Back to Journals » OncoTargets and Therapy » Volume 13
Long Noncoding RNA LINC00551 Suppresses Glycolysis and Tumor Progression by Regulating c-Myc-Mediated PKM2 Expression in Lung Adenocarcinoma
Authors Wang L ![]() , Wang H, Wu B
, Wang H, Wu B ![]() , Zhang C, Yu H, Li X
, Zhang C, Yu H, Li X ![]() , Wang Q
, Wang Q ![]() , Shi X
, Shi X ![]() , Fan C
, Fan C ![]() , Wang D, Luo J, Yang J
, Wang D, Luo J, Yang J ![]()
Received 19 August 2020
Accepted for publication 7 October 2020
Published 9 November 2020 Volume 2020:13 Pages 11459—11470
DOI https://doi.org/10.2147/OTT.S273797
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Federico Perche
Li Wang,1,* Huishan Wang,2,* Bining Wu,3,* Chun Zhang,1 Hualin Yu,1 Xueyan Li,1 Qinjue Wang,4 Xiaoli Shi,5 Chengfeng Fan,1 Dayu Wang,6 Jing Luo,7 Jinsong Yang1
1Department of Oncology, Nanjing First Hospital, Nanjing Medical University, Nanjing, People’s Republic of China; 2Department of Gastroenterology, Shanghai Songjiang District Central Hospital, Shanghai, People’s Republic of China; 3Respiratory Department, Nanjing Yuhua Hospital, Yuhua Branch of Nanjing First Hospital, Nanjing, People’s Republic of China; 4Department of Orthopedics, Nanjing First Hospital, Nanjing Medical University, Nanjing, People’s Republic of China; 5Department of Hepatology Surgery, First Affiliated Hospital of Nanjing Medical University, Nanjing, People’s Republic of China; 6Department of Obstetrics and Gynecology, Drum Tower Hospital, Medical School of Nanjing University, Nanjing, People’s Republic of China; 7Department of Cardiothoracic Surgery, Jinling Hospital, Medical School of Nanjing University, Nanjing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jinsong Yang
Department of Oncology, Nanjing First Hospital, Nanjing Medical University, 68 Changle Road, Nanjing City, Jiangsu Province, People’s Republic of China
Tel +86 18951670329
Fax +86 25-86621776
Email [email protected]
Jing Luo
Department of Cardiothoracic Surgery, Jinling Hospital, Medical School of Nanjing University, 305 East Zhongshan Road, Nanjing City, Jiangsu Province, People’s Republic of China
Tel +86 15996230938
Email [email protected]
Background: Lung adenocarcinoma (LUAD) is a leading cause of mortality associated with cancer globally. Thus, it is essential to elucidate its tumorigenesis and prognosis. Accumulating evidence shows that long noncoding RNAs (lncRNAs) play important roles in the occurrence and progression of tumors by regulating their glucose metabolism.
Methods: Bioinformatics analysis was performed to explore the expression of LINC00551 in LUAD. The level of LINC00551 in LUAD cells and tissues was detected by RT-qPCR. CCK-8, colony formation, EDU and transwell assays were conducted to evaluate the cell growth and migration of LUAD cells (A549 and PC9). High throughput sequencing was used to discover the downstream genes of LINC00551. The metabolic function of LUAD cells was identified by glucose uptake and lactate production assays. Furthermore, tumor xenografts were established to investigate the effects of LINC00551 on tumor growth in vivo.
Results: Herein, we found that LINC00551 was low-expressed in LUAD, and its level correlated with clinical prognosis. Ectopic expression of LINC00551 inhibited the proliferation and migration of LUAD cells (A549 and PC9). High throughput sequencing and gene enrichment analysis revealed that LINC0551 may be involved in metabolic pathway. Glucose uptake and lactate production assays suggested that LINC00551 suppressed glycolysis of LUAD cells. Mechanistically, our work revealed that LINC00551 inhibited glycolysis in LUAD cells by impairing c-Myc-mediated transcription of an important glycolysis-related enzyme PKM2.
Conclusion: In summary, our study identifies LINC00551 as a tumor suppressor in LUAD and implicates the LINC00551/c-Myc/PKM2 axis in the glycolytic remodeling of LUAD.
Keywords: lung adenocarcinoma, LINC00551, c-Myc, PKM2, glycolysis
Introduction
Globally, lung cancer is the leading cause of cancer-associated mortality, with less than 20% survival rate in 5 years.1 Lung adenocarcinoma (LUAD) accounts for 40% of all diagnosed lung cancers.2 Although great advances have been made in the clinical treatment of LUAD,3 overall survival time of patients with LUAD has not improved dramatically, principally due to a lack of reliable molecular biomarkers. Thus, a better understanding of the molecular mechanisms underlying LUAD initiation and progression may improve the diagnosis and therapy of this lethal disease.
Due to the advancement of sequencing technology, numerous long noncoding RNAs (lncRNAs) have been identified. LncRNAs are defined as transcripts with more than 200 nucleotides in length that usually lack protein-coding capacity.4 A growing body of literature implicates lncRNAs in every step of physiological and pathological processes, at the genome level, transcriptional level, posttranscriptional level, translational level and posttranslational level.5
Metabolic remodeling is a defining hallmark of cancer and its common feature is the preference for the glycolytic pathway for ATP generation even in the presence of oxygen to meet the high energy demands for growth, invasion, and metastasis.6 This phenomenon, also known as the Warburg effect or aerobic glycolysis,7 results in increased glucose uptake, elevated lactate levels, decreased ATP production, and more readily available energy supply.8 LncRNAs regulate glucose metabolism in tumor cells by modulating metabolic enzymes, regulatory molecules, signaling pathways, and related oncogenes.9,10 A study by Song et al revealed that lncRNA PVT1 is a molecular sponge that suppresses the miR-497 expression, thus promoting glucose metabolism and cell growth in osteosarcoma via the PVT1/miR-497/HK2axis.11 Zhao et al found that LINC00092 is highly expressed in ovarian carcinoma where it binds glycolysis pathway enzyme PKKFB2 (fructose-2,6-bisphosphatase) to facilitate glucose breakdown and thereby accelerating tumor metastasis and sustains localized cancer-related fibroblasts reinforcement.12 Although the effects of lncRNAs on the glucose metabolism have been described, there is still a far way to clarify and discover the mechanisms underlying lncRNA regulation of glucose metabolism. Specifically, it is necessary to determine which other glycolytic enzymes and metabolic pathways lncRNAs are associated with. In the present research, we identified that LINC00551 was downregulated in LUAD tissues and exerted suppressive roles in the progression and glycolysis of LUAD, partially by regulating the expression of c-Myc-mediated PKM2. Our study enhanced understanding on the complex networks of glucose metabolism in cancer and offered a possible molecular biomarker for clinical diagnosis and therapy of diseases.
