Back to Journals » OncoTargets and Therapy » Volume 12
Long noncoding RNA SNHG16 induces sorafenib resistance in hepatocellular carcinoma cells through sponging miR-140-5p
Authors Ye J, Zhang R, Du X, Chai W, Zhou Q
Received 24 May 2018
Accepted for publication 7 November 2018
Published 4 January 2019 Volume 2019:12 Pages 415—422
DOI https://doi.org/10.2147/OTT.S175176
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Junfeng Ye,1 Ruoyan Zhang,1 Xiaohong Du,1 Wengang Chai,1 Qiang Zhou2
1Department of Hepato-Biliary-Pancreatic Surgery, The First Hospital of Jilin University, Changchun City, Jilin Province, PR China; 2Department of Hepatology, The First Hospital of Jilin University, Changchun City, Jilin Province, PR China
Background: Sorafenib is widely used for treatment of hepatocellular carcinoma (HCC), but the acquired resistance remains a major obstacle for its application. Thus it is of critical importance to elucidate the molecular mechanisms underlying sorafenib resistance in HCC. This study aimed to determine the roles of long noncoding RNA SNHG16 in sorafenib-resistant HCC cells.
Methods: HCC and matched adjacent normal liver tissue samples were obtained from 103 HCC patients. Sorafenib-resistant HepG2/SOR cell line was established from its parental HepG2 cells by exposure to increasing concentrations of sorafenib. SNHG16 and miR-140-5p expression levels in tissue samples and cells were detected by RT-qPCR analysis. The sensitivity of cells to sorafenib in vitro was evaluated by MTT assay, and the sensitivity of HepG2/SOR cells to sorafenib in vivo was estimated using the nude mouse-based xenograft model. The potential binding relation between SNHG16 and miR-140-5p was validated by dual luciferase reporter assay and biotinylated RNA pull-down assay.
Results: The results showed that SNHG16 expression was remarkably increased in HCC tissues and cell lines, and its high expression was closely associated with aggressive clinicopathological features and poor prognosis of HCC patients. Further experiments showed that SNHG16 is upregulated in HepG2/SOR cells, whereas knockdown of SNHG16 increases the sensitivity of HepG2/SOR cells to sorafenib in vitro and in vivo. Further mechanistic study identified that SNHG16 functions as an endogenous sponge for miR-140-5p in HepG2 cells, and in HCC tissues, the expression of miR-140-5p is negatively correlated with SNHG16 expression. Moreover, miR-140-5p overexpression also increases the sensitivity of HepG2/SOR cells to sorafenib, and the effects of SNHG16 knockdown on sorafenib resistance could be blocked by miR-140-5p inhibitor.
Conclusion: Collectively, our findings demonstrated that knockdown of SNHG16 attenuated sorafenib resistance in HCC through sponging miR-140-5p, indicating that SNHG16 might be as a promising therapeutic target to boost the effectiveness of chemotherapy for HCC patients.
Keywords: long noncoding RNA, SNHG16, hepatocellular carcinoma, sorafenib resistance, miR-140-5p
Introduction
Hepatocellular carcinoma (HCC) is the sixth most common cancer and the third leading cause of cancer-related death.1 Although great progress in diagnostic and therapeutic methods has been made, the prognosis of HCC patients after hepatectomy remains dismal.2 Sorafenib, a multikinase inhibitor, is a common chemotherapeutic drug for patients with advanced HCC, but the clinical application of sorafenib is largely limited by chemoresistance.3,4 Accordingly, it is of critical importance to figure out the molecular mechanisms underlying sorafenib resistance in HCC, which might be helpful for improving the therapeutic efficacy of HCC patients.
Long noncoding RNAs (lncRNAs) stand for a class of transcribed RNA molecules with length of more than 200 nucleotides. Up to now, many lncRNAs have been identified, and they have gained widespread attention because they are frequently highlighted as critical regulators of human tumorigenesis.5,6 For example, small nucleolar RNA host gene 16 (SNHG16), a newly identified lncRNA, has been found to be highly expressed in many kinds of malignancies, including breast cancer,7 cervical cancer,8 ovarian cancer,9 and glioma.10 But the exact role of SNHG16 in HCC remains to be further elucidated.
Till now, limited studies reported the functions of lncRNAs in the sorafenib resistance of HCC cells. In the present study, we aimed to validate the role of SNHG16 in sorafenib-resistant HCC cells and investigate the potential mechanisms. We believed that our findings might provide a novel therapeutic strategy for HCC.
