Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 13
Long-term evolution of lung function in individuals with alpha-1 antitrypsin deficiency from the Spanish registry (REDAAT)
Authors Esquinas C, Serreri S, Barrecheguren M, Rodríguez E, Nuñez A ![]() , Casas-Maldonado F
, Casas-Maldonado F ![]() , Blanco I
, Blanco I ![]() , Pirina P
, Pirina P ![]() , Lara B
, Lara B ![]() , Miravitlles M
, Miravitlles M ![]()
Received 26 October 2017
Accepted for publication 29 January 2018
Published 23 March 2018 Volume 2018:13 Pages 1001—1007
DOI https://doi.org/10.2147/COPD.S155226
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Cristina Esquinas,1,2,* Sonia Serreri,3,* Miriam Barrecheguren,1 Esther Rodriguez,1 Alexa Nuñez,1 Francisco Casas-Maldonado,4 Ignacio Blanco,5 Pietro Pirina,3 Beatriz Lara,6 Marc Miravitlles1,7
1Pneumology Department, University Hospital Vall d’Hebron, Barcelona, Spain; 2Public Health, Mental, Maternal and Child Health Nursing Department, Faculty of Medicine and Health Sciences, University of Barcelona, Barcelona, Spain; 3Università di Sassari, Sassari, Italy; 4Pneumology Department, University Hospital San Cecilio, Granada, Spain; 5Alpha-1 Antitrypsin Deficiency Spanish Registry (REDAAT), Spanish Society of Pneumology (SEPAR), Barcelona, Spain; 6Coventry and Warwickshire University Hospital, Coventry, UK; 7CIBER de Enfermedades Respiratorias (CIBERES), Spain
*These authors contributed equally to this work
Background: The clinical course of alpha-1 antitrypsin deficiency (AATD) is very heterogeneous. It is estimated that 60% of individuals with severe AATD (Pi*ZZ) develop emphysema. The main objective of this study was to describe the outcomes of long-term lung function in individuals with AATD-associated emphysema after at least 8 years of follow-up.
Materials and methods: We performed a retrospective analysis of longitudinal follow-up data of AATD PiZZ patients from the Spanish registry (AATD Spanish Registry [REDAAT]). The main follow-up outcome was the annual rate of decline in forced expiratory volume in 1 second (FEV1) calculated using the FEV1 values at baseline and in the last post-bronchodilator spirometry available.
Results: One hundred and twenty-two AATD PiZZ patients were analyzed. The median follow-up was 11 years (interquartile range =9–14). The mean FEV1 decline was 28 mL/year (SD=54), with a median of 33 mL/year. Tobacco consumption (β=19.8, p<0.001), previous pneumonia (β=27.8, p=0.026) and higher baseline FEV1% (β=0.798, p=0.016) were independently related to a faster FEV1 decline.
Conclusion: In this large cohort with a long follow-up, we observed a very variable decline of FEV1. However, the mean FEV1 decline was similar to that observed in large cohorts of smoking-related COPD. Tobacco consumption, previous pneumonia and better lung function at baseline were related to a faster decline in FEV1. These results highlight the importance of early diagnosis and effective treatment.
Keywords: alpha-1 antitrypsin deficiency, lung function, registers
Introduction
Alpha-1 antitrypsin deficiency (AATD) is the most frequent hereditary condition in adults and is associated with an increased risk of pulmonary emphysema and liver disease. Alpha-1 antitrypsin is a highly pleomorphic glycoprotein belonging to the serpin superfamily, with >100 varieties and with antiprotease function, especially anti-neutrophil elastase, being its main characteristic.1
More than 90% of the population have a normal allele called PI*M. The most frequent deficient allelic variants are PI*S and PI*Z, which are responsible for the production of abnormal proteins that polymerize within the hepatocytes; thus, the plasma levels of these proteins are markedly reduced in subjects who are carriers of at least one, and especially two, Z deficiency alleles.2,3
Pi*ZZ is the most frequent genotype associated with poor pulmonary and hepatic prognosis. AATD is the main genetic factor contributing to the development of pulmonary emphysema in adults, as the absence of alpha-1 antitrypsin causes an imbalance favoring proteases, which cause tissue damage.4
The evolution of emphysema due to AATD in clinical terms and life expectancy are both directly related to the accelerated loss of forced expiratory volume in 1 second (FEV1), and it has been described that the decline in lung function is faster in COPD patients with AATD compared to those without AATD.5–7 However, the clinical and functional impact of AATD is not homogeneous in all individuals, and the studies in the literature include very diverse populations with different and usually short follow-up periods. The aim of this study was to describe the long-term functional evolution of individuals with AATD after at least 8 years of follow-up based on the Spanish registry of AATD.