Materials and Methods
Patient Tissue Samples
A total of 32 paired LUAD and adjacent healthy tissues were obtained from the Department of Cardiothoracic Surgery, Nanjing First Hospital, Nanjing Medical University between 2016 and 2019. After excision, the samples were rapidly frozen in liquid nitrogen. Comprehensive data concerning clinical features, tumor stage, and histopathological features were obtained under authorization by the Ethics Committee of Nanjing First Hospital. An informed consent for participation was obtained from patients prior to analysis. The clinical information of these 32 patients were shown in Table S1.
Cell Culture
Here, 4 human lung adenocarcinoma lines, A549, H1299, H1975, and PC9, and one normal bronchial epithelial cell, HBE were utilized. A549 and H1299 cell lines were obtained from Jiangsu Cancer Hospital, while PC9, H1975, and HBE cell lines were obtained from Jinling Hospital. All cell lines were approved by Nanjing Hospital affiliated to Nanjing Medical University. HBE, A549, and H1299 cells were cultivated in RPMI-1640 medium (Gibco, China) supplemented with 10% fetal bovine serum (FBS, Science Cell), 100 U/mL penicillin-streptomycin mixture, L-glutamine and, sodium pyruvate, in 5% CO2 presence at 37 °C. H1975 and PC9 cells were maintained under the same conditions, in Dulbecco’s Modified Eagle Medium (DMEM; Gibco, China).
Plasmid Transfection
The full-length complementary DNA (cDNA) of LINC00551 and c-Myc were cloned into the pcDNA3.1+ expression vector. DNA Midiprep kits (OMEGA, Shanghai, China) was used to extract the plasmid vectors. Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) and P3000 (Invitrogen) were utilized to transfect the plasmids into LUAD cell lines (A549 and PC9 cells) in a six-well plate according to the manufacture’s protocols. Subsequent experiments were performed after 24 or 48 h transfection. The transfection efficacy was confirmed through quantitative PCR (RT-qPCR) prior to biological function experiments.
Total Extraction of RNA and RT-qPCR
Total RNA was extracted from LUAD tissue samples and cultured cells using Trizol reagent (Invitrogen, USA) according to the manufacture’s protocol. The RNA was reverse transcribed into cDNA using PrimerScript Reverse Transcription Kit (Takara, RR047A) and amplified by qRT-PCR with TB Green™ Premix Ex Taq (Takara RR420A) in an ABI 7500 System (Applied Biosystems, Foster City, CA, USA). Actin housekeeping gene acted as the internal control. The 2−ΔΔCt method quantified the expression of the target gene. The primers used in this study were acquired from Invitrogen Shanghai Sangon Biotech (Invitrogen, China). The primers sequences were as follows: LINC00551 Forward: 5ʹ-TCAGCAGCCTTCAGTTGGAG-3ʹ Reverse: 5ʹ-AGGCTCTGTTCAGCTGGTTC-3ʹ; c-Myc Forward: 5ʹ-GGCTCCTGGCAAAAGGTCA-3ʹ Reverse: 5ʹ-CTGCGTAGTTGTGCTGATGT-3ʹ; Actin Forward: 5ʹ-AGCGAGCATCCCCCAAAGTT-3ʹ Reverse: 5ʹ- GGGCACGAAGGCTCATCATT-3ʹ; PKM2 Forward: 5ʹ-ATGTCGAAGCCCCATAGTGAA-3ʹ Reverse: 5ʹ-TGGGTGGTGAATCAATGTCC-3ʹ.
The Cell Proliferation Assays
LUAD cells, A549 and PC9, were transfected with plasmids or small interfering RNAs (siRNAs) prior to cell proliferation assays. For the EDU (5-ethynyl-2′-deoxyuridine) assay (EdU Apollo®488 In Vitro Imaging Kit, RiboBio, Guangzhou, China), 15◊103 cells/100 µL/well were seeded into a 96-well plate. Once the cells adhered to the plate, 100 µL reagent A was added in the wells and allowed to incubate at 37 ºC for 2 h. Afterward, the cells were fixed with 4% paraformaldehyde (Biosharp), treated with 0.5% TritonX-100 (100 µL), then subjected to Apollo and Hoechst staining. After washing with phosphate-buffered saline (PBS) three times, the cells were photographed using a fluorescence microscope to calculate the proliferation rates. In CCK-8 assay, 2◊103 cells/well were seeded into 96-well plates and 10 µL CCK-8 reagent was added. And then the plates were incubated for 2 h under 5% CO2 environment. After 0-, 24-, 48-, 72-, and 96-h incubation, absorbance at 450 nm was recorded. To evaluate colony-formation, 400 cells/well were seeded into 6-well plate and cultivated for 10–14 days. Following incubation, the cells were fixed in 4% paraformaldehyde for 30 min, stained with 0.1% crystal violet solution, washed with PBS, and then photographed.
Transwell Migration Assay
The migrate potential of LUAD cells (A549 and PC9) was assessed using the Millicell Hanging Cell Culture Insert (Millicell MCEP24H48). According to c recommendations, 4 ◊ 104 cells in 200 μL of serum-free medium were put in the upper chamber while lower chamber was filled with 800 µL 10% FBS supplemented medium. After incubating for 24-h at 37 °C, non-migrated cells were scraped out using cotton swabs. Subsequently, migrated cells were fixed, stained with 10% crystal violet solution, washed with PBS, and then photographed.
Analysis of Nuclear and Cytoplasmic RNA
Cytoplasmic and nuclear RNA was isolated using the PARIS Kit (Invitrogen), following the provided procedure. Briefly, LUAD cells (A549 and PC9) were trypsinized and washed twice with PBS. After cell pellets lysis using buffer I, and the cytoplasmic lysate was collected as supernatant. Nuclear pellets were further lysed in buffer II and the supernatants were collected as nuclear lysates following centrifugation. The cytoplasmic and nuclear portions were divided to extract RNA and for qRT-PCR. The GAPDH and U6 were applied as corresponding cytoplasmic and nuclear markers, respectively.
In vivo Tumorigenesis Assay
To assess LINC00551 effect in vivo, male mice (age 6–8 weeks) were randomly classified into 2 groups of 5 mice. The mice were subcutaneously injected with 2 ◊ 106 A549 cells/100 µL sterile PBS. The LUAD cells had been transfected with plasmids expressing LINC00551 cDNA, or an empty pcDNA3.1+ vector (control). Starting one week after the injection, the mice were observed three times a week for six weeks, and tumor growth was evaluated by measuring harvested tumor nodules volume and mass. All animal studies were performed in accordance with NIH animal use guidelines. The study design was approved by Experimental Animal Ethics Committee of Nanjing Hospital affiliated to Nanjing Medical University.
