Back to Journals » OncoTargets and Therapy » Volume 13
Long Non-Coding RNA SNHG16 Activates USP22 Expression to Promote Colorectal Cancer Progression by Sponging miR-132-3p
Authors He X, Ma J, Zhang M, Cui J, Yang H
Received 4 January 2020
Accepted for publication 21 April 2020
Published 18 May 2020 Volume 2020:13 Pages 4283—4294
DOI https://doi.org/10.2147/OTT.S244778
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Carlos E Vigil
Xiaowen He, Jun Ma, Mingming Zhang, Jianhua Cui, Hao Yang
Department of General Surgery, Liu Zhou People’s Hospital, Liuzhou, Guangxi 545006, People’s Republic of China
Correspondence: Xiaowen He
Department of General Surgery, Liu Zhou People’s Hospital, No. 8, Wenchang Road, Liuzhou, Guangxi 545006, People’s Republic of China
Tel +86-0772-2662012
Email [email protected]
Background: Colorectal cancer (CRC) is the most common cause of cancer-related mortality in the world. Long non-coding RNAs (lncRNAs) are involved in the development of many cancers. However, studies on the effect of lncRNA small nucleolar RNA host gene 16 (SNHG16) on the proliferation, metastasis and apoptosis of CRC are still few.
Methods: Quantitative real-time polymerase chain reaction (qRT-PCR) was performed to determine the expression levels of SNHG16, microRNA-132-3p (miR-132-3p) and ubiquitin specific peptidase 22 (USP22). The proliferation, apoptosis, migration and invasion of CRC cells were evaluated by the 3-(4,5-dimethyl-2 thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay, flow cytometry and transwell assay, respectively. Dual-luciferase reporter assay was used to verify the interactions among SNHG16, miR-132-3p and USP22. Also, Western blot analysis was used to assess the protein levels of USP22 and metastasis-related markers. Moreover, mice xenograft models were used to determine the effect of SNHG16 on CRC tumor growth in vivo.
Results: SNHG16 was highly expressed in CRC tissues and cells. Knockdown of SNHG16 reduced the proliferation, migration, invasion, and promoted the apoptosis of CRC cells. MiR-132-3p could interact with SNHG16, and its inhibitor recovered the suppression effect of silenced SNHG16 on CRC cell progression. Besides, USP22 was a target of miR-132-3p, and its overexpression restored the inhibition effect of miR-132-3p mimic on CRC cell progression. In addition, interference of SNHG16 reduced CRC tumor growth in vivo.
Conclusion: LncRNA SNHG16 might act as an oncogene in CRC. The discovery of the SNHG16/miR-132-3p/USP22 pathway provided new thinking for the treatment of CRC.
Keywords: colorectal cancer, progression, SNHG16, miR-132-3p, USP22
Introduction
Colorectal cancer (CRC) has become a common type of cancer worldwide and progresses rapidly.1,2 According to statistics, there were more than 1.8 million new CRC cases worldwide in 2018, ranking the third and second respectively in morbidity and mortality.3 In recent decades, significant progress has been made in the prevention, diagnosis and treatment of CRC.4,5 However, due to the complex pathogenesis of CRC, the 5-year survival rate of CRC was still very low.6 Therefore, it is urgent to elucidate the molecular mechanism of CRC and explore new approaches to treat CRC.
Studies have shown that many pro-cancer and anti-cancer genes, long non-coding RNAs (lncRNAs) and microRNAs (miRNAs) are correlated with the occurrence of CRC.7,8 Both lncRNAs and miRNAs belong to non-coding RNAs, with a length of about 20 to 200 nucleotides (nts), respectively.9 Also, lncRNAs and miRNAs could function as the oncogenes or tumor suppressor genes in CRC development process by regulating cell proliferation, invasion, apoptosis and prognosis.10–12 For instance, lncRNA Malat1 induced CRC cell proliferation and inhibited apoptosis through sponging miR-101, hence promoted the CRC progression.13 Du et al discovered that lncRNA DUXAP8 served as competing endogenous RNA (ceRNA) to modulate miR-577 to promote the metastasis of CRC cells.14 Besides, Zhao et al reported that lncRNA LINC02418 regulated CRC tumorigenesis by absorbing miR-1273g-3p.15 Therefore, to explore the mechanism of lncRNAs in CRC was beneficial to provide more new evidence for CRC genomic therapy.
Small nucleolar RNA host gene 16 (SNHG16) has been shown to be highly expressed in many cancers, including hepatocellular carcinoma, breast cancer and glioma.16–18 Similarly, Li et al found that SNHG16 expression was increased in CRC and was correlated with the poor prognosis of CRC patients.19 Furthermore, Christensen et al confirmed that SNHG16 was related to the lipid metabolism of CRC.20 However, few studies have been conducted on the regulation of CRC proliferation and metastasis by SNHG16 at present.
In our research, we investigated the role and mechanism of SNHG16 in CRC progression. The discovery of the SNHG16/miR-132-3p/USP22 axis established the effect of SNHG16 as an oncogene in CRC, and provided a new possibility for targeted therapy of CRC.
Materials and Methods
Tissue Samples Collection
Tumor tissues and adjacent normal tissues of 50 CRC patients admitted to Liu Zhou People’s Hospital from July 2016 to January 2018 without any treatment were collected. All patients signed informed consent, and the study plans were authorized by the Ethics Committee of Liu Zhou People’s Hospital.
Cell Culture and Transfection
CRC cell lines (SW480 and SW620) and normal human colonic epithelial cells (CCD841 CON) were bought from the American Type Culture Collection (ATCC, Manassas, VA, USA). Culture medium was contained with RPMI-1640 (Gibco, Waltham, MA, USA), 10% fetal bovine serum (FBS; Gibco), 100 U/mL penicillin and 0.1 mg/mL streptomycin (Invitrogen, Carlsbad, CA, USA), and all cells were cultured in a 5% CO2 at 37°C incubator. Small interfering RNA (siRNA) against SNHG16 and ubiquitin specific peptidase 22 (USP22) (si-SNHG16 and si-USP22) or negative control (si-NC), SNHG16 and USP22 overexpression plasmids (pcDNA-SNHG16 and pcDNA-USP22) or negative control (pcDNA), miR-132-3p mimic (miR-132-3p), miR-132-3p inhibitor and their negative controls (miR-NC and inhibitor-NC) were purchased from RiboBio (Guangzhou, China). Lentiviral short hairpin RNA (shRNA) targeting SNHG16 (sh-SNHG16) and negative control (sh-NC) were synthesized by Genechem (Shanghai, China). All oligonucleotides or plasmid vectors were transfected into SW480 and SW620 cells using Lipofectamine 3000 (Invitrogen).