Materials and methods
Patients and tissue samples
HCC and corresponding adjacent normal liver tissue samples were obtained from 103 HCC patients who had undergone surgery at The First Hospital of Jilin University (Changchun, PR China). The clinicopathological characteristics of these patients were listed in Table 1. None of these patients had received chemotherapy or radiotherapy before surgery. The collected tissue samples were confirmed by histopathological examination, immediately snap frozen in liquid nitrogen and stored at −80°C until required. The present study was approved by the Research Ethics Committee of the First Hospital of Jilin University, and written informed consent forms were acquired from all patients or their relatives.
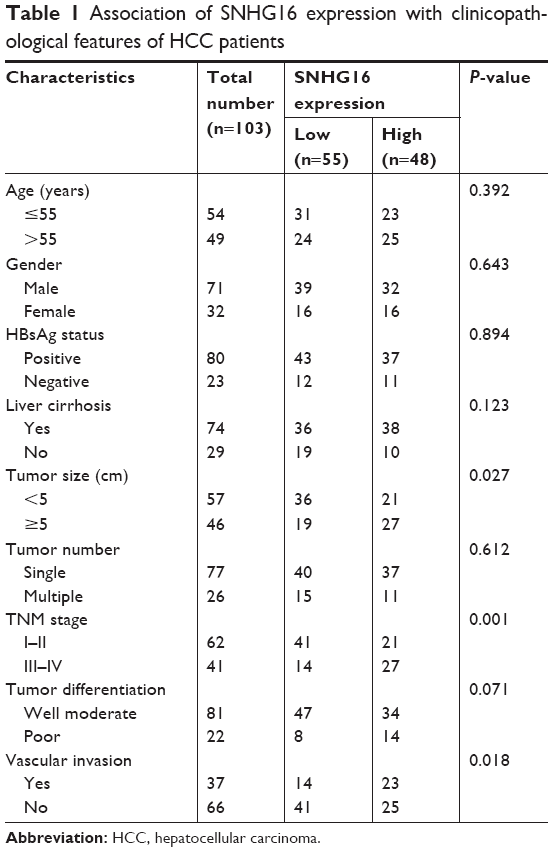 | Table 1 Association of SNHG16 expression with clinicopathological features of HCC patients |
Cell culture and treatments
The human HCC cells, HepG2, SK-hep1, Huh7, and HCCLM3, and normal liver cell line LO2 were obtained from the American Type Culture Collection (Manassas, VA, USA). HepG2/SOR cell line was established from its parental HepG2 cells by exposure to increasing concentrations ranging from 1 to 10 μM of sorafenib (Sigma-Aldrich, St Louis, MO, USA) for 6 months in our laboratory. Cells were maintained in DMEM (Invitrogen, Carlsbad, CA, USA) supplemented with 10% FBS (HyClone, Logan, USA) at 37°C in a humidified atmosphere containing 5% CO2. The culture medium for HepG2/SOR cells contained 10 μM sorafenib to maintain its drug resistance. Before the experiments, HepG2/SOR cells were cultured without sorafenib for 1 week.
The siRNA targeting SNHG16 (si-SNHG16) and negative control (si-NC) were designed and chemically synthesized by GenePharma (Shanghai, PR China). To overexpress SNHG16, the full-length human SNHG16 cDNA was inserted into pcDNA3.1 vector (Invitrogen). The miR-140-5p mimics, mimics control, miR-140-5p inhibitor, and inhibitor control were obtained from Guangzhou RiboBio (RiboBio, PR China). Cells were seeded into 6-well plates and transfection was performed using Lipofectamine 2000 (Invitrogen). Forty-eight hours after transfection, the transfection efficiency was evaluated by RT-qPCR analysis.
MTT assay
Cell viability was detected by MTT assay. The cells were seeded in a 96-well plate at a density of 3×103 cells/well. Sorafenib to a concentration of 1, 5, 10, or 20 μM was added to each well. After incubation for 48 hours, 20 μL MTT solution (5 mg/mL; Sigma-Aldrich) was added to each well and incubated for another 4 hours, followed by the precipitate dissolving in 100 μL DMSO (Sigma-Aldrich). Absorbance levels were measured at a wavelength of 450 nm using an ELISA reader (MultiskanEX, Lab systems, Helsinki, Finland). Experiments were performed in triplicate. The IC50 (drug concentration producing 50% growth inhibition) was calculated using SPSS 19 software package (IBM SPSS Inc, Chicago, IL, USA).
RNA extraction and RT-qPCR analysis
Total RNA samples were isolated from tissues or cells using Trizol Reagent (Invitrogen). Nuclear and cytoplasmic fractions were separated from cells using PARIS™ Kit (Invitrogen) following the manufacturer’s instruction. cDNA was synthesized using the PrimeScript RT reagent Kit (TaKaRa), and qPCR was performed using the Real-Time Quantitative PCR SYBR Green kit (TaKaRa) on the ABI 7500 fast real-time PCR system (Applied Biosystems, Foster City, CA, USA). The relative expression of individual genes was determined by 2−ΔΔCt methods,11 and GAPDH or U6 was used as an endogenous control.