Bioinformatic Study
Human exon arrays for LUAD and adjacent healthy tissues were downloaded from the NCBI’s Gene Expression Omnibus (GEO, GSE19804 https://www.ncbi.nlm.nih.gov/gds/?term=GSE19804). Correlation analysis between expression of c-Myc and glycolysis enzymes was performed using GEPIA (http://gepia.cancer-pku.cn/index.html). The protein-coding potential of LINC00551 was assessed using CPAT (http://lilab.research.bcm.edu/cpat/index.php) and LNCipedia (https://lncipedia.org/db). Survival analysis of LINC00551 and PKM2 was performed using GEPIA. Cancer Cell Line Encyclopedia (https://portals.broadinstitute.org/ccle/about) was queried for LINC00551 expression in different cancer cell lines.
RNA Sequencing Assay
To further illustrate the molecular mechanism of LINC00551 function in LUAD, we conducted high-throughput RNA sequencing in control and LINC00551-overexpressing A549 cells. The threshold set for upregulated and downregulated genes was a fold change >2.0 and a P value < 0.05. Differentially expressed genes were subjected to enrichment analysis to determine the biological processes that LINC00551 may participate in.
Measurement of Glucose Uptake
Glucose levels were quantified with a Standard glucose assay kit (Thermo Fisher Scientific). In brief, the LINC00551 overexpression plasmid and pCDNA3.1+ vector control plasmid was used to transfect A549 and PC9 cells for 24 h and the harvested cells were seeded into a 96-well plate (5 x 104 cells/well). Once the cells attached to the plate, the culture medium was removed and replaced by serum-free medium for 6 h to overnight. Subsequently, 50 µL of fluorescent 2-DG (Molecular Probes, Invitrogen) was added and the cells were incubated at 37 °C under 5% CO2 environment for 30 min. The cells were washed twice with cold PBS and lysed before Assay Mixture (100 µL/well) was added, and kept at room temperature for 30–120 min. Cellular 2-DG uptake was measured by monitoring optical density ratio increase at 570/610 nm using a microplate reader.
Quantification of Lactate Levels
LUAD Cells (A549 and PC9) were seeded in a 6-well plate and transfected with a control plasmid or LINC00551-overexpressing plasmid. The lactate assay kit (Nanjing Jiancheng Bioengineering, China) was utilized to quantify the lactate levels in the culture medium based on the manufacturer’s guidelines. Absorbance at 570 nm was recorded via a plate reader. The outcomes were standardized to the total protein concentration of each sample.
Statistical Analyses
All statistical studies were completed in GraphPad Prism (version 7.0, GraphPad, USA). Student’s t-test was employed to compare group means while Log rank test and Kaplan-Meier plots were used to evaluate the survival differences. The correlation among the different genes’ expression was assessed through Pearson correlation analysis. P values < 0.05 were considered statistically significant. All data are expressed as the mean ± standard deviation (SD).
Results
LINC00551 Was Downregulated in LUAD
Since the function of LINC00551 has not been studied in LUAD, we sought to determine whether LINC00551 could play a role in LUAD tumorigenesis and development. LINC00551 is located at 13q33.3, with 4 exons and 1967 bp in length (Figure S1). According to CPAT and LNCipedia, LINC00551 has almost no protein-coding potential (coding probability: 0.11) (Figure 1A and B). Analysis of the GTEx dataset and Cancer Cell Line Encyclopedia revealed that LINC00551 is highly expressed in normal lung tissue (Figure S2), but relatively low expressed in non-small-cell lung cancer cells (Figure S3). In GEPIA, LINC00551 was downregulated in LUAD (Figure 1C), and correlated with tumor stage of LUAD patients. (Figure 1D). The overall survival analysis using GEPIA showed that low LINC00551 expression predicted a poorer prognosis in LUAD patients (Figure 1E). In line with these results, LINC00551 expression was markedly lower in LUAD cell lines (A549, H1299, H1975, and PC9) and tumor tissue than in normal lung epithelial cell line HBE (Figure 1F) and adjacent normal tissue, respectively (Figure 1G). Since the subcellular localization of lncRNAs largely influences their mechanism of action,13 we analyzed the nuclear and cytoplasmic RNA fractions isolated from A549 and PC9 cells to estimate the localization of LINC00551. Approximately 75% of LINC00551 located in the nucleus of LUAD cells (Figure 1H). These results implied that LINC00551was downregulated in LUAD cells and tissues and might exert its function in LUAD progression.
