Back to Journals » OncoTargets and Therapy » Volume 12
Long Non-Coding RNA TUG1 Modulates Proliferation, Migration, And Invasion Of Acute Myeloid Leukemia Cells Via Regulating miR-370-3p/MAPK1/ERK
Authors Li G, Zheng P ![]() , Wang H, Ai Y, Mao X
, Wang H, Ai Y, Mao X ![]()
Received 30 May 2019
Accepted for publication 4 October 2019
Published 29 November 2019 Volume 2019:12 Pages 10375—10388
DOI https://doi.org/10.2147/OTT.S217795
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Takuya Aoki
Gang Li, Peiming Zheng, Huiling Wang, Yushu Ai, Xiaohuan Mao
Department of Clinical Laboratory, Henan Provincial People’s Hospital, People’s Hospital of Zhengzhou University, Zhengzhou 450003, Henan, People’s Republic of China
Correspondence: Xiaohuan Mao
Department of Clinical Laboratory, Henan Provincial People’s Hospital, People’s Hospital of Zhengzhou University, Weiwu Road, No. 7, Zhengzhou 450003, Henan, People’s Republic of China
Email [email protected]
Background: Acute myeloid leukemia (AML) is the most common form of acute leukemia in adults. Long non-coding RNA taurine-upregulated gene 1 (lncRNA TUG1) has been discovered to participate in multiple cancers including AML. However, the detailed mechanism of TUG1 in AML remains obscure.
Materials and methods: AML cell lines HL-60 and Kasumi-1 were taken as cell models. TUG1 knockdown or overexpression cell lines were generated. Then, the biological influence of TUG1 on cancer cells was studied using CCK-8 assay, transwell assay and Western blot in vitro. Interaction between TUG1 and miR-370-3p was determined by bioinformatics analysis, RT-PCR, and luciferase assay. Western blot, RT-PCR, and luciferase assay were carried out to validate the interaction between miR-370-3p and its target gene Mitogen-Activated Protein Kinase 1 (MAPK1).
Results: Knockdown of TUG1 markedly reduced viability and metastasis of AML cells, while its overexpression had the opposite effect. MAPK1 was verified as a target gene of miR-370-3p. TUG1 could reduce the level of functional miR-370-3p, facilitate MAPK1 expression, and in turn activate ERK1/2 signaling.
Conclusion: TUG1 could modulate malignant phenotypes of AML cells via miR-370-3p/MAPK1/ERK signaling. Our study would help to clarify the mechanism of AML tumorigenesis and progression.
Keywords: LncRNA, TUG1, AML, miR-370-3p, MAPK1
Introduction
Acute myeloid leukemia (AML) is one of the most frequently diagnosed hematological malignancies among adults. AML patients are characterized by uncontrolled proliferation of immature myeloid precursor cells, which ultimately leads to hematopoietic impairment and even failure of bone marrow.1–3 The overall survival rate of patients under 60 years old (including 60 years old) was 30–40% and patients over 60 years old were less than 10%.4 Notwithstanding the tremendous achievements in radiotherapy and chemotherapy, these approaches only provide very limited survival advantages.5–7 Obviously, there was an urgent need to further dig out the mechanism of AML to explore novel treatment strategies.
Long non-coding RNA (lncRNA) is a class of endogenous non-protein coding RNA with 200 nucleotides in length or larger.8 Accumulating studies have pointed out that lncRNAs were involved in the tumorigenesis of multiple cancers.9,10 LncRNAs have been found to act as oncogenes or tumor suppressors in a wide range of tumors; thus, its aberrant expression can be used as an indicator to evaluate the tumorigenesis, metastasis, and prognosis of cancers.11,12 Recent studies have indicated that lncRNA was a crucial regulator for genes or signaling pathways. For instance, LINC01354 promoted colorectal cancer progression via activating Wnt/β-catenin signaling pathway.13 LncRNA NKILA can impede the metastasis of breast cancer and tongue cancer cells via inhibiting NF-κB signaling pathway.14 In addition, the high expression of lncRNA taurine upregulated gene 1 (TUG1) could be observed in bone marrow tissues and tumor cells of AML patients, and the high expression level of TUG-1 has been validated to be closely tied to patients’ poor prognosis.15–18 Unfortunately, its mechanism of facilitating AML progress has not been fully clarified.
MicroRNA (miRNA) is a kind of single-stranded non-coding RNA with the function of post-transcriptional regulation, which results in translation inhibition or degradation by binding to the target gene 3ʹ-untranslated region (3ʹ-UTR).19,20 A line of evidences validated that miRNA was involved in a variety of biological processes, including cell cycle process, apoptosis, differentiation, and hematopoiesis.21–23 Over 50% miRNA was well acknowledged to be located in the cancer-related gene region, which played a carcinogenic or anti-cancer role.24 In addition, miRNA is a key factor in hematogenesis, and its abnormal expression would lead to leukemia.25 For example, miR-125b enhanced AML tumorigenesis.26 Differently, miR-370 played an inhibitory role in AML.27 Mechanically in AML, miR-370 can target NF1 and FOXM1 to play an inhibitory role.28,29 As a member of miR-370 family, miR-370-3p has been shown to inhibit mounting tumors, such as glioma, ovarian cancer, and bladder cancer.30–32 The impact of miR-370-3p on AML and its regulatory relationship remain to be fully clarified.
Mitogen-activated protein kinase (MAPK) pathway serves as a crucial signal transduction component, which converts extracellular stimulation into intracellular signal by connecting cell surface receptors with transcription factors.33 MAPK has been proved to control the differentiation, proliferation, survival, and migration of multiple cancer cells.33,34 It was pointed out that MAPK signaling pathway was of increasing importance in tumorigenesis and progression of AML. MAPK signaling pathway imposed a significant effect on AML.35,36 Nonetheless, the role of MAPK in AML and its regulatory mechanism remain far from being thoroughly elucidated.
Bioinformatics suggested that there were potential binding sites between TUG1 and miR-370-3p. The roles of TUG1, miR-370-3p, and MAPK 1 in AML and their regulatory relationships need further clarification. This study aimed to explore the function of TUG1/miR-370-3p/MAPK1/ERK in AML and provide theoretical basis to elucidate the molecular mechanism of AML, thus rendering potential therapeutic schemes for AML patients.
Materials And Methods
Clinical Data
Bone marrow tissue samples from 23 newly diagnosed AML patients were selected. All the patients had complete clinical and pathological data. All patients knew the purpose of the study and signed a written informed consent. Our study was consistent with the Declaration of Helsinki. This research was conducted under the guidance of the Ethics Committee of Henan Provincial People’s Hospital.
Cell Culture
Human AML cell lines (HL-60 and Kasumi-1) were purchased from Shanghai Cell Library of the Chinese Academy of Sciences. All cells were cultured in RPMI-1640 (Gibco, Grand Island, NY, USA) medium containing 10% FBS (Gibco, Grand Island, NY, USA) and 1% double antibody (penicillin/streptomycin Gibco, Thermo Fisher Scientific, USA). The culture environment was 5% CO2 at 37°C. Cells in the logarithmic growth phase were prepared for subsequent experiments. Overexpressed TUG1 plasmid (pcDNA-TUG1), TUG1 shRNA, no-load plasmid (pcDNA-NC), and no-load shRNA were constructed by GENECHEM (Shanghai, China). miRNA mimics and miRNA inhibitors were purchased from RiboBio (Guangzhou, China).
qRT-PCR Assay
The total RNA in tissues and cells was extracted by TRIzol (Invitrogen, Carlsbad, CA, USA). Complementary DNA was then synthesized from RNA using prime Script RT kit (Takara, Bio, Inc., Otsu, Japan). After that, we adopted ABI 7500 Real-Time PCR system (Applied Biosystems, USA) to detect expressions of miR-370-3p, TUG1, and MAPK1 with U6 or β-actin as an internal reference. PCR primers were synthesized by Thermo Fisher Scientific Co., Ltd. (Shanghai, China). The primer sequences are listed in Table 1. The relative expressions were calculated using the 2−ΔΔCt method.
 |
Table 1 Primers For qRT-PCR |
Cell Transfection
Overexpressed TUG1 plasmid (pcDNA-TUG1), TUG1 shRNA, control plasmid (pcDNA-NC), and control shRNA were constructed by GeneChem (Shanghai, China). miR-370-3p mimics, miRNA 370-3p inhibitors, and miRNA mimics control were purchased from RiboBio (Guangzhou, China). By applying LipofectamineTM 2000 (Invitrogen, Carlsbad, CA, US) according to the instructions, we transfected plasmid, shRNA or microRNA mimics into cells.