Dual luciferase reporter assay
The fragment from SNHG16 containing the putative binding sites for miR-140-5p was amplified by PCR and cloned into the psiCHECK-2 vector (Promega, Madison, WI, USA), which was named SNHG16-WT. Point mutations in the binding seed regions were created by Quick Change Site-Directed Mutagenesis Kit (Agilent, Roseville City, CA, USA), and the resultant product was named SNHG16-MUT. A total of 3×104 HepG2 cells were seeded on 12-well plates and then cotransfected with SNHG16-WT or SNHG16-MUT, together with the Renilla luciferase expression vector pRL-TK (Promega) and miR-140-5p mimics or mimics control using Lipofectamine 2000. The activities of firefly luciferase and Renilla luciferase were measured 48 hours after transfection using the dual luciferase reporter assay system (Promega). Renilla luciferase was detected for data normalization.
Biotinylated RNA pull-down assay
HepG2 cells with SNHG16 overexpression were transfected with biotinylated miR-140-5p mimics or its MUT mimics using Lipofectamine 2000. Two days after transfection, the cells were harvested, lysed, sonicated, and incubated with M-280 streptavidin magnetic beads (Invitrogen). The abundance of SNHG16 in the bound fractions was evaluated by RT-qPCR analysis.
Animal tumor xenograft model
Forty male nude mice (Athymic nu/nu, 5–6 weeks, 20–25 g) were purchased from the Shanghai Lab Animal Research Center (Shanghai, PR China) and maintained under specific pathogen-free condition. The mice were allocated to four groups (n=10 each group): sh-NC+saline group, sh-NC+sorafenib group, sh-SNHG16+saline group, and sh-SNHG16+sorafenib group. A total of 1×106 HepG2/SOR cells stably transfected with sh-NC or sh-SNHG16 were harvested and resuspended in 100 μL of serum-free medium, and then subcutaneously injected into the right flank of nude mice, and tumor volume was measured using an external caliper every 3 days and calculated as follows: volume=(length×width2×0.5). One week after cell inoculation, mice were orally administered with 10 mg/kg sorafenib or the same volume of saline once daily. The mice were killed on 20 days after cell inoculation, and the tumors were excised and weighed. All animal procedures were approved by the Research Ethics Committee of The First Hospital of Jilin University, and all efforts were made to minimize animal suffering according to the NIH Guide for the Care and Use of Laboratory Animals.
Statistical analysis
All experimental data were presented as the mean ± SD. Statistical analysis was performed using GraphPad Prism 6.0 software (GraphPad Software, San Diego, CA, USA) and SPSS 19 software package. Student’s t-test (two-tailed) and one-way ANOVA were used for the continuous data analysis. The association between SNHG16 expression and clinicopathological features of HCC patients was evaluated using the chi-squared test. P<0.05 was considered to be statistically significant.
Results
SNHG16 is upregulated in HCC tissues and cell lines
To understand the role of SNHG16 in HCC, the expression levels of SNHG16 in 103 pairs of HCC tissues and their corresponding adjacent normal liver tissues were examined by RT-qPCR analysis. We observed that SNHG16 expression levels were significantly increased in HCC tissues compared to that in their normal counterparts (Figure 1A). Also, as shown in Figure 1B, SNHG16 expression levels were significantly higher in all four HCC cell lines than that in LO2 cells.
