")
Back to Journals » Infection and Drug Resistance » Volume 7
Long-term efficacy and safety of raltegravir in the management of HIV infection
Authors Liedtke MD, Tomlin CR, Lockhart SM, Miller MM, Rathbun RC
Received 19 November 2013
Accepted for publication 8 January 2014
Published 18 March 2014 Volume 2014:7 Pages 73—84
DOI https://doi.org/10.2147/IDR.S40168
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Michelle D Liedtke, C Ryan Tomlin, Staci M Lockhart, Misty M Miller, R Chris Rathbun
Department of Clinical and Administrative Sciences, College of Pharmacy, University of Oklahoma Health Sciences Center, Oklahoma City, OK, USA
Abstract: Raltegravir is an integrase strand-transfer inhibitor approved for the treatment of HIV infection. It was the first medication in a novel class of antiretroviral agents to be approved for use in the United States in 2007. Raltegravir exhibits potent activity against wild-type HIV-1, but resistance development has been noted through three different pathways. It is metabolized primarily through uridine diphosphate glucuronosyltransferase 1A1 and has a single inactive glucuronide metabolite. Raltegravir is not a substrate, inhibitor, or inducer of cytochrome P450 enzymes and exhibits low potential for drug–drug interactions; however, strong uridine diphosphate glucuronosyltransferase 1A1 inhibitors or inducers can alter the pharmacokinetics of raltegravir. It is well tolerated, and the most commonly reported adverse effects include headache, nausea, and diarrhea. Serious adverse effects with raltegravir are rare but include rhabdomyolysis and severe skin and hypersensitivity reactions. It has been approved for use in both treatment-naïve and treatment-experienced patients and is a preferred first-line agent in both United States and European HIV treatment guidelines. Although initial approval was granted on 48-week data, 5-year clinical data have recently been published. This article reviews the data supporting long-term efficacy and safety of raltegravir in the treatment of HIV infection.
Keywords: antiretroviral, integrase inhibitor, INSTI, Isentress®
Introduction
Raltegravir (RAL), brand name Isentress® (Merck and Co., Inc., Whitehouse Station, NJ, USA), was approved by the United States Food and Drug Administration (FDA) for the treatment of human immunodeficiency virus (HIV) infection in antiretroviral treatment-experienced patients in October 2007.1 It was the first medication approved in a novel class of antiretrovirals known as integrase strand-transfer inhibitors (INSTIs) or integrase inhibitors. Since the approval of RAL, two other agents have been added to the integrase inhibitor class. Elvitegravir (EVG), currently only available in the fixed-dose combination tablet Stribild® (EVG/cobicistat/tenofovir [TDF]/emtricitabine [FTC]; Gilead Sciences, Inc., Foster City, CA, USA) was approved in August 2012, and dolutegravir (Tivicay®, ViiV Healthcare, Brentford Middlesex, UK) was approved in August 2013.2,3 Following its initial approval, RAL’s indication was expanded in 2009 to include initial treatment of antiretroviral treatment-naïve patients, and, in 2011, it was approved for use in children and adolescents 2–18 years of age.4,5
As with most new antiretroviral agents, initial FDA approval of RAL was based on 48-week clinical data.6,7 HIV-infection is now treated as a chronic disease, and affected patients will potentially be on life-long therapy; 48-week studies leave the questions of viral suppression durability and long-term safety and tolerability unanswered. RAL has now been in clinical use for over 6 years; in 2013, 5-year clinical trial data was published on its use.8,9 This review will summarize the available data supporting the safety and efficacy of RAL in long-term use.
Pharmacology
RAL is a hydroxypyrimidinone carboxamide that functions as an INSTI, similar to other HIV integrase inhibitors.10–12 Integrase catalyzes two consecutive steps for integration of proviral sequences into host nuclear DNA: 3′-processing, in which proviral DNA sequences are cleaved in the cytoplasm, and strand transfer of the pre-integration complex, in which 3′ ends are attached to the host chromosome in the nucleus.13 RAL inhibits integration of the pre-integration complex by binding to magnesium or manganese cofactors in the integrase enzyme, leading to inhibition of the strand-transfer reaction.10
RAL is considered a first-generation INSTI based on its resistance profile. It exhibits potent activity against wild-type HIV-1, with an in vitro 95% inhibitory concentration of 15 ng/mL in 50% human serum.14 Resistance to RAL has been noted through one of three different pathways: Q148H/K/R ± G140S/A, N155H ± E92Q, and Y143C/R ± T97A. Acquisition of Q148H/K/R, Y143C/R, or N155H is associated with a greater than ten-fold decline in susceptibility to RAL and is frequently accompanied by one or more resistance-associated mutations that further reduce susceptibility.15
Pharmacokinetics
RAL is rapidly absorbed from the gastrointestinal tract, with peak plasma concentration achieved within a median of 0.5 to 1.3 hours in the fasting state.14 Administration with a high-fat meal increases area under the serum concentration–time curve (AUC) and minimum serum concentration (Cmin) by 19% and 7.5-fold, respectively; however, RAL may be given without regard to food.16 Serum concentrations decline in a biphasic manner with a terminal elimination half-life of 7–12 hours in HIV-negative, healthy volunteers.14 RAL exhibits high inter- and intrapatient pharmacokinetic variability, with 212% and 122% variance in trough concentrations, respectively.17 RAL undergoes hepatic metabolism primarily by uridine diphosphate glucuronosyltransferase 1A1 (UGT1A1) and has a single inactive glucuronide metabolite.18 Subjects with polymorphic UGT1A1 have higher AUC and Cmin (40% and 91%, respectively) but do not require dosage adjustment.19 Renal elimination accounts for approximately 10% of the oral dose as unchanged drug. Protein binding to plasma proteins is 83%.14
