Back to Journals » Journal of Inflammation Research » Volume 19
LncRNA TLR8-AS1 Restricts HIV-1 Infection and Inflammation in Macrophages by Suppressing Arachidonic Acid Metabolism Through NFAT1
Authors Wang F, Luo S ![]() , Zhang J, Zhang Y
, Zhang J, Zhang Y ![]() , Wei H
, Wei H ![]() , Liu J, Mo C, Jiang J, Ye L
, Liu J, Mo C, Jiang J, Ye L ![]() , Yuan Z
, Yuan Z ![]()
Received 10 January 2026
Accepted for publication 17 March 2026
Published 30 March 2026 Volume 2026:19 591629
DOI https://doi.org/10.2147/JIR.S591629
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Shouya Feng
Fengyi Wang,1,2,* Shengrui Luo,1,2,* Junhan Zhang,1,2 Yukai Zhang,1,2 Hailun Wei,1,2 Jie Liu,1– 3 Chuye Mo,1,2 Junjun Jiang,1– 3 Li Ye,1– 3 Zongxiang Yuan1,2
1Guangxi Key Laboratory of AIDS Prevention and Treatment, School of Public Health, Guangxi Medical University, Nanning, Guangxi, People’s Republic of China; 2Joint Laboratory for Emerging Infectious Diseases in China (Guangxi)-ASEAN, Life Sciences Institute, Guangxi Medical University, Nanning, Guangxi, People’s Republic of China; 3Guangxi Collaborative Innovation Center for Biomedicine, Life Science Institute, Guangxi Medical University, Nanning, Guangxi, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zongxiang Yuan; Li Ye, Email [email protected]; [email protected]
Background: Long non-coding RNA (lncRNA) TLR8-AS1 has been implicated in immune regulation, but its role in HIV-1 infection remains unexplored.
Methods: TLR8-AS1 expression was assessed in PBMCs and primary monocyte-derived macrophages (MDMs) from HIV-1/AIDS patients and healthy controls. Its subcellular localization was determined via bioinformatics, FISH, and nucleocytoplasmic fractionation. In THP-1-derived macrophages, the functional impact of TLR8-AS1 was evaluated using TLR8-AS1 overexpression and NFAT1-knockdown models; viral replication, inflammatory cytokines, and arachidonic acid (AA) metabolism were analyzed by qPCR, ELISA, and Western blot.
Results: TLR8-AS1 expression levels were positively correlated with CD4+ T cell counts (r=0.439, P < 0.05), suggesting a potential association with immune status. In THP-1-derived macrophages, TLR8-AS1 overexpression significantly inhibited HIV-1 p24 production, viral gene (Pol, Vif, Nef, LTR, and Gag) expression, and secretion of IL-1β, TNF-α, and AA. Mechanistically, cytoplasmic TLR8-AS1 downregulated NFAT1 and PTGS2 (COX-2) expression, selectively suppressing the prostaglandin pathway while leaving the lipoxygenase branch (ALOX5, ALOX15) unaffected. NFAT1 knockdown reproduced the antiviral and anti-inflammatory effects of TLR8-AS1, confirming NFAT1 as a key downstream mediator. In contrast, TLR8-AS1 did not alter TLR8 or its downstream signaling molecules (MyD88 and IRF7), suggesting a TLR8-independent mechanism.
Conclusion: TLR8-AS1 restricts HIV-1-induced inflammation and viral replication through the NFAT1-AA axis. These findings identify TLR8-AS1 as a potential therapeutic target for mitigating chronic inflammation and viral persistence in HIV-1 infection.
Keywords: HIV-1, LncRNA, TLR8-AS1, NFAT1, arachidonic acid, inflammation
Introduction
Acquired immunodeficiency syndrome (AIDS), caused by infection with the human immunodeficiency virus (HIV), remains a major global public health challenge, with an estimated 40.8 million people living with HIV and 630,000 HIV-related deaths worldwide in 2024.1 Despite significant advances in treatment through combination antiretroviral therapy (cART), persistent low-level viremia and chronic inflammation have yet to be addressed.2 In HIV-1-infected individuals, macrophages serve as key long-lived viral reservoirs owing to their inherent longevity.3 Furthermore, sustained low-level transcription of viral RNAs in these cells contributes to ongoing immune activation.3,4 Therefore, elucidating the mechanisms underlying viral persistence and immune activation in macrophages is crucial for advancing HIV cure research.
Long non-coding RNAs (lncRNAs) are transcripts longer than 200 nucleotides that generally do not encode proteins, but play key roles in regulating gene expression at the epigenetic, transcriptional, and post-transcriptional levels.5,6 Several LncRNAs, such as LncNRON, regulate HIV-1 replication through an NFAT-dependent mechanism, while others, like LncNEAT1, serve as diagnostic markers for HIV-1 infection.7,8 Among these regulatory lncRNAs, Toll-like receptor 8 antisense RNA 1 (TLR8-AS1) has attracted our attention. As its nomenclature indicates, this lncRNA is transcribed from the antisense strand opposite to the TLR8 protein-coding gene.9,10 In our previous study, TLR8-AS1 was found to be up-regulated by 8.91-fold in highly exposed persistently seronegative (HEPS) individuals compared with healthy controls.11 This finding suggests that TLR8-AS1 may play an essential role in this particular population with natural HIV-1 resistance. Given the central role of macrophages in HIV-1 persistence and immune dysregulation, TLR8-AS1 represents a particularly compelling candidate for further investigation. However, its role in HIV-1 infection and the underlying molecular mechanisms remain unknown.