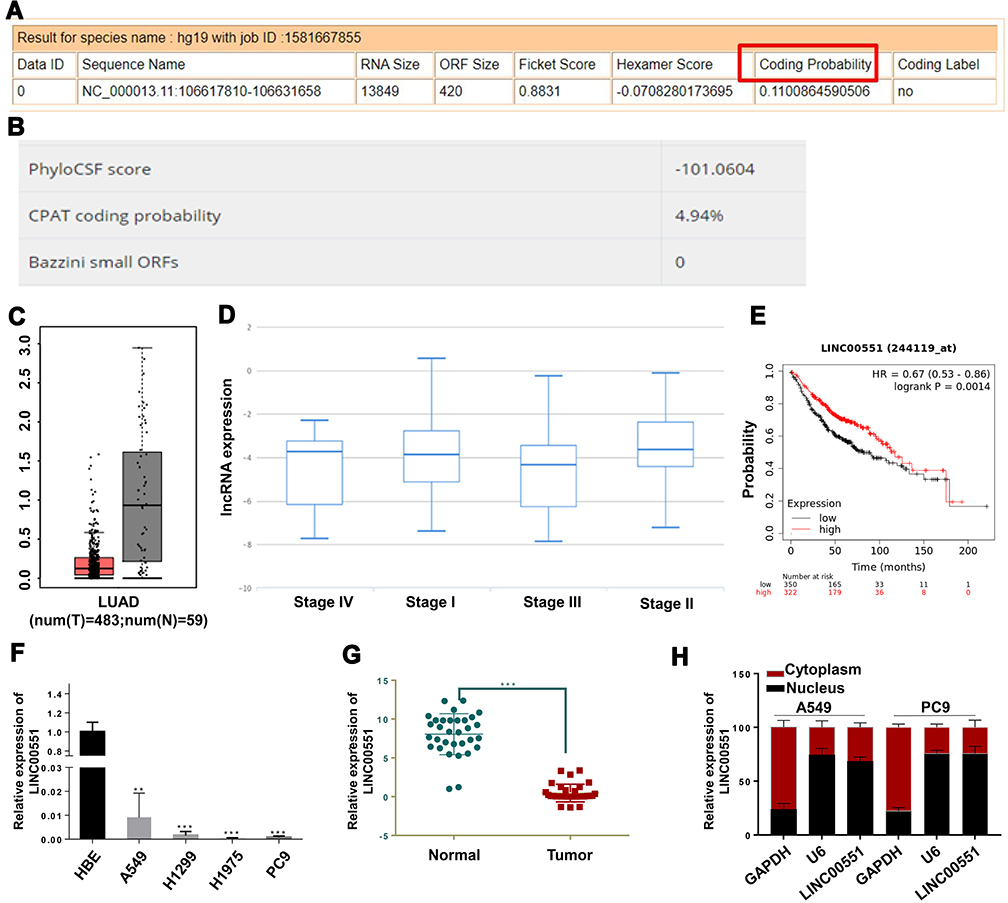 |
Figure 1 LINC00551 was downregulated in lung adenocarcinoma and correlated with clinical prognosis. (A and B). Computational assessment of lncRNAs in CAPT and Incipedia database revealed that LINC00551 was of limited protein-coding capacity (coding probability marked with red box). (C). LINC00551 was downregulated in LUAD tissues compared with adjacent normal tissues in TCGA dataset. (D). LINC00551 correlated with tumor stages of LUAD in TANRIC database. (E). Kaplan-Meier survival analysis showed that low expression of LINC00551 was associated with a worse overall survival of lung cancer patients. (F–G). Expression profiles of LINC00551 in human lung adenocarcinoma cell lines relative to one normal lung epithelial cell line (HBE) and 32 paired clinical samples of LUAD. 1h. Subcellular localization studies indicated that LINC00551 was mainly located in nucleus. (**p < 0.01; ***p < 0.001). |
LINC00551 Overexpression Suppressed Proliferation, Colony Formation, and Migration of LUAD Cells in vitro
To further explore the biological effects of LINC00551 on LUAD development, we overexpressed LINC00551 in A549 and PC9 cells by plasmid transfection (Figure 2A). Overexpression of LINC00551 inhibited cell proliferation (Figure 2B) and decreased colony formation (Figure 2C and B) and migration in both cell lines (Figure 2E–F). EDU staining confirmed that LINC00551 impaired the proliferation of LUAD cells (Figure 2G–H).
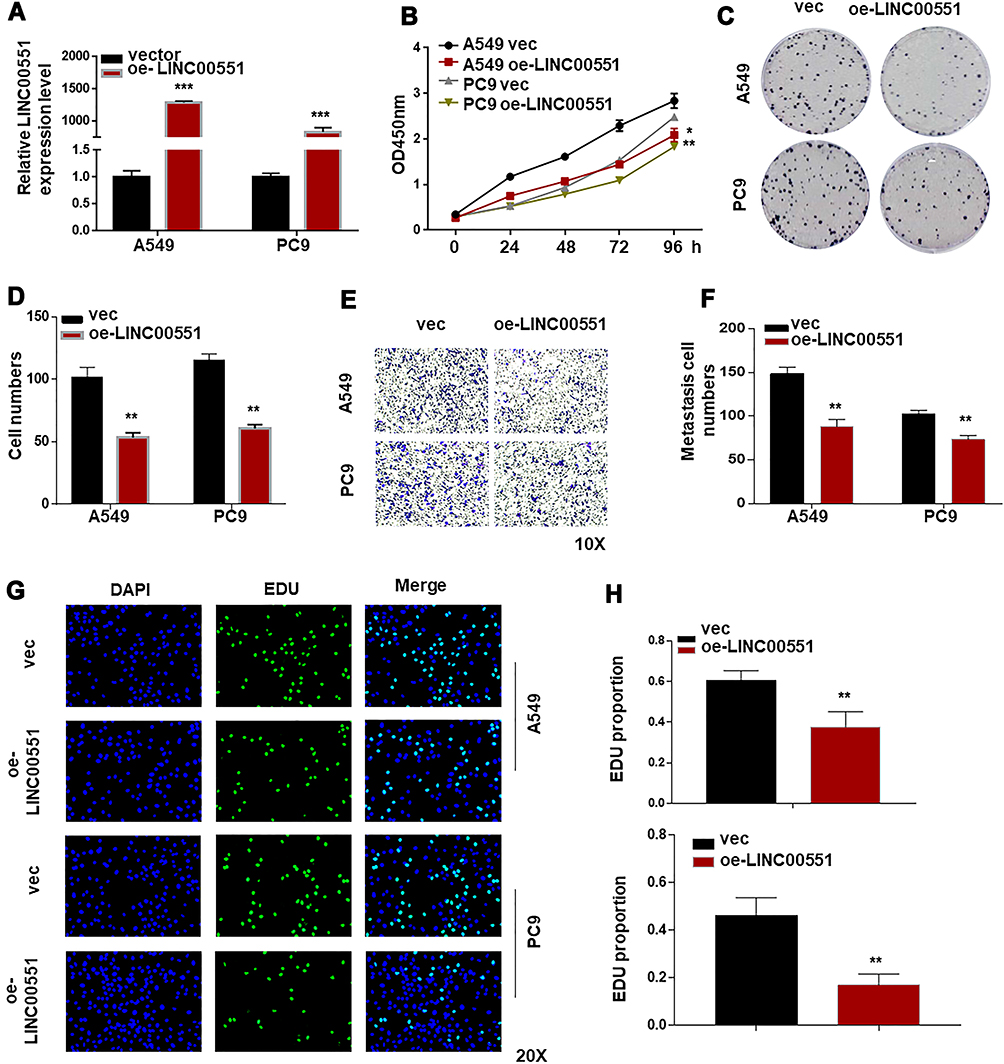 |
Figure 2 LINC00551 suppressed the progression of LUAD cells. (A). Verification of LINC00551 over-expression efficiency in A549 and PC9 cells by qRT-PCR. (B). CCK-8 assay demonstrated that LINC00551 inhibited the proliferation of A549 and PC9 cells. (C–D). Overexpression of LINC00551 inhibited the ability of colony formation of A549 and PC9 cells. (E–F). Transwell assay showed that LINC00551 could inhibit the migration ability of A549 and PC9 cells. (G–H). Overexpression of LINC00551 decreased cell proliferation of A549 and PC9 cells, as detected by EdU staining. The data were represented as the means ± SD of at least three independent experiments. (*p < 0.05; **p < 0.01; ***p < 0.001). |
LINC00551 Overexpression Reduced c-Myc Levels and Affected Aerobic Glycolysis via PKM2
To clarify the underlying mechanisms of LINC00551 in LUAD, we performed high-throughput RNA sequencing of LINC00551-overexpressing A549 cells (Figure 3A). Gene enrichment analysis of differentially expressed genes revealed that LINC0551 may be involved in metabolic pathways (Figure 3B). Since altered glucose metabolism is a distinct tumor cell feature, we hypothesized that LINC00551 participates in glucose metabolism. Indeed, overexpression of LINC00551 decreased the levels of glycolytic enzymes, especially PKM2 (Figure 3C), inhibited cellular uptake of glucose (Figure 3D), and reduced synthesis of lactate (Figure 3E) in the A549 and PC9 cells. To further elucidate the mechanisms of LINC00551 in LUAD glycolysis, we found MYC mRNA level was significantly decreased among multiple differentially expressed genes as shown in Figure 3A consistent with the result of RT-qPCR (Figure 3F). MYC is a well-known oncogene encoding c-Myc transcription factor,7 which enhances cell development and progression of14 and controls the transcription of many of the genes involved in glucose metabolism, including PKM2.15 Furthermore, the expression of c-Myc positively correlated with PKM in TCGA dataset (Figure 3G). Considering that LINC00551 was mainly located in the nucleus (Figure 1H), we hypothesized that LINC00551 regulates glucose metabolism through c-Myc-mediated transcription of PKM2.