RAL is not a substrate, inhibitor, or inducer of cytochrome P450 enzymes and exhibits low potential for drug–drug interactions; however, strong UGT1A1 inhibitors or inducers can alter the pharmacokinetics of RAL.18 Coadministration of RAL and atazanavir with or without ritonavir results in modest increases in RAL systemic exposure (45%–75%) in healthy subjects that are not considered to be clinically significant.20 Comparable increases in RAL AUC have been observed in HIV-infected subjects with atazanavir.21 Rifampin reduces RAL serum concentrations by 40%–60% and requires an increase in RAL dose to 800 mg twice daily to achieve comparable systemic exposures.22 Coadministration of RAL with efavirenz, etravirine, or maraviroc results in modest reductions (21%–34%) in RAL Cmin in healthy volunteers but is not considered to be clinically significant.23–26 TDF elevates RAL serum concentrations by 69%–78% but is not associated with safety concerns.27 RAL is a substrate for p-glycoprotein and exhibits lower serum concentrations with tipranavir/ritonavir coadministration (AUC and Cmin decreased by 24% and 54%, respectively) in healthy volunteers.18,28 Viral suppression (HIV-1 RNA below 50 copies/mL) was achieved in 55% of antiretroviral-experienced patients with this combination in Phase III studies, so no dosage adjustment is deemed to be necessary.29
Acid suppressants have mixed effects on RAL serum concentrations. Omeprazole increases RAL systemic exposure by three-fold and trough concentrations by 45% in healthy volunteers, presumably due to improved RAL bioavailability at higher gastric pH. The effect of acid suppressants on RAL trough concentrations in HIV-infected subjects is less pronounced (~20%) and does not warrant dosage adjustment.30,31 Conversely, administration of RAL with an aluminum-, magnesium-, and simethicone-containing antacid reduces RAL trough concentrations by 67%. The clinical significance of this reduction in RAL concentration is unknown, but patients should be advised to separate RAL and antacid coadministration by a minimum of 2 hours, and preferably by 4 hours, to avoid an interaction.32
Antiretroviral treatment-naïve studies
The long-term efficacy of RAL as an initial regimen for HIV treatment has been studied in two major clinical trials: the STARTMRK trials and Protocol 004 (Table 1). In both of these trials, RAL is paired with two nucleoside reverse-transcriptase inhibitors (NRTIs) and efficacy is compared to that of efavirenz coupled with the same NRTI backbone. These two studies allow for analysis of long-term efficacy of RAL as they were conducted over the course of 5 years.6,8,33,34
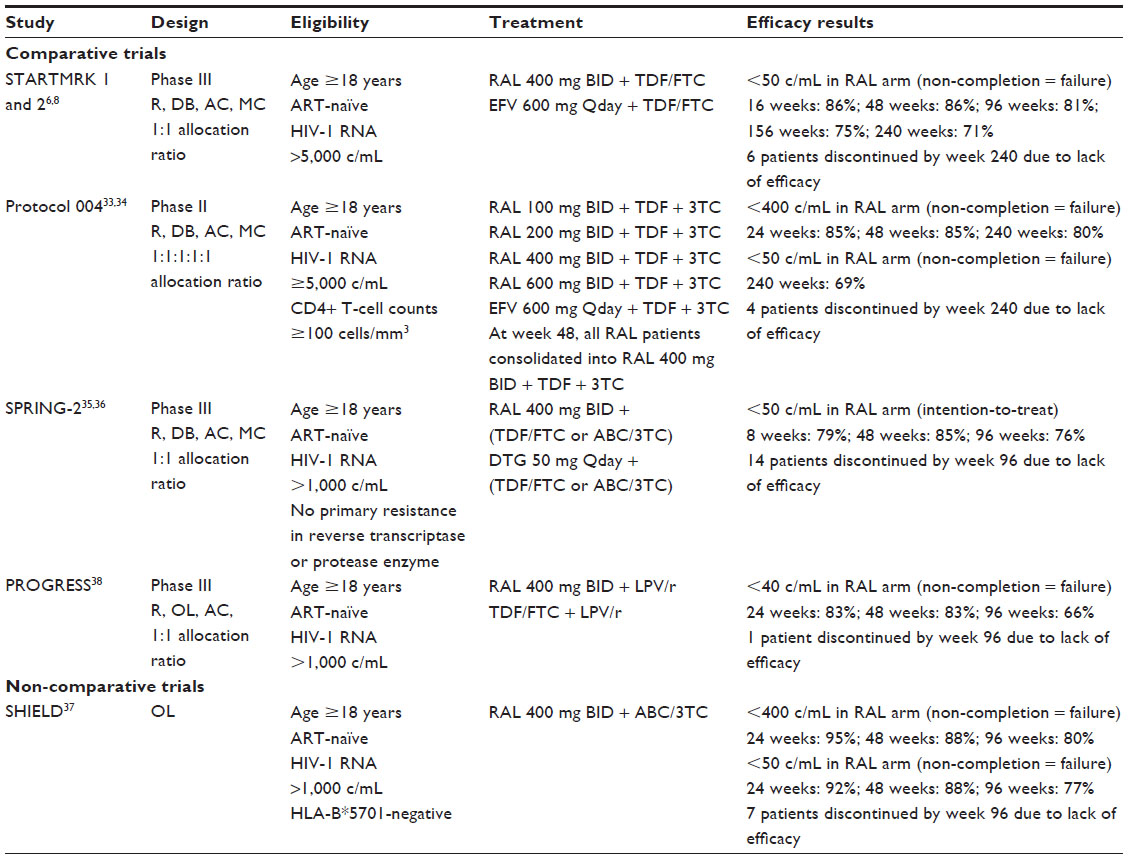 | Table 1 Summary of raltegravir efficacy studies with antiretroviral-naïve patients |
STARTMRK 1 and 2
The STARTMRK 1 and 2 trials were blinded, double-dummy, randomized, Phase III clinical trials comparing RAL to efavirenz with an NRTI backbone of TDF and FTC. Efficacy outcomes were planned for 96 weeks but extended to 240 weeks with blinding maintained throughout the study duration.6,8 Participants had to have an initial viral load >5,000 copies/mL and no documented resistance to any study medications. The primary efficacy end point was the percentage of patients with viral RNA levels <50 copies/mL, utilizing a non-completer equals failure approach.
Overall, 281 and 282 patients were enrolled in the RAL and efavirenz arms, respectively. In regard to the primary outcome at week 48, 241 of 280 (86.1%) RAL patients had a viral load <50 copies/mL, with 81% remaining suppressed through week 96. To support long-term efficacy, 71% (198/279) of RAL patients remained suppressed at week 240. This compared to 171 of 279 (61.3%) efavirenz recipients. The authors discuss the long-term efficacy of RAL, pointing out the relatively low number of discontinuations throughout the 5-year study period. Seventy-one (25%) patients on RAL discontinued therapy prior to the end of the study period. Of those 71 patients, only six discontinued due to lack of efficacy. The remaining 65 patients discontinued therapy due to adverse effects or withdrawal of consent, or were lost to follow-up. Of those six patients, four discontinued RAL within the first 48 weeks. Thus, of the 257 RAL patients who continued the study past 48 weeks, discontinuation due to efficacy was only of concern for two (0.8%).
Protocol 004
A second trial, Protocol 004, also compared previously untreated patients on an initial regimen of either RAL or efavirenz paired with TDF and lamivudine.33,34 This multicenter, double-blind, randomized controlled study included patients with HIV-1 viral RNA ≥5,000 copies/mL. After a 48-week period looking at efficacy of RAL doses ranging from 100 mg to 600 mg twice a day, all patients in the RAL group were consolidated and received RAL 400 mg twice daily for the remainder of the 5-year study.
A total of 160 patients received a RAL containing regimen and 38 received an efavirenz containing regimen. By the end of 5 years, 44 (27%) patients in the RAL group and 12 (31%) in the efavirenz group had discontinued the study. Of the 44 in the RAL group, only four stopped treatment due to lack of efficacy. Two of the four patients who discontinued due to lack of efficacy failed within the first 48 weeks. In regard to primary outcomes, with study discontinuation considered failure, 85% of patients on RAL had viral loads <400 copies/mL at both 24 and 48 weeks. This percentage dropped to only 80% by the end of 5 years. Viral load <50 copies/mL was a secondary end point, with 110 (69%) of the patients on RAL compared to 24 (63%) in the efavirenz group achieving this goal at 240 weeks.33,34
SPRING-2
The most recently published comparative trial reporting 96-week data, SPRING-2, is a comparison of RAL with the newest INSTI, dolutegravir (DTG), each combined with an NRTI backbone.35,36 This study is ongoing and is a Phase III trial looking at noninferiority of DTG compared to RAL in a treatment-naïve population. Although the study is designed to look at DTG outcomes, the 96-week results lend support to long-term efficacy of RAL, showing 76% of patients with viral loads <50 copies/mL.36 In contrast to previously reported studies, 14 patients were reported as discontinued due to lack of efficacy, but little detail was given.36
SHIELD
A final, smaller study looking at the long-term efficacy of RAL with NRTIs paired the medication with abacavir and lamivudine.37 This study, the SHIELD trial, was a noncomparative, open-label trial that enrolled patients with HIV-1 RNA >1,000 copies/mL within 21 days prior to study enrollment. At week 96, HIV-1 RNA was <50 copies/mL in 77% (27/35) of patients using a non-completion equals failure analysis. While the majority of the patients who discontinued the study did so after week 48 (five patients), only one was discontinued due to lack of efficacy.