Materials and methods
Study design
This was a retrospective analysis of longitudinal follow-up data of individuals with AATD under real-life management in Spain. The patients included in the analysis were selected from the Spanish registry of individuals with AATD (REDAAT). The REDAAT was established in 1992 and includes patients with severe AATD (Pi*ZZ, Pi*SZ and carriers of rare deficient variants) with and without augmentation therapy from 153 centers in Spain.8,9 Clinical and spirometric data from the follow-up of registered individuals are included in this registry.
This study included individuals with the following: 1) severe AATD, demonstrated by serum AATD levels <50 mg/dL (2.8 mmol/L) with the Pi*ZZ phenotype and 2) at least two post-bronchodilator spirometries available over a period of no less than 8 years. The exclusion criteria were the presence of other deficient phenotypes, such as Pi*SZ, carriers of rare deficient variants and patients who had undergone lung transplant or lung volume reduction surgery. The study was done in accordance with the principles of the Declaration of Helsinki, the guidelines for good practice in epidemiological research and local regulations for the use of study databases. All patients provided written informed consent prior to inclusion in the REDAAT, and were followed with clinical visits every 6 or 12 months and were treated at the discretion of their attending physician.
Study variables
Sociodemographic, anthropometric, clinical (phenotype, age of diagnosis, year of onset of symptoms, comorbidities and treatments) and lung function data were collected at baseline. Lung function tests were performed in tertiary hospitals by respiratory technicians according to standardized recommendations and national guidelines.10 Repeated measurements for each patient were always performed in the same center.
The yearly post-bronchodilator FEV1 decline in mL/year was the main outcome of the study and was calculated as ((final FEV1 (mL) − baseline FEV1 (mL)/months of follow up) ×12)).
Data management and statistical analysis
REDAAT can be accessed through its web page (www.redaat.es). The data obtained were included anonymously in a database in Access format, and the quality was checked, as was the frequency of absent data.9
The frequency and valid percentage of the qualitative variables were determined. Regarding the quantitative variables, measurements of central tendency and dispersion were used (mean and SD).
The decline in FEV1 (mL/year) was transformed into a qualitative variable according to quartile values. In the case of quantitative variables, comparisons of patient characteristics according to FEV1 decline quartiles were carried out with the use of the analysis of variance (Kruskal–Wallis if normality was not assumed) with linear trend analysis. The chi-squared test (Fisher’s test for frequencies <5) was used for comparison of categorical variables with the Kendall’s Tau linear trend test.
A multivariate analysis was performed using a back stepwise linear regression model in order to identify the variables related to FEV1 decline (mL/year). Relevant clinical variables with a p-value <0.2 in the univariate analysis with outcome were included as independent variables in the stepwise models.
For all the tests, a difference with a p-value <0.05 was determined to be significant. The statistical package SPSS (V 23) was used.
Results
A total of 650 AATD individuals were registered in the REDAAT in 2016, with 10% corresponding to individuals <18 years old. Among the adult cohort, 450 were Pi*ZZ (75%), and of them, 122 Pi*ZZ (27% of the adult Pi*ZZ individuals) had at least 8 years of follow-up data on lung function on record and were included in this analysis.
Individuals with 8 years or more of follow-up had a mean age of 61.6 (16.2) years at baseline; 76 (58.5%) were men and 8 (6.2%) were current smokers. The mean age at diagnosis was 54.2 (11.2) years, and the age at the onset of symptoms was 38.6 years (SD=12.2). The most common clinical presentation was emphysema (83.1%, n=108), and the mean baseline FEV1% was 60.8% (SD=30.6%). A previous episode of pneumonia had been presented by 30.8% (n=40). Long-term oxygen therapy was used by 16% (n=21), and 59.2% of patients (n=77) received augmentation therapy (Table 1).