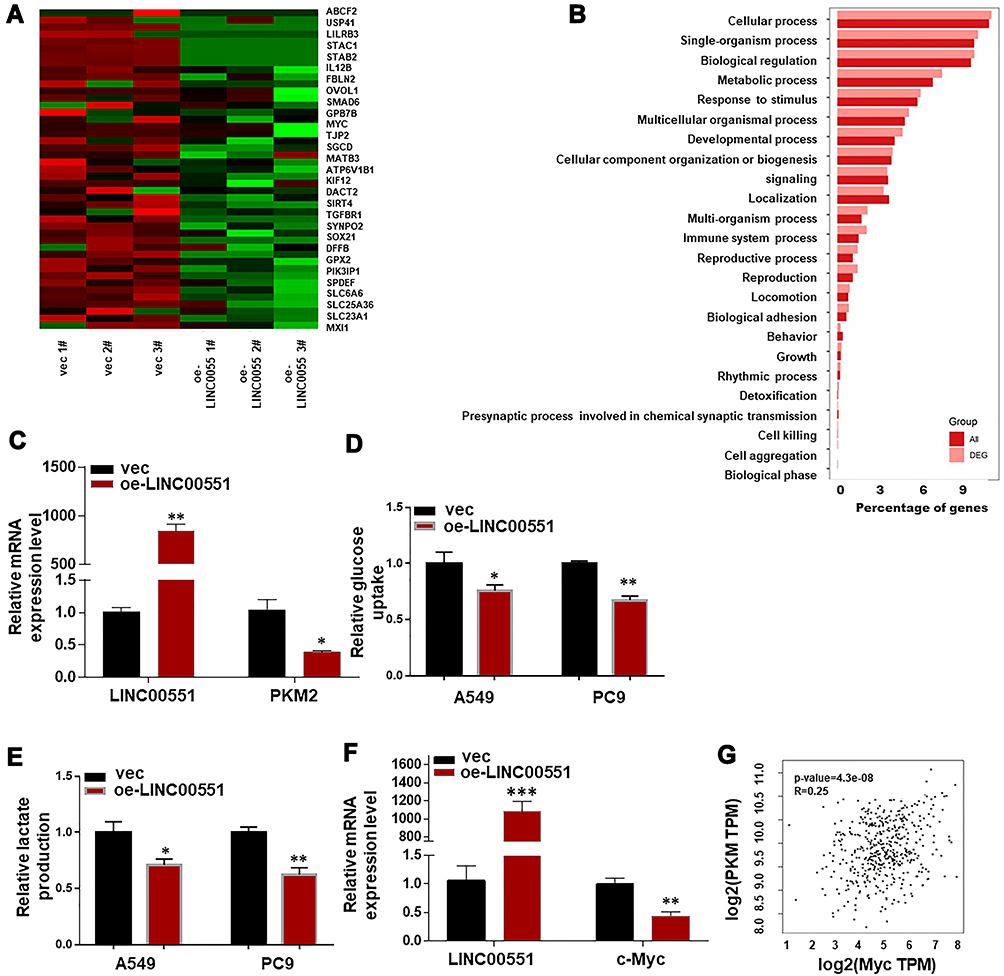 |
Figure 3 LINC00551 overexpression reduced c-Myc expression and affected aerobic glycolysis via PKM2. (A). Differential expressed genes were compared between control and LINC00551-overexpression A549 cells. (B). Enrichment analysis of DEGs (Differential Expressed Genes) in biological process (BP) sets hinted that LINC00551 might participate in metabolic process. (C). Overexpression of LINC00551 decreased expression of PKM2. (D–E). LINC00551 inhibited the aerobic glycolysis of LUAD cells (A549 and PC9), confirmed by glucose uptake and lactate production. (F). Overexpression of LINC00551 down-regulated the expression of c-Myc. (G). The expression of c-Mycpositively correlated with PKM in TCGA dataset. The data are represented as the means ± SD of at least three independent experiments. (*p < 0.05; **p < 0.01; ***p < 0.001). |
c-Myc Overexpression in LINC00551-Overexpressing Cells Restored PKM2 Expression and Warburg Effect, and Inhibited LUAD Progression
To test the hypothesis that the effect of LINC00551 on glycolysis is mediated by c-Myc, we performed further experiments. The results showed that PKM2 mRNA level could be restored by c-Myc in LINC00551-overexpressing A549 cells (Figure 4A). c-Myccould also rescue the inhibitory effect of LINC00551 overexpression on glucose uptake, lactate production (Figure 4B and C), cell proliferation (Figure 4D-G), and metastasis in A549 and PC9 cells (Figure 4H-I). Overall, these results indicated that LINC00551 inhibited development of LUAD cells by affecting tumor cell glycolysis via c-Myc-mediated transcription of PKM2.