PROGRESS
In comparison to the first three trials, long-term, 96-week efficacy and safety of RAL in treatment-naïve patients has also been analyzed when used as part of an NRTI sparing regimen.38 In the PROGRESS study,38 patients were antiretroviral naïve, and had viral loads >1,000 copies/mL at screening. These patients were randomized to lopinavir/ritonavir with either RAL or TDF/FTC. The efficacy was evaluated as the proportion of subjects with plasma HIV-1 RNA <40 copies/mL at 96 weeks.
In this study, 101 patients were assigned to lopinavir/ritonavir with RAL and 105 to lopinavir/ritonavir with TDF/FTC. In the RAL group, 19 (19%) discontinued the study versus 15 (14%) in the TDF/FTC group. Of the 19 patients assigned to RAL who discontinued therapy, only one was noted as due to virologic failure. At 96 weeks, 67 (66.3%) patients receiving RAL and 72 (68.6%) of those receiving TDF/FTC had a viral load <40 copies/mL. In addition, the authors note the long-term durability of the RAL + lopinavir/ritonavir regimen by citing that 63% of all patients suppressed on the regimen maintained undetectable viral loads throughout the remainder of the study, with only another 21% having one viral blip reported before returning to undetectable.
Antiretroviral treatment-experienced studies
BENCHMRK 1 and 2
The initial approval of RAL for use in HIV-infected, treatment-experienced patients was based on the BENCHMRK 1 and 2 trials; each was a large, multicenter, international trial with 699 patients combined (Table 2).9 The BENCHMRK studies were Phase III, randomized, double-blind, placebo-controlled trials in HIV-infected patients with documented 3-class resistance (resistance to at least one NRTI, one non-NRTI, and one protease inhibitor [PI]) and failing current antiretroviral therapy. The primary end point was viral suppression at week 48, but the trial remained blinded to week 156. Upon completion of the double-blind portion, approximately 50% of patients in the original RAL treatment arms entered an open-label extension arm that reported outcomes at 240 weeks. These 240-week outcome data provide the most compelling evidence for the durability of RAL-containing regimens.
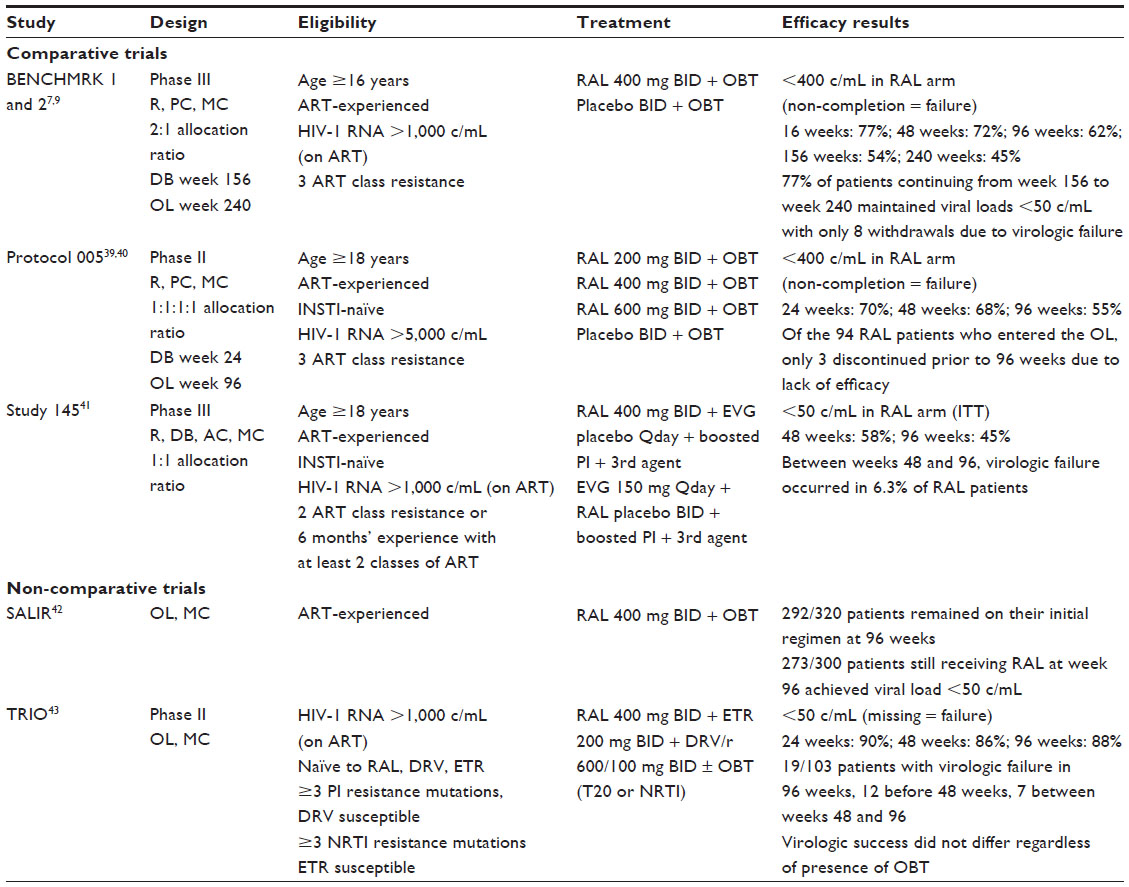 | Table 2 Summary of raltegravir efficacy studies with antiretroviral-experienced patients |
The studies compared RAL against placebo, both paired with optimized background therapy (OBT) selected by the investigator based on the patient’s treatment history and any available genotypic or phenotypic resistance results. Although darunavir was still investigational at the time of enrollment, it was allowed to be included in the OBT. Efficacy was measured by the percentage of patients achieving HIV viral loads <400 copies/mL and <50 copies/mL using a non-completer equals failure approach.
Long-term efficacy was demonstrated by the continued response rate throughout follow-up. At week 48, 62.1% (285/459) of RAL recipients had a viral load <50 copies/mL.7 The response rate continued to be strong, with 51% (233/461) of RAL patients maintaining a viral load <50 copies/mL at week 156. To further support the durability of RAL-containing regimens, the open-label continuation arm reported 42% (193/462) of patients with viral loads <50 copies/mL at 240 weeks. This shows that 77% of RAL treated patients entering the open-label study maintained viral suppression of <50 copies/mL between weeks 156 and 240. Of the 30 patients who did not complete the 240 weeks, only eight (3%) of them were identified as dropping out due to poor efficacy. The authors noted that most virologic failure in those patients treated with RAL occurred in the first 48 weeks. The reasons for failure were likely multifactorial, including adherence, adverse effects, and resistance development; however, most patients who achieved suppression at week 48 continued to maintain suppression throughout follow-up, supporting long-term efficacy of RAL-containing regimens.9
Protocol 005
The only other placebo-controlled RAL trial that has published longer-term outcomes is Protocol 005, a Phase II, dose-ranging study started prior to the BENCHMRK trials reporting 96-week data.39,40 This study consisted of four arms, three with differing RAL doses (200 mg, 400 mg, or 600 mg, administered twice daily) and one placebo comparator arm, each paired with OBT selected on the basis of resistance and antiretroviral treatment history. The patients in this study were also multi-drug resistant with documented 3-class resistance. Neither tipranavir nor darunavir were allowed to be used as part of the OBT.