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
TRIzol reagent (Invitrogen) was utilized for extracting the total RNA. The Reverse Transcription Kit (Takara, Dalian, China) was used to synthesize cDNA. SYBR Green (Takara) was used to quantify the levels of SNHG16, miR-132-3p and USP22 using ABI7500 system (Applied Biosystems, Foster City, CA, USA). The amplification process was as below: denaturation at 95°C for 5 min, followed by 40 cycles of at 95°C for 15 s, annealing at 55°C for 30 s, and extension at 60°C for 1 min. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and U6 were used as internal controls. The primers were listed as below: SNHG16: F, 5ʹ-CAGAATGCCATGGTTTCCCC-3ʹ, R, 5ʹ-TGGCAAGAGACTTCCTGAGG-3ʹ; USP22: F, 5ʹ-GGCGGAAGATCACCACGTAT-3ʹ, R, 5ʹ-TTGTTGAGACTGTCCGTGGG-3ʹ; GAPDH: F, 5ʹ-CCACATCGCTCAGACACCAT-3ʹ, R, 5ʹ-ACCAGGCGCCCAATACG-3ʹ; miR-132-3p: F, 5ʹ-GCGCGCGTAACAGTCTACAGG-3ʹ, R, 5ʹ-GTCGTATCCAGTGCAGGGTCC-3ʹ; U6: F, 5ʹ-CTCGCTTCGGCAGCACATATACT-3ʹ, R, 5ʹ-CGCTTCACGAATTTGCGTGT-3ʹ. The fold-change was calculated using the 2−ΔΔCt method.
Cell Vitality Assay
The proliferation of CRC cells was determined using 3-(4, 5-dimethyl-2 thiazolyl)-2, 5-diphenyl-2-H-tetrazolium bromide (MTT) Assay Kit (Beyotime, Shanghai, China). The transfected SW480 and SW620 cells were cultured with MTT solution for 4 h. After the cells were washed with phosphate buffer saline (PBS), dimethylsulfoxide (DMSO) was added for shock dissolution for 10 min, and the absorbance at 490 nm was tested using a spectrophotometer.
Flow Cytometry
Annexin V-FITC/PI Apoptosis Detection Kit (Yeasen, Shanghai, China) was used for detecting cell apoptosis. In brief, after digestion with trypsin, SW480 and SW620 cells were re-suspended with PBS and collected into the centrifuge tube. Then, the cells were stained with Annexin V-FITC and PI away from light for 20 min. Flow cytometer (Merck KGaA, Darmstadt, Germany) was used to detect the fluorescence signals of cells to evaluate the number of apoptotic cells.
Transwell Assay
Transwell assay was performed using 8 μm polycarbonate membrane of chambers (Corning Inc., Corning, NY, USA). The upper chambers were pre-coated with Matrigel (BD Biosciences, San Jose, CA, USA) to detect cell invasion, while uncoated to detect cell migration. The transfected SW480 and SW620 cells were collected and seeded into the upper chambers. The upper chambers were filled with serum-free medium, and the lower chambers were filled with serum medium. After 24 h, the lower chamber cells were fixed with paraformaldehyde and stained with crystal violet. Finally, images were taken under a microscope (Shoif, Shanghai, China) to observe and count the number of migrated and invaded cells (× 200).
Dual-Luciferase Reporter Assay
The SNHG16 wild-type (SNHG16-WT) and mutant-type (SNHG16-MUT) reporter vectors containing the miR-132-3p binding sites and mutant binding sites were synthesized by General Biosystems (Anhui, China). Similarly, USP22 3ʹUTR-WT/MUT reporter vectors were constructed in the same way. The above reporter vectors were co-transfected with miR-132-3p mimic and miR-NC into SW480 and SW620 cells using Lipofectamine 3000. After 48 h, Dual-luciferase Reporter Assay Kit (Promega, Madison, WI, USA) was performed to detect the luciferase reaction intensity of Firefly and Renilla to evaluate the luciferase activities of cells (Firefly luciferase reaction intensity/Renilla luciferase reaction intensity).
Western Blot (WB) Analysis
SW480 and SW620 cells were lysed with lysis buffer (Beyotime). Protein concentration was measured using the BCA Assay Kit (Yeasen). Protein samples were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel and then transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA, USA). After blockage with 5% non-fat milk, the membranes were cultured with the primary antibodies against USP22 (1:2000, Invitrogen), MMP2 (1:800, Invitrogen), MMP9 (1:1000, Invitrogen) or β-actin (1:5000, Invitrogen) overnight at 4°C. Then, the membranes were cultured with horseradish peroxidase-coupled secondary antibody (1:2000, Invitrogen). Protein signals were observed with an enhanced chemiluminescence (ECL) solution (Beyotime).
Mice Xenograft Models
Twelve male BALB/c-nude mice (5-week-old) were bought from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). SW620 cells transfected with sh-SNHG16 or sh-NC (n = 6 per group) were subcutaneously injected into the mice. From d 7, the tumor length and width were recorded to calculate tumor volume every 5 d. After 27 d, the tumor was removed for weight measurement and qRT-PCR, and the lung, liver and brain tissues were isolated to count the number of metastatic nodules. All experiments were authorized by the Animal Care Committee of Liu Zhou People’s Hospital and performed in compliance with the ARRIVE guidelines and the Basel Declaration.
Statistical Analysis
Data were presented as mean ± standard deviation (SD). Statistical analysis was performed using GraphPad 5.0 software (GraphPad Software, La Jolla, CA, USA). The Student’s t-test and one-way analysis of variance (ANOVA) were used to analyze the differences between the paired groups and multiple groups, respectively. The significant difference was defined as P < 0.05.
Results
SNHG16 Was Highly Expressed in CRC
To investigate the effect of SNHG16 on CRC, we first detected the expression of SNHG16 in CRC. As shown in Figure 1A, compared with adjacent normal tissues, SNHG16 expression was markedly increased in CRC tumor tissues. Besides, compared with CCD841 CON cells, the level of SNHG16 was also up-regulated in SW480 and SW620 cells (Figure 1B). These indicated that SNHG16 had an abnormal expression in CRC.