Dual-Luciferase Reporter Gene Assay
StarBase (http://starbase.sysu.edu.cn) and TargetScan (http://www.targetscan.org/vert_72/) were used to predict the binding sequence between TUG1 and miR-370-3p, miR-370-3p and MAPK1 3ʹ-UTR, respectively. The sequences of wild-type TUG1 (TUG1-WT), mutant TUG1 (TUG1-Mut), wild-type MAPK1 3ʹ-UTR (MAPK1-WT), and mutant MAPK1 3ʹ-UTR (MAPK1-Mut) containing the presumed binding sites were, respectively, amplified and cloned into pGL3 Basic vector (Promega, Madison, WI, USA). Lipofectamine 2000 (Invitrogen) was used to co-transfect the reporter vectors of miR-370-3p or miR-NC into HL-60 and Kasumi-1 cells. After 48 hrs of culture, the cells were lysed and the luciferase activity was detected by double luciferase reporter gene detection system (Promega, Madison, WI, USA).
CCK-8 Assay
HL-60 and Kasumi-1 cells in logarithmic phase were selected and inoculated into 96-well plates with 1×103 cells per well. A total of 10 μL CCK 8 solution (Hubei Biossci Biotechnology Co., Ltd.) was added into the plate. Afterwards, the absorbance was measured at a wavelength of 450 nm to indicate the proliferation ability of cells. After that, the absorbance of cells was measured at 24 hrs, 48 hrs, 72 hrs, and 96 hrs, respectively.
Apoptosis Assay
HL-60 and Kasumi-1 cells were collected, washed twice with PBS, and then suspended in buffer. FITC Annexin V cell apoptosis detection kit (Ruibo, Guangzhou, China) was applied to stain for 30 mins in the dark at room temperature. Moreover, cell apoptosis was detected by flow cytometry (Becton Dickinson, Mountain View, USA).
Transwell Assay
The migration assay was carried out in the culture chamber using polycarbonate filter membrane with a specification of 8 μm. Each group of cells was a cell suspension with a density of 1×105/mL. A total of 200 μL cell suspension was inoculated into Transwell upper chamber and the medium containing 10% fetal bovine serum was added into lower chamber. Each group was equipped with 3 compound wells. After 24 hrs of culture, the migrated cells were counted under a microscope. In invasion assays, the matrix gel was covered on the lower surface of the chamber, and the other steps were the same as the migration experiment.
Western Blot
Cells are washed with PBS and then lysed by RIPA lysis buffer (Thermo Science, Rockford, IL, USA) containing protease inhibitor. After high-speed centrifugation, the supernatant was collected and then heated in a water bath to denature the protein. Following that, the protein quantified by BCA method was separated by SDS-PAGE gel electrophoresis and transferred to nitrocellulose membrane (Millipore, MA, USA). Then, under the conditions of 37°C and 5% CO2, the mixture was sealed with skim milk for 30 mins. The cells were incubated overnight at 4°C with the primary antibody (1:500). After rinsed with TBST, the PVDF membrane was incubated with the secondary antibody (Goat Anti-Rabbit IgG, 1:2000) at room temperature for 1 hr. Ultimately, the chemiluminescence and development were performed using hypersensitive ECL (Hubei Biossci Biotechnology Co., Ltd.). The primary antibodies used were Anti-Bax antibody (ab53154), Anti-Bcl-2 antibody (ab196495), Anti-N-Cadherin antibody (ab18203), Anti-E-Cadherin antibody (ab15148), Anti-Vimentin antibody (ab137321), Anti-MAPK1 antibody (ab102930), and Anti-ERK antibody (ab131438). The Second antibody: Goat Anti-Rabbit IgG H&L (HRP) (ab205718).
Statistical Analysis
All statistical analysis was carried out using Graphpad prism 7 software and Service Solutions (SPSS) 16.0 software (SPSS Inc., Chicago, IL, USA). All data were expressed as mean ± standard deviation. The differences between two or more groups were analyzed by Student’s test or One-way ANOVA. Differences with p<0.05 were considered to be statistically significant.
Result
TUG1 Expression Has A Close Connection With The Expressions Of miR-370-3p And ERK In Bone Marrow Tissues
Previous researches have confirmed that TUG1 expression was markedly upregulated in bone marrow tissues of patients with AML.15–18 In addition, by analyzing the TCGA data, we found that TUG1 expression in AML tissues was significantly upregulated compared with that in normal bone marrow tissues (Supplementary Figure 1). Furthermore, by qRT-PCR, we also found that compared with normal bone marrow cells, the expression of TUG1 and MAPK1 in AML cells was significantly upregulated, while the expression of miR-370-3p was significantly downregulated (Supplementary Figure 2). To figure out the relations among TUG1, miR-370-3p, and MAPK-1, we firstly detected their expressions in 23 cases with AML by qRT-PCR. Then, the correlation analysis informed us that there was a negative correlation between TUG1 and miR-370-3p (Figure 1A, R=−0.5715, p<0.01). Furthermore, miR-370-3p expression was negatively correlated with MAPK 1 (Figure 1B, R=−0.5236, p<0.05), while there was a positive correlation between TUG1 and MAPK 1 (Figure 1C, R=0.6695, p<0.001). These data above demonstrated that there was a potential regulatory relationship between TUG1, miR-370-3p, and MAPK 1.
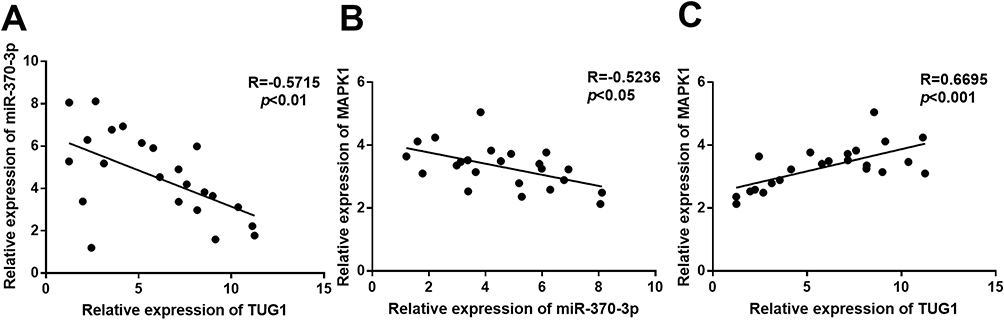 |
Figure 1 The correlation between the expression levels of TUG1, miR-370-3p, and MAPK1. (A) The expression level of TUG1 was negatively correlated with that of miR-370-3p in bone marrow tissues of 23 cases with AML. (B) The expression level of MAPK1 was negatively correlated with that of miR-370-3p in bone marrow tissues of 23 cases with AML. (C) The expression level of MAPK1 was positively correlated with that of TUG1 in bone marrow tissues of 23 cases with AML. |
TUG1 Sponged miR-370-3p
Subsequently, we resorted to Starbase (http://starbase.sysu.edu.cn) to discover miR-370-3p as one of the candidate targets of TUG1, and their binding sites are shown in Figure 2A. qRT-PCR indicated that overexpressed TUG1 notably decreased miR-370-3p expression in HL-60 cells, whereas TUG1 knockdown promoted miR-370-3p expression in Kasumi-1 cells (Figure 2B). On top of that, luciferase reporter gene analysis verified that there was a binding site between TUG1 sequence and miR-370-3p, which could play a role as a “sponge” (Figure 2C).