The study protocol specified a blinded design to week 24, at which time patients were allowed to continue or join in an open-label extension with RAL 400 mg twice daily for a total of 96 weeks. Ninety-four of the original 133 RAL patients continued into the open-label portion of the trial; 86 patients completed the 96 weeks of therapy. At the end of 24 weeks, approximately 60% of patients in the combined RAL arms achieved a viral load <50 copies/mL. At week 48, this percentage fell to 55%, and, at week 96, 48% remained suppressed. Nine patients who achieved <50 copies/mL at week 48 experienced viral rebound prior to week 96. Seven of the nine patients had a genotypic sensitivity score of 0, indicating extensive antiretroviral resistance. Considering durable virologic suppression, 92% of patients achieving <50 copies/mL at week 24 remained suppressed at week 96. This also supports the hypothesis that failure generally occurs earlier in therapy, and that, once suppression is achieved, the effect is durable.39,40
Study 145
The final comparative trial with published 96-week data, Study 145, is a recently published study comparing virologic outcomes of RAL with the more recently approved integrase inhibitor EVG.41 This study was a double-blind, randomized, active-controlled trial conducted in treatment-experienced patients. The two study arms were RAL or EVG in combination with a ritonavir-boosted PI and a third agent chosen at the discretion of the study clinician. Noninferiority was evaluated at week 48, but the study remained blinded until week 96. Using a modified intent-to-treat design, patients with viral loads <50 copies/mL at week 48 for EVG and RAL were 59% (207/351) and 58% (203/351), respectively. At week 96, 45% (158/351) of the RAL patients remained suppressed at <50 copies/mL. Again, looking at durability of the RAL regimen, 78% of those patients achieving an outcome of <50 copies/mL maintained suppression to week 96. Of the 45 patients not achieving the viral load goal, only 22 were identified as discontinuing due to virologic failure. The remaining 23 patients discontinued due to adverse events or other reasons unrelated to the study drug.41
Noncomparative studies
Two noncomparative trials looking at RAL as part of salvage regimens in patients with extensive antiretroviral resistance have also published 96-week data.42,43 A multicenter, observational study conducted in Italy included 320 patients initiated on RAL in combination with a background regimen based on genotypic sensitivity and treatment history. At week 96, 292 (91.25%) patients were still receiving their initial therapy. Eight patients who were not on their baseline regimen were still on a RAL-containing regimen with an alteration to the background regimen. Ninety-one percent of patients (273/300) who were still receiving RAL at 96 weeks had a viral load <50 copies/mL.42
In 2012, French researchers published the 96-week data from the ANRS 139 TRIO study.43 This study was a Phase II, multicenter, noncomparative trial looking at outcomes in multi-drug resistant patients on a regimen of RAL, ritonavir-boosted darunavir, and etravirine in combination with an optimized background regimen consisting of NRTIs or enfuvirtide. Patients had to be naïve to RAL, darunavir, and etravirine to meet inclusion criteria. At week 48, the study reported that 86% of patients had achieved a viral load <50 copies/mL. Of these patients, 100 agreed to participate in extended follow-up, and, at week 96, 88% of those patients had a viral load <50 copies/mL with a 95% confidence interval of 82%–94%. Furthermore, the authors stated that the inclusion of an optimized background regimen did not significantly affect the results, reporting 81% success without an OBT and 85% with an OBT. The authors also concluded that baseline HIV RNA, CD4, and genotypic sensitivity score were not related to success at 96 weeks. Only seven patients experienced virologic failure between week 48 and week 96; this included two patients with missing data who were classified as failure.43
These trials demonstrate long-term efficacy and viral suppression durability with RAL that ranged from 96 to 240 weeks. Most failures appear to occur in the first 24 to 48 weeks of therapy. This is likely not due to drug efficacy, but rather adherence and other factors.
Switch studies
Providers often find themselves needing to change antiretroviral therapy for reasons other than virologic failure, most commonly adverse effects, drug interactions, and patient preference. There have been several studies looking at substituting RAL for another agent in a patient virologically suppressed (Table 3). These trials have examined substitutions for enfuvirtide, ritonavir-boosted PIs, and efavirenz. Outcomes from these trials tend to be shorter and range anywhere from 4 to 48 weeks.44–49
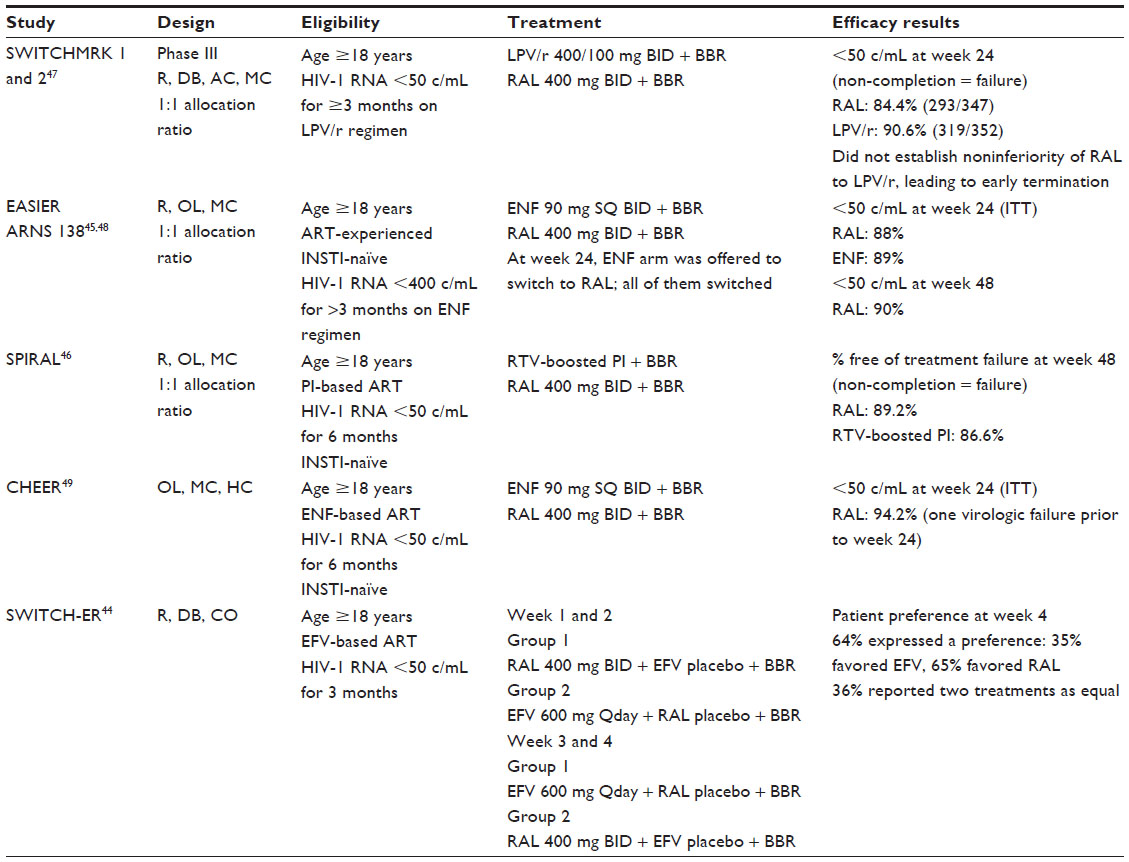 | Table 3 Summary of raltegravir switch studies |
Two studies have reported outcomes at 48 weeks.45,46,48 One study examined RAL substituted for enfuvirtide, and the second study looked at using RAL in place of a ritonavir-boosted PI.45,46,48 The EASIER ARNS 138 trial was an open-label, randomized study looking at patients with 3-class drug failure or intolerance on enfuvirtide-based regimens with viral loads <400 copies/mL for >3 months.45,48 All patients were naïve to integrase inhibitor therapy. Patients were randomized 1:1 to either stay on their current antiretroviral therapy or substitute RAL for enfuvirtide, maintaining their current backbone therapy. The study group remaining on their original regimen was offered the option to switch to a RAL-based regimen at week 24 with all patients switching at that time.