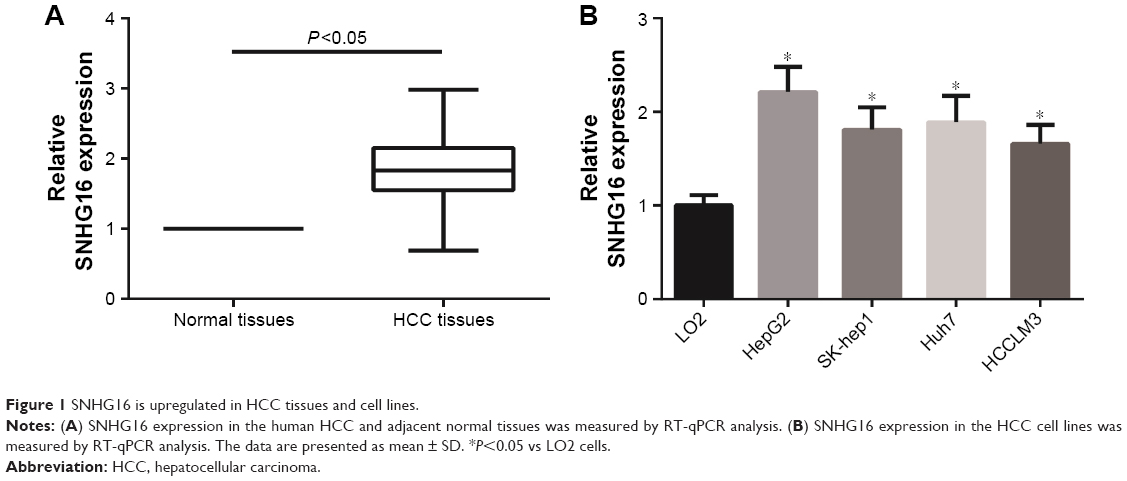 | Figure 1 SNHG16 is upregulated in HCC tissues and cell lines. |
Next, we examined the potential clinical significance of SNHG16 in HCC. A total of 103 HCC patients were divided into a low-expression group (<mean, n=55) and a high-expression group (≥mean, n=48) according to the cutoff value of SNHG16 expression. As listed in Table 1, high SNHG16 expression in HCC tissues was closely associated with tumor size (P=0.027), TNM stage (P=0.001), and vascular invasion (P=0.018) of HCC patients.
SNHG16 is upregulated in sorafenib-resistant HCC cells
As indicated by RT-qPCR analysis, HepG2/SOR cells showed more powerful resistance to sorafenib than HepG2 cells, and the half maximal inhibitory concentration (IC50) values of HepG2 cells and HepG2/SOR cells to sorafenib were 4.1 and 22.8 μM, respectively (Figure 2A). In addition, the expression of SNHG16 in HepG2/SOR cells was remarkably higher than that in HepG2 cells (Figure 2B).
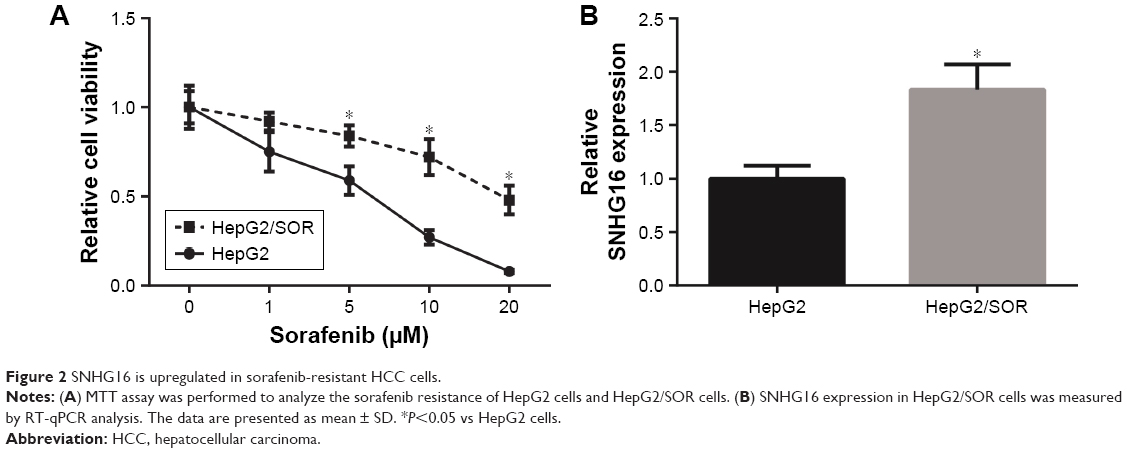 | Figure 2 SNHG16 is upregulated in sorafenib-resistant HCC cells. |
Knockdown of SNHG16 attenuates sorafenib resistance in vitro and in vivo
To further determine the critical role of SNHG16 in sorafenib resistance, si-SNHG16 was transfected into HepG2/SOR cells to knockdown the expression of SNHG16, and the transfection was successful, as confirmed by RT-qPCR analysis (Figure 3A). The results of MTT assay indicated that knockdown of SNHG16 reduced the IC50 of HepG2/SOR cells to sorafenib to 7.5 μM (Figure 3B).