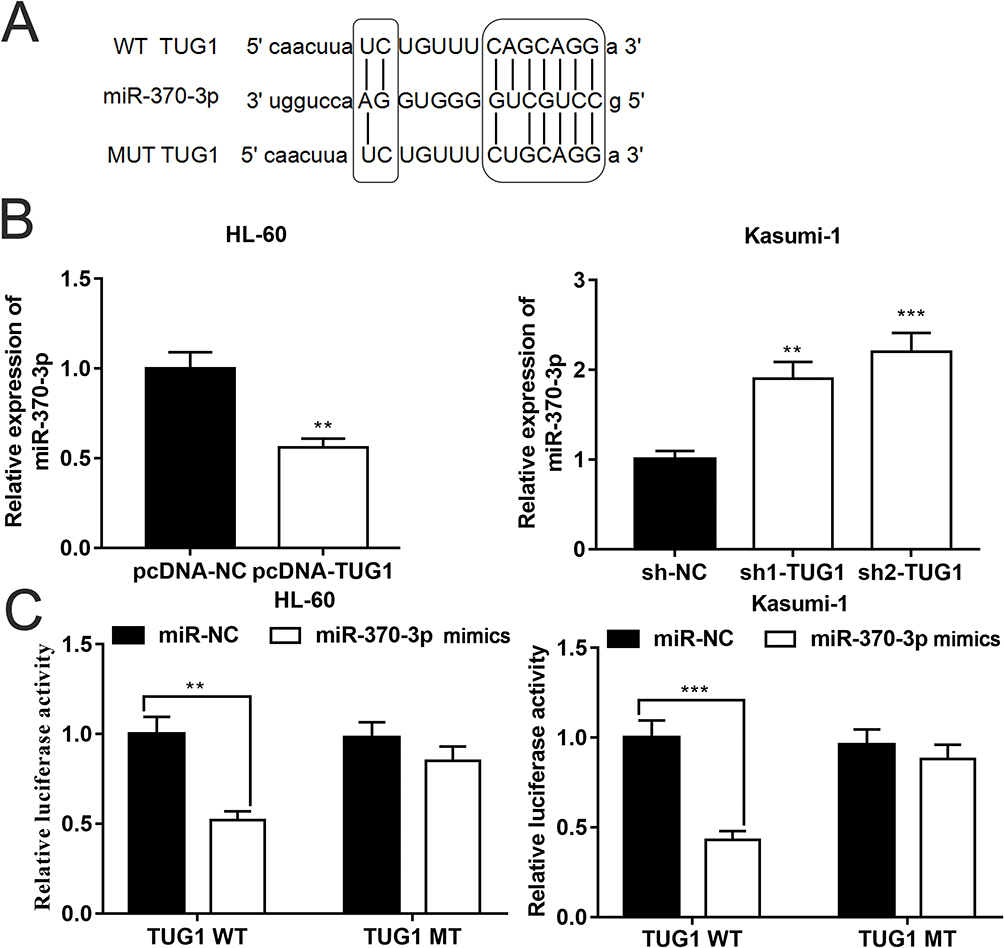 |
Figure 2 TUG1 downregulated miR-370-3p expression in AML. (A) The binding sequence of TUG1 with miR-370-3p. (B) TUG1 regulated miR-370-3p expression in HL-60 and Kasumi-1 cells. (C) MiR-370-3p exerted an inhibitory effect on the luciferase activity of wild-type TUG1 reporter gene but had no significant impact on that of mutant type TUG1. ** and *** represent p<0.01 and p<0.001, respectively. |
TUG1 Promoted The Proliferation Of AML Cells Via miR-370-3p
In order to further clarify effects of TUG1 and miR-370-3p on AML cells, we transfected TUG-1 plasmid into HL-60 cells and successfully established the overexpression model of TUG-1. Meanwhile, the low-expression model of TUG1 was successfully set up by transfecting shRNA into Kasumi-1 cells (Figure 3A). On this basis, the proliferation of HL-60 and Kasumi-1 cells was monitored by CCK 8 method. We observed that HL-60 cells with overexpressed TUG1 proliferated at a quicker rate when compared with the control group. Besides, its proliferation ability was blocked after the transfection of miR-370-3p mimics (Figure 3B). The proliferation rate of Kasumi-1 cells with low expression of TUG1 was significantly lower than that of the control group. After transfection with miR-370-3p inhibitor, the proliferation of Kasumi-1 cells with low expression of TUG1 was significantly enhanced (Figure 3B).
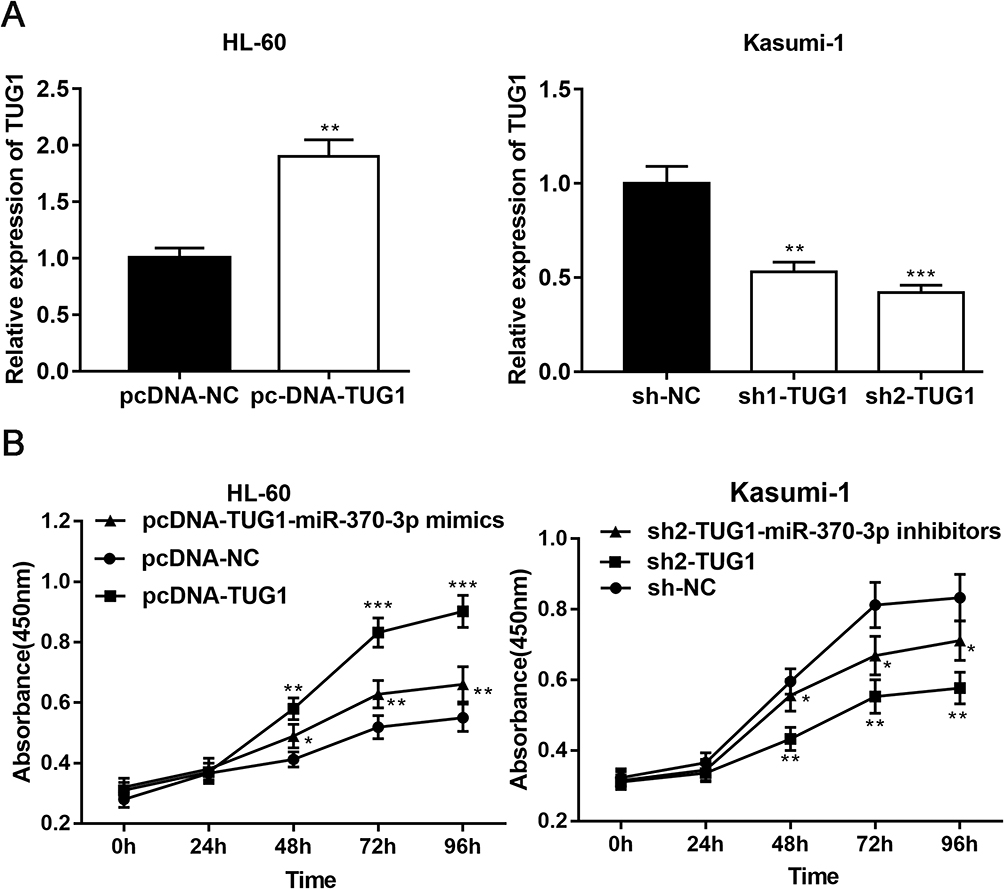 |
Figure 3 TUG1 can promote the proliferation of AML cells via modulating miR-370-3p. (A) TUG1 overexpression plasmid and TUG1 shRNAs were transfected into HL-60 and Kasumi-1 cells and the overexpression model and low-expression model of TUG1 were successfully established, which is validated by qRT-PCR. (B) Overexpressed TUG1 promoted HL-60 cells proliferation and the transfection of miR-370-3p mimics into HL-60 reversed the effect of TUG1 on the proliferation (left). Knockdown of TUG1 inhibited Kasumi-1 cell proliferation, and miR-370-3p inhibitor offset the effect of inhibiting cell proliferation (right). *, ** and *** represent p<0.05, p<0.01, and p< 0.001, respectively. |
TUG1 Inhibited AML Cell Apoptosis Via miR-370-3p
After confirming that TUG1 could facilitate the proliferation of AML, we measured the apoptosis rate of AML cells by flow cytometry. In line with our expectation, the apoptosis rate of HL-60 cells with overexpressed TUG1 was markedly lower than the control group. After miR-370-3p mimics transfection, the apoptosis rate was markedly increased. On the other side, the apoptosis rate of Kasumi with TUG1 knockdown was significantly higher than the control group (p < 0 05). The activity of cells transfected with miR-370-3p inhibitor decreased (Figure 4A and B). Afterwards, we further analyzed the expression level of apoptosis-related proteins in AML cells by Western blot. We then found that the expression level of Bcl-2 in HL-60 with overexpressed TUG1 was higher than the control group, while Bax expression was lower (Figure 4C). What’s more, the increase of Bcl-2 expression as well as the decrease of Bax expression could be reversed by miR-370-3p mimics. Additionally, Kasumi with TUG1 knock-down or transfected with miR-370-3p inhibitor showed opposite effects (Figure 4C). The data above suggested that TUG-1 could arrest the apoptosis of AML cells.