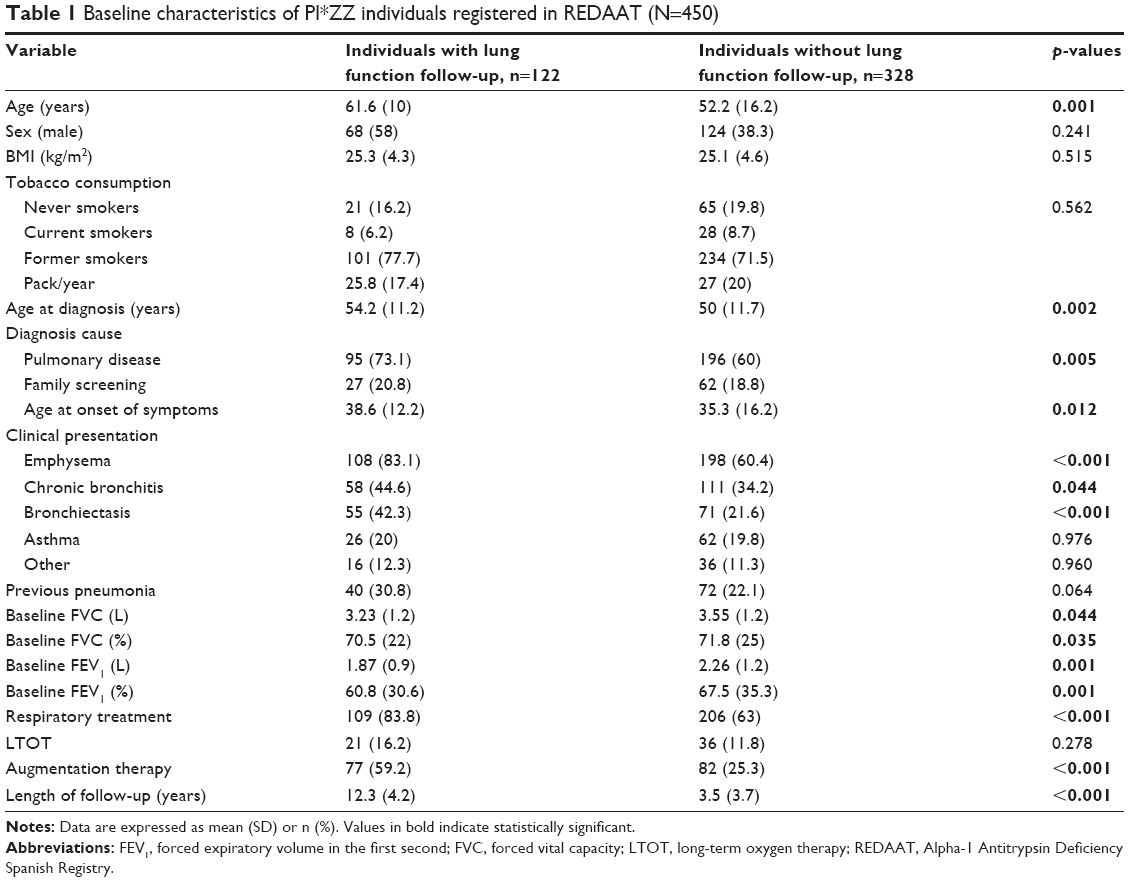 | Table 1 Baseline characteristics of PI*ZZ individuals registered in REDAAT (N=450) |
Patients with 8 years or more of follow-up were similar in terms of sociodemographic or/and clinical variables to those with a shorter follow-up. Nevertheless, individuals with >8 years of follow-up were older at diagnosis, compared to those with <8 years of follow-up (54.2 [11.2] vs 50 [11.7] years; p=0.002). They also presented a higher frequency of emphysema (83% vs 60.4%; p<0.001) and bronchiectasis (44% vs 34%; p<0.001) a lower FEV1% (60.8 [30.6] vs 67.5 [35.3]; p=0.001), and more frequently received respiratory treatment (84% vs 63%; p<0.001) compared to those with <8 years of follow-up (Table 1).
Follow-up and evolution of lung function
The median length of follow-up of the study group was 11 (interquartile range =9–14) years, and 30 (23.1%) patients died.
The mean decline in FEV1 was 28 mL/year (SD=54) and the median was 33 mL/year (interquartile range =8–55). When FEV1 decline was transformed into an ordinal variable according to quartile values, individuals corresponding to the fourth quartile lost a mean of 83.5 mL/year (SD=32.3), in contrast to some patients in whom FEV1 improved during follow-up (quartile 1) with a mean improvement of 35.1 mL/year (SD=20.7), as shown in Figure 1.