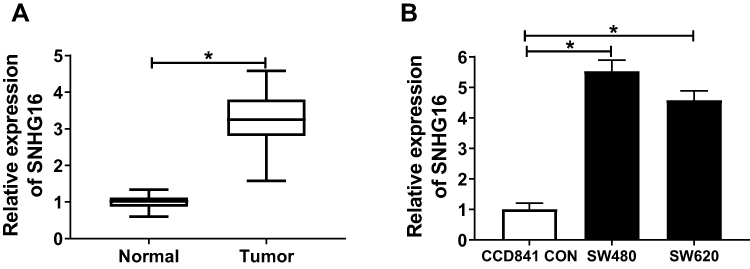 |
Figure 1 The expression of SNHG16 in CRC tissues and cells. (A) QRT-PCR revealed that the expression of SNHG16 was highly expressed in CRC tumor tissues (Tumor) (n = 50) compared to adjacent normal tissues (Normal) (n = 50). (B) The results of qRT-PCR indicated that SNHG16 expression was up-regulated in CRC cells (SW480 and SW620) compared with that in colonic epithelial cells (CCD841 CON). *P < 0.05. |
Silencing of SNHG16 Suppressed the Proliferation, Migration, Invasion and Promoted the Apoptosis of CRC Cells
Subsequently, we verified the effect of SNHG16 on CRC cell progression by reducing the expression of SNHG16. The efficiency test results showed that si-SNHG16 had an excellent inhibitory effect on SNHG16 expression, which could be used for follow-up experiments (Figure 2A and B). MTT assay revealed that knockdown of SNHG16 impaired the proliferation ability of SW480 and SW620 cells (Figure 2C and D). Flow cytometry confirmed that si-SNHG16 accelerated the number of apoptotic CRC cells (Figure 2E). Besides, transwell assay results further demonstrated that the migration and invasion capacities of SW480 and SW620 cells were decreased by SNHG16 silencing (Figure 2F and G). In addition, we also measured the effect of SNHG16 overexpression on the proliferation, migration and invasion of CRC cells. Contrary to the results of SNHG16 knockdown, we discovered that overexpression of SNHG16 could promote the proliferation, migration and invasion of SW480 and SW620 cells (Supplement Figure 1). These results confirmed that SNHG16 played a pro-cancer role in CRC progression.
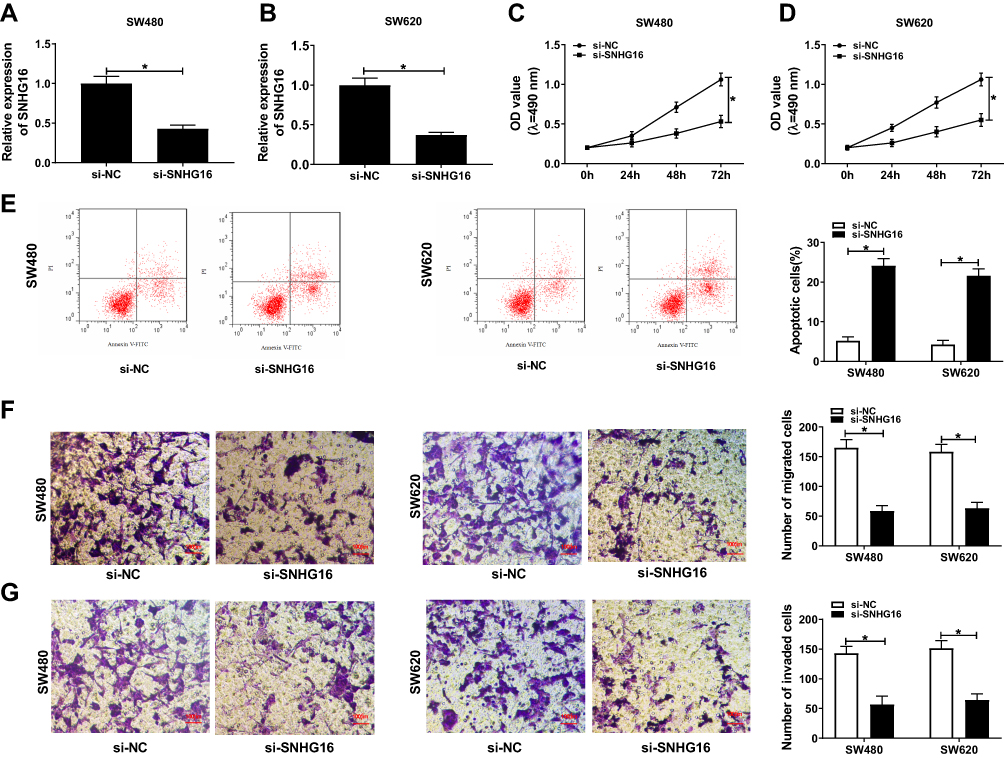 |
Figure 2 Silenced-SNHG16 regulated the progression of CRC cells. SW480 and SW620 cells were transfected with si-SNHG16 and si-NC. (A–B) The expression of SNHG16 was detected by qRT-PCR to evaluate the transfection efficiency of si-SNHG16. The results of MTT assay (C–D), flow cytometry (E) and transwell assay (F–G) suggested that SNHG16 knockdown inhibited the proliferation, promote the apoptosis and suppressed the migration and invasion of CRC cells. *P < 0.05. |
MiR-132-3p Had a Targeting Relationship with SNHG16 in CRC
To investigate the effect of SNHG16 on the proliferation and metastasis of CRC cells, we used the Starbase3.1 tool to predict the targets of SNHG16 and found that miR-132-3p had complementary binding sites with SNHG16. Subsequently, we constructed the SNHG16-WT and SNHG16-MUT reporter vectors to verify the functional relationship between SNHG16 and miR-132-3p (Figure 3A). Dual-luciferase reporter assay demonstrated that miR-132-3p mimic could significantly repress the luciferase activity of SNHG16-WT, while had no effect on SNHG16-MUT (Figure 3B and C). Besides, miR-132-3p expression was increased by SNHG16 knockdown, and reduced by SNHG16 overexpression, indicating that miR-132-3p expression was regulated by SNHG16 in CRC (Figure 3D and E). Through qRT-PCR, we found that the expression of miR-132-3p was decreased in CRC tumor tissues and cells (Figure 3F and G). Also, correlation analysis revealed that the expression of miR-132-3p was negatively correlated with SNHG16 in CRC tissues (Figure 3H). Therefore, we confirmed that miR-132-3p could interact with SNHG16 in CRC.