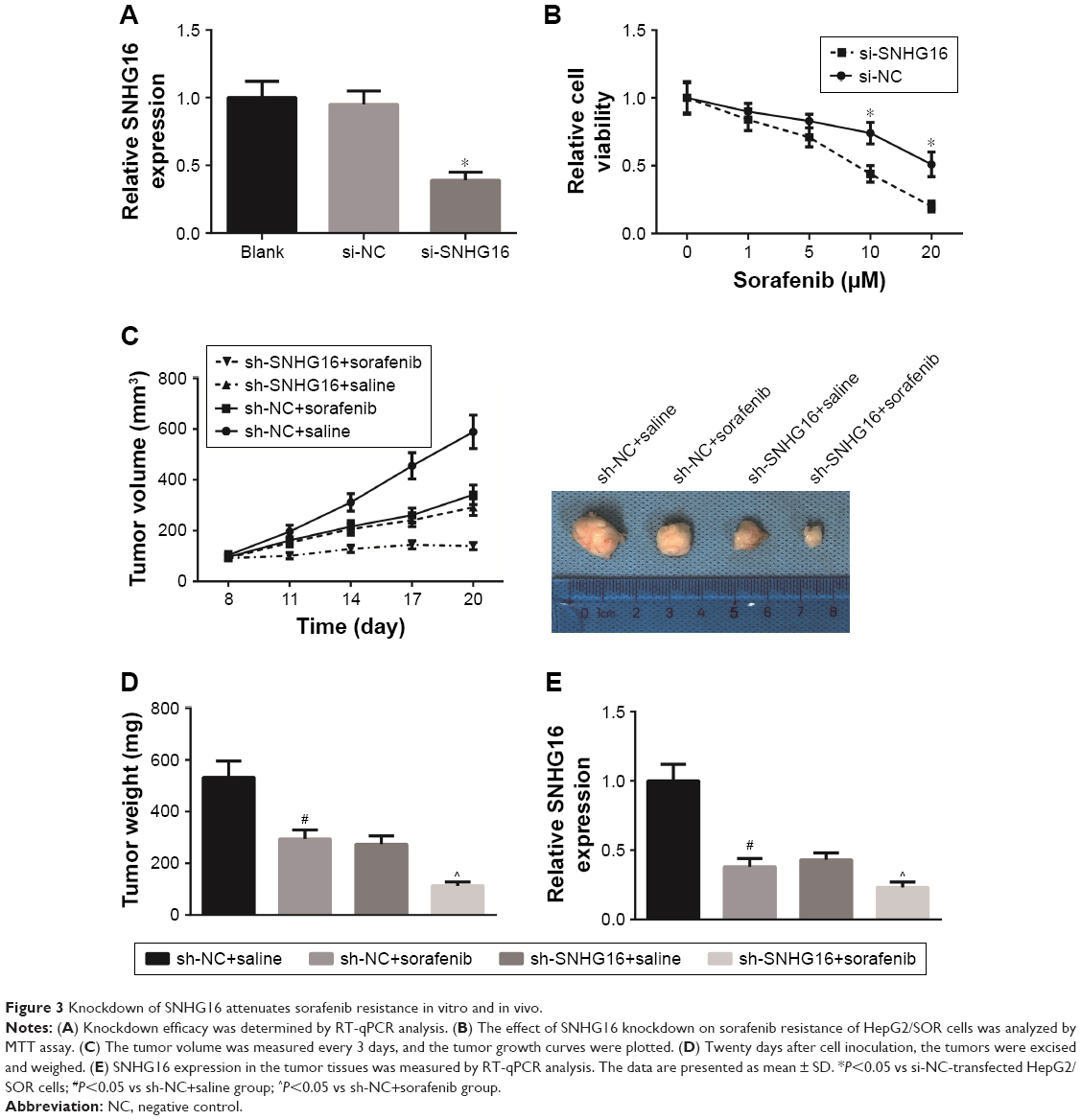 | Figure 3 Knockdown of SNHG16 attenuates sorafenib resistance in vitro and in vivo. |
Next a nude mouse-based xenograft model was established. As shown in Figure 3C, sorafenib treatment significantly reduced the tumor sizes, but cotreatment of sh-SNHG16 and sorafenib provided the most obvious suppressive effect on the tumor growth. Moreover, the average weight of tumors and the expression of SNHG16 were all lowest in sh-SNHG16+sorafenib group (Figure 3D and E).
SNHG16 directly interacts with miR-140-5p
Next, the nuclear and cytoplasmic fractions of HepG2 cells were separated, and we found that SNHG16 mostly distributed in the cytoplasm of HepG2 cells (Figure 4A). Furthermore, our bioinformatics analysis (http://starbase.sysu.edu.cn/mirLncRNA.php) revealed a binding site between miR-140-5p and SNHG16 (Figure 4B), and the results of dual luciferase reporter assay demonstrated a dramatically decreased relative luciferase activity in HepG2 cells cotransfected with SNHG16-WT and miR-140-5p mimics (Figure 4C). Then biotin-labeled miR-140-5p and its MUT mimics were used to pull-down SNHG16 in HepG2 cells with SNHG16 overexpression. As shown in Figure 4D, miR-140-5p-WT captured more SNHG16 compared with its MUT mimics. In addition, we found that SNHG16 knockdown could increase the expression of miR-140-5p in HepG2 cells (Figure 4E). We also found that compared with adjacent normal tissues, the expression of miR-140-5p was significantly reduced in HCC tissues (Figure 4F), and miR-140-5p expression was negatively correlated with SNHG16 expression in HCC tissues (P=0.007; Figure 4G).