LncRNAs are also increasingly recognized as important regulators of immunometabolic homeostasis. In macrophages, HIV-1 infection severely disrupts this immunometabolic balance.12 Crucially, this dysregulation is characterized by a significant shift in lipid metabolism.13,14 A key component of this process is dysregulated arachidonic acid (AA)metabolism, with AA, a key precursor of proinflammatory lipid mediators, playing a central role.15 AA is primarily metabolized through three key enzyme systems: cyclooxygenase (COX), lipoxygenase (LOX), and cytochrome P450 (CYP450). In the context of HIV-1 infection, the viral envelope glycoprotein gp120 has been shown to upregulate AA-metabolizing enzymes, including COX-2 and 5-LOX, thereby contributing to neuroinflammation.16–18 Notably, elevated levels of inflammatory mediators such as IL-1β and TNF-α can, in turn, enhance HIV-1 replication in macrophages. Beyond its role in inflammation, AA can also directly activate the HIV-1 long terminal repeat (LTR) through both the LOX and COX pathways, further promoting viral gene expression.19 Therefore, dysregulated AA metabolism contributes to both neuroinflammation and enhanced HIV-1 replication in macrophages. However, whether host regulatory lncRNAs participate in this process remains largely unclear.
Nuclear factor of activated T cells 1 (NFAT1) is a key member of the NFAT transcription factor family, expressed in both immune and non-immune cells. It plays a central role in innate and adaptive immune responses by regulating the transcription of immune-related genes.20 Beyond its classical immunoregulatory functions, NFAT1 has also been implicated in the control of inflammation-associated gene expression and may thereby influence metabolic pathways linked to immune activation.21 Notably, although lncRNA-mediated regulation of NFAT family members has been described in some contexts, the relationship between TLR8-AS1 and NFAT1 in HIV-1 infection has not been defined.
Therefore, this study focuses on the long non-coding RNA TLR8-AS1 to elucidate its potential role in regulating AA associated metabolic and inflammatory pathways during HIV-1 infection. Considering the dual involvement of macrophages in viral persistence and immune activation, we hypothesize that TLR8-AS1 suppresses HIV-1 replication and inflammation by modulating the NFAT1-AA axis. Elucidating this mechanism may provide new insight into the interplay between host immune metabolism and viral persistence, and identify TLR8-AS1 as a potential target for combined antiviral and anti-inflammatory therapy in HIV-1 infection.
Materials and Methods
Ethical Statement
The study protocol was reviewed and approved by Ethics Committee of Guangxi Medical University (approval number: 2019-SB-080) in accordance with the ethical standard of the Declaration of Helsinki. Written informed consent was obtained from all participating patients. Peripheral blood mononuclear cells (PBMCs) used in this study were collected specifically for the dual purposes of HIV-1Bal virion production and the subsequent detection of target lncRNA expression.
Study Subjects
We recruited a total of 45 HIV-1/AIDS individuals who met the predefined inclusion and exclusion criteria.22 The inclusion criteria were as follows: (1) age ≥ 18 years; (2) confirmed HIV-positive status; (3) received ART treatment. The exclusion criteria included: (1) missing baseline CD4+ T cell counts; (2) major chronic diseases such as cardiac, renal, pulmonary, endocrine, metabolic, or autoimmune diseases; (3) concurrent infection with hepatitis B/C virus, syphilis, or tuberculosis; and (4) pregnancy or lactation. Additionally, we recruited 20 HIV-seronegative healthy controls to establish the baseline expression of TLR8-AS1. By comparing TLR8-AS1 levels between this control group and the ART-treated cohort, we aimed to evaluate its expression dynamics in the context of HIV infection and ART. These control subjects were confirmed HIV-negative and met the inclusion criterion (1) and the general exclusion criteria described above.
Cell Culture and Differentiation
The THP-1 cell line (ATCC number: TIB-202) was employed as an in vitro model to investigate the effect of TLR8-AS1 on HIV-1 infection. Cells were maintained at 37°C in a humidified atmosphere with 5% CO2 in RPMI-1640 medium (Gibco, USA), supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco, USA) and 1% penicillin–streptomycin solution (Solarbio, China). For differentiation, THP-1 cells were seeded into 6-well plates (NEST, China) at a density of 1×106 cells per well in DMEM (Gibco, USA) containing 10% FBS (Gibco, USA) and 1% penicillin-streptomycin solution (Solarbio, China), and subsequently stimulated with 50 ng/mL phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich, USA) for 48 hours to generate THP-1-derived macrophages, followed by a 24-hour rest period in fresh medium without PMA before infection.23
Isolation of Human PBMCs by Ficoll-Paque Density Gradient Centrifugation
As referenced in Ethical Statement, the collection and utilization of PBMCs in the present study complied with the approved ethical protocol. PBMCs were isolated using the following procedure: Venous blood was centrifuged at 800×g for 10 minutes to remove cellular debris and plasma components. The residual fraction was diluted with an equal volume of room-temperature PBS (Solarbio, China). Subsequently, the diluted sample was subjected to Ficoll-Paque density gradient centrifugation (Cytiva, Sweden) at room temperature (20–25°C) at 400×g for 45 minutes with the acceleration set to 3 and deceleration set to 1. PBMCs were collected from the interface between the Ficoll-Paque and plasma layers into 50mL centrifuge tubes (NEST, China), washed with PBS containing 10% EDTA (500×g, 5 minutes, 4°C), and resuspended in RPMI-1640 medium supplemented with 10% FBS. The number and viability of PBMCs were evaluated using trypan blue staining (Beyotime, China) and a hemocytometer.