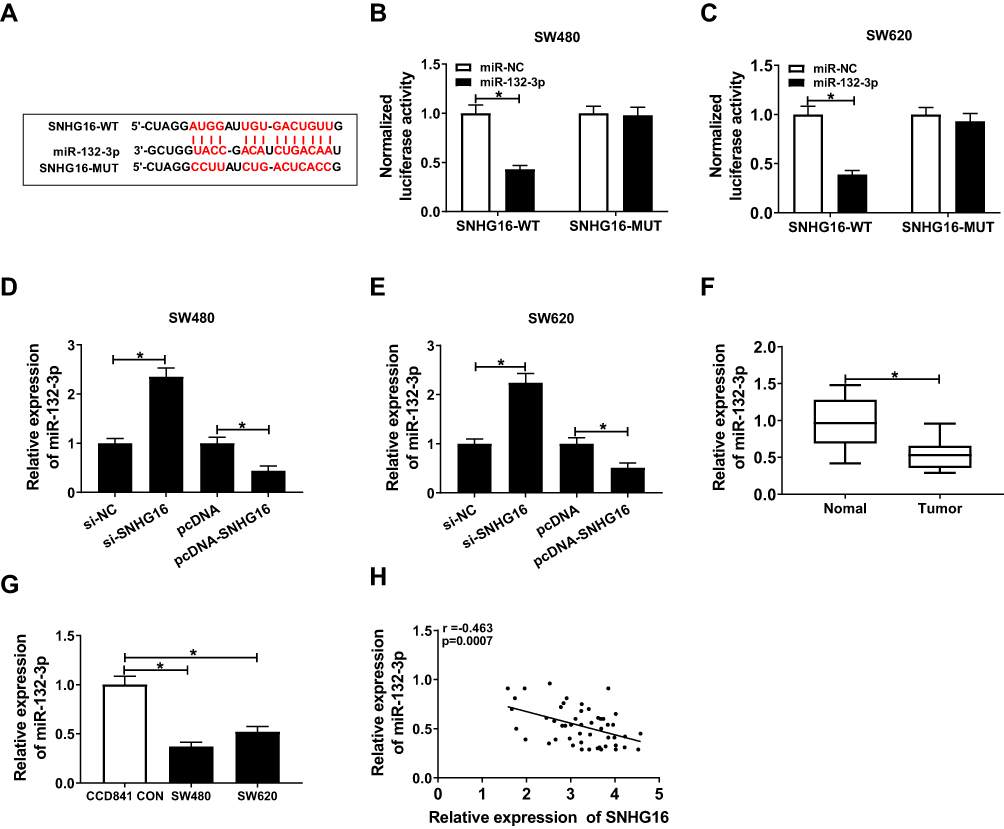 |
Figure 3 SNHG16 acted as a sponge of miR-132-3p. (A) The sequences of SNHG16 containing miR-132-3p binding sites (SNHG16-WT) and mutant binding sites (SNHG16-MUT) were shown. (B–C) Dual-luciferase reporter assay indicated that miR-132-3p interacted with SNHG16 in SW480 and SW620 cells. (D–E) QRT-PCR results indicated that the expression of miR-132-3p was regulated by SNHG16 in SW480 and SW620 cells. (F–G) The expression of miR-132-3p was decreased in CRC tumor tissues and cells detected by qRT-PCR. (H) Pearson correlation coefficient analysis revealed that miR-132-3p expression was negatively correlated with SNHG16 expression in CRC tissues. *P < 0.05. |
MiR-132-3p Inhibitor Recovered the Suppression Effect of SNHG16 Knockdown on CRC Cell Progression
To verify whether SNHG16 regulating CRC cell progression by sponging miR-132-3p, SW480 and SW620 cells were co-transfected with si-SNHG16 and miR-132-3p inhibitor. The efficiency experiment results showed that the miR-132-3p inhibitor significantly blocked miR-132-3p expression (Figure 4A). Through the test results of MTT, flow cytometry and transwell assays, we concluded that miR-132-3p inhibitor could reverse the inhibition effect of silenced-SNHG16 on the proliferation, migration and invasion of SW480 and SW620 cells, as well as the promotion effect on apoptosis (Figure 4B–F). These results indicated that SNHG16 regulated the proliferation, apoptosis, migration and invasion of CRC cells through sponging miR-132-3p.
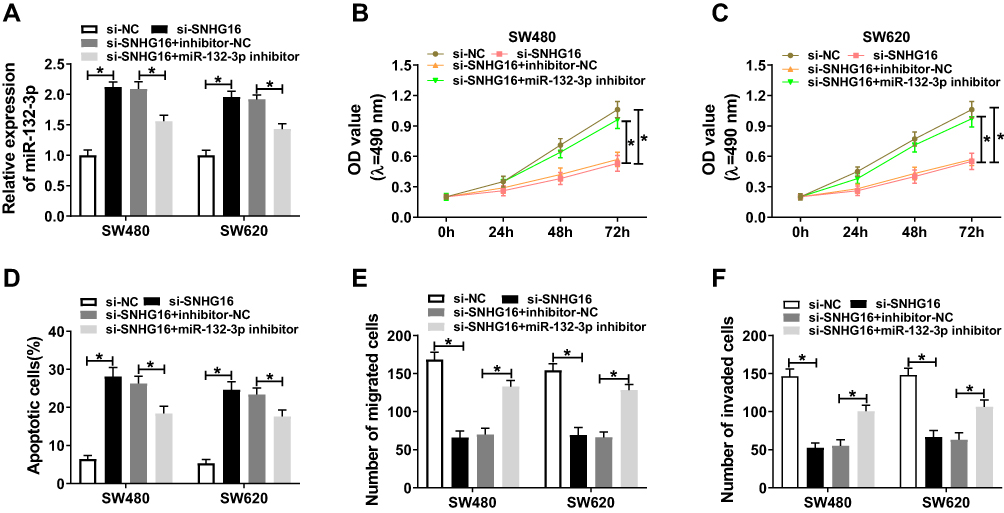 |
Figure 4 Knockdown of SNHG16 and miR-132-3p regulated the progression of CRC cells. SW480 and SW620 cells were co-transfected with si-SNHG16 and miR-132-3p inhibitor or their corresponding negative controls (si-NC and inhibitor-NC). (A) The expression of miR-132-3p was evaluated by qRT-PCR to evaluate the transfection efficiency of si-SNHG16 and miR-132-3p inhibitor. The results of MTT assay (B–C), flow cytometry (D) and transwell assay (E–F) indicated that miR-132-3p inhibitor could reverse the suppression effect of SNHG16 knockdown on the proliferation, migration and invasion, and the promotion effect of it on the apoptosis of SW480 and SW620 cells. * P < 0.05. |
USP22 Was a Target of miR-132-3p in CRC
To perfect the mechanism of SNHG16, we used the TargetScanHuman tool to predict the downstream target genes of miR-132-3p, and the results showed that USP22 had the complementary binding sites with miR-132-3p, so we constructed USP22 3ʹUTR-WT and USP22 3ʹUTR-MUT reporter vectors for the validation of dual-luciferase reporter assay (Figure 5A). The results showed that overexpression of miR-132-3p remarkably inhibited the luciferase activity of USP22 3ʹUTR-WT in SW480 and SW620 cells, without affecting the luciferase activity of USP22 3ʹUTR-MUT (Figure 5B and C). Then, the mRNA and protein expression levels of USP22 were down-regulated in SW480 and SW620 cells transfected with miR-132-3p mimic (Figure 5D and E). Besides, through detecting the expression of USP22, we found that UCSP22 was increased in CRC tumor tissues and cells (Figure 5F and G), and was negatively correlated with miR-132-3p level in CRC tissues (Figure 5H). These results confirmed the targeting relationship between USP22 and miR-132-3p in CRC.