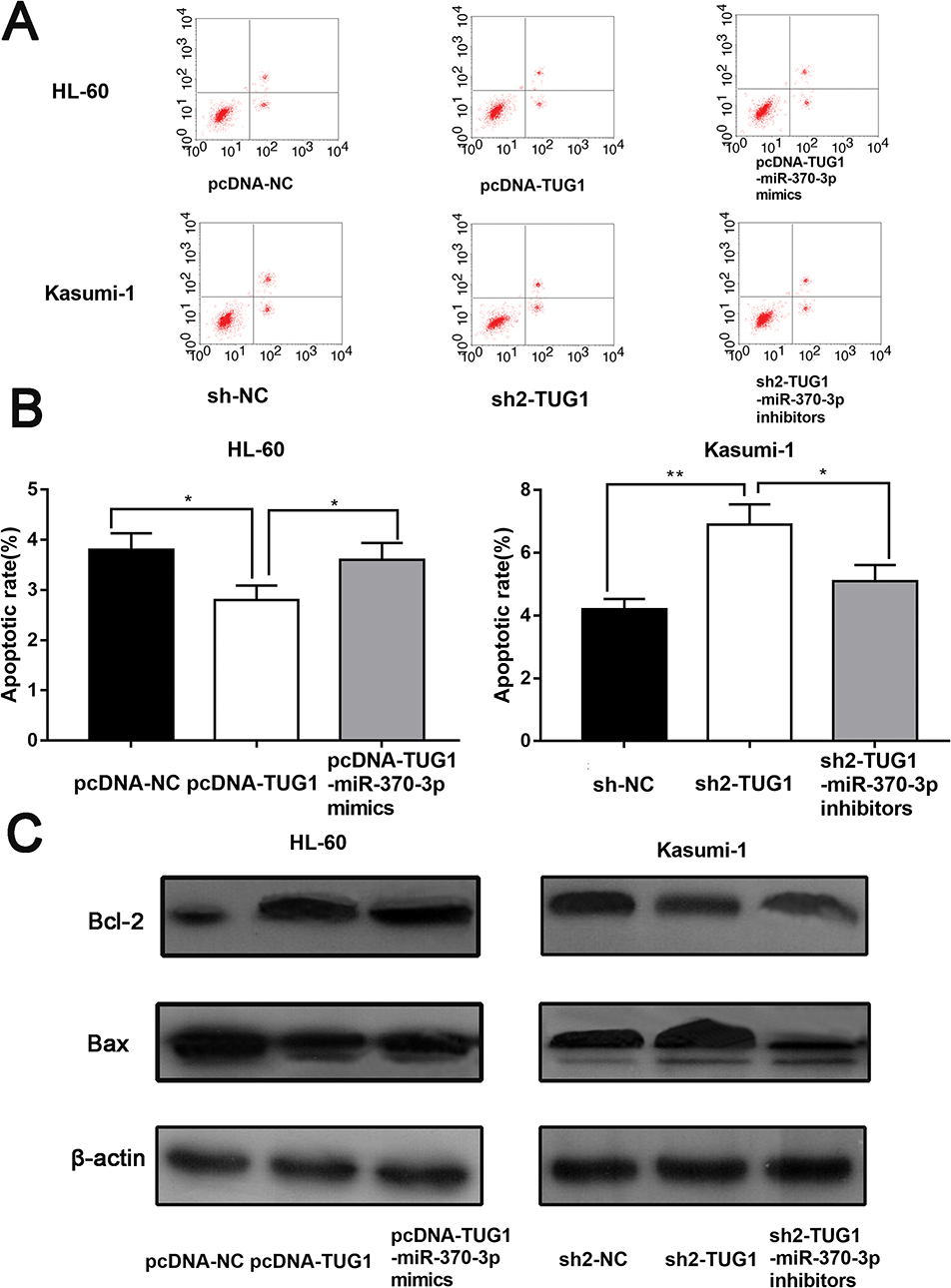 |
Figure 4 TUG1 can inhibit the apoptosis of AML cells via mediating miR-370-3p. (A) The role of overexpression and knockdown of TUG1 in AML apoptosis was determined by flow cytometry. (B) Overexpressed TUG1 impeded the apoptosis of HL-60 cells, while miR-370-3p mimics transfected into HL-60 induced the apoptosis (left). Knockdown of TUG1 facilitated the apoptosis of Kasumi-1 cells, whereas miR-370-3p inhibitor was transfected into Kasumi-1 to arrest the apoptosis (right). (C) Western blotting was performed to detect the expression levels of Bcl-2 and Bax in HL-60 and Kasumi-1 cells.*, * and *** represent p<0.05, p<0.01, and p<0.001, respectively. |
TUG-1 Facilitated The Migration And Invasion Of AML Cells Via miR-370-3p
Then, we studied the role of TUG1 on regulating the migration and invasion of AML cells by transwell assay. In HL-60 cells, the number of cells migrated and invaded with overexpressed TUG1 was notably higher than that in the control group. MiR-370-3p mimics transfection could partially neutralize the function of TUG1, which was significantly different from that in the control group. The metastatic ability of Kasumi-1 cells with knockdown TUG1 was blocked, while Kasumi-1 metastasis was enhanced after transfected with miR-370-3p inhibitor (Figure 5A and B). Following that, we detected the expressions of EMT marker molecules by Western blot. The results indicated that the expression levels of N-cadherin and vimentin increased, while E-cadherin expression appeared to decrease in HL-60 cells with overexpressed TUG1. Moreover, the expression changes of these EMT marker proteins can be weakened by miR-370-3p. As expected, the expression levels of N-cadherin and Vimentin were downregulated. On the contrary, E-cadherin expression was upregulated in Kasumi-1 cells with TUG1 knockdown, and these changes could be offset by miR-370-3p inhibitors (Figure 5C).