HIV-1 Production and Infection
HIV-1Bal virions were originally from the NIH AIDS Reagent Program and reproduced in our laboratory. The detailed procedures were described in the methods section of our previously published study24 and are as follows: PBMCs were stimulated with 5 μg/mL phytohemagglutinin (PHA, Sigma-Aldrich, USA) in RPMI-1640 complete medium supplemented with 100 U/mL IL-2 (Sigma-Aldrich, USA) for 3 days at 37°C under a 5% CO2. Subsequently, 2 μg/mL Polybrene (Beyotime, China) was added to the culture to facilitate subsequent viral infection.24 THP-1-derived macrophages were infected with HIV-1Bal virions at an MOI of 1 for 6 hours. Subsequently, the cells were washed with PBS to remove unbound viruses. And then supplemented with fresh DMEM medium and cultivated until harvest at subsequent experiment time.
Lentiviral Construction and Transduction
The lentiviral vector carrying human TLR8-AS1 (LV-TLR8-AS1, 5′-GTGTCACGCCCACTTTGCCAG-3′) and the negative control lentivirus (LV-NC, 5′-CGTCGCCGTCCAGCTCGACCAG-3′), as well as lentiviral particles containing specific shRNAs against NFAT1 (sh-NFAT1, 5′-GAACTTCTACGTCATCAAT-3′) and its corresponding negative control (sh-NC, 5′-TTCTCCGAACGTGTCACGT-3′), were purchased from GeneChem (China). Target cells in the logarithmic growth phase were seeded into 24-well plates at an optimal density of 3×105 cells/well. The cells were then transduced with lentiviral particles at an MOI of 30 in the presence of HitransG A (at a final concentration of 1×) by adding 20 µL of the 25× stock solution to the culture medium. After 24 hours of incubation, the virus-containing medium was replaced with fresh complete RPMI-1640 medium. To establish stably transduced THP-1 cell lines, puromycin (Sigma-Aldrich, USA) was added to the culture medium at a final concentration of 2 μg/mL at 48 hours post-transduction.
Determination of HIV-1 p24 and Inflammatory Cytokines
The concentrations of HIV-1 p24 levels and inflammatory cytokines (IL-1β and TNF-α) in the cell culture supernatants were quantified using commercial ELISA kits (HIV-1 p24 kit from BD PMG, China; IL-1β and TNF-α kits from NOVUS, USA) according to the manufacturers’ protocols. Briefly, culture supernatants were harvested at 48 hours, centrifuged to remove debris, and generated the standard curve for each target protein was generated using the serially diluted standards provided in the kits. Samples were appropriately diluted and added to the antibody-precoated wells. Incubation was performed for 1 hour at room temperature for the HIV-1 p24 assay, and for 2 hours at room temperature for the cytokine assays. After sequential incubation, plate washing, binding with enzyme conjugate, and substrate reaction were further processed. The enzymatic reaction was stopped by adding a stop solution, which changed the color from blue to yellow. The absorbance was measured at 450 nm with a reference wavelength of 630 nm for the HIV-1 p24 assay, and at 450 nm with a wavelength correction at 540 nm for cytokine assays, using a microplate reader (BioTek, USA). Concentrations were calculated from the respective standard curves. All experiments were performed in at least three independent biological replicates (n≥3).
Fluorescence in Situ Hybridization (FISH)
The Fluorescence in situ hybridization was performed to determine the subcellular localization. Specific probes targeting TLR8-AS1 were synthesized using PCR products generated with the following primers: TLR8-AS1-F (5′-cagggagagcacgtgtgtt-3′), TLR8-AS1-R (5′-gatgtgttctacattcctttattgg-3′), TLR8-AS1-F-T7 (5′-AAAATAATACGACTCACTATAGGGcagggagagcacgtgtgtt-3′), TLR8-AS1-R-T7 (5′-AAAATAATACGACTCACTATAGGGgatgtgttctacattcctttattgg-3′). Briefly, THP-1 cells were seeded on glass slides and differentiated into macrophages, followed by fixation with 4% paraformaldehyde (Solarbio, China), permeabilization, and pre-hybridization. Fluorescence-labeled probes (Red) were then hybridized according to the manufacturer’s instructions (RiboBio, China). After washing gently three times, nuclei were counterstained with DAPI (Blue). Fluorescence signals were observed and recorded under a fluorescence microscope (EVOS FL Auto2, USA).
Isolation of Nuclear and Cytoplasmic RNA from THP-1-Derived Macrophages
The subcellular distribution of the TLR8-AS1 was assessed by separating nuclear and cytoplasmic fractions from the THP-1 macrophages, using the Cytoplasmic & Nuclear RNA Purification Kit (Aidlab, China). Briefly, harvested cells were lysed with ice-cold Buffer RLN (supplemented with 1 mM DTT and RNase inhibitor) and incubated on ice. Following centrifugation at 13,000 g for 3 min, the supernatant (cytoplasmic fraction) was transferred to a new tube, and the pellet (nuclear fraction) was retained for further processing. RNA from each fraction was purified through RA columns according to the kit protocol. For the cytoplasmic fraction, the supernatant was mixed with Buffer RLT Plus and ethanol before column purification. For the nuclear fraction, the pellet was dissolved in Buffer RLT Plus, and the lysate was passed through a column; the flow-through was then mixed with ethanol for a second round of column purification. After washing steps, RNA was eluted in RNase-free water. Fractionation efficiency was validated by RT-qPCR using GAPDH (cytoplasmic) and MALAT1 (nuclear) as compartment-specific markers (Table S1).