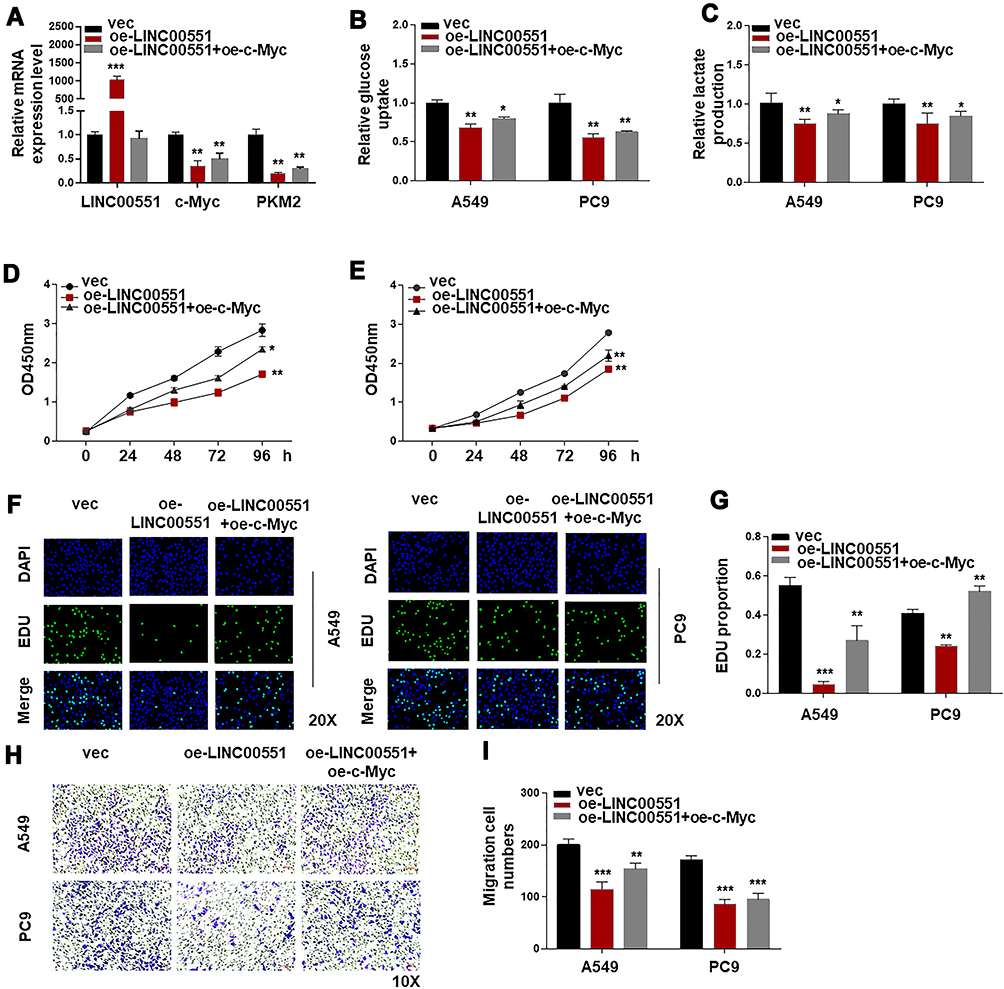 |
Figure 4 LINC00551 regulated glucose metabolism of LUAD partly by controlling the expression of c-Myc. (A). c-Myc complementation in LINC00551-overexpressed cells (A549 and PC9) restored PKM2 expression. (B–C). Ectopic overexpression of c-Mycrescued the influence of LINC00551 on glucose metabolism of LUAD cells (A549 and PC9). (D–I). c-Myc partially reversed the effect of LINC00551 on proliferation and migration of LUAD cells (A549 and PC9). (*p < 0.05; **p < 0.01; ***p < 0.001). |
LINC00551 Overexpression Suppressed LUAD Development in vivo
To examine the function of LINC00551 in vivo, xenograft tumors were utilized (Figure 5A). Overexpression of LINC00551 markedly reduced tumor volume and weight (Figure 5B and C). We also testified that LINC00551 was low expressed in harvested tumor nodules and plasma (Figure 5D-E). Additionally, high level of PKM2 correlated with poor clinical prognosis in GEPIA dataset (Figure 5F). Altogether, these outcomes reveal that LINC00551 suppresses the development of LUAD through c-Myc mediated glycolysis enzyme PKM2 (Figure 5G).
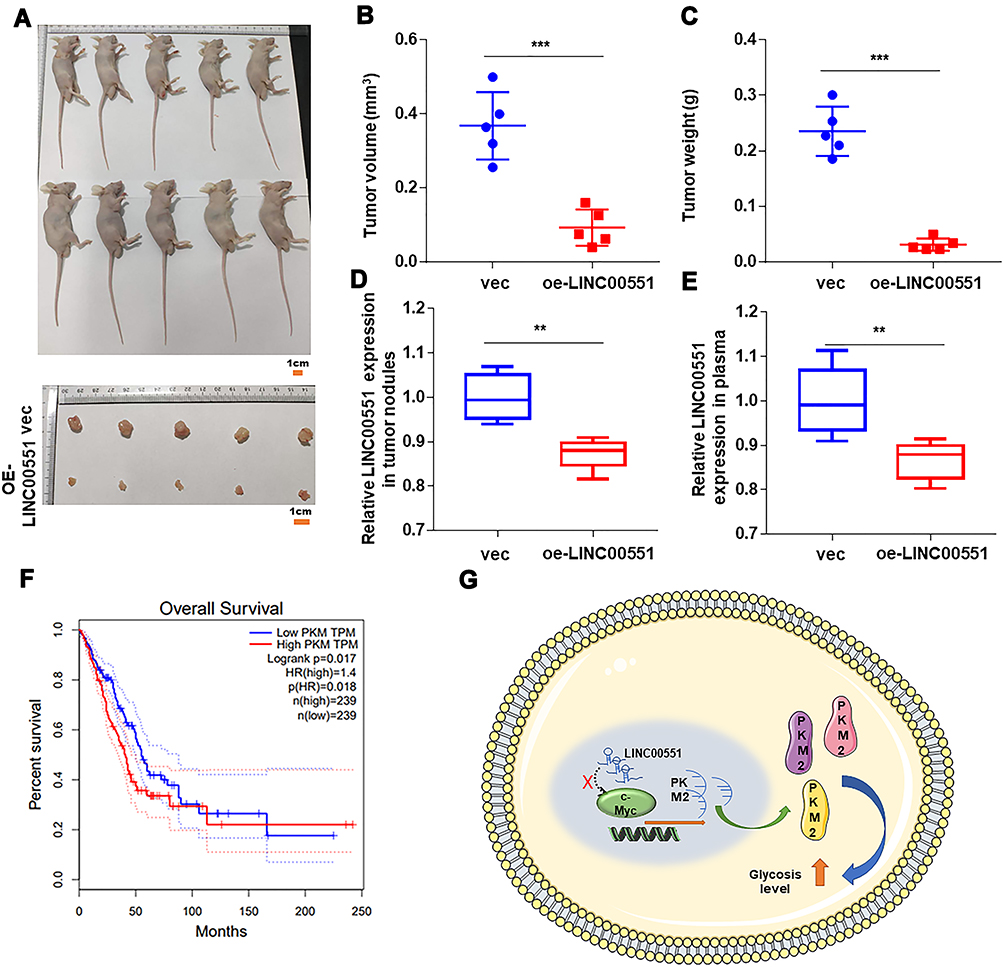 |
Figure 5 LINC00551 repressed LUAD proliferation in vivo. (A). Harvested tumor nodules were shown. (B–C). The volume and weight of tumor nodules of each group are measured and the results revealed that LINC00551 inhibited proliferation of LUAD. (D–E). LINC00551 also low expressed in harvested tumor nodules and plasma. (F). Patients with high expression of PKM have a poor prognosis in GEPIA database. (G). Schematic diagram of LINC00551 affects tumor progression through c-Myc regulation of PKM. The data are represented as the means ± SD (**p < 0.01 ***p < 0.001). |
Discussion
The late diagnosis and the complex processes involved in the development of cancer lead to poor LUAD prognosis.16 Although the past decades saw significant progress in the treatment of the disease,17 drug resistance and side effects are significant obstacles to effective targeted therapy.18,19 Thus, there is an urgent need to identify key molecular drivers of LUAD to yield new targets for its therapy. Recent studies revealed an aberrant expression of lncRNAs in many cancers which promotes tumor initiation and progression.20
Previously LINC00551 has been reported to be downregulated in esophageal squamous cancer and inhibited cancer proliferation and invasion by increase in HSP27phosphorylation.21 In the present study, we showed that LINC00551 suppressed LUAD cell proliferation, and abnormally expressed LINC00551 are related with clinical pathological characteristics and overall survival of LUAD patients. Moreover, we revealed that LINC00551 participates in metabolic processes and thus affects tumor development. Altered glucose metabolism is a hallmark of cancer that is essential for the progression of many tumors.22 Growing evidence suggests that lncRNAs regulate glucose metabolism in cancer cells23–25 by modulating enzymes, regulatory molecules, metabolism-related signaling pathways, and oncogenes.22,26–30 In agreement with these findings, our results suggest that LINC00551 exerts its anticancer effects by inhibiting glycolysis via repression of c-Myc-mediated transcription of PKM2. Most glucose metabolic pathway associated genes are transcriptional targets of Myc. For instance, Myc increases the expression of genes coding hexokinase and glucose transporters enhancing glucose uptake, and stimulates various glycolytic genes expression, such as that encoding phosphoglucose isomerase, phosphofructokinase, glyceraldehyde-3-phosphate dehydrogenase, phosphoglycerate kinase, enolase, as well as lactate dehydrogenase A.15,29,31,32 Myc also regulates the alternative pyruvate kinase (PK) splicing, a key determiner of the glycolytic pathway, by increasing the heterogeneous nuclear ribonucleoproteins (hnRNP) expression. PK is composed of two different isoforms: PKM M1, which is expressed in most adult tissues, and PKM M2, which is expressed mainly during embryonic development. While aerobic glycolysis activated by PKM M2, oxidative phosphorylation is endorsed by PKM M1. By upregulating the expression of hnRNPA1 and hnRNPA2, Myc preserves an ultimate PKM M2/PKM M1 ratio by increasing hnRNPA1 and hnRNPA2 expression, ensuring peak glycolysis when oxygen is available.33