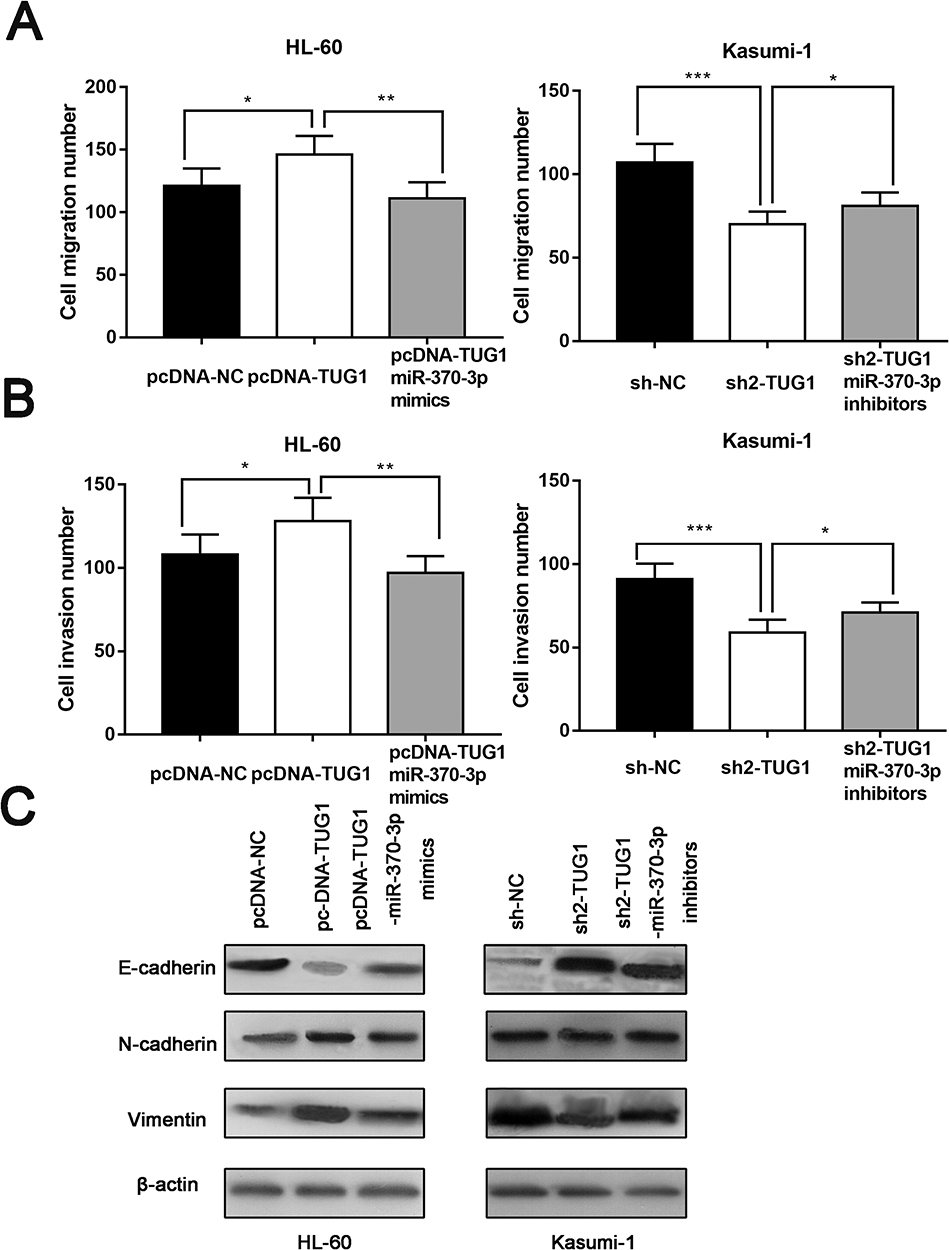 |
Figure 5 TUG1 enhanced the migration and invasion of AML cells via regulating miR-370-3p. (A) Transwell assay indicated that TUG1 overexpression promoted HL-60 cells migration, and such an effect can be partially neutralized by miR-370-3p mimics. TUG1 gene knockdown can arrest the migration of Kasumi-1 cells, and miR-370-3p inhibitor can partially weaken its function (right). (B) Transwell assay demonstrated that TUG1 overexpression enhanced HL-60 cells invasion, and miR-370-3p mimics transfected into HL-60 reversed its function (left). TUG1 gene knock-down can suppress Kasumi-1 cells invasion, and miR-370-3p inhibitor can partially weaken this inhibition (right). (C) Western blot was carried out to detect EMT marker molecules, such as E-cadherin, N-cadherin, and vimentin.*, ** and *** represent p<0.05, p<0.01, and p<0.001, respectively. |
MiR-370-3p Directly Bound To 3ʹ-UTR Of MAPK 1
Next we predicted the target gene of miR-370-3p by TargetScan. MAPK1 was a candidate target gene of miR-370-3p, and the binding site is shown in Figure 6A. Subsequently, luciferase reporter gene assay showed that miR-370-3p could bind specifically to 3ʹ-UTR of MAPK1 (Figure 6B). Furthermore, qRT-PCR demonstrated that MAPK 1 and ERK mRNA decreased significantly after transfection of miR-370-3p mimics into HL-60 and Kasumi-1 cells, while increased after transfection of miR-370-3p inhibitors (Figure 6C). Nevertheless, the expression of MKK3 and MKK4 did not change significantly (Figure 6D).
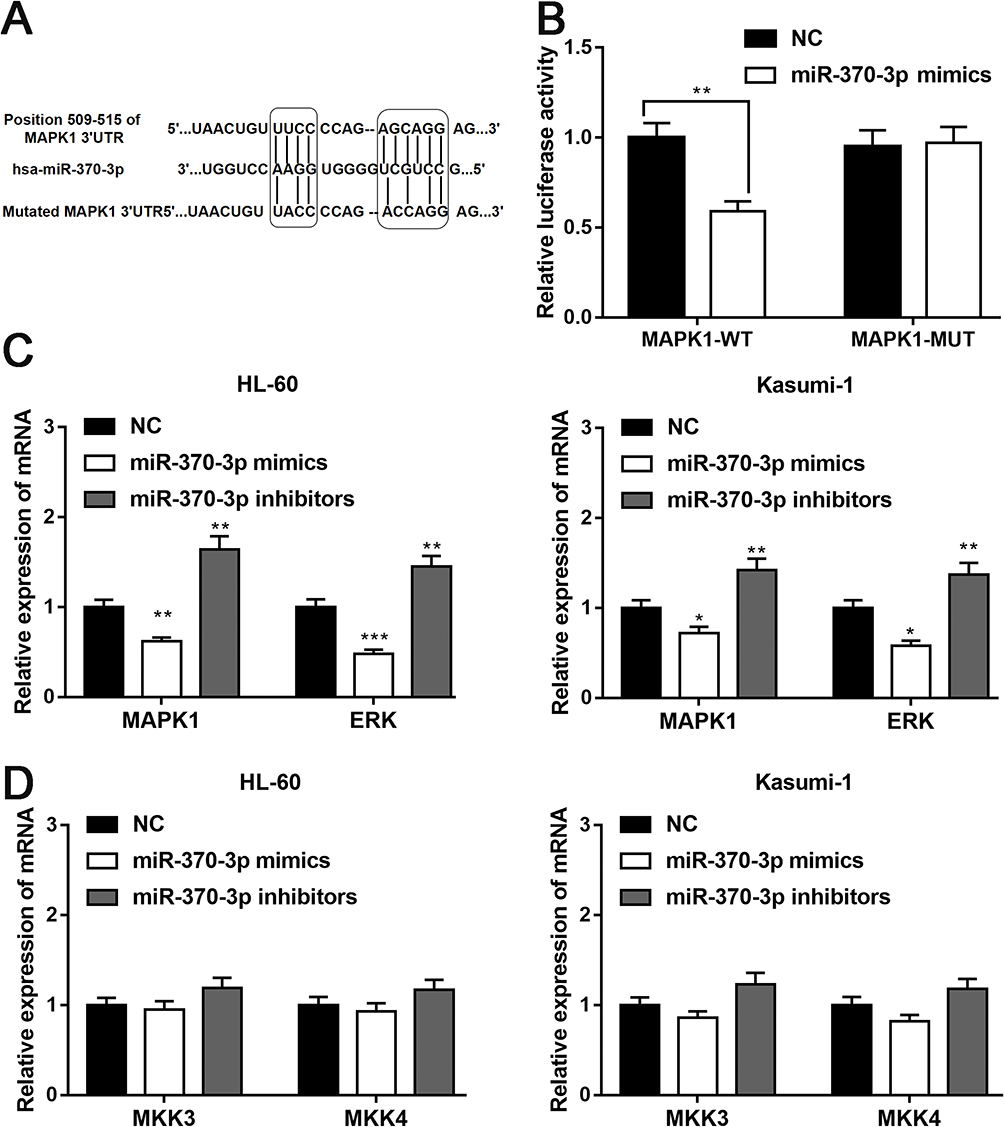 |
Figure 6 MAPK1 is the target gene of miR-370-3p. (A) The binding sequence of MAPK 1 3ʹ-UTR and miR-370-3p was shown in the figure. (B) MiR-370-3p can markedly hold back the luciferase activity of wild-type MAPK1 3ʹ-UTR but has no significant effect on the luciferase activity of HL-60 cells after MAPK 1 3ʹ-UTR mutation. (C and D) qRT-PCR showed that the transfection of miR-370-3p mimics can notably reduce the mRNA expression of MAPK1 and ERK. On the contrary, the transfection of miR-370-3p inhibitor can increase the expression of these mRNA. However, the changes of MKK3 mRNA and MKK4 mRNA after the transfection of miR-370-3p mimics and inhibitor were not significant. *, ** and *** represent p<0.05, p<0.01, and p<0.001, respectively. |
In AML Cells, TUG1 Was Considered As A Promoting Factor Of MAPK1/ERK Signaling Pathway, While miR-370-3p Was An Inhibitor
Then, we did Western blot to detect the expressions of MAPK1, ERK, MAPK1, and ERK in transfected HL-60 and Kasumi-1 cells. The substantial increase in expressions of the above proteins was observed in AML cells transfected with pcDNA-TUG1 and miR-370-3p inhibitors. Nevertheless, sh-TUG-1 and miR-370-3p mimics transfected can inhibit the expression of these proteins (Figure 7A and B). In the next step, we detect the expressions of MKK3 and MKK4 in AML cells treated with the same manner by Western blot, but the expression levels of MKK3 and MKK4 did not change markedly, implying that the change of p-MAPK1 was due to the change of baseline expression level of MAPK1, but not phosphorylation activation (Figure 7C and D).