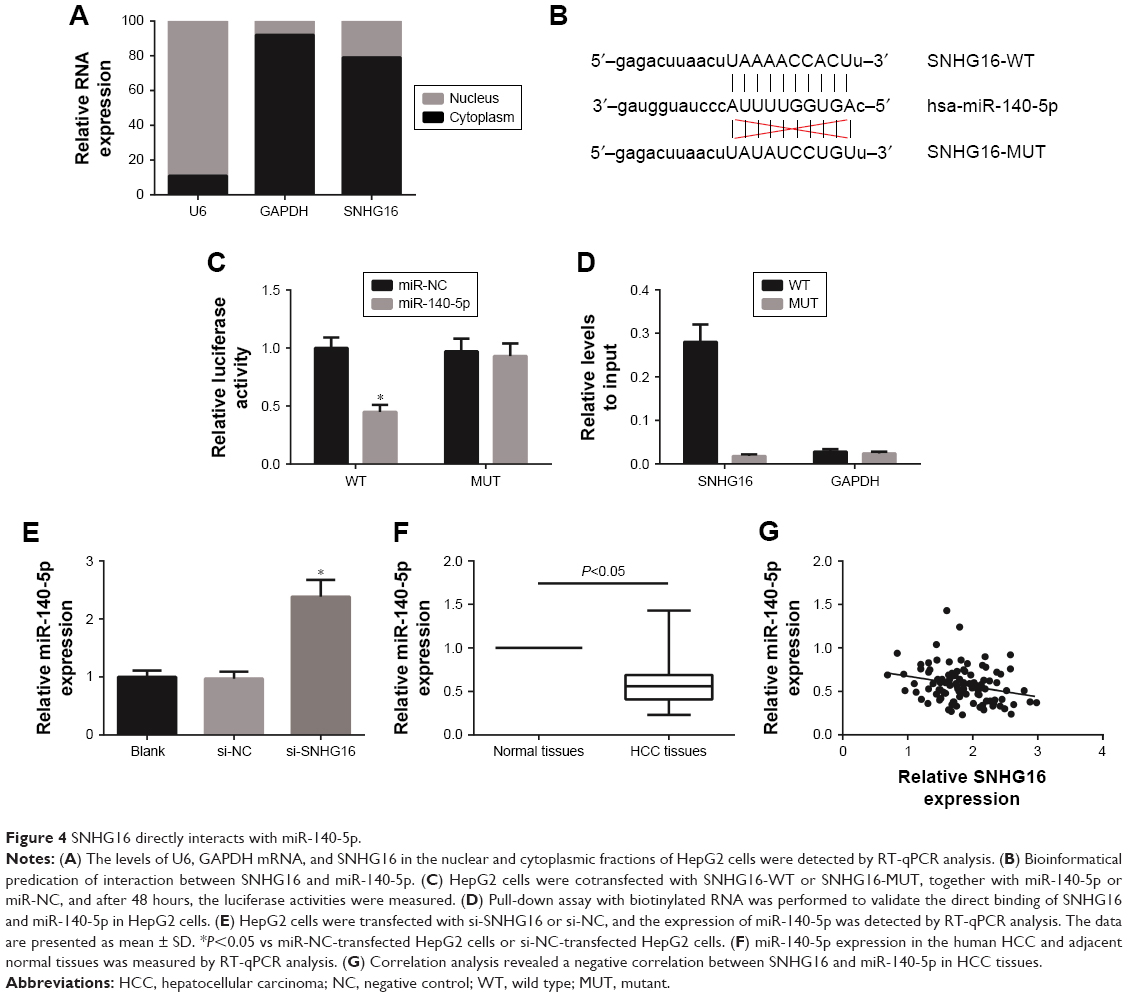 | Figure 4 SNHG16 directly interacts with miR-140-5p. |
MiR-140-5p inhibitor abrogates the effects of SNHG16 knockdown on sorafenib resistance
We further investigated the role of miR-140-5p on sorafenib resistance. We found that the expression of miR-140-5p in HepG2/SOR cells was significantly lower than that in HepG2 cells (Figure 5A). Besides, as indicated by MTT assay, miR-140-5p overexpression reduced the IC50 of HepG2/SOR cells to sorafenib to 9.0 μM (Figure 5B), and cotransfection with miR-140-5p inhibitor restored the IC50 of si-SNHG16-transfected HepG2/SOR cells to sorafenib to 13.3 μM (Figure 5C).