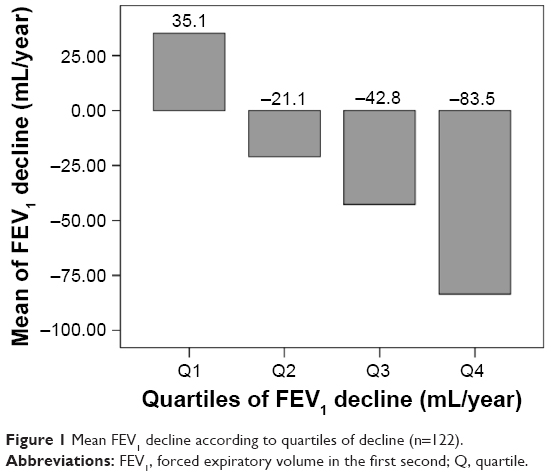 | Figure 1 Mean FEV1 decline according to quartiles of decline (n=122). |
Table 2 shows the baseline characteristics according to quartiles of FEV1 decline. Linear trends were observed in lung function variables, and thus, patients having faster FEV1 decline (quartile 4) presented a more preserved lung function at baseline. No differences were found in other clinical variables according to quartiles of FEV1 decline.
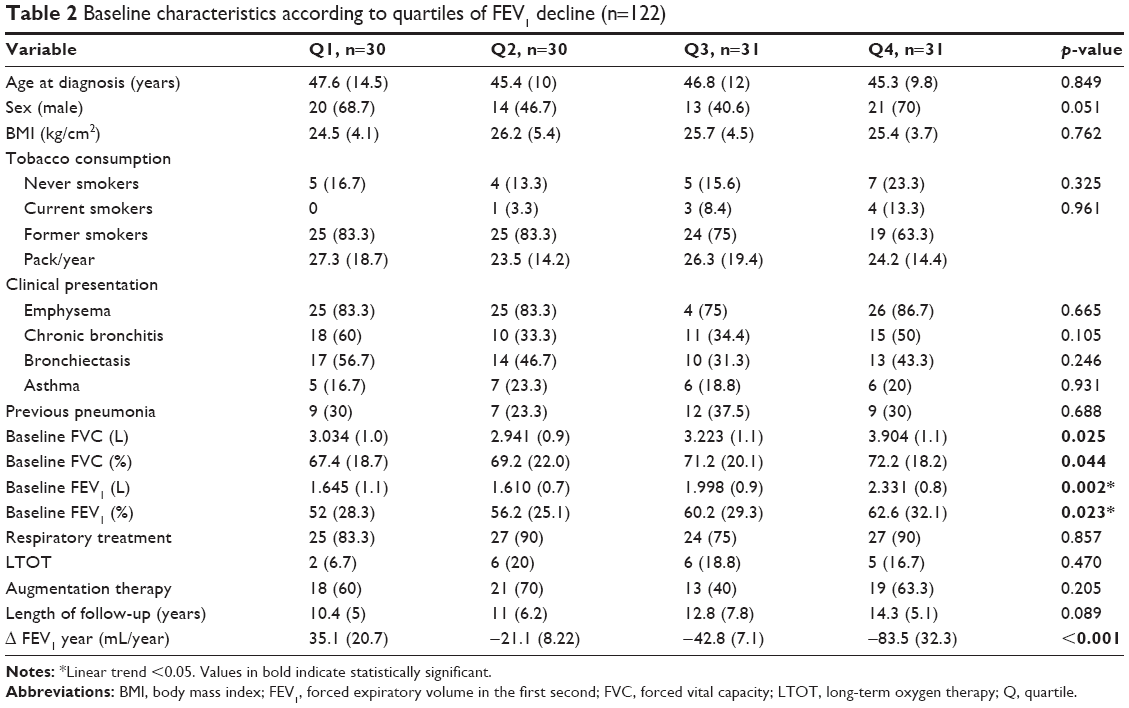 | Table 2 Baseline characteristics according to quartiles of FEV1 decline (n=122) |
Variables related to FEV1 decline on multivariate analysis
In the multivariate analysis, tobacco consumption (β=19.8, p=0.001), previous pneumonia (β=27.9, p=0.026) and higher baseline FEV1% (β=0.798, p=0.010) were independently related to faster FEV1 decline (Table 3).