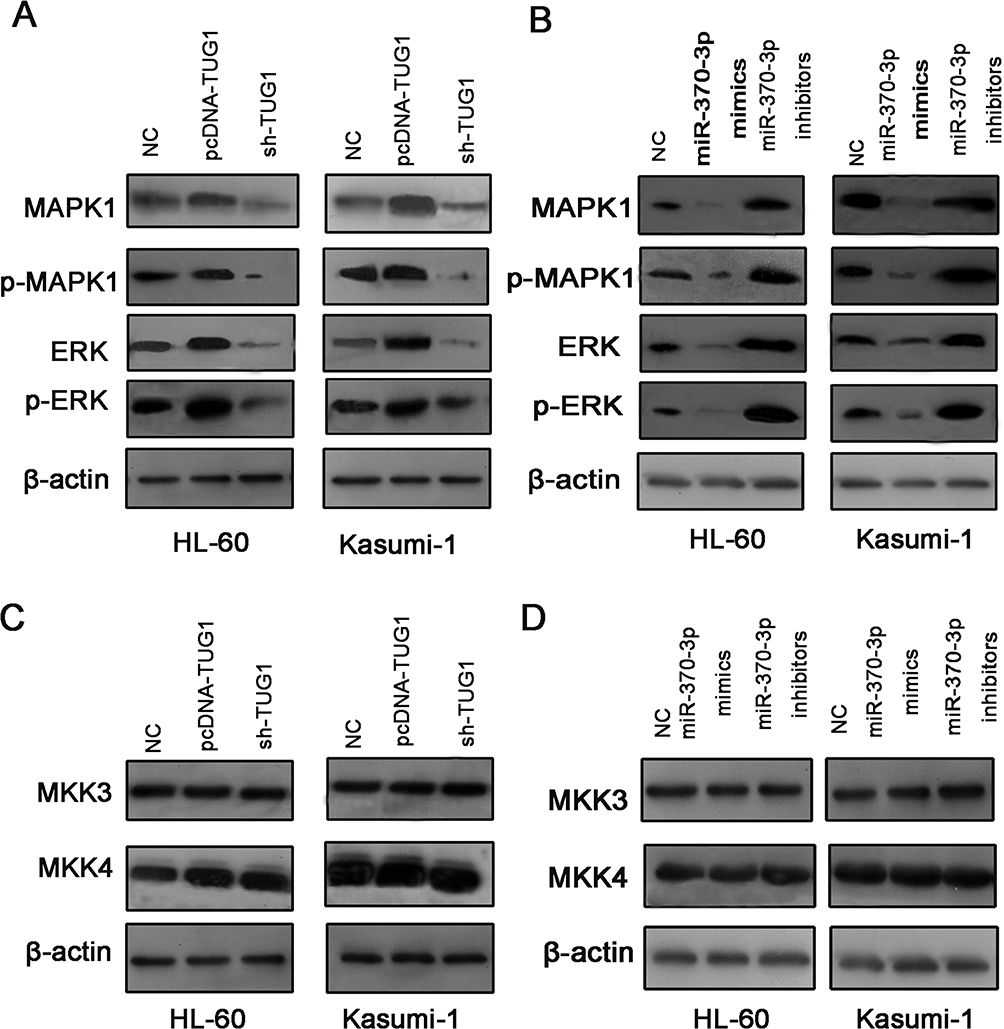 |
Figure 7 The expressions of related proteins were detected after transfection of HL-60 and Kasumi-1 cells. (A) Western blot was used to detect the expression levels of MAPK 1, p-MAPK 1, ERK, and p-ERK after pcDNA-TUG1 and sh2-TUG1 were transfected into HL-60 and Kasumi-1. (B) Western blot was carried out to monitor the expression levels of MAPK 1, p-MAPK 1, ERK, and p-ERK after the transfection of miR-370-3p mimics and miR-370-3p inhibitors into HL-60 and Kasumi-1. (C) The expressions of MKK3 and MKK4 transfected with pcDNA-TUG1 and sh2-TUG1 were detected. (D) The expression levels of MKK3 and MKK4 were detected after the transfection of miR-370-3p mimics and miR-370-3p inhibitors into HL-60 and Kasumi-1. |
Discussion
At present, AML features a low rate of early diagnosis, quick development, and poor therapeutic effect, which contributes to poor prognosis of patients.1,2,4,36,37 Correspondingly, it is imperative to probe into the biological mechanism of AML. In this study, we found that TUG-1 promoted biological behaviors of AML cells via miR-370-3p/MAPK1/ERK signaling pathway, which is expected to provide a potential treatment for AML patients.
Mounting studies supported that TUG1 expression was upregulated in multiple cancers.38–40 For instance, TUG1 promoted the development of renal clear cells, and its high expression level was correlated with the shorter overall survival time of patients.41,42 The high expression level of TUG1 was related to the tumor volume, clinical stage, differentiation, and lymph node metastasis in cervical carcinoma.43 It is also reported that TUG1 was highly expressed in small cell lung cancer (SCLC) tissues, and its expression level is associated with clinical stage and short survival time.44 However, a recent study discovered that TUG1 was generally downregulated in non-small cell lung cancer tissues, and the low expression of TUG1 was related to TNM stage, tumor size, and overall survival time.45 This indicated that the function of TUG1 in tumorigenesis was correlated to the specificity of cells and tissues. Some scholars demonstrated that TUG1 was highly expressed in tissues and cell lines of AML patients, and the high expression of TUG1 was also closely related to poor prognosis of AML.16 TUG1 induced cell proliferation and represses cell apoptosis via targeting AURKA in AML.18 In this study, we successfully constructed the overexpressed and low-expressed TUG1 cell models, respectively. Through CCK-8 and transwell assay, we tested the effect of TUG1 on the biological behavior of AML cells. Overexpressed TUG1 significantly enhanced cell proliferation, migration, and invasion, while cells with knockdown TUG1 showed the opposite effect. The data demonstrated that TUG1 was a tumor-promoting lncRNA for AML, which is consistent with the conclusions of previous studies.15–18
Currently, it has been proved that miR-370 family played an anti-cancer role in a variety of tumors.46–48 MiR-370 repressed cell proliferation and migration by reducing the expression of Epidermal growth factor receptor.47 MiR-370 induced apoptosis of hepatoma cells by activating Akt/FoxO3a signal transduction pathway.49 In addition, miRNA-370 played an anti-cancer role in leukemia.27 From the perspective of mechanism, miR-370 arrested AML progress by targeting FoxM1.28 MiR-370-3p belongs to miR-370 family, which has also been confirmed as a tumor suppressor.50 Overexpression of miR-370-3p can not only block G0/G1 phase of cell cycle but also directly bind to β-catenin mRNA 3ʹ-UTR, thus impeding expressions of cyclin D1 and c-Myc, and in turn suppressed the proliferation of glioma cells.30 MiR-370-3p is reported to be regulated by the mechanism of ceRNA. For example, LNC00707 sponged miR-370-3p to promote bone marrow mesenchymal osteogenesis.51 In this study, we learned from Starbase database (http://starbase.sysu.edu.cn/) that TUG1 was a potential sponge of miR-370-3p. By dual-luciferase reporter gene assays, we confirmed the direct binding site of miR-370-3p and TUG1 sequence. What’s more, after miR-370-3p mimics was transfected into AML cells, the cancer-promoting effect of TUG1 was reversed. Our data suggested that TUG1 sponged miR-370-3p, thus affecting the ability of miR-370-3p to bind to downstream targets.