The primary end point was the proportion of patients with failure up to week 48. Secondarily, the percentage of patients with viral loads <50 copies/mL was also reported. One-hundred and seventy patients were randomized; 168 patients completed 48 weeks of therapy (two were not included due to withdrawal prior to study or for non-medication-related reasons). Ninety-eight percent of patients at time of enrollment were receiving at least three antiretrovirals in addition to enfuvirtide. At baseline, 86% of patients had a viral load <50 copies/mL and, at 48 weeks, 90% of patients in the switch arm achieved a viral load of <50 copies/mL. In the RAL arm, only one patient developed virologic failure, with no emergence of RAL resistance mutations. This patient was highly treatment experienced, with a genotypic sensitivity score of 0. In the intent-to-treat analysis, the failure rate for RAL was 1.2% (this included a non-drug-related treatment failure). In all but one patient, RAL regimens also contained a ritonavir-boosted PI. Among the two arms, there was no significant change in virologic suppression and no change in CD4 count.45
The SPIRAL study was a multicenter, randomized, open-label trial conducted in Spain that looked at substituting RAL for the ritonavir-boosted PI component of a suppressive antiretroviral regimen.46 Two hundred and eighty-two patients were randomized, with 273 receiving at least one dose of medication. Patients included in the trial were stable on current antiretroviral therapy consisting of a ritonavir-boosted PI with at least two other agents and a viral load <50 copies/mL for at least 6 months. The primary end point was the proportion of patients who were not considered treatment failures at 48 weeks, utilizing a non-completer equals failure approach. At week 48, 89.2% (124/139) of patients switched to RAL and 86.6% (116/134) patients maintaining their ritonavir-boosted PI regimen were not considered treatment failures. When looking at virologic failure specifically, 96.9% and 95.1% of RAL and ritonavir-boosted PI patients were non-failures, respectively. Of the four RAL virologic failures, three had failed by week 16 and only one at week 48. Though not significant, this shows a trend to early failure with RAL switch, supporting long-term efficacy with those patients not experiencing early failure.46
In two identical parallel studies, SWITCHMRK 1 and 2, patients controlled on twice-daily lopinavir/ritonavir with a two NRTI backbone were randomized to either continue or to substitute RAL 400 mg twice daily for lopinavir/ritonavir.47 The primary efficacy end point was the proportion of patients with a viral load <50 copies/mL. Although this study only has 24-week data, it is important in that it was terminated early when an advisory committee recommended study discontinuation due to increased rates of virologic failure in the group that switched to RAL.47
Using an intent-to-treat analysis, in SWITCHMRK 1, 80.8% of patients on RAL (n=139) and 87.4% on lopinavir/ritonavir (n=152) had suppressed virus, while 88.0% of RAL patients (n=154) and 93.8% of lopinavir/ritonavir patients (n=167) were suppressed in SWITCHMRK 2. Combining the studies leads to a treatment difference of –6.2% (–1.3% to –11.2%) in favor of lopinavir/ritonavir. The difference in outcomes between the previously discussed switch studies and these two may be partially explained by patient selection. A post hoc analysis determined that those patients on their first antiretroviral regimen or without previous virologic failure had similar suppression rates whether on RAL or lopinavir/ritonavir. Researchers therefore theorized that patients on RAL who experienced virologic failure may have been on an NRTI backbone that was not fully susceptible and therefore less effective.47
These switch studies highlight the need for careful and complete examination of patients’ prior antiretroviral exposure before making any changes to therapy. With appropriate patient selection, the SWITCHMRK studies likely would have shown equal efficacy among the RAL and lopinavir/ritonavir regimens. As RAL does have a lower genetic barrier to resistance than ritonavir-boosted PI-based regimens, it is likely not the best choice in patients with few background options; however, with careful consideration and evaluation of patient antiretroviral therapy history, RAL substitutions for whatever reason (tolerance, drug interactions, patient preference) are likely to be successful.
Long-term safety and tolerability
RAL is considered one of the better-tolerated antiretroviral medications, due to limited side effects and few long-term safety concerns. Adverse effects from clinical trials have been consistent in both treatment-naïve and treatment-experienced patients. The most common adverse effects reported were headache, nausea, and diarrhea.50 Although central nervous system adverse effects have been described, they most often occur in the presence of elevated RAL serum concentrations and are less common compared to non-NRTIs.8,34,51 Laboratory changes that have been noted in clinical trials include mild increases in aspartate aminotransferase and alanine aminotransferase; however, these lab value increases are infrequent and, in many studies, not shown to be statistically significant.9,34,47
Serious adverse effects with RAL are rare, and include rhabdomyolysis and severe skin and hypersensitivity reactions.52,53 Trials that focused on the safety and efficacy of RAL described several reports of increased creatine kinase (CK), including a few patients that progressed to rhabdomyolysis.34,36 In an attempt to determine the incidence of this adverse effect, a recent cross-sectional study evaluated the occurrence of skeletal muscle toxicity in HIV-infected patients on RAL compared to patients on other antiretroviral therapy.54 Skeletal muscle toxicity was defined as the presence of at least one of the following: asymptomatic, isolated CK elevation; diffuse myalgia without weakness; proximal myopathy (proximal weakness on examination); or rhabdomyolysis. Of the 318 patients enrolled, skeletal muscle toxicity was present in 37% of the RAL group versus 19% in the control group (P<0.001); however, no patients developed rhabdomyolysis.