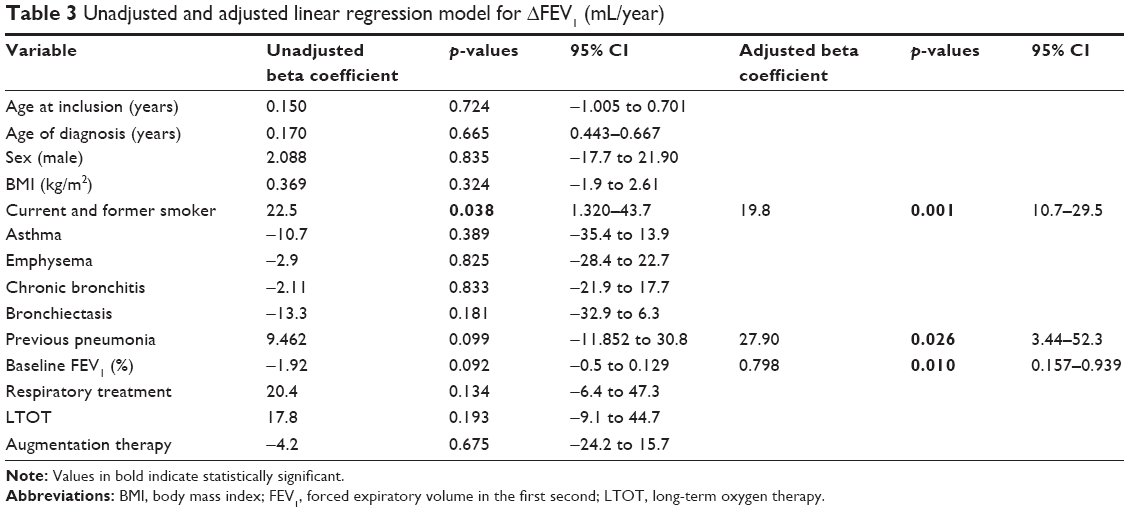 | Table 3 Unadjusted and adjusted linear regression model for ΔFEV1 (mL/year) |
Discussion
Our study showed a great variability in the annual rate of decline in FEV1 in patients with severe AATD followed for >8 years. The analysis showed that tobacco consumption, previous pneumonia and more preserved lung function at baseline were independently related to a more rapid decline in FEV1 in patients with AATD.
The REDAAT was founded in 1992 with the aim of increasing the scientific knowledge about the natural history of this deficiency and its treatment through basic and clinical research, to raise awareness about the deficiency and to provide support for the diagnosis of the AATD through phenotyping and genotyping.8,11 The REDAAT currently includes >650 AATD patients, and prospective analysis of the data from REDAAT has helped to understand the clinical evolution and epidemiology of AATD in Spain.7,9,12 Previous studies investigating the natural course of lung function have usually followed patients for shorter periods. One of the largest follow-up studies derived from the Danish AATD Registry included 347 PI*ZZ individuals with a median follow-up of 6 years. The authors of this study found that a lower baseline FEV1 was an independent predictor of mortality, but this study did not analyze changes in FEV1 during follow-up.13 The American Registry of AATD followed patients over a mean of 5 years and observed a mean decline in FEV1 of 54 mL/year. This decline was increased in males, patients aged 30–44 years, active smokers and those with baseline FEV1 values between 35% and 79%.14 More recently, a marked variation in individual rates of FEV1 decline was also found in a 3-year follow-up of AATD patients from the UK. These data indicate that physiological decline differs among subjects at all stages of lung function impairment for both index and non-index patients as well as for never smokers and ex-smokers.15
Previous data derived from the REDAAT observed a mean decline of 26.9 mL/year ranging from −101 to +106.4 mL/year over a mean follow-up of 5 years.7 Similarly, our current analysis on long-term lung function data with a median follow-up of 11 years found a mean FEV1 decline of 28 mL/year with a great variability, and FEV1 even improved in some patients during this long follow-up. This heterogeneous progression in lung function has also been observed in COPD cohorts. For instance, in the ECLIPSE study, the mean rate of decline in FEV1 was 33 mL/year with a large between-patient SD (59 mL/year).16 In a longitudinal analysis reported by Casanova et al, the decline in FEV1 also ranged between 38 and 86 mL/year.17 In a recent study performed in 607 COPD patients, a similar range of FEV1 decline of between 26 and 78 mL/year was reported. Despite the progressive nature of emphysema, all these studies described an unexpectedly large number of patients showing improvement in FEV1 over time.18 This improvement could be explained by random variation or by the potential effects of respiratory treatment in some patients. In fact, the mean decline observed in our study is very similar to that observed in non-deficient, smoking-related COPD. This is not surprising because most of our patients were not smoking anymore and were treated in reference respiratory clinics with up-to-date respiratory medications, and even a significant number were receiving intravenous augmentation therapy with AATD.