What’s more, the significance of MAPK signaling pathway in tumors was increasingly prominent.52,53 For example, miR-508 accelerated the progression of ovarian cancer by activating MAPK1/ERK signaling pathway.54 In AML patients, abnormally activated MAPK signaling pathway was associated with worse prognosis.55 In this study, the dual-luciferase assay indicated that MAPK1 was a target gene of miR-370-3p, and the expression levels of MAPK1, p-MAPK1, ERK, and p-ERK were significantly reduced after AML cells transfected with miR-370-3p mimics. However, cells transfected with miR-370-3p inhibitors turned out to have the opposite effect. We also demonstrated that TUG1 could modulate the expression level of MAPK1, p-MAPK1, ERK, and p-ERK, indicating the crucial role of TUG1/miR-370-3p/MAPK1 in AML progression.
To sum up, this study confirmed that TUG-1 facilitated AML cells via miR-370-3p/MAPK1/ERK signal transduction pathway. During this process, MAPK1/ERK inactivation signaling pathway suppressed EMT, thus holding back migration and invasion of AML cells. The results from this study not only expand our understanding of AML mechanisms but may also contribute to the development of drugs targeting TUG1/miR-370-3p/MAPK/ERK axis.
Ethics Statement
Our study was approved by the Medical College Review Board of Henan Provincial People’s Hospital.
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon request.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Leisch M, Jansko B, Zaborsky N, Greil R, Pleyer L. Next generation sequencing in AML-on the way to becoming a new standard for treatment initiation and/or modulation? Cancers (Basel). 2019;11(2):252. Published online 2019 Feb 21. doi:10.3390/cancers11020252
2. Huang Y, Su R, Sheng Y, et al. Small-molecule targeting of oncogenic FTO demethylase in acute myeloid leukemia. Cancer Cell. 2019;35(4):677–691. e10. doi:10.1016/j.ccell.2019.03.006
3. Van Der W, Jamieson C. The Yin and Yang of RNA Methylation: an imbalance of erasers enhances sensitivity to FTO demethylase small-molecule targeting in leukemia stem cells. Cancer Cell. 2019;35(4):540–541. doi:10.1016/j.ccell.2019.03.011
4. Estey EH. Treatment of acute myeloid leukemia. Haematologica. 2009;94(1):10–16. doi:10.3324/haematol.2008.001263
5. Qidong Y, Pan F, Dou J, Wang N .Downregulation of PDIA3 inhibits proliferation and invasion of human acute myeloid leukemia cells. Onco Targets Ther.2018;11:2925–2935. Published online 2018 May 17. doi:10.2147/OTT.S162407
6. Ganapule A, Nemani S, Korula A, et al. Allogeneic stem cell transplant for acute myeloid leukemia: evolution of an effective strategy in India. J Glob Oncol. 2017;3(6):773–781. doi:10.1200/JGO.2016.006650
7. Fontana MC, Marconi G, Feenstra JDM, et al. Chromothripsis in acute myeloid leukemia: biological features and impact on survival. Leukemia. 2017. doi:10.1038/s41375-018-0035-y
8. Gugnoni M, Ciarrocchi A. Long noncoding RNA and epithelial mesenchymal transition in cancer. Int J Mol Sci. 2019;20(8):
9. Oliva-Rico D, Herrera LA. Regulated expression of the lncRNA TERRA and its impact on telomere biology. Mech Ageing Dev. 2017;167:16–23. doi:10.1016/j.mad.2017.09.001
10. Hosseini ES, Meryet-Figuiere M, Sabzalipoor H, Kashani HH, Nikzad H, Asemi Z. Dysregulated expression of long noncoding RNAs in gynecologic cancers. Mol Cancer. 2017;16(1):107. doi:10.1186/s12943-017-0671-2
11. Xu K, Zhang Z, Qian J, et al. LncRNA FOXD2-AS1 plays an oncogenic role in hepatocellular carcinoma through epigenetically silencing CDKN1B(p27) via EZH2. Exp Cell Res. 2019. pii: S0014-4827(19)30190-9. doi:10.1016/j.yexcr.2019.04.016
12. Zhang Y, Tang B, Song J, et al. .Lnc-PDZD7 contributes to stemness properties and chemosensitivity in hepatocellular carcinoma through EZH2-mediated ATOH8 transcriptional repression. J Exp Clin Cancer Res.2019;38:92. Published online 2019 Feb 20. doi:10.1186/s13046-019-1106-2
13. Li J, He M, Xu W, Huang S. LINC01354 interacting with hnRNP-D contributes to the proliferation and metastasis in colorectal cancer through activating Wnt/β-catenin signaling pathway. J Exp Clin Cancer Res. 2019;38(1):161. doi:10.1186/s13046-019-1150-y
14. Ke S, Li RC, Meng FK, Fang MH. NKILA inhibits NF-κB signaling and suppresses tumor metastasis. Aging (Albany NY). 2018;10(1):56–71. doi:10.18632/aging.101359
15. Li Q, Song W, Wang J. TUG1 confers Adriamycin resistance in acute myeloid leukemia by epigenetically suppressing miR-34a expression via EZH2. Biomed Pharmacother. 2019;109:1793–1801. doi:10.1016/j.biopha.2018.11.003
16. Qin J, Bao H, Li H. Correlation of long non-coding RNA taurine-upregulated gene 1 with disease conditions and prognosis, as well as its effect on cell activities in acute myeloid leukemia. Cancer Biomark. 2018;23(4):569–577. doi:10.3233/CBM-181834
17. Luo W, Yu H, Zou X, Ni X, Wei J. Long non-coding RNA taurine-upregulated gene 1 correlates with unfavorable prognosis in patients with refractory or relapsed acute myeloid leukemia treated by purine analogue based chemotherapy regimens. Cancer Biomark. 2018;23(4):485–494. doi:10.3233/CBM-181405
18. Wang X, Zhang L, Zhao F, et al. Long non-coding RNA taurine-upregulated gene 1 correlates with poor prognosis, induces cell proliferation, and represses cell apoptosis via targeting aurora kinase A in adult acute myeloid leukemia. Ann Hematol. 2018;97(8):1375–1389. doi:10.1007/s00277-018-3315-8
19. Liu X, Cai H, Sheng W, Huang H, Long Z, Wang Y .microRNAs expression profile related with response to preoperative radiochemotherapy in patients with locally advanced gastric cancer. BMC Cancer.2018;18:1048. Published online 2018 Oct 29. doi:10.1186/s12885-018-4967-4
20. Valdmanis PN, Kim HK, Chu K, et al. miR-122 removal in the liver activates imprinted microRNAs and enables more effective microRNA-mediated gene repression. Nat Commun. 2018;9:5321. Published online 2018 Dec 14. doi:10.1038/s41467-018-07786-7
21. Zhao Y, Yang J, Liu Y, Fan J, Yang H. HSV-2-encoded miRNA-H4 Regulates Cell Cycle Progression and Act-D-induced Apoptosis in HeLa Cells by Targeting CDKL2 and CDKN2A. Virol Sin. 2019. doi:10.1007/s12250-019-00101-8
22. Wu D, Chen X, Wang L, Chen F, Cen H, Shi L. Hypoxia-induced microRNA-141 regulates trophoblast apoptosis, invasion, and vascularization by blocking CXCL12β/CXCR2/4 signal transduction. Biomed Pharmacother. 2019;116:108836. doi:10.1016/j.biopha.2019.108836
23. Kim M, Civin CI, Kingsbury TJ. MicroRNAs as regulators and effectors of hematopoietic transcription factors. Wiley Interdiscip Rev RNA. 2019;21:e1537. doi:10.1002/wrna.1537
24. Tutar L, Özgür A, Tutar Y. Involvement of miRNAs and Pseudogenes in Cancer. Methods Mol Biol. 2018;1699:45–66.
25. Starczynowski DT, Morin R, McPherson A, et al. Genome-wide identification of human microRNAs located in leukemia-associated genomic alterations. Blood. 2011;117(2):595–607. doi:10.1182/blood-2010-03-277012
26. Zhang H, Luo XQ, Feng DD, et al. Upregulation of microRNA-125b contributes to leukemogenesis and increases drug resistance in pediatric acute promyelocytic leukemia. Mol Cancer. 2011;10:108.