LncRNAs, as regulators of metabolism, may constitute attractive targets for cancer therapy. Goldberg and Sharp found that siRNA-mediated knockdown of PKM2 inhibited tumor development and increased cell apoptosis in vitro. Besides, this induced tumor regression in mouse xenograft model (in vivo).34 Pusapati and colleagues showed that inhibition of glycolysis in combination with mTOR inhibitors prevented metabolic reprogramming and induced cancer cell apoptosis.34 These antiglycolytic mediators might exhibit high efficacy when combined.
In conclusion, a better understanding of the mechanisms regulating aerobic glycolysis can help develop glycolytic inhibitors as anticancer agents. Our study showed that overexpression of LINC00551 repressed c-Myc-mediated transcription of PKM2, thereby impairing the progression and glucose metabolism of LUAD cells. Thus, the novel lncRNA LINC00551 is a possible biomarker as well as a prospective treatment target for LUAD.
Conclusions
In this study, we found lncRNA LINC00551, worked as a suppressor in regulating LUAD cells glucose metabolism through c-Myc-mediated transcription of PKM2, can be a possible treatment target for LUAD.
Abbreviations
LUAD, lung adenocarcinoma; lncRNA, long noncoding RNA; NSCLC, non-small-cell lung cancer.
Data Sharing Statement
All data involved in this study are contained in this published paper and its supplementary materials.
Ethics Statement
This study was complied with the Declaration of Helsinki. All human-based research in the study was approved by Nanjing Hospital affiliated to Nanjing Medical University. And All animal studies were conducted in accordance with NIH animal use guidelines and approved by Experimental Animal Ethics Committee of Nanjing Hospital affiliated to Nanjing Medical University.
Acknowledgment
The authors thank Nanjing Xinjia Medical Technology Co. Ltd for providing technical platform support.
Author Contributions
Jinsong Yang and Jing Luo designed and supervised the study. Li Wang, Huishan Wang and Bining Wu performed most of the experiments. Li Wang, Bining Wu, Hualin Yu and Xueyan Li participated in acquisition of data. Chengfeng Fan, Xiaoli Shi, and Chun Zhang denoted to analyze and interpret data. Jinsong Yang and Jing Luo agreed to submit to the current journal. All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work. Consent for publication: All authors read and approved the final version of the manuscript. These authors contributed equally to the work and should be regarded as joint first authors: Li Wang, Huishan Wang, and Bining Wu.
Funding
This research was supported by the Medical Science Development Subject in Science and Technology Project of Nanjing (Grant No. ZKX13017) the Natural Science Foundation of Jiangsu province (No. BK20151086), the National Nature Scientific Foundation of China (No.81673030). National Science Foundation for Natural Youth Grant (No. 81902354).
Disclosure
The authors report no conflicts of interest for this work.
References
1. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132.
2. Kerdidani D, Chouvardas P, Arjo AR, et al. Wnt1 silences chemokine genes in dendritic cells and induces adaptive immune resistance in lung adenocarcinoma. Nat Commun. 2019;10(1):1405.