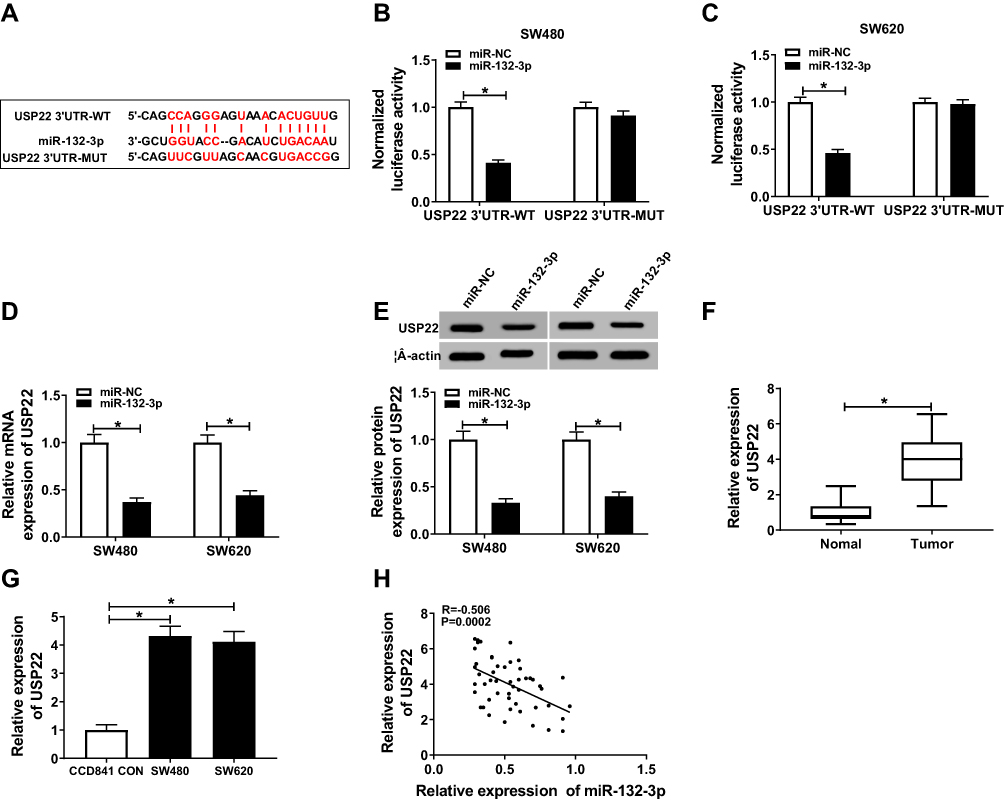 |
Figure 5 USP22 was a target of miR-132-3p. (A) Wild-type and mutant-type USP22 3ʹUTR reporter vectors (USP22 3ʹUTR-WT and USP22 3ʹUTR-MUT) were established according to the complementary sequences of miR-132-3p. (B–C) Dual-luciferase reporter assay confirmed that USP22 could interact with miR-132-3p in SW480 and SW620 cells. (D–E) QRT-PCR and WB analysis results revealed that miR-132-3p overexpression could suppress the mRNA and protein levels of USP22 in SW480 and SW620 cells. (F–G) The expression of USP22 was increased in CRC tumor tissues and cells detected by qRT-PCR. (H) Pearson correlation coefficient analysis suggested that USP22 expression was negatively correlated with miR-132-3p expression in CRC tissues. * P < 0.05. |
Overexpressed USP22 Restored the Inhibition Effects of miR-132-3p on CRC Cell Progression
Subsequently, we determined the effect of USP22 knockdown and overexpression on CRC cell progression. Through measuring the proliferation, migration and invasion of SW480 and SW620 cells, we uncovered that USP22 knockdown could suppress the proliferation, migration and invasion of CRC cells, while USP22 overexpression had an opposite effect (Supplement Figure 2). To confirm whether miR-132-3p regulated CRC cell progression through targeting USP22, we co-transfected with USP22 overexpression plasmid and miR-132-3p mimic into SW480 and SW620 cells. BY detecting the mRNA and protein expression of USP22, we discovered that USP22 overexpression could markedly restore the inhibition effect of miR-132-3p mimic on USP22 expression, indicating that the transfection efficiency of miR-132-3p mimic and pcDNA-USP22 was good (Figure 6A and B). Further experimental verification showed that overexpression of USP22 reversed the suppression effects of miR-132-3p overexpression on the proliferation, migration and invasion of SW480 and SW620 cells, as well as the acceleration effect of apoptosis (Figure 6C–G), further confirming that miR-132-3p regulated the progression of CRC by targeting USP22.