RNA Extraction and RT-qPCR
Total cellular RNA was extracted using TRIzol reagent (Sigma-Aldrich, USA). Cells were lysed with TRIzol, and chloroform (Tedia, USA) was added for phase separation. The upper aqueous phase was collected after high-speed centrifugation and RNA was precipitated with isopropanol (Tedia, USA). The purified RNA was then further purified with 75% ethanol (Tedia, USA). RNA concentration and purity were determined using a NanoDrop 2000C spectrophotometer (Thermo Fisher Scientific, USA). Reverse transcription was performed using 1 μg of RNA and PrimeScript RT Master Mix (Takara, Japan) to synthesize cDNA. Quantitative real-time PCR (qPCR) was performed on cDNA samples using SYBR Premix Ex TaqTM (Takara, Japan) according to the manufacturer’s protocol. Primer sequences are listed in Table S1. Relative mRNA expression levels were normalized to the housekeeping gene GAPDH using the 2−ΔΔCt method. All experiments were performed in at least three independent biological replicates (n≥3).
Western Blot
Protein expression was analyzed by Western blot. Cellular protein were extracted using RIPA lysis buffer with a 1:100 dilution of protease inhibitor (Solarbio, China). The total protein concentration of the lysates was quantified using a bicinchoninic acid (BCA) assay kit (Beyotime, China). Equal amounts of protein were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and subsequently transferred onto polyvinylidene fluoride (PVDF) membranes. After blocking, the membranes incubated overnight at 4°C with primary antibodies: anti-NFAT1 (Cat#5861), anti-COX-2 (Cat#12282), anti-ALOX5 (Cat#3289), anti-NF-κB (Cat#8242), anti-TLR8 (Cat#11886), anti-MyD88 (Cat#4283) and anti-β-actin (Cat#3700) were obtained from Cell Signaling Technology, Inc. (USA). Additionally, anti-ALOX15 (Cat#ab244205) and anti-IRF7 (Cat#ab109255) were obtained from Abcam (UK). Following incubation with fluorescently labeled secondary antibodies, protein bands were visualized and quantified using the LI-COR Odyssey CLx imaging system.
Statistical Analysis
Data collection and organization were performed using Microsoft Excel 2019. Statistical analysis was conducted using IBM SPSS Statistics 25 and GraphPad Prism 8.0.2, while figures were generated using GraphPad Prism 8.0.2. Descriptive statistical analysis was used to examine the general characteristics of the study population, with the characteristics of HIV-1/AIDS cases described as median and interquartile range (IQR). The relationship between TLR8-AS1 and CD4+ T cell counts was investigated using Spearman’s rank correlation. All experiments were performed in at least three independent biological replicates (n≥3). P-values were determined by an independent samples t-test. A significance level of P < 0.05 was considered statistically significant.
Results
Association Between TLR8-AS1 Expression and Immune Status in HIV-1/AIDS Patients
Our preliminary transcriptomic analysis revealed that TLR8-AS1 was significantly upregulated in HEPS compared to matched healthy controls (Table S2). Specifically, subjects included strictly screened HEPS individuals and age, sex, and ethnicity matched healthy controls. And TLR8-AS1 expression in PBMCs from HEPS individuals was elevated by 8.91-fold relative to controls (Table S2). Subsequently, we assessed the expression levels of TLR8-AS1 across different population groups. The demographic characteristics of the participants are summarized in Table 1. Compared with the healthy controls, TLR8-AS1 expression in PBMCs was significantly up-regulated in HIV-1/AIDS patients receiving ART (P < 0.05) (Figure 1A). Subsequently, the ART group was further stratified by CD4+ T cell counts: < 200 cells/μL (severe immunodeficiency), 200–500 cells/μL (mild to moderate immunodeficiency), and > 500 cells/μL (non-immunocompromised).25 A significant positive correlation was observed between CD4+ T cell counts and TLR8-AS1 expression (r = 0.439, P < 0.05), with higher CD4+ counts corresponding to higher expression levels (Figure 1B). Notably, both the 200–500 cells/μL and >500 cells/μL subgroups showed significantly higher TLR8-AS1 expression compared to the <200 cells/μL subgroup (Figure 1B, P < 0.05). These results indicate a significant positive correlation between TLR8-AS1 expression and CD4+ T cell counts, highlighting its potential as a marker of immune status in HIV-1/AIDS patients. T cells, B cells, and monocytes were isolated from the peripheral blood of healthy donors, and monocytes were induced to differentiate into monocyte-derived macrophages (MDMs). TLR8-AS1 expression was predominantly observed in MDMs compared to other cell types (Figure 1C, P < 0.05). Given the predominant expression of TLR8-AS1 in MDMs and the crucial role of macrophages in HIV infection, we utilized a macrophage-based model for subsequent in vitro functional validation.
