin Ther. 2007;29(2):261–278. | ||
Calverley PM, Anderson JA, Celli B, et al. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med. 2007;356(8):775–789. | ||
Brook RD, Anderson JA, Calverley PM, et al. Cardiovascular outcomes with an inhaled beta2-agonist/corticosteroid in patients with COPD at high cardiovascular risk. Heart. 2017;103(19):1536–1542. | ||
Donohue JF, Hanania NA, Make B, et al. One-year safety and efficacy study of arformoterol tartrate in patients with moderate to severe COPD. Chest. 2014;146(6):1531–1542. | ||
Donohue JF, Hanania NA, Sciarappa KA, et al. Arformoterol and salmeterol in the treatment of chronic obstructive pulmonary disease: a one year evaluation of safety and tolerance. Ther Adv Respir Dis. 2008;2(2):37–48. | ||
Tashkin DP, Celli B, Senn S, et al. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med. 2008;359(15):1543–1554. |
Supplementary materials
Ethical review boards participating in the clinical trial
Three ethical review boards were used for the trial. All investigator sites used a Central Institutional Review Board (New England Institutional Review Board [NEIRB]) except for three sites, which used their local institutional review board (IRB).
The details of the IRBs are as follows:
 | Table S1 Central IRB |
 | Table S2 Local IRBs |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.