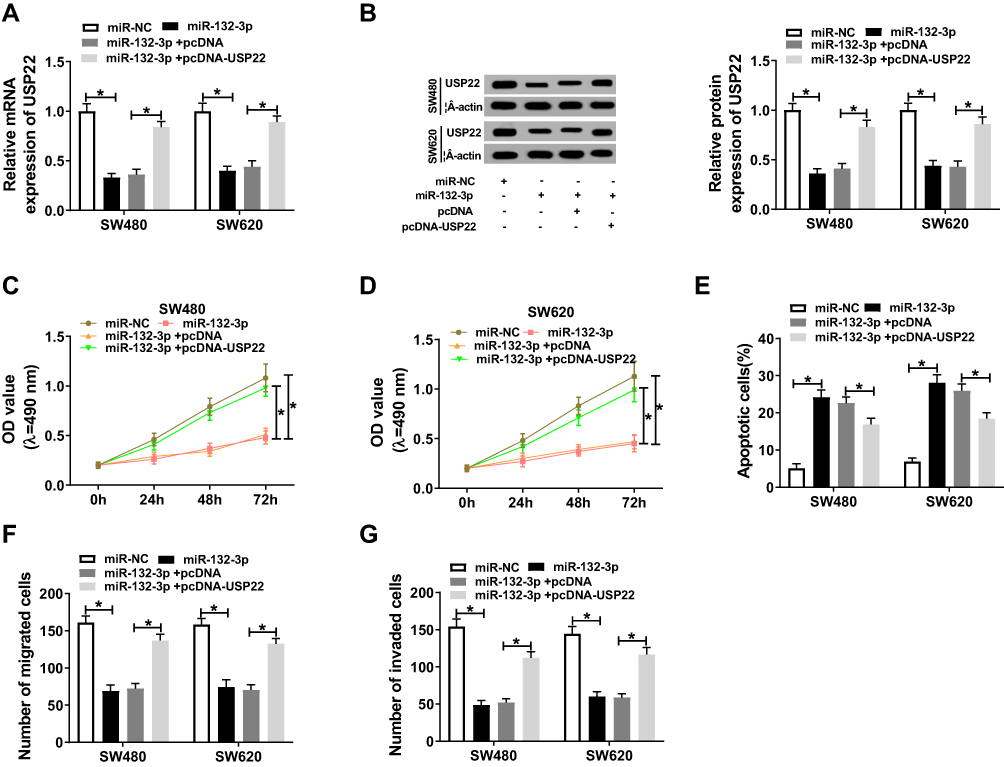 |
Figure 6 Overexpression of miR-132-3p and USP22 regulated the progression of CRC cells. MiR-132-3p mimic and pcDNA-USP22 or their negative controls (miR-NC and pcDNA) were co-transfected into SW480 and SW620 cells. (A–B) The mRNA and protein expression of USP22 was evaluated by qRT-PCR and WB analysis to evaluate the transfection efficiency of miR-132-3p mimic and pcDNA-USP22. The results of MTT assay (C–D), flow cytometry (E) and transwell assay (F–G) showed that USP22 overexpression could reverse the inhibition effect of miR-132-3p overexpression on the proliferation, migration and invasion, and the increasing effect of it on the apoptosis of were used to detect the abilities of SW480 and SW620 cells. * P < 0.05. |
SNHG16 Improved USP22 Expression via Sponging miR-132-3p in CRC Cells
Meanwhile, we examined the expression of USP22 under the premise of knocking down SNHG16 and miR-132-3p. QRT-PCR results showed that the SNHG16 knockdown could inhibit the expression of USP22 in SW480 and SW620 cells, while the addition of miR-132-3p inhibitor could reverse this effect (Figure 7A and B). Also, WB results confirmed that the inhibition of si-SNHG16 on the expression of USP22 could be alleviated by miR-132-3p inhibitor in SW480 and SW620 cells (Figure 7C and D). Hence, our data suggested that SNHG16 regulated USP22 level through sponging miR-132-3p in CRC.
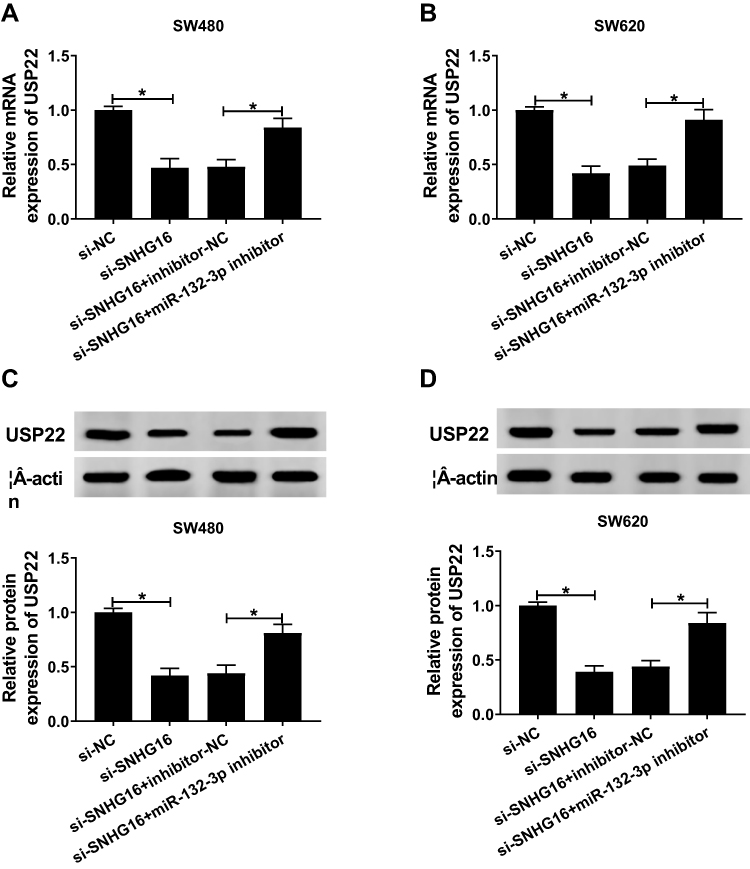 |
Figure 7 Inhibition of SNHG16 and miR-132-3p regulated the expression of USP22. SW480 and SW620 cells were co-transfected with si-SNHG16 and miR-132-3p inhibitor or their negative controls (si-NC and inhibitor-NC). (A–B) QRT-PCR results revealed that miR-132-3p inhibitor could reverse the suppression effect of SNHG16 silencing on the mRNA expression of USP22 in SW480 and SW620 cells. (C–D) WB analysis showed that miR-132-3p inhibitor could invert the inhibition effect of SNHG16 knockdown on the protein level of USP22 in SW480 and SW620 cells. *P < 0.05. |
Interference of SNHG16 Reduced Tumor Growth in vivo
We also constructed the mice xenograft models to further verify the effect of SNHG16 on CRC tumor growth in vivo. As shown in Figure 8A, the tumor volume of the sh-SNHG16 group was significantly smaller than that of the sh-NC group with an increase of transplantation days. After tumor weighing, we found that the tumor weight in the SNHG16 knockdown group was obviously decreased (Figure 8B). According to qRT-PCR analysis, silenced-SNHG16 blocked the expression levels of SNHG16 and USP22 and promoted miR-132-3p level in tumors (Figure 8C). In addition, in isolated lung, liver and brain tissues, metastatic nodules were observed only in isolated lung tissues, and by calculating the number of metastatic nodules, we found that knockdown of SNHG16 significantly decreased the number of metastasis nodules (Figure 8D). Furthermore, the protein levels of metastasis-related markers (MMP2 and MMP9) were detected to evaluate the metastasis ability of CRC tumors in vivo. As shown in Figure 8E, the results revealed that the protein levels of MMP2 and MMP9 in the sh-SNHG16 group were obviously suppressed compared with the sh-NC group, indicating that SNHG16 knockdown could inhibit the metastasis ability of CRC tumors in vivo. These data suggested that silencing of SNHG16 impeded the tumor growth of CRC through regulating the expression levels of miR-132-3p and USP22 in vivo.