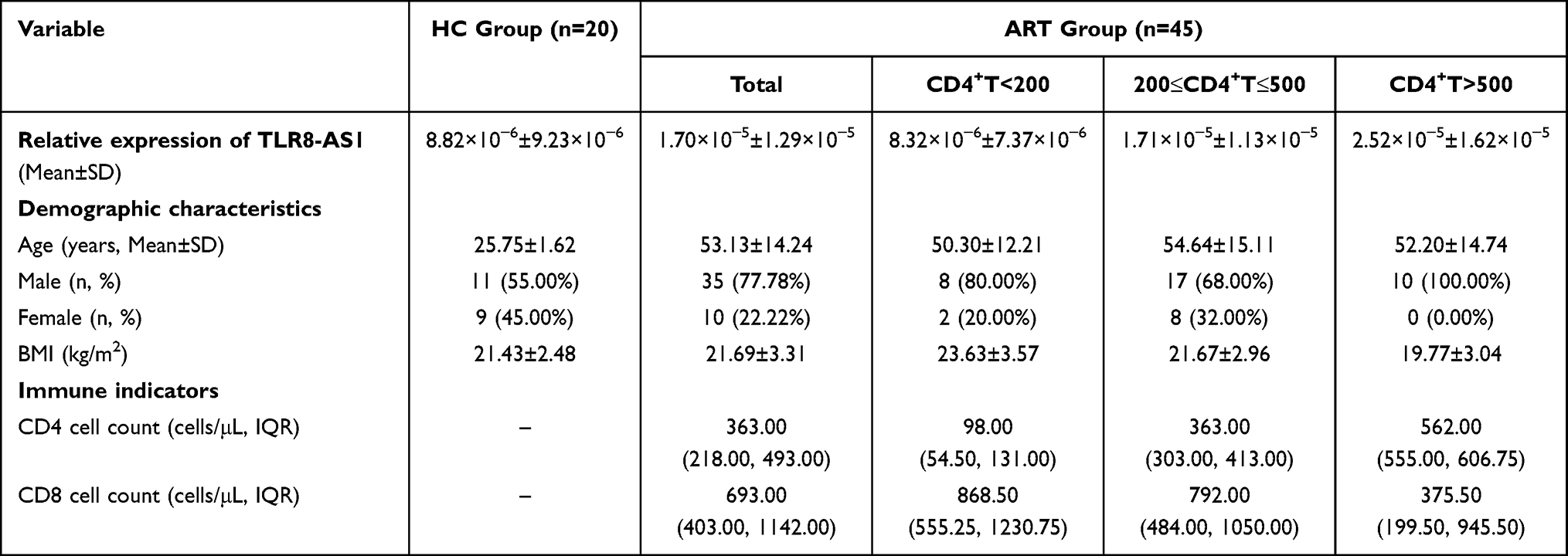 |
Table 1 Comparison of Demographic Characteristics and Immune Indicators Between the HIV-1/AIDS Antiviral Treatment Group and the Healthy Control Group |
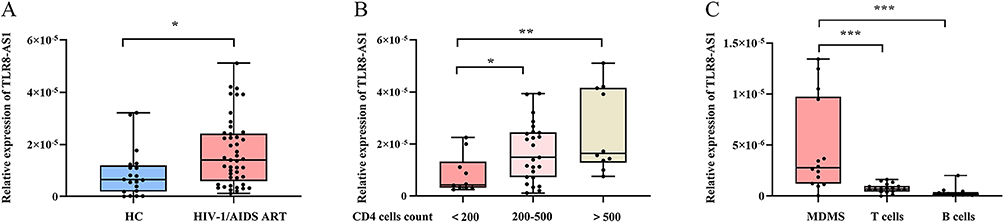 |
Figure 1 The expression levels of TLR8-AS1 in different populations. (A) In PBMCs, the expression levels of TLR8-AS1 were significantly elevated in the HIV-1/AIDS antiretroviral therapy (ART) group compared to the healthy control (HC) group. (B) Compared to the group with severe immunodeficiency (less than 200 cells/μL), both the moderate immunodeficiency group (200–500 cells/μL) and the non-immunodeficient group (more than 500 cells/μL) exhibited significantly elevated expression levels of TLR8-AS1. (C) TLR8-AS1 expression is enriched in monocyte-derived macrophages (MDMs). The relative expression levels of TLR8-AS1 were analyzed in primary human T cells, B cells, monocytes, and MDMs (differentiated from monocytes of healthy donors). Data are presented as mean ± SD (n ≥ 3 independent biological cell cultures) *P < 0.05; **P < 0.01; ***P < 0.001. |
The Overexpression of TLR8-AS1 Inhibits HIV-1 Replication and Attenuates Inflammatory Responses
To examine the association between elevated TLR8-AS1 expression and immune status, we generated THP-1 cells stably overexpressing TLR8-AS1 (Figure 2A), and subsequently differentiated them into macrophages in vitro. Compared with the control macrophages transfected with an empty vector, TLR8-AS1-overexpressing cells exhibited a significant reduction in HIV-1 p24 protein production (Figure 2B, P < 0.05). qPCR analysis further revealed that TLR8-AS1 overexpression significantly reduced the mRNA levels of multiple key HIV-1 genes, including Pol, Vif, Nef, long terminal repeat (LTR), and Gag, relative to controls (Figure 2B, P < 0.05). In addition, the protein levels of inflammatory factors were measured by enzyme-linked immunosorbent assay (ELISA). ELISA results revealed that, following HIV-1 infection, the levels of IL-1β and TNF-α were significantly decreased in the TLR8-AS1-overexpression group (Figure 2C, P < 0.05). These findings suggest that TLR8-AS1 may suppress HIV-1 replication and alleviate virus-induced inflammatory responses.