In this study, it was found that a better preserved lung function at baseline, previous pneumonia and smoking were related to a faster decline in FEV1. These findings concur with previous AATD cohort studies. Dawkins et al observed that the greatest FEV1 decline occurred in the group with moderate severity (FEV1=50%–80% predicted) compared with patients with very severe decrease. However, the annual decline in transfer coefficient of the lung for carbon monoxide was greatest in patients with severe and very severe disease.19
There are a few studies on the impact of pneumonia on the evolution of lung disease in AATD. The prevalence of previous pneumonia found in our cohort (31%) was similar to that observed in the American AATD Registry analysis, which was 32%.20 In another analysis based on AATD patients from the REDAAT data, pneumonia was associated with a lower FEV1,7 and this relationship was also observed in non-AATD-associated COPD.21
Studies in COPD patients have described the impact of pneumonia on the deterioration of lung function. In a recent study by Koskela et al, a previous episode of pneumonia increased the risk of being a rapid FEV1 decliner.18 Respiratory infections, in general, may modulate the deterioration of lung function over time. In this line, the study performed by Dransfield et al observed that exacerbations were associated with an excess FEV1 decline, in particular, in mild COPD, in which each exacerbation was associated with an additional decline of 23 mL/year and each severe exacerbation was associated with an additional decline of 87 mL/year. Statistically significant but smaller effects were observed in more severe COPD patients.22 It has long been theorized that the increased burden of neutrophil elastase associated with pulmonary infections may lead to increased lung destruction in patients with compromised antiprotease defenses.23
In this study, 6% of AATD individuals were current smokers and 78% were former smokers. Similar prevalences were observed in the Alpha One International Registry24 and in the Danish Registry.13 These two studies reported that the prevalence of current smokers among AATD individuals was lower compared with that found in COPD cohorts, being around 20%–25%.25,26 This suggests that AATD individuals are amenable to smoking cessation at the time of AATD diagnosis and indicates how successful antismoking programs could be in this population. Indeed, these programs are essential in these patients due to the impact of smoking on the natural history of lung disease in AATD.27
A total of 60% of the PI*ZZ patients included in the study were receiving augmentation therapy. Several studies have shown that augmentation treatment can slow the rate of decline of lung function,14,28 reduce the loss of lung tissue and the progression of emphysema29,30 and may increase the survival of patients with AATD.14 The results of this study, however, did not show any significant difference in the rate of decline of FEV1 according to whether the patients received augmentation treatment. In relation to treatment, it is of note that the decision to prescribe augmentation treatment is made by the physician depending on the severity and the prognosis of each patient, leading to an indication bias, with this type of treatment being preferentially prescribed to more severe patients or faster decliners.31
Our study has several limitations. It is important to take into account that the REDAAT is not a population-based study, and our findings may not be generalizable to the universe of individuals with severe AATD. Our study is also limited by the collection of spirometry at only two time points. Although analysis of FEV1 decline has traditionally relied on more frequent collection of data, in large studies such as the Lung Health Study, the slopes of FEV1 decline estimated with the results of five annual tests did not differ from those measured at only two time points.32 The method of initial and final FEV1 measurements was also used for the main outcome calculation in a clinical trial with intravenous AATD.33 Stockley et al observed that the slope of decline calculated with a few data points was a good predictor of long-term lung function decline.15 Although the quality of spirometry was not formally evaluated, all measurements were conducted in reference hospitals by trained and certified staff following national/international guidelines.10
Conclusion
Our findings provide valuable information about the natural history of AATD and suggest that tobacco consumption, pneumonia and better lung function at baseline have potential predictive values for the detection of rapid deterioration of the lung function in affected individuals. Future clinical studies are needed to fully understand the factors associated with accelerated decline in FEV1 and to provide personalized treatment to high-risk individuals. In this line, genetic studies of rapid decliners might unveil new insights into the underlying mechanisms of disease progression in AATD.34
Acknowledgments
The Alpha-1 Antitrypsin Deficiency Spanish Registry (REDAAT) is supported in part by an unrestricted grant from Grifols (Barcelona, Spain). Part of the results of this study have been presented in the form of an abstract to the European Respiratory Society (ERS) International Congress 2015 (Amsterdam, the Netherlands).