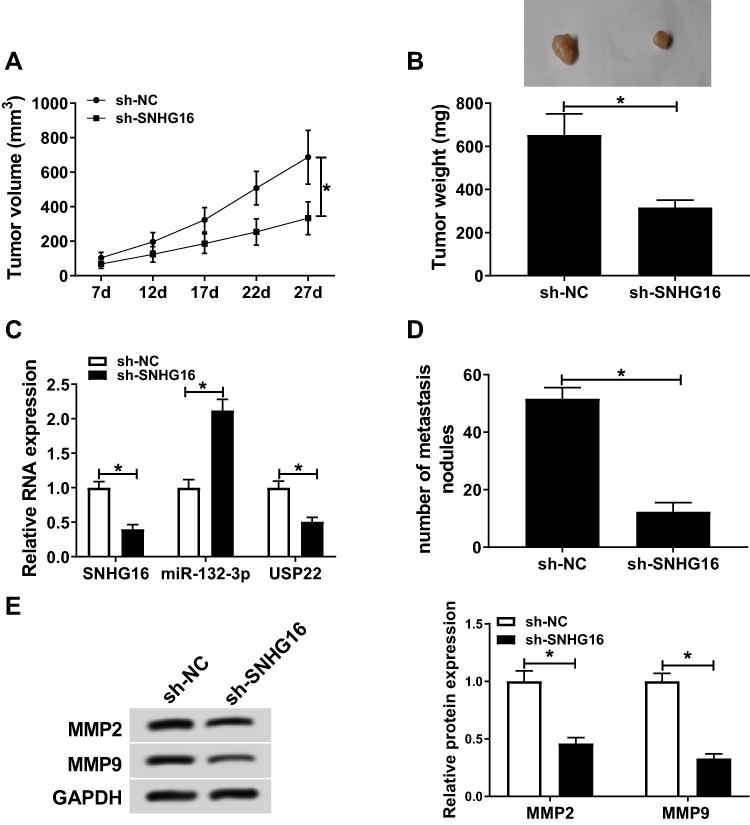 |
Figure 8 SNHG16 knockdown regulated the tumor growth of CRC in vivo. SW620 cells transfected with sh-SNHG16 or sh-NC were subcutaneously injected into the mice to construct the xenograft models. The tumor volume (A) and tumor weight (B) in the sh-SNHG16 group was remarkably reduced compared to the sh-NC group. (C) QRT-PCR results revealed that SNHG16 and USP22 expression was decreased, while miR-132-3p expression was increased in the tumor tissues of the sh-SNHG16 group compared to the sh-NC group. (D) The number of metastasis nodules in the tumor was counted. (E) WB analysis results showed that the protein levels of MMP2 and MMP9 were inhibited by SNHG16 knockdown in the tumor tissues. *P < 0.05. |
Discussion
Recent studies have shown that SNHG16 was a potential oncogene in many cancers. In CRC, Li et al reported that SNHG16 improved CRC progression and tumor growth.19 In our system, consistent with published findings, SNHG16 was enriched in CRC tumor tissues and cells. Functional experiments confirmed that SNHG16 knockdown restrained the progression of CRC cells in vitro and reduced CRC tumor growth in vivo. Consistent with the role of SNHG16 on other cancers,16–18 our results revealed that SNHG16 played an active role in the development of CRC. Therefore, elucidating the molecular mechanism of SNHG16 in CRC can help us develop new methods to treat CRC.
MiR-132-3p was a tumor suppressor miRNA in multiple cancers, including mesothelioma, bladder carcinoma and breast cancer.21–23 Previous studies have covered the low level of miR-132-3p in CRC compared with normal samples, and overexpression of miR-132-3p restrained the proliferation and metastasis of CRC cells.24 Song et al suggested that miR-132-3p might involve in the regulation of XIST on CRC cell proliferation.25 In our researches, we proved that SNHG16 could sponge miR-132-3p in CRC, and miR-132-3p expression was regulated by SNHG16 in vitro and in vivo. Consistent with the previous studies, our results revealed that miR-132-3p expression was reduced in CRC. Additionally, the results of rescue experiments suggested that SNHG16 regulated CRC proliferation, apoptosis, migration and invasion by sponging miR-132-3p. Therefore, we confirmed that miR-132-3p also could act as a tumor suppressor in CRC. In addition, the anti-cancer effect of miR-132-3p also helps us better understand the potential causes of SNHG16 promoting CRC progression.
USP22 is a member of the deubiquitinating enzyme (DUB) family, which is related to the occurrence and development of various tumor types, including CRC.26–28 Depletion of USP22 led to the accumulation in G1 phase and blocked the proliferation of CRC cells.29 It has been identified that high USP22 expression was markedly associated with the poor prognosis of CRC.30 Consistent with previous research results, here, we found that USP22 expression was improved in CRC, and it could play a pro-cancer role in CRC progression. In terms of mechanism, our results revealed that USP22 was a target of miR-132-3p. Moreover, the abnormal expression of USP22 recovered the detraction function of miR-132-3p overexpression on proliferation and metastasis of CRC cells to promote the CRC progression. To improve the functional relationship of SNHG16/miR-132-3p/USP22 axis, we also tested the effects of SNHG16 and miR-132-3p on USP22 level. Our results confirmed that SNHG16 regulated USP22 expression by sponging miR-132-3p. These results perfected the hypothesis that lncRNA SNHG16 served as a ceRNA to regulate the expression of downstream target genes through sponging miRNAs,31 and enriched the study of SNHG16 on CRC progression.
Of course, the current study has some limitations. For the proliferation, apoptosis and metastasis of cells, the detection results of relevant protein levels may further verify our experimental results and enrich our experimental content. In addition, our study found that the reversal effect of miR-132-3p inhibitor on SNHG16 knockdown was only partial, suggesting that there may be other miRNAs involved in the regulation of SNHG16 on CRC progression, so further confirmation is needed.
Conclusion
In summary, we concluded that lncRNA SNHG16 regulated USP22 expression to improve CRC progression through absorbing miR-132-3p. Our results provided a clear recognizing of the relationship between lncRNA SNHG16 and CRC molecular pathogenesis, providing a potential pathway for the treatment of CRC.
Disclosure
The authors declare that they have no financial conflicts of interest.