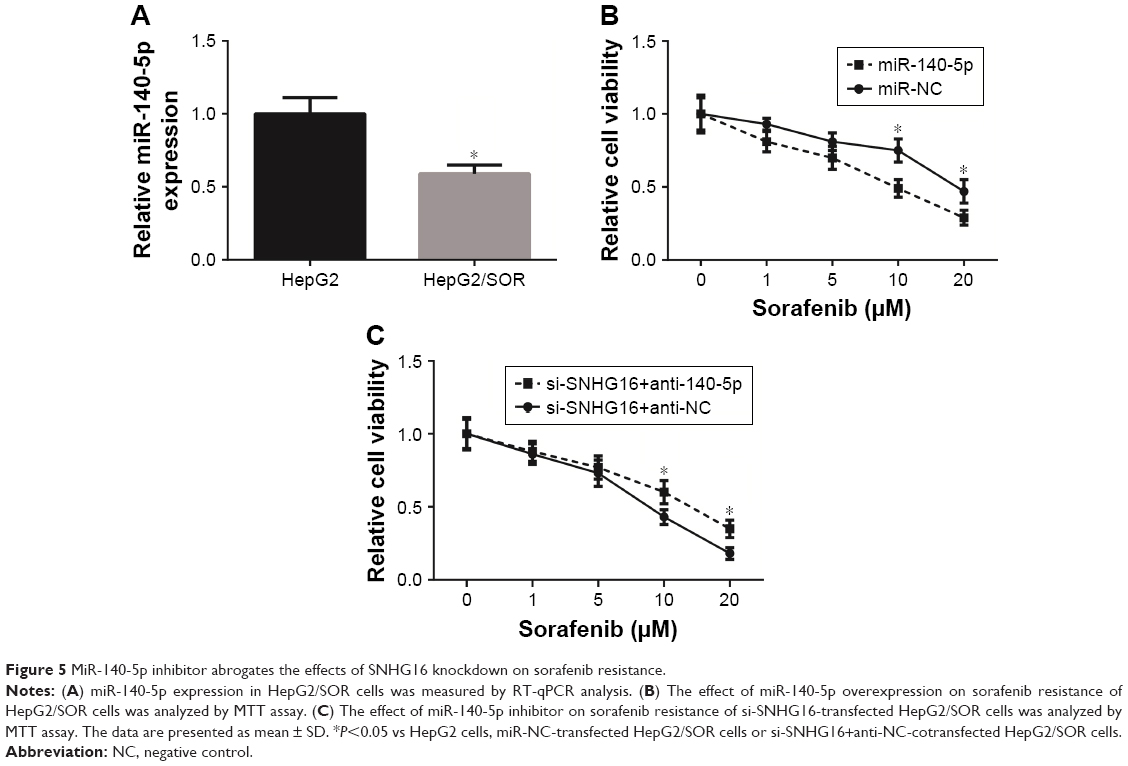 | Figure 5 MiR-140-5p inhibitor abrogates the effects of SNHG16 knockdown on sorafenib resistance. |
Discussion
Currently, HCC poses a big socioeconomic burden worldwide, and chemotherapy is widely used in the treatment of HCC.12 But chemoresistance, either intrinsic or acquired, still remains one of the major obstacles. Some lncRNAs are implicated in the development of sorafenib resistance in HCC. For example, forced expression of SNHG3 induced sorafenib resistance in HCC,13 and knockdown of TUC338 in HCC cells sensitized its reaction to sorafenib treatment.14 Therefore, lncRNAs provide an opportunity to identify effective therapeutic strategies for sorafenib resistance.
The oncogenic role of SNHG16 in other tumors has been frequently reported. In the present study, SNHG16 was found to be significantly upregulated in HCC tissues and cell lines, indicating the potential oncogenic role of SNHG16 in HCC. High SNHG16 expression in HCC tissues was closely associated with unfavorable clinicopathological features of HCC patients. Further experimental studies uncovered that SNHG16 was upregulated in sorafenib-resistant HCC cells, and SNHG16 knockdown significantly enhanced the sensitivity to sorafenib treatment in sorafenib-resistant HCC cells both in vitro and in vivo. These findings convincingly indicated that upregulation of SNHG16 might be one of the reasons for sorafenib resistance in HCC, but the underlying molecular mechanisms are required to be further elucidated.