A second study from Spain evaluated CK elevation retrospectively in a cohort of 475 patients on RAL-containing regimens.55 The investigators utilized the World Health Organization recommendations for the classification of acute and subacute toxic effects. CK elevations were seen in 53 patients (11.2%), with an incidence of 3.8/100 person-years. Symptoms were reported in seven patients, all of whom had only a grade 1 or 2 elevation in CK. No cases of rhabdomyolysis were reported. Based on a multivariate analysis, the authors stated that evidence of CK elevation prior to RAL therapy and abnormal baseline CK were associated with higher risk of CK elevation. Although there was no control group for comparison, the cohort revealed CK elevations to be not uncommon, yet associated with few symptoms or clinical outcomes.55
In 2011, an FDA MedWatch report updated the adverse effect section of the package insert to include severe skin and hypersensitivity reactions.56 The report cites cases of Stevens–Johnson syndrome and toxic epidermal necrolysis and encourages close monitoring for these adverse events. Additional postmarketing adverse reactions reported to MedWatch include depression, anxiety, thrombocytopenia, increased aspartate aminotransferase and alanine aminotransferase, and rhabdomyolysis. Furthermore, a recent meta-analysis of 48 studies that reviewed the safety and efficacy of RAL noted similar findings of nausea, diarrhea, skin rash, CK elevation, muscle weakness, rhabdomyolysis, transient elevation of serum transaminase levels, and hypersensitivity reactions.57
Several trials have evaluated the reduction in adverse effects in patients switched from various antiretroviral agents to RAL. Treatment-naïve studies have demonstrated a lipid-neutral effect in patients on RAL-containing regimens.6,8,33,34 When transitioning patients from a ritonavir-boosted PI regimen, statistically significant decreases in total plasma cholesterol, low-density lipoprotein, and triglycerides were demonstrated.46,47 The SPIRAL study, as discussed previously, was a multicenter study looking at substituting RAL for the ritonavir-boosted PI component of a suppressive antiretroviral regimen.46 A defined secondary outcome was the change in fasting plasma lipids. A substudy was performed on this data that showed a significant decrease at 48 weeks from baseline in total cholesterol (15.02%), low-density lipoprotein cholesterol (12.66%), and triglycerides (34.17%).58 Similarly, at 24 weeks in the SWITCHMRK study, patients who were changed to RAL experienced a mean decrease in total cholesterol from baseline of 12.8% and a decrease in triglycerides of 41.5%. The decrease in low-density lipoprotein was reported as 2.4% and was not found to be statistically significant.47 Patients transitioned from an efavirenz-based regimen indicated a preference for RAL-based regimens due to a reduction in CNS adverse effects; most notably, anxiety and insomnia.44,45,49
Place in therapy
Due to its relative lack of drug interactions and generally low adverse effect profile, RAL has become a useful addition in the treatment of HIV infection. Its novel mechanism of action at time of approval also made RAL extremely beneficial in the treatment of patients with extensive drug resistance. It is a preferred/recommended agent for initial therapy by the United States Department of Health and Human Services adult and adolescent treatment guidelines, the International AIDS Society-USA treatment guidelines, and the European AIDS Clinical Society treatment guidelines.59–61 Currently, it is still considered an alternate agent by the United States Department of Health and Human Services pediatric treatment guidelines simply due to lack of use and data.62
Describing the place in therapy for antiretroviral agents is complicated by the personalized nature of antiretroviral selection. Although there are treatment guidelines in place, each patient should be viewed individually and therapy should be tailored, based on patient characteristics. For treatment-experienced patients, resistance-testing strongly guides antiretroviral choice. The one evident drawback to RAL when compared to the other preferred agents is the need for twice-daily administration. RAL has been evaluated at a dose of 800 mg once daily compared to the approved twice-daily dosing, but failed to meet noninferiority criteria.63,64 It may be that the virologic outcomes are so strong with twice-daily RAL and, if compared to other preferred regimens, once-daily dosing would be found noninferior; however, this discussion is outside the scope of this article.63,64 Currently, the approved and recommended dosing for RAL is 400 mg twice daily.50
The advantages of a RAL-based regimen include the limited drug interactions, relative lack of adverse effects, and anticipated low de novo resistance rates. Nonetheless, with the approval of newer integrase inhibitors such as DTG, several of these advantages are lost, given similar metabolism and adverse effect profiles combined with the advantage of once-daily administration.65 In light of these similarities, sequencing of antiretroviral medications remains an important consideration when selecting therapy. As DTG retains activity against some RAL-resistant viruses, it may be prudent to consider RAL earlier in therapy despite twice-daily administration. If virologic failure were to occur due to RAL resistance mutations, a second integrase inhibitor remains a treatment option.65
Conclusion
RAL was the first INSTI approved for treatment of HIV infection. Five-year clinical trial outcomes and clinical experience have demonstrated durable virologic suppression in both treatment-naïve and treatment-experienced patients alike, including patients with extensive antiretroviral history and documented antiretroviral resistance. Studies have also exhibited low adverse effect rates and reliable long-term safety lending to improved tolerance. Given its negligible interaction with the cytochrome P450 system, RAL displays minimal drug–drug interactions, making it a good option for patients on multiple medications. RAL is currently a preferred agent for first-line therapy in both United States and European treatment guidelines. Over the past 6 years of clinical use, it has proven an effective and beneficial addition to antiretroviral therapy.
Disclosure
The authors report no conflicts of interest in this work.
References
NME drug and new biologic approvals in 2007 [webpage on the Internet]. Silver Spring, MD: US Food and Drug Administration [updated February 10, 2009]. Available from: http://www.fda.gov/drugs/developmentapprovalprocess/howdrugsaredevelopedandapproved/drugandbiologicapprovalreports/ucm081690.htm. Accessed October 31, 2013. | |
US Food and Drug Administration. FDA approves new combination pill for HIV treatment for some patients [press release]. Silver Spring, MD: US Food and Drug Administration; 2012 [August 27; updated August 29, 2012]. Available from: http://www.fda.gov/newsevents/newsroom/pressannouncements/ucm317004.htm. Accessed October 31, 2013. | |