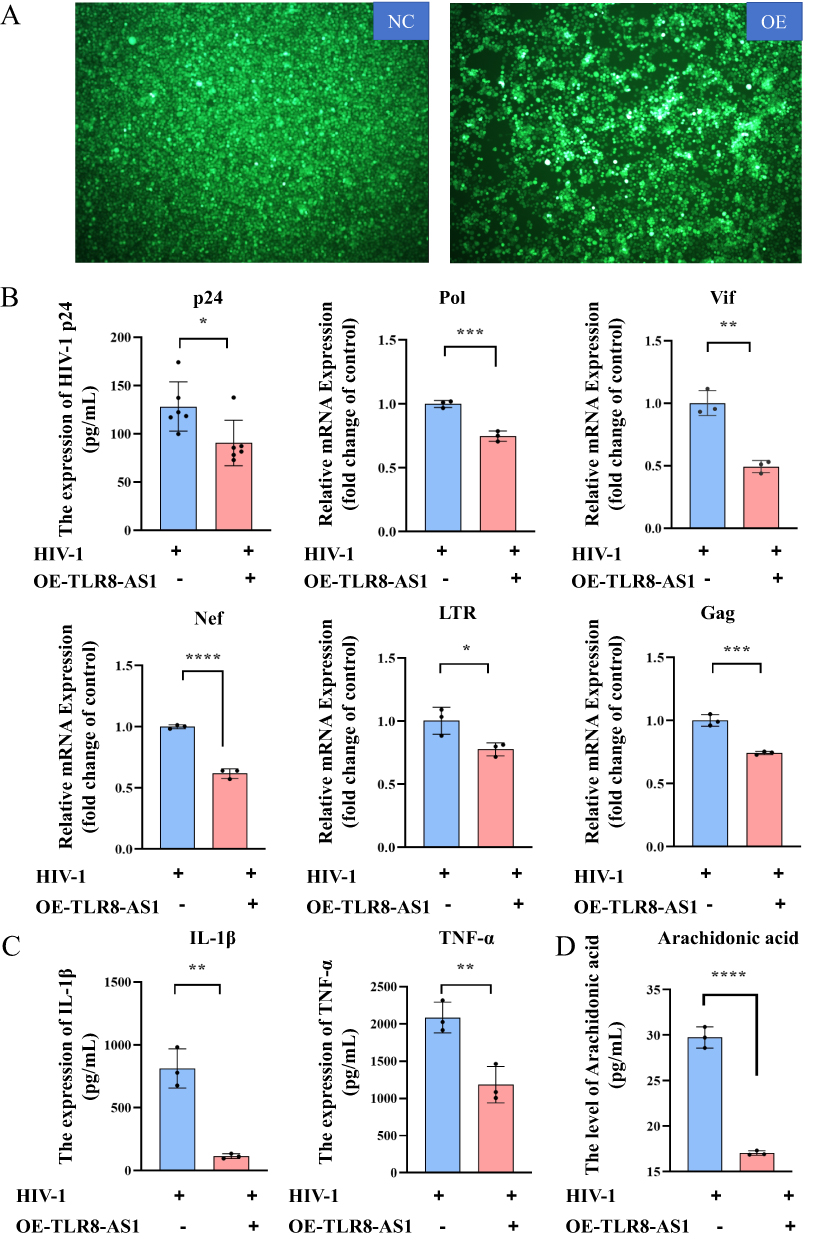 |
Figure 2 The impact of TLR8-AS1 on HIV-1 replication and cellular inflammatory factors. (A)THP-1 cells were transfected with a TLR8-AS1 overexpression plasmid or an empty vector control prior to HIV-1 infection. And TLR8-AS1 overexpression was successfully constructed in THP-1-derived macrophages. (B) Overexpression of TLR8-AS1 in THP-1 cells resulted in the inhibition of HIV-1 p24 protein expression, as measured by ELISA. Additionally, qPCR analysis further revealed that TLR8-AS1 overexpression led to the repression in the mRNA levels of multiple key HIV-1 genes, including Pol, Vif, Nef, LTR, and Gag (Student’s t-test). (C) Downregulation of inflammatory factors (IL-1β and TNF-α) at the protein level in cells overexpressing TLR8-AS1 in HIV-1 infection. (D) After the overexpression of TLR8-AS1, the level of arachidonic acid was significantly reduced in HIV-1 infection. Data are presented as mean ± SD (n ≥ 3 independent biological cell cultures). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. |
Cytoplasmic Localization of TLR8-AS1 in THP-1 Macrophages
We employed two computational tools, lncLocator26 (http://www.csbio.sjtu.edu.cn/bioinf/lncLocator/) and iLoc-LncRNA(2.0)27 (http://lin-group.cn/server/iLoc-LncRNA(2.0)/home.php), to predict the subcellular localization of TLR8-AS1. Both methods consistently indicated that TLR8-AS1 is predominantly localized in the cytoplasm (Figure 3A). To experimentally validate this prediction, we conducted RNA fluorescence in situ hybridization (FISH), which clearly demonstrated cytoplasmic distribution of TLR8-AS1 in differentiated THP-1 macrophages (Figure 3B). Subsequently, cytoplasmic and nuclear RNA fractions were isolated from THP-1 macrophages to further verify the localization pattern. As expected, GAPDH mRNA was highly enriched in the cytoplasmic fraction, whereas the well-characterized nuclear-retained lncRNA MALAT1 was predominantly detected in the nuclear fraction (Figure 3C). Consistently, RT-qPCR analysis confirmed that TLR8-AS1 was primarily localized in the cytoplasm of THP-1 macrophage cells (Figure 3C).