3. Hirsch FR, Scagliotti GV, Mulshine JL, et al. Lung cancer: current therapies and new targeted treatments. Lancet. 2017;389(10066):299–311. doi:10.1016/S0140-6736(16)30958-8
4. Lin C, Yang L. Long Noncoding RNA in Cancer: wiring Signaling Circuitry. Trends Cell Biol. 2018;28(4):287–301. doi:10.1016/j.tcb.2017.11.008
5. Yang B, Zhang L, Cao Y, et al. Overexpression of lncRNA IGFBP4–1 reprograms energy metabolism to promote lung cancer progression. Mol Cancer. 2017;16(1):145. doi:10.1186/s12943-017-0722-8
6. Valle-Mendiola A, Soto-Cruz I. Energy Metabolism in Cancer: the Roles of STAT3 and STAT5 in the Regulation of Metabolism-Related Genes. Cancers. 2020;12(1):1203. doi:10.3390/cancers12010124
7. Liu H, Luo J, Luan S, He C, Li Z. Long non-coding RNAs involved in cancer metabolic reprogramming. Cell Mol Life Sci. 2019;76(3):495–504. doi:10.1007/s00018-018-2946-1
8. Shankaraiah RC, Veronese A, Sabbioni S, Negrini M. Non-coding RNAs in the reprogramming of glucose metabolism in cancer. Cancer Lett. 2018;419:167–174. doi:10.1016/j.canlet.2018.01.048
9. Fan C, Tang Y, Wang J, et al. Role of long non-coding RNAs in glucose metabolism in cancer. Mol Cancer. 2017;16(1):130. doi:10.1186/s12943-017-0699-3
10. Lu W, Cao F, Wang S, Sheng X, Ma J. LncRNAs: the Regulator of Glucose and Lipid Metabolism in Tumor Cells. Front Oncol. 2019;9:1099. doi:10.3389/fonc.2019.01099
11. Song J, Wu X, Liu F, et al. Long non-coding RNA PVT1 promotes glycolysis and tumor progression by regulating miR-497/HK2 axis in osteosarcoma. Biochem Biophys Res Commun. 2017;490(2):217–224. doi:10.1016/j.bbrc.2017.06.024
12. Evans JR, Feng FY, Chinnaiyan AM. The bright side of dark matter: lncRNAs in cancer. J Clin Invest. 2016;126(8):2775–2782. doi:10.1172/JCI84421
13. Tsagakis I, Douka K, Birds I, Aspden JL. Long non-coding RNAs in development and disease: conservation to mechanisms. J Pathol. 2020;250(5):480–495. doi:10.1002/path.5405
14. Panda S, Banerjee N, Chatterjee S. Solute carrier proteins and c-Myc: a strong connection in cancer progression. Drug Discov Today. 2020;25(5):891–900. doi:10.1016/j.drudis.2020.02.007
15. Gupta A, Ajith A, Singh S, Panday RK, Samaiya A, Shukla S. PAK2–c-Myc–PKM2 axis plays an essential role in head and neck oncogenesis via regulating Warburg effect. Cell Death Dis. 2018;9(8):8. doi:10.1038/s41419-018-0887-0
16. Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong -K-K. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer. 2014;14(8):535–546. doi:10.1038/nrc3775
17. Lim SM, Choi JW, Hong MH, et al. Indoor radon exposure increases tumor mutation burden in never-smoker patients with lung adenocarcinoma. Lung Cancer. 2019;131:139–146. doi:10.1016/j.lungcan.2019.04.002
18. Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol. 2014;11(8):473–481. doi:10.1038/nrclinonc.2014.104
19. Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26. doi:10.1126/scitranslmed.3002003
20. Kim J-W, Gao P, Liu Y-C, Semenza GL, Dang CV. Hypoxia-Inducible Factor 1 and Dysregulated c-Myc Cooperatively Induce Vascular Endothelial Growth Factor and Metabolic Switches Hexokinase 2 and Pyruvate Dehydrogenase Kinase 1. Mol Cell Biol. 2007;27(21):7381–7393. doi:10.1128/MCB.00440-07
21. Peng X, Zhou Y, Chen Y, et al. Reduced LINC00551 expression promotes proliferation and invasion of esophageal squamous cancer by increase in HSP27 phosphorylation. J Cell Physiol. 2020;1–14.
22. Abdel-Wahab AF, Mahmoud W, Al-Harizy RM. Targeting glucose metabolism to suppress cancer progression: prospective of anti-glycolytic cancer therapy. Pharmacol Res. 2019;150:104511. doi:10.1016/j.phrs.2019.104511
23. Liu X, Gan B. lncRNA NBR2 modulates cancer cell sensitivity to phenformin through GLUT1. Cell Cycle. 2016;15(24):3471–3481. doi:10.1080/15384101.2016.1249545
24. Malakar P, Stein I, Saragovi A, et al. Long Noncoding RNA MALAT1 Regulates Cancer Glucose Metabolism by Enhancing mTOR-Mediated Translation of TCF7L2. Cancer Res. 2019;79(10):2480–2493. doi:10.1158/0008-5472.CAN-18-1432
25. Matouk IJ, DeGroot N, Mezan S, et al. The H19 non-coding RNA is essential for human tumor growth. PLoS One. 2007;2(9):e845. doi:10.1371/journal.pone.0000845
26. Guan Y, Ai Y-L, et al. Nur77-activated lncRNA WFDC21P attenuates hepatocarcinogenesis via modulating glycolysis. Oncogene. 2020;39(11):2408–2423. doi:10.1038/s41388-020-1158-y
27. Tang J, Yan T, Bao Y, et al. LncRNA GLCC1 promotes colorectal carcinogenesis and glucose metabolism by stabilizing c-Myc. Nat Commun. 2019;1:10.
28. Liu X, Xiao Z-D, Han L, et al. LncRNA NBR2 engages a metabolic checkpoint by regulating AMPK under energy stress. Nat Cell Biol. 2016;18(4):431–442. doi:10.1038/ncb3328
29. Zhang P, Cao L, Fan P, Mei Y, Wu M. Lnc RNA - MIF, a c-Myc-activated long non-coding RNA, suppresses glycolysis by promoting Fbxw7-mediated c-Myc degradation. EMBO Reports. 2016;17(8):1204–1220. doi:10.15252/embr.201642067
30. Kino T, Hurt DE, Ichijo T, Nader N, Chrousos GP. Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci Signal. 2010;3(107):ra8. doi:10.1126/scisignal.2000568
31. Osthus RC, Shim H, Kim S, et al. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem. 2000;275(29):21797–21800. doi:10.1074/jbc.C000023200
32. Le A, Cooper CR, Gouw AM, et al. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A. 2010;107(5):2037–2042. doi:10.1073/pnas.0914433107
33. David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010;463(7279):364–368. doi:10.1038/nature08697
34. Pusapati RV, Daemen A, Wilson C, et al. mTORC1-Dependent Metabolic Reprogramming Underlies Escape from Glycolysis Addiction in Cancer Cells. Cancer Cell. 2016;29(4):548–562. doi:10.1016/j.ccell.2016.02.018
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.