Disclosure
Sonia Serreri was supported by an Erasmus placement 2014–2015 from Università di Sassari (Sassari, Italy). Marc Miravitlles has received speaker fees from Boehringer Ingelheim, Chiesi, Cipla, Menarini, Grifols and Novartis, and consulting fees from Boehringer Ingelheim, GlaxoSmithKline, Gebro Pharma, CSL Behring, Novartis and Grifols. Miriam Barecheguren has received speaker fees from Menarini, GlaxoSmithKline and Gebro Pharma. The authors report no other conflicts of interest in this work.
References
Carrell RW, Lomas DA, Sidhar S, Foreman R. Alpha1-antitrypsin deficiency. A conformational disease. Chest. 1996;110:243–247. | ||
Lomas DA, Mahadeva R. Alpha-1-antitrypsin polymerization and the serpinopathies: pathobiology and prospects for therapy. J Clin Invest. 2002;110:1585–1590. | ||
Casas F, Blanco I, Martínez MT, et al. Indications for active case searches and intravenous alpha-1 antitrypsin treatment for patients with alpha-1 antitrypsin deficiency chronic obstructive pulmonary disease: an update. Arch Bronconeumol. 2015;51:185–192. | ||
Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α-1-antitrypsin deficiency. Eur Respir J. 2017;50(5):pii:1700610. | ||
Seersholm N, Kok-Jensen A, Dirksen A. Survival of patients with severe AATD with special reference to non-index cases. Thorax. 1994;49:695–698. | ||
Tanash HA, Nilsson PM, Nilsson JA, Pituilainen E. Clinical course and prognosis of never smokers with severe AATD (PiZ). Thorax. 2008;63:1091–1095. | ||
Tirado-Conde G, Lara B, Casas F, et al. Factors associated with the evolution of lung function in patients with alpha-1 antitrypsin deficiency in the Spanish registry. Arch Bronconeumol. 2011;47:495–503. | ||
Miravitlles M, Vidal R, Barros-Tizón JC, et al. Usefulness of a national registry of alpha-1 antitrypsin deficiency. The Spanish experience. Respir Med. 1998;92:1181–1187. | ||
Lara B, Blanco I, Martínez MT, et al. Spanish registry of patients with alpha-1 antitrypsin deficiency: database evaluation and population analysis. Arch Bronconeumol. 2017;53:13–18. | ||
García-Río F, Calle M, Burgos F, et al; Spanish Society of Pulmonology and Thoracic Surgery (SEPAR). Spirometry. Spanish Society of Pulmonology and Thoracic Surgery (SEPAR). Arch Bronconeumol. 2013;49:388–401. | ||
Vidal R, Miravitlles M. A report on the Spanish registry of patients with alpha-1 antitrypsin deficiency. The alpha-1 antitrypsin deficiency study group. Arch Bronconeumol. 1995;31:299–302. | ||
Lara B, Miravitlles M. Spanish registry of patients with alpha-1 antitrypsin deficiency; comparison of the characteristics of PiSZ and PiZZ individuals. COPD. 2015;12(S1):27–31. | ||
Seersholm N, Kok-Jensen A. Survival in relation to lung function and smoking cessation in patients with severe hederatary alpha 1-antritrypsin deficiency. Am J Respir Crit Care Med. 1995;151:369–373. | ||
Survival and FEV1 decline in individuals with severe deficiency of alpha1-antitrypsin. The Alpha 1 Antitrypsin Deficiency Group. Am J Respir Crit Care Med. 1998;185:44–49. | ||
Stockley RA, Edgar RG, Pillai A, Turner AM. Individualized lung function trends in alpha-1-antitrypsin deficiency: a need for patience in order to provide patient centered management? Int J Chron Obst Pulm Dis. 2016;11:1745–1756. | ||
Vestbo J, Edwards LD, Scanlon PD, et al. Changes in forced expiratory volume in 1 second over time in COPD. N Engl J Med. 2011;365:1184–1192. | ||
Casanova C, de Torres JP, Aguirre-Jaíme A, et al. The progression of chronic obstructive pulmonary disease is heterogeneous: the experience of the BODE cohort. Am J Respir Crit Care Med. 2011;184:1015–1021. | ||