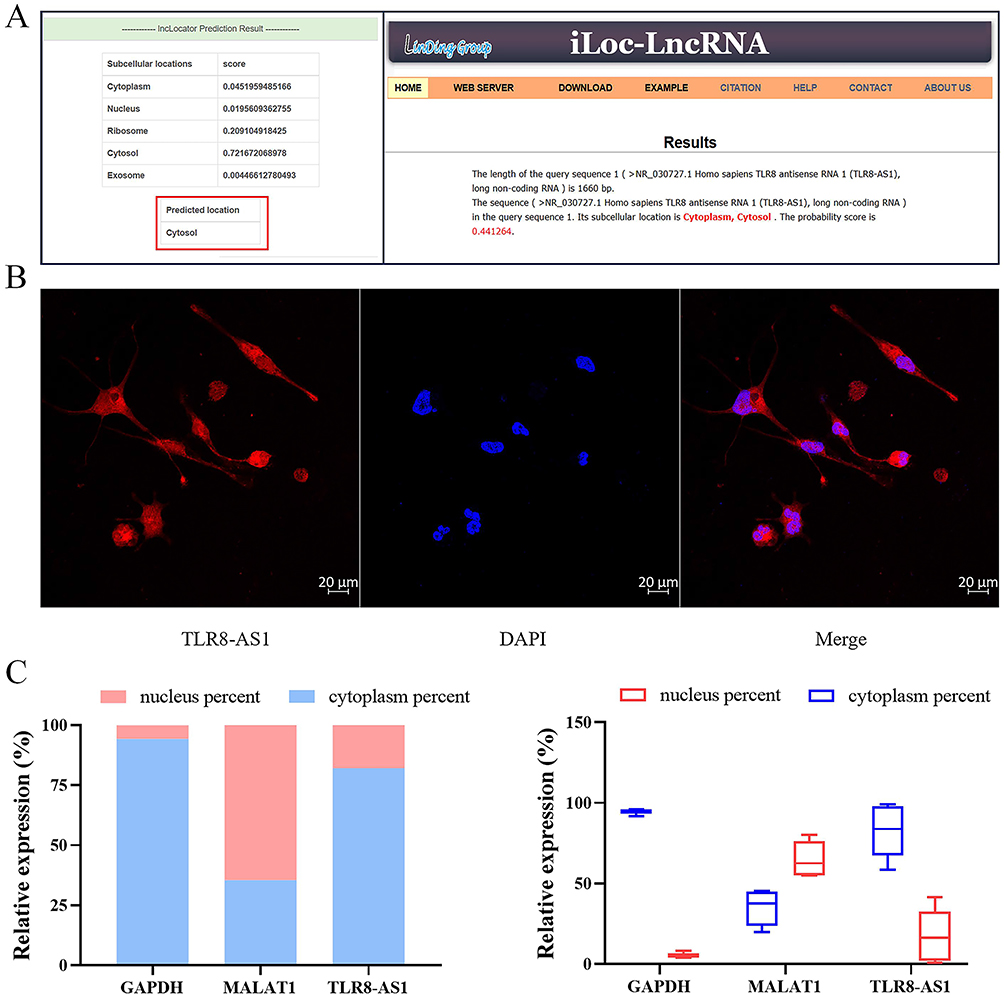 |
Figure 3 Cytoplasmic Localization of TLR8-AS1 in THP-1 Macrophages. (A) Bioinformatics predicts (using lncLocator and iLoc-LncRNA) that TLR8-AS1 is located in the cytoplasm and cytosol. (B) The subcellular localization of TLR8-AS1 was determined by RNA FISH in THP-1 cells. And TLR8-AS1 was primarily located in the cytoplasm. Red: TLR8-AS1; Blue: DAPI. (C) The relative abundance of the lncRNA TLR8-AS1, the cytoplasmic mRNA marker GAPDH, and the nuclear lncRNA marker MALAT1 in the cytoplasmic and nuclear fractions of THP-1 cells was determined by RT-qPCR. The results confirm the efficacy of the cellular fractionation and demonstrate the predominant cytoplasmic localization of TLR8-AS1. Data are presented as mean ± SD (n ≥ 3 independent biological cell cultures). |
TLR8-AS1 Attenuates AA Pathway Activity Through PTGS2 (COX-2) Downregulation
Previous studies have identified that the AA pathway has emerged as a central hub for the production of inflammation mediators.28,29 To explore the role of TLR8-AS1 in this pathway, we constructed THP-1-derived macrophages overexpressing TLR8-AS1 and examined their effects on AA levels and the expression of key metabolic genes during HIV-1 infection.
Compared with the controls, TLR8-AS1 overexpression significantly reduced AA release as measured by ELISA (Figure 2D). And the mRNA expression levels of PTGS2 (COX-2), PTGDS, PTGES, and LTC4S were also significantly reduced by TLR8-AS1 overexpression (Figure 4A, P < 0.05). At the protein level, TLR8-AS1 overexpression significantly downregulated COX-2 (Figure 4B, P < 0.05), whereas ALOX5 and ALOX15 remained unaffected at both the mRNA and protein levels (P > 0.05, Figure 4A and B). Therefore, we speculate that TLR8-AS1 selectively modulates the AA metabolic pathway through the downregulation of PTGS2 during HIV-1 infection.