The mechanisms of lncRNAs affecting tumorigenesis and chemoresistance vary with circumstance, and in recent years, a great deal of evidence has suggested that lncRNAs competitively bind to targeted miRNAs through serving as miRNA molecular sponges.15 In esophageal squamous cell carcinoma, SNHG16 has been reported to function as a molecular sponge of miR-140-5p.16 MiR-140-5p was previously reported to act as a tumor suppressor in HCC,17,18 and in this study, we found that SNHG16 directly interacted with miR-140-5p in HCC cells, and SNHG16 expression was negatively correlated with miR-140-5p expression in human HCC tissues. More importantly, miR-140-5p inhibitor abrogated the effects of SNHG16 knockdown on sorafenib resistance. These results disclosed an inverse relationship between SNHG16 and miR-140-5p in HCC and their pivotal role in sorafenib resistance.
In summary, our data showed that SNHG16 is upregulated in sorafenib-resistant HCC cells, and knockdown of SNHG16 attenuated sorafenib resistance partly through sponging miR-140-5p. To our knowledge, this is the first systematic study to indicate that the SNHG16/miR-140-5p regulatory axis might be a potential therapeutic target for HCC patients with sorafenib resistance.
Acknowledgment
The work of our research was supported by grant from Wu Jieping Medical Foundation (320.6750.17217).
Disclosure
The authors report no conflicts of interest in this work.
References
Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379(9822):1245–1255. | ||
Fan ST, Mau Lo C, Poon RT, et al. Continuous improvement of survival outcomes of resection of hepatocellular carcinoma: a 20-year experience. Ann Surg. 2011;253(4):745–758. | ||
Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. | ||
Avila M, Berasain C. Making sorafenib irresistible: In vivo screening for mechanisms of therapy resistance in hepatocellular carcinoma hits on Mapk14. Hepatology. 2015;61(5):1755–1757. | ||
Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43(6):904–914. | ||
Gutschner T, Diederichs S. The hallmarks of cancer: a long non-coding RNA point of view. RNA Biol. 2012;9(6):703–719. | ||
Cai C, Huo Q, Wang X, Chen B, Yang Q. SNHG16 contributes to breast cancer cell migration by competitively binding miR-98 with E2F5. Biochem Biophys Res Commun. 2017;485(2):272–278. | ||
Zhu H, Zeng Y, Zhou CC, Ye W. SNHG16/miR-216-5p/ZEB1 signal pathway contributes to the tumorigenesis of cervical cancer cells. Arch Biochem Biophys. 2018;637:1–8. | ||
Yang XS, Wang GX, Luo L. Long non-coding RNA SNHG16 promotes cell growth and metastasis in ovarian cancer. Eur Rev Med Pharmacol Sci. 2018;22(3):616–622. | ||
Lu YF, Cai XL, Li ZZ, et al. LncRNA SNHG16 functions as an oncogene by sponging MiR-4518 and up-regulating PRMT5 expression in glioma. Cell Physiol Biochem. 2018;45(5):1975–1985. | ||
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods. 2001;25(4):402–408. | ||
Devita VT, Chu E. A history of cancer chemotherapy. Cancer Res. 2008;68(21):8643–8653. | ||
Zhang PF, Wang F, Wu J, Wu Y, Huang W, Liu D, et al. LncRNA SNHG3 induces EMT and sorafenib resistance by modulating the miR-128/CD151 pathway in hepatocellular carcinoma. Journal of cellular physiology. 2019;234(3):2788–2794. | ||
Jin W, Chen L, Cai X, et al. Long non-coding RNA TUC338 is functionally involved in sorafenib-sensitized hepatocarcinoma cells by targeting RASAL1. Oncol Rep. 2017;37(1):273–280. | ||
du Z, Sun T, Hacisuleyman E, et al. Integrative analyses reveal a long noncoding RNA-mediated sponge regulatory network in prostate cancer. Nat Commun. 2016;7:10982. | ||
Zhang K, Chen J, Song H, Chen LB. SNHG16/miR-140-5p axis promotes esophagus cancer cell proliferation, migration and EMT formation through regulating ZEB1. Oncotarget. 2018;9(1):1028–1040. | ||
Yang H, Fang F, Chang R, Yang L. MicroRNA-140-5p suppresses tumor growth and metastasis by targeting transforming growth factor β receptor 1 and fibroblast growth factor 9 in hepatocellular carcinoma. Hepatology. 2013;58(1):205–217. | ||
Yan X, Zhu Z, Xu S, et al. MicroRNA-140-5p inhibits hepatocellular carcinoma by directly targeting the unique isomerase Pin1 to block multiple cancer-driving pathways. Sci Rep. 2017;7:45915. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.