Koskela J, Katajisto M, Kallio A, Kilpeläinen M, Lindqvist A, Laitinen T. Individual FEV1 trajectories can be identified from a COPD cohort. COPD. 2016;13:425–430. | ||
Dawkins PA, Dawkins CL, Wood AM, Nightingale PG, Stockley JA, Stockley RA. Rate of progression of lung function impairment in alpha 1-antitrypsin deficiency. Eur Respir J. 2009;33:1338–1344. | ||
McElvaney NG, Stoller JK, Buist AS, et al. Baseline characteristics of enrollees in the National Heart, Lung and Blood Institute Registry of alpha 1-antitrypsin deficiency. Alpha 1-Antitrypsin Deficiency Registry Study Group. Chest. 1997;111:394–403. | ||
Crim C, Dransfield MT, Bourbeau J, et al. Pneumonia risk with inhaled fluticasone furoate and vilanterol compared with vilanterol alone in patients with COPD. Ann Am Thorac Soc. 2015;12:27–34. | ||
Dransfield MT, Kunisaki KM, Strand MJ, et al. Acute exacerbations and lung function loss in smokers with and without chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2017;195:324–330. | ||
Gramegna A, Amati F, Terranova L, et al. Neutrophil elastase in bronchiectasis. Respir Res. 2017;18:211. | ||
Stockley RA, Luisetti M, Miravitlles M, Piitulainen E, Fernandez P, on behalf of the Alpha One International Registry (AIR) group. Ongoing research in Europe: Alpha One International Registry (AIR) objectives and development. Eur Respir J. 2007;29:582–586. | ||
Nishimura M, Makita H, Nagai K, et al. Annual change in pulmonary function and clinical phenotype in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;185:44–52. | ||
Burrows B, Bloom JW, Traver GA, Cline MG. The course and prognosis of different forms of chronic airways obstruction in a sample from the general population. N Engl J Med. 1987;317:1309–1314. | ||
Tanash HA, Ekström M, Rönmark E, Lindberg A, Piitulainen E. Survival in individuals with severe alpha 1-antitrypsin deficiency (PiZZ) in comparison to a general population with known smoking habits. Eur Respir J. 2017;50:1700198. | ||
Seersholm N, Wencker M, Banik N, et al. Does alpha-1 antitrypsin augmentation therapy slow the annual decline in FEV1 in patients with severe hereditary AAT deficiency? WATL alpha-1 study group. Eur Respir J. 1997;10:2260–2263. | ||
Stockley RA, Parr DG, Piitulainen E, Stolk J, Stoel BC, Dirksen A. Therapeutic efficacy of alpha-1 antitrypsin augmentation therapy on the loss of lung tissue: an integrated analysis of 2 randomised clinical trials using computed tomography densitometry. Respir Res. 2010;11:136. | ||
Chapman KR, Burdon JG, Piitulainen E, et al. Intravenous augmentation treatment and lung density in severe α1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;386:360–368. | ||
Stockley RA, Miravitlles M, Vogelmeier C. Augmentation therapy for alpha-1 antitrypsin deficiency: towards a personalised approach. Orphanet J Rare Dis. 2013;8:149. | ||
Anthonisen NR, Connett JE, Kiley JP, et al. Effects of smoking intervention and the use of an inhaled anticholinergic bronchodilator on the rate of decline of FEV1. The Lung Health Study. JAMA. 1994;272:1497–1505. | ||
Dirksen A, Piitulainen E, Parr DG, et al. Exploring the role of CT densitometry: a randomised study of augmentation therapy in alpha1-antitrypsin deficiency. Eur Respir J. 2009;33:1345–1353. | ||
Esquinas C, Janciauskiene S, Gonzalo R, et al. Gene and miRNA expression profiles in PBMCs from patients with severe and mild emphysema with PiZZ alpha1-antitrypsin deficiency. Int J Chron Obst Pulmon Dis. 2017;12:3381–3390. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.