References
1. Siegel RL, Miller KD, Fedewa SA, et al. Colorectal cancer statistics, 2017. CA Cancer J Clin. 2017;67(3):177–193. doi:10.3322/caac.21395
2. Brenner H, Kloor M, Pox CP. Colorectal cancer. Lancet. 2014;383(9927):1490–1502. doi:10.1016/S0140-6736(13)61649-9
3. Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
4. Lee RM, Cardona K, Russell MC. Historical perspective: two decades of progress in treating metastatic colorectal cancer. J Surg Oncol. 2019;119(5):549–563. doi:10.1002/jso.25431
5. Parizadeh SM, Jafarzadeh-Esfehani R, Hassanian SM, et al. Targeting cancer stem cells as therapeutic approach in the treatment of colorectal cancer. Int J Biochem Cell Biol. 2019;110:75–83. doi:10.1016/j.biocel.2019.02.010
6. Mody K, Baldeo C, Bekaii-Saab T. Antiangiogenic Therapy in Colorectal Cancer. Cancer J. 2018;24(4):165–170. doi:10.1097/PPO.0000000000000328
7. Slattery ML, Herrick JS, Mullany LE, et al. The co-regulatory networks of tumor suppressor genes, oncogenes, and miRNAs in colorectal cancer. Genes Chromosomes Cancer. 2017;56(11):769–787. doi:10.1002/gcc.22481
8. Wei L, Wang X, Lv L, Zheng Y, Zhang N, Yang M. The emerging role of noncoding RNAs in colorectal cancer chemoresistance. Cell Oncol (Dordr). 2019;42(6):757–768. doi:10.1007/s13402-019-00466-8
9. Beermann J, Piccoli MT, Viereck J, Thum T. Non-coding RNAs in development and disease: background, mechanisms, and therapeutic approaches. Physiol Rev. 2016;96(4):1297–1325. doi:10.1152/physrev.00041.2015
10. Xu J, Meng Q, Li X, et al. Long non-coding RNA MIR17HG promotes colorectal cancer progression via miR-17-5p. Cancer Res. 2019;79(19):4882–4895. doi:10.1158/0008-5472.CAN-18-3880
11. Liu L, Wang HJ, Meng T, et al. lncRNA GAS5 inhibits cell migration and invasion and promotes autophagy by targeting mir-222-3p via the gas5/pten-signaling pathway in CRC. Mol Ther Nucleic Acids. 2019;17:644–656. doi:10.1016/j.omtn.2019.06.009
12. Tang J, Yan T, Bao Y, et al. LncRNA GLCC1 promotes colorectal carcinogenesis and glucose metabolism by stabilizing c-Myc. Nat Commun. 2019;10(1):3499. doi:10.1038/s41467-019-11447-8
13. Si Y, Yang Z, Ge Q, et al. Long non-coding RNA Malat1 activated autophagy, hence promoting cell proliferation and inhibiting apoptosis by sponging miR-101 in colorectal cancer. Cell Mol Biol Lett. 2019;24(1):50. doi:10.1186/s11658-019-0175-8
14. Du C, Wang HX, Chen P, Chen CH. STAT3-induced upregulation of lncRNA DUXAP8 functions as ceRNA for miR-577 to promote the migration and invasion in colorectal cancer through the regulation of RAB14. Eur Rev Med Pharmacol Sci. 2019;23(14):6105–6118. doi:10.26355/eurrev_201907_18424
15. Zhao Y, Du T, Du L, et al. Long noncoding RNA LINC02418 regulates MELK expression by acting as a ceRNA and may serve as a diagnostic marker for colorectal cancer. Cell Death Dis. 2019;10(8):568. doi:10.1038/s41419-019-1804-x
16. Cai C, Huo Q, Wang X, Chen B, Yang Q. SNHG16 contributes to breast cancer cell migration by competitively binding miR-98 with E2F5. Biochem Biophys Res Commun. 2017;485(2):272–278. doi:10.1016/j.bbrc.2017.02.094
17. Chen H, Li M, Huang P. LncRNA SNHG16 promotes hepatocellular carcinoma proliferation, migration and invasion by regulating mir-186 expression. J Cancer. 2019;10(15):3571–3581. doi:10.7150/jca.28428
18. Lu YF, Cai XL, Li ZZ, et al. LncRNA SNHG16 functions as an oncogene by sponging MiR-4518 and up-regulating PRMT5 expression in glioma. Cell Physiol Biochem. 2018;45(5):1975–1985. doi:10.1159/000487974
19. Li Y, Lu Y, Chen Y. Long non-coding RNA SNHG16 affects cell proliferation and predicts a poor prognosis in patients with colorectal cancer via sponging miR-200a-3p. Biosci Rep. 2019;39(5):BSR20182498. doi:10.1042/BSR20182498
20. Christensen LL, True K, Hamilton MP, et al. SNHG16 is regulated by the Wnt pathway in colorectal cancer and affects genes involved in lipid metabolism. Mol Oncol. 2016;10(8):1266–1282. doi:10.1016/j.molonc.2016.06.003
21. Liu P, Li X, Guo X, et al. Circular RNA DOCK1 promotes bladder carcinoma progression via modulating circDOCK1/hsa-miR-132-3p/Sox5 signalling pathway. Cell Prolif. 2019;52(4):e12614. doi:10.1111/cpr.12614
22. Li S, Xu JJ, Zhang QY. miR-132-3p inhibits tumor malignant progression by regulating LAPTM4B in breast cancer. Cancer Sci. 2019;110(10):3098–3109. doi:10.1111/cas.14164
23. Weber DG, Gawrych K, Casjens S, et al. Circulating miR-132-3p as a candidate diagnostic biomarker for malignant mesothelioma. Dis Markers. 2017;2017:9280170. doi:10.1155/2017/9280170
24. Zhang M, Li Y, Wang H, Yu W, Lin S, Guo J. LncRNA SNHG5 affects cell proliferation, metastasis and migration of colorectal cancer through regulating miR-132-3p/CREB5. Cancer Biol Ther. 2019;20(4):524–536. doi:10.1080/15384047.2018.1537579
25. Song H, He P, Shao T, Li Y, Li J, Zhang Y. Long non-coding RNA XIST functions as an oncogene in human colorectal cancer by targeting miR-132-3p. J BUON. 2017;22(3):696–703.
26. Liao Y, Liang X, Liang W, et al. High expression of ubiquitin carboxyl-terminal hydrolase 22 is associated with poor prognosis in hepatitis B virus-associated liver cancer. Oncol Lett. 2019;17(6):5159–5168. doi:10.3892/ol.2019.10154
27. Liu T, Liu J, Chen Q, et al. Expression of USP22 and the chromosomal passenger complex is an indicator of malignant progression in oral squamous cell carcinoma. Oncol Lett. 2019;17(2):2040–2046. doi:10.3892/ol.2018.9837
28. Jiang S, Miao D, Wang M, Lv J, Wang Y, Tong J. MiR-30-5p suppresses cell chemoresistance and stemness in colorectal cancer through USP22/Wnt/beta-catenin signaling axis. J Cell Mol Med. 2019;23(1):630–640. doi:10.1111/jcmm.13968
29. Liu YL, Jiang SX, Yang YM, Xu H, Liu JL, Wang XS. USP22 acts as an oncogene by the activation of BMI-1-mediated INK4a/ARF pathway and Akt pathway. Cell Biochem Biophys. 2012;62(1):229–235. doi:10.1007/s12013-011-9287-0
30. Liu YL, Yang YM, Xu H, Dong XS. Aberrant expression of USP22 is associated with liver metastasis and poor prognosis of colorectal cancer. J Surg Oncol. 2011;103(3):283–289. doi:10.1002/jso.21802
31. Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505(7483):344–352. doi:10.1038/nature12986
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.