27. Lin X, Wang Z, Wang Y, Feng W.Serum MicroRNA-370 as a potential diagnostic and prognostic biomarker for pediatric acute myeloid leukemia. Int J Clin Exp Pathol. 2015;8(11):14658–14666. eCollection 2015.
28. Zhang X, Zeng J, Zhou M, et al. The tumor suppressive role of miRNA-370 by targeting FoxM1 in acute myeloid leukemia. Mol Cancer. 2012;11:56. doi:10.1186/1476-4598-11-56
29. García-Ortí L, Cristóbal I, Cirauqui C, et al. Integration of SNP and mRNA arrays with microRNA profiling reveals that MiR-370 is upregulated and targets NF1 in acute myeloid leukemia. PLoS One. 2012;7(10):e47717. doi:10.1371/journal.pone.0047717
30. Peng Z, Wu T, Li Y, et al. MicroRNA-370-3p inhibits human glioma cell proliferation and induces cell cycle arrest by directly targeting β-catenin. Brain Res. 2016;1644:53–61. doi:10.1016/j.brainres.2016.04.066
31. Li J, Huang Y, Deng X, et al. Long noncoding RNA H19 promotes transforming growth factor-β-induced epithelial-mesenchymal transition by acting as a competing endogenous RNA of miR-370-3p in ovarian cancer cells. Onco Targets Ther. 2018;11:427–440. doi:10.2147/OTT.S149908
32. Huang X, Zhu H, Gao Z, et al. Wnt7a activates canonical Wnt signaling, promotes bladder cancer cell invasion, and is suppressed by miR-370-3p. J Biol Chem. 2018;293(18):6693–6706. doi:10.1074/jbc.RA118.001689
33. Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9(8):537–549. doi:10.1038/nrc2694
34. Xu Y, Zheng Y, Liu H, Li T. Modulation of IGF2BP1 by long non-coding RNA HCG11 suppresses apoptosis of hepatocellular carcinoma cells via MAPK signaling transduction. Int J Oncol. 2017;51(3):791–800. doi:10.3892/ijo.2017.4066
35. Abdelbaset-Ismail A, Cymer M, Borkowska-Rzeszotek S, et al. Bioactive phospholipids enhance migration and adhesion of human leukemic cells by inhibiting heme oxygenase 1 (HO-1) and Inducible Nitric Oxygenase Synthase (iNOS) in a p38 MAPK-dependent manner. Stem Cell Rev. 2019;15(1):139–154. doi:10.1007/s12015-018-9853-6
36. Kampen KR, Scherpen FJG, Mahmud H, et al. VEGFC antibody therapy drives differentiation of AML. Cancer Res. 2018;78(20):5940–5948. doi:10.1158/0008-5472.CAN-18-0250
37. Ma H, Padmanabhan IS, Parmar S, Gong Y. Targeting CLL-1 for acute myeloid leukemia therapy. J Hematol Oncol. 2019;12(1):41. doi:10.1186/s13045-019-0726-5
38. Ren K, Li Z, Li Y, Zhang W, Han X. Long noncoding RNA taurine-upregulated gene 1 promotes cell proliferation and invasion in gastric cancer via negatively modulating miRNA-145-5p. Oncol Res. 2017;25(5):789–798. doi:10.3727/096504016X14783677992682
39. Katsushima K, Natsume A, Ohka F, et al. Targeting the Notch-regulated non-coding RNA TUG1 for glioma treatment. Nat Commun. 2016;7:13616. doi:10.1038/ncomms13616
40. Yang XL, Wei C, Zhang YB, Guo HQ. Long noncoding RNA TUG1 promotes progression via upregulating DGCR8 in prostate cancer. Eur Rev Med Pharmacol Sci. 2019;23(6):2391–2398. doi:10.26355/eurrev_201903_17385
41. Wang PQ, Wu YX, Zhong XD, Liu B, Qiao G. Prognostic significance of overexpressed long non-coding RNA TUG1 in patients with clear cell renal cell carcinoma. Eur Rev Med Pharmacol Sci. 2017;21(1):82–86.
42. Li N, Shi K, Kang X, Li W. Prognostic value of long non-coding RNA TUG1 in various tumors. Oncotarget. 2017;8(39):65659–65667. doi:10.18632/oncotarget.20025
43. Hu Y, Sun X, Mao C, et al. Upregulation of long noncoding RNA TUG1 promotes cervical cancer cell proliferation and migration. Cancer Med. 2017;6(2):471–482. doi:10.1002/cam4.2017.6.issue-2
44. Niu Y, Ma F, Huang W, et al. Long non-coding RNA TUG1 is involved in cell growth and chemoresistance of small cell lung cancer by regulating LIMK2b via EZH2. Mol Cancer. 2017;16(1):5. doi:10.1186/s12943-016-0575-6
45. Zhang EB, Yin DD, Sun M, et al. P53-regulated long non-coding RNA TUG1 affects cell proliferation in human non-small cell lung cancer, partly through epigenetically regulating HOXB7 expression. Cell Death Dis. 2014;5:e1243. doi:10.1038/cddis.2014.201
46. Yongkang H, Xiaofeng H. MicroRNA-370 regulates cell epithelial-mesenchymal transition, migration, invasion, and prognosis of hepatocellular carcinoma by targeting GUCD1. Yonsei Med J. 2019;60(3):267–276. Published online 2019 Feb 18. doi:10.3349/ymj.2019.60.3.267
47. Pan X, Wang H, Tong D, et al.Physcion induces apoptosis in hepatocellular carcinoma by modulating miR-370. Am J Cancer Res. 2016;6(12):2919–2931. Published online 2016 Dec 1.
48. Ning T, Zhang H, Wang X, et al. miR-370 regulates cell proliferation and migration by targeting EGFR in gastric cancer. Oncol Rep. 2017;38(1):384–392.
49. Sun G, Hou YB, Jia HY, et al. MiR-370 promotes cell death of liver cancer cells by Akt/FoxO3a signalling pathway. Eur Rev Med Pharmacol Sci. 2016;20(10):2011–2019.
50. Cai F, Dai C, Chen S, et al. CXCL12-regulated miR-370-3p functions as a tumor suppressor gene by targeting HMGA2 in nonfunctional pituitary adenomas. Mol Cell Endocrinol. 2019;15(488):25–35. doi:10.1016/j.mce.2019.02.020
51. Jia B, Wang Z, Sun X, Chen J, Zhao J, Qiu X .Long noncoding RNA LINC00707 sponges miR-370-3p to promote osteogenesis of human bone marrow-derived mesenchymal stem cells through upregulating WNT2B. Stem Cell Res Ther.2019;10:67. Published online 2019 Feb 22. doi:10.1186/s13287-019-1161-9
52. Zhu Y, Yang T, Duan J, Ninghui M, Zhang T. MALAT1/miR-15b-5p/MAPK1 mediates endothelial progenitor cells autophagy and affects coronary atherosclerotic heart disease via mTOR signaling pathway. Aging (Albany NY). 2019;11(4):1089–1109. Published online 2019 Feb 21. doi:10.18632/aging.101766
53. Wengong S, Shen J, Chengyong D, et al. A miR-20a/MAPK1/c-Myc regulatory feedback loop regulates breast carcinogenesis and chemoresistance. Cell Death Differ. 2018;25(2):406–420. Published online 2017 Nov 10. doi:10.1038/cdd.2017.176
54. Hong L, Wang Y, Chen W, Yang S. MicroRNA-508 suppresses epithelial-mesenchymal transition, migration, and invasion of ovarian cancer cells through the MAPK1/ERK signaling pathway. J Cell Biochem. 2018;119(9):7431–7440. doi:10.1002/jcb.v119.9
55. Mak PY, Mak DH, Mu H, et al. Apoptosis repressor with caspase recruitment domain is regulated by MAPK/PI3K and confers drug resistance and survival advantage to AML. Apoptosis. 2014;19(4):698–707. doi:10.1007/s10495-013-0954-z
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.