US Food and Drug Administration. FDA approves new drug to treat HIV infection [press release]. Silver Spring, MD: US Food and Drug Administration; 2013 [August 12; updated August 13, 2013]. Available from: http://www.fda.gov/newsevents/newsroom/pressannouncements/ucm364744.htm. Accessed October 31, 2013. | |
US Food and Drug Administration. Isentress (raltegravir) indication extended for the treatment of HIV-1 infection in treatment-naïve patients [press release]. Silver Spring, MD: US Food and Drug Administration [updated July 13, 2009]. Available from: http://www.fda.gov/forconsumers/byaudience/forpatientadvocates/hivandaidsactivities/ucm171317.htm. Accessed October 31, 2013. | |
US Food and Drug Administration. FDA expands use of HIV drug Isentress to children and adolescents [press release]. Silver Spring, MD: US Food and Drug Administration; 2011 [December 21]. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm284473.htm. Accessed October 31, 2013. | |
Lennox JL, DeJesus E, Lazzarin A; STARTMRK investigators. Safety and efficacy of raltegravir-based versus efavirenz-based combination therapy in treatment-naive patients with HIV-1 infection: a multicentre, double-blind randomised controlled trial. Lancet. 2009;374(9692):796–806. | |
Steigbigel AR, Cooper DA, Kumar PN; BENCHMRK Study Teams. Raltegravir with optimized background therapy for resistant HIV-1 infection. N Engl J Med. 2008;359(4):339–354. | |
Rockstroh JK, DeJesus E, Lennox JL; STARTMRK Investigators. Durable efficacy and safety of raltegravir versus efavirenz when combined with tenofovir/emtricitabine in treatment-naive HIV-1-infected patients: final 5-year results from STARTMRK. J Acquir Immune Defic Syndr. 2013;63(1):77–85. | |
Eron JJ, Cooper DA, Steigbigel RT; BENCHMRK Study Teams. Efficacy and safety of raltegravir for treatment of HIV for 5 years in the BENCHMRK studies: final results of two randomized placebo-controlled trials. Lancet Infect Dis. 2013;13(7):587–596. | |
Summa V, Petrocchi A, Bonelli F, et al. Discovery of raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection. J Med Chem. 2008;51(18):5843–5855. | |
Shimura K, Kodama E, Sakagami Y, et al. Broad antiretroviral activity and resistance profile of the novel human immunodeficiency virus integrase inhibitor elvitegravir (JTK-303/GS-9137). J Virol. 2008;82(2):764–774. | |
Johns BA, Kawasuji T, Weatherhead JG, et al. Carbamoyl pyridine HIV-1 integrase inhibitors 3. A diastereomeric approach to chiral nonracemic tricyclic ring systems and the discovery of dolutegravir (S/GSK1349572) and (S/GSK1265744). J Med Chem. 2013;56(14):5901–5916. | |
Pommier Y, Johnson AA, Marchand C. Integrase inhibitors to treat HIV/AIDS. Nat Rev Drug Discov. 2005;4(3):236–248. | |
Iwamoto M, Wenning LA, Petry AS, et al. Safety, tolerability, and pharmacokinetics of raltegravir after single and multiple doses in healthy subjects. Clin Pharmacol Ther. 2008;83(2):293–299. | |
Blanco JL, Varghese V, Rhee SY, Gatell JM, Shafer RW. HIV-1 integrase inhibitor resistance and its clinical implications. J Infect Dis. 2011;203(9):1204–1214. | |
Wenning L, Anderson M, Petry A, et al. Raltegravir (RAL) dose proportionality and effect of food (abstract H-1046). 47th Interscience Conference on Antimicrobial Agents and Chemotherapy; San Francisco, CA; September 17–20, 2007. | |
Cattaneo D, Gervasoni C, Meraviglia P, et al. Inter- and intra-patient variability of raltegravir pharmacokinetics in HIV-1-infected subjects. J Antimicrob Chemother. 2012;67(2);460–464. | |
Kassahun M, MacIntosh I, Cui D, et al. Metabolism and disposition in humans of raltegravir (MK-0518), an anti-AIDS drug targeting the human immunodeficiency virus 1 integrase enzyme. Drug Metab Dispos. 2007;35(9):1657–1663. | |
Wenning LA, Petry AS, Kost JT, et al. Pharmacokinetics of raltegravir in individuals with UGT1A1 polymorphisms. Clin Pharmacol Ther. 2009;85(6):623–627. | |
Iwamoto M, Wenning LA, Mistry GC, et al. Atazanavir modestly increases plasma levels of raltegravir in healthy subjects. Clin Infect Dis. 2008;47(1):137–140. | |
Cattaneo D, Ripamonti D, Baldelli S, Cozzi V, Conti F, Clementi E. Exposure-related effects of atazanavir on the pharmacokinetics of raltegravir in HIV-1-infected patients. Ther Drug Monit. 2010;32(6):782–786. | |
Wenning LA, Hanley WD, Brainard DM, et al. Effect of rifampin, a potent inducer of drug-metabolizing enzymes, on the pharmacokinetics of raltegravir. Antimicrob Agents Chemother. 2009;53(7):2852–2856. | |
Iwamoto M, Wenning LA, Petry AS, et al. Minimal effects of ritonavir and efavirenz on the pharmacokinetics of raltegravir. Antimicrob Agents Chemother. 2008;52(12):4338–4343. | |
Anderson MS, Kakuda TN, Hanley W, et al. Minimal pharmacokinetic interaction between the human immunodeficiency virus nonnucleoside reverse transcriptase inhibitor etravirine and the integrase inhibitor raltegravir in healthy subjects. Antimicrob Agents Chemother. 2008;52(12):4228–4232. | |
Barrail-Tran A, Yazdanpanah Y, Fagard C, et al. Lack of Interaction between etravirine and raltegravir plus darunavir/ritonavir when combined in treatment-experienced patients: a substudy of the ANRS 139 TRIO trial (abstract 606). 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA; February 16–19, 2010. | |
Andrews E, Glue P, Fang J, Crownover P, Tressler R, Damle B. Assessment of the pharmacokinetics of co-administered maraviroc and raltegravir. Br J Clin Pharmacol. 2010;69(1):51–57. | |
Wenning LA, Friedman EJ, Kost JT, et al. Lack of a significant drug interaction between raltegravir and tenofovir. Antimicrob Agents Chemother. 2008;52(9):3253–3258. | |
Hanley WD, Wenning LA, Moreau A, et al. Effect of tipranavir-ritonavir on pharmacokinetics of raltegravir. Antimicrob Agents Chemother. 2009;53(7):2752–2755. | |
Cooper DA, Steigbigel RT, Gatell JM; BENCHMRK Study Teams. Subgroup and resistance analyses of raltegravir for resistant HIV-1 infection. N Engl J Med. 2008;359(4):355–365. | |
Iwamoto M, Wenning LA, Nguyen BY, et al. Effects of omeprazole on plasma levels of raltegravir. Clin Infect Dis. 2009;48(4):489–492. | |
Rhame F, Matson M, Wood D, et al. Effects of famotidine and omeprazole on raltegravir pharmacokinetics in HIV infected persons (abstract PE4.1/1). 12th Europeans AIDS Conference; Cologne; November 11–14, 2009. | |
Kiser JJ, Bumpass JB, Meditz AL, et al. Effect of antacids on the pharmacokinetics of raltegravir in human immunodeficiency virus-seronegative volunteers. Antimicrob Agents Chemother. 2010;54(12):4999–5003. | |
Markowitz M, Nguyen BY, Gotuzzo E; Protocol 004 Part II Study Team. Rapid and durable antiretroviral effect of the HIV-1 Integrase inhibitor raltegravir as part of combination therapy in treatment-naive patients with HIV-1 infection: results of a 48-week controlled study. J Acquir Immune Defic Syndr. 2007;46(2):125–133. | |
Gotuzzo E, Markowitz M, Ratanasuwan W; Protocol 004 Study Team. Sustained efficacy and safety of raltegravir after 5 years of combination antiretroviral therapy as initial treatment of HIV-1 infection: final results of a randomized, controlled, phase II study (Protocol 004). J Acquir Immune Defic Syndr. 2012;61(1):73–77. | |
Raffi F, Rachlis A, Stellbrink HJ; SPRING-2 Study Group. Once-daily dolutegravir versus raltegravir in antiretroviral-naive adults with HIV-1 infection: 48 week results from the randomized, double-blind, non-inferiority SPRING-2 study. Lancet. 2013;381(9868):735–743. | |
Raffi F, Jaeger H, Quiros-Roldan E; extended SPRING-2 Study Group. Once-daily dolutegravir versus twice-daily raltegravir in antiretroviral-naive adults with HIV-1 infection (SPRING-2 study): 96 week results from a randomized, double-blind, non-inferiority trial. Lancet Infect Dis. 2013;13(11):927–935. | |
Young B, Vanig T, DeJesus E; SHIELD Study Team. 96-week results of a pilot study of abacavir/lamivudine and raltegravir in antiretroviral-naïve HIV-1-infected patients: the SHIELD trial. HIV Clin Trials. 2011;12(4):228–233. | |
Reynes J, Trinh R, Pulido F, et al. Lopinavir/ritonavir combined with raltegravir or tenofovir/emtricitabine in antiretroviral-naive subjects: 96-week results of the PROGRESS study. AIDS Res Hum Retroviruses. 2013;29(2):256–265. | |
Gatell JM, Katlama C, Grinsztejn B; Protocol 005 Team. Long-term efficacy and safety of the HIV integrase inhibitor raltegravir in patients with limited treatment options in a Phase II study. J Acquir Immune Defic Syndr. 2010;53(4):456–463. | |
Grinsztejn B, Nguyen AY, Katlama C; Protocol 005 Team. Safety and efficacy of the HIV-1 integrase inhibitor raltegravir (MK-0518) in treatment-experienced patients with multidrug-resistant virus: a phase II randomised controlled trial. Lancet. 2007;369(9569):1261–1269. | |
Elion R, Molina JM, Ramón Arribas López J; Study 145 Team. A randomized phase 3 study comparing once-daily elvitegravir with twice-daily raltegravir in treatment-experienced subjects with HIV-1 infection: 96-week results. J Acquir Immune Defic Syndr. 2013;63(4):494–497. | |
Capetti A, Landonio S, Meraviglia P, et al. 96 week follow-up of HIV-infected patients in rescue with raltegravir plus optimized backbone regimens: a multicenter Italian experience. PLoS One. 2012;7:e39222. | |
Fagard C, Colin C, Charpentier C; ANRS 139 TRIO Trial Group. Long-term efficacy and safety of raltegravir, etravirine, and darunavir/ritonavir in treatment-experienced patients: week 96 results from the ANRS 139 TRIO trial. J Acquir Immune Defic Syndr. 2012;59(5):489–493. | |
Nguyen A, Calmy A, Delhumeau C, et al. A randomized cross-over study to compare raltegravir and efavirenz (SWITCH-ER) study. AIDS. 2011;25(12):1481–1487. | |
Gallien S, Braun J, Delaugerre C; EASIER ANRS 138 Study Group. Efficacy and safety of raltegravir in treatment-experienced HIV-1-infected patients switching from enfuvirtide-based regimens: 48 week results of the randomized EASIER ANRS 138 trial. J Antimicrob Chemother. 2011;66(9):2099–2106. | |
Martinez E, Larrousse M, Llibre JM; SPIRAL Study Group. Substitution of raltegravir for ritonavir-boosted protease inhibitors in HIV-infected patients: the SPIRAL study. AIDS. 2010;24(11):1697–1707. | |
Eron JJ, Young B, Cooper DA; SWITCHMRK 1 and 2 investigators. Switch to a raltegravir-based regimen versus continuation of a lopinavir-ritonavir-based regimen in stable HIV-infected patients with suppressed viraemia (SWITCHMRK 1 and 2): two multicenter, double-blind, randomized controlled trials. Lancet 2010;375(9712):396–407. | |
De Castro N, Braun J, Charreau I; EASIER ANRS 138 Study Group. Switch from enfuvirtide to raltegravir in virologically suppressed multidrug-resistant HIV-1-infected patients: a randomized open-label trial. Clin Infect Dis. 2009;49(8):1259–1267. | |
Towner W, Klein D, Kerrigan HL, Follansbee S, Yu K, Horberg M. Virologic outcomes of changing enfuvirtide to raltegravir in HIV-1 patients well controlled on an enfuvirtide based regimen: 24-week results of the CHEER study. J Acquir Immune Defic Syndr. 2009;51(4):367–373. | |
Isentress® (raltegravir) [package insert]. Whitehouse Station, NJ: Merck Sharp and Dohme Corp.; 2011. | |
Madeddu G, Menzaghi B, Ricci E; CISAI Group. Raltegravir central nervous system tolerability in clinical practice: results from a multicenter observational study. AIDS. 2012;26(18):2412–2415. | |
Tsai WJ, Lee SS, Tsai HC, et al. Rapid onset of rhabdomyolysis after switching to a raltegravir-based antiretroviral regimen. J Microbiol Immunol Infect. Epub April 20, 2013. | |
Croce F, Vitello P, Dalla Pria A, Riva A, Galli M, Antinori S. Severe raltegravir-associated rhabdomyolysis: a case report and review of the literature. Int J STD AIDS. 2010;21(11):783–785. | |
Lee FJ, Amin J, Bloch M, Pett SL, Marriott D, Carr A. Skeletal muscle toxicity associated with raltegravir-based combination antiretroviral therapy in HIV-infected adults. J Acquir Immune Defic Syndr. 2013;62(5):525–533. | |
Monteiro P, Perez I Pich J, Gatell JM, Martinez E. Creatine kinase elevation in HIV-1-infected patients receiving raltegravir-containing antiretroviral therapy: a cohort study. J Antimicrob Chemother. 2013;68(2):404–408. | |
Isentress (raltegravir potassium) tablet and chewable tablet [webpage on the Internet]. Silver Spring, MD: US Food and Drug Administration; 2013 [updated November 14, 2013]. Available from: http://www.fda.gov/safety/medwatch/safetyinformation/ucm360369.htm. Accessed September 20, 2013. | |
Messiaen P, Wensing AM, Fun A, Nijhuis M, Brusselaers N, Vandekerckhove L. Clinical use of HIV integrase inhibitors: a systematic review and meta-analysis. PLoS One. 2013;8(1):e52562. | |
Saumoy M, Sánchez-Quesada JL, Martínez E, et al. LDL subclasses and lipoprotein-phospholipase A2 activity in suppressed HIV-infected patients switching to raltegravir: Spiral substudy. Atherosclerosis. 2012;225(1):200–207. | |
Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents. Department of Health and Human Services [updated February 12, 2013]. Available from: http://aidsinfo.nih.gov/contentfiles/lvguidelines/AdultandAdolescentGL.pdf. Accessed October 10, 2013. | |
Thompson MA, Aberg JA, Hoy JF, et al. Antiretroviral treatment of adult HIV infection: 2012 recommendations of the International Antiviral Society-USA panel. JAMA. 2012;308(4):387–402. | |
European AIDS Clinical Society Guidelines. Version 7.0. European AIDS Clinical Society; 2013. Available from https://www.eacsociety.org/Portals/0/Guidelines_Online_131014.pdf. Accessed January 29, 2014. | |
Guidelines for the Use of Antiretroviral Agents in Pediatric HIV Infection. Panel on Antiretroviral Therapy and Medical Management of HIV-Infected Children [updated November 5, 2012]. Available from: http://aidsinfo.nih.gov/contentfiles/lvguidelines/pediatricguidelines.pdf. Accessed October 10, 2013. | |
Eron JJ Jr, Rockstroh JK, Reynes J: QDMRK Investigators. Raltegravir once daily or twice daily in previously untreated patients with HIV-1: a randomised, active-controlled, phase 3 non-inferiority trial. Lancet Infect Dis. 2011;11(12):907–915. | |
Rizk ML, Hang Y, Luo WL, et al. Pharmacokinetics and pharmacodynamics of once-daily versus twice-daily raltegravir in treatment-naive HIV-infected patients. Antimicrob Agents Chemother. 2012;56(6):3101–3106. | |
Tivicay® (dolutegravir) [package insert]. Research Triangle Park, NC: ViiV Healthcare; 2013. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.