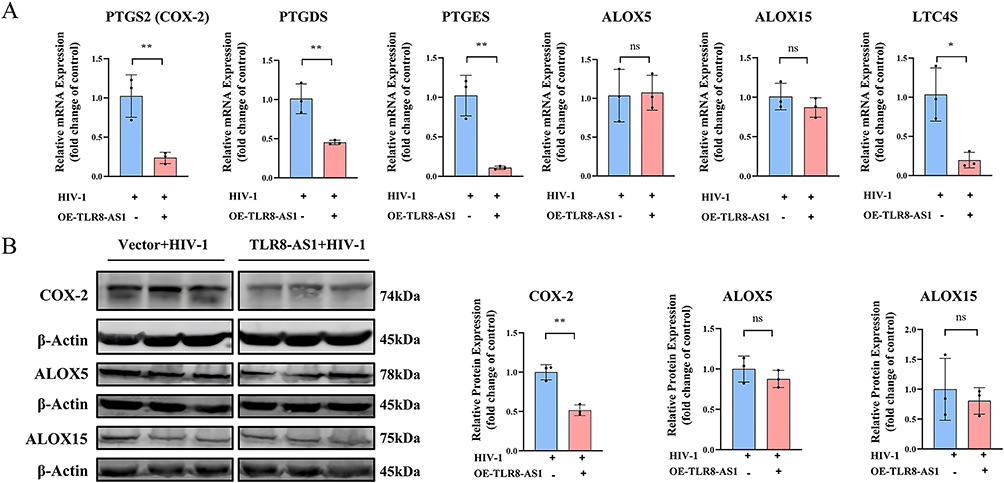 |
Figure 4 TLR8-AS1 exerts its biological function by down-regulating the arachidonic acid metabolic pathway. (A) The mRNA expression levels of key AA metabolic genes (PTGS2, PTGDS, PTGES, ALOX5, ALOX15, and LTC4S) were measured by RT-qPCR in TLR8-AS1 overexpressing cells under HIV-1 infection. (B) Protein levels of COX-2, ALOX5, and ALOX15 were analyzed by Western blotting. And bar graph depicting the quantification of proteins expression levels, and the relative expression levels were normalized to β-Actin protein. TLR8-AS1 overexpression selectively suppressed the expression of PTGS2/COX-2 and other key enzymes in the prostaglandin branch, but not the lipoxygenase branch, of the AA metabolic pathway. Data are presented as mean ± SD (n ≥ 3 independent biological cell cultures).*P < 0.05; **P < 0.01; ns, P > 0.05. |
TLR8-AS1 Suppresses NFAT1 Expression to Modulate AA Metabolism and Inflammatory Responses
Next, to explore the mechanism by which TLR8-AS1 regulates the AA pathway in HIV-1-infected macrophages, we first evaluated two candidate transcription factors: NFAT1 and NF-κB. The results showed that in HIV-1-infected macrophages, TLR8-AS1 overexpression specifically downregulated NFAT1 at both the protein and mRNA levels (Figure 5A and B, P < 0.05), while showing no significant effect on protein for NF-κB (Figure 6A and B, P > 0.05). To further confirm the role of NFAT1, we constructed NFAT1-silenced THP-1-derived macrophages (Figure 5C, P < 0.05). Compared with the controls, NFAT1 knockdown during HIV-1 infection resulted in a significant reduction in the mRNA levels of PTGS2, PTGDS, PTGES, and LTC4S (Figure 5D, P < 0.05), while no significant changes were observed in ALOX5 and ALOX15 (Figure 5D, P > 0.05). Furthermore, the levels of AA were also significantly decreased in the NFAT1 knockdown group (Figure 5E, P < 0.05), as well as the mRNA levels of inflammatory factors (IL-1β and TNF-α) and HIV-1 genes (Pol, and Vif), together with the protein expression of HIV-1 p24 (Figure 5F–H, P < 0.05). These findings identify NFAT1 as a critical downstream mediator through which TLR8-AS1 regulates AA metabolism, inflammatory responses, and HIV-1 replication.
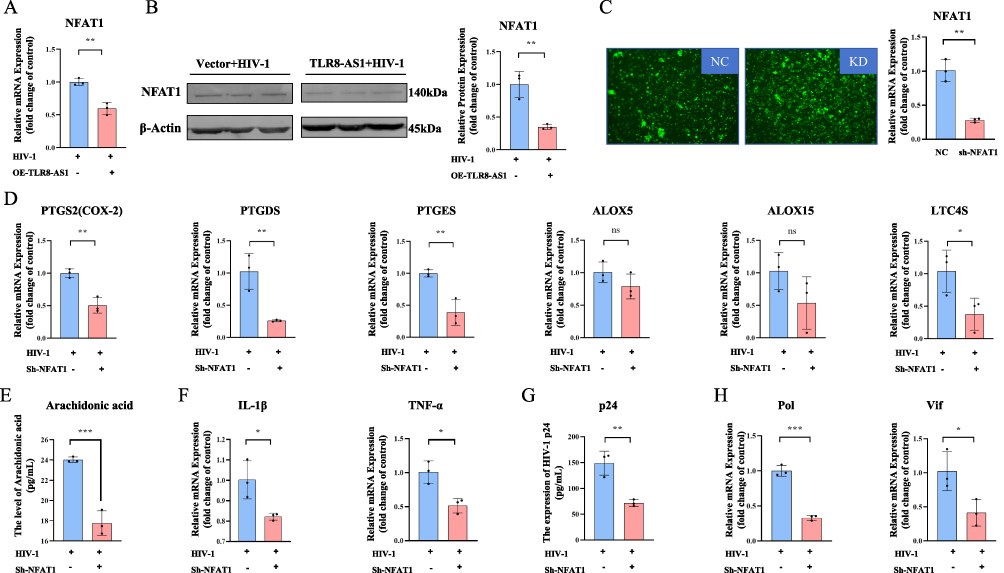 |
Figure 5 TLR8-AS1 modulates AA metabolism and inflammatory responses by suppressing NFAT1 in HIV-1-infected macrophages. (A and B) The effect of TLR8-AS1 overexpression on NFAT1 expression in HIV-1-infected THP-1-derived macrophages was assessed by (A) RT-qPCR and (B) Western blot analysis. Overexpression of TLR8-AS1 significantly downregulated the expression of NFAT1 both in mRNA level (A) and protein level (B). And the NFAT1 protein relative expression level was normalized to β-Actin protein. (C) NFAT1 knockdown was achieved in THP-1-derived macrophages, as confirmed by RT-qPCR. (D) The mRNA expression levels of key gene LTC4S, PTGDS, PTGES, and PTGS2 (COX-2) in the arachidonic acid metabolic pathway were measured by RT-qPCR and exhibited a significant decrease, whereas the change in ALOX5 and ALOX15 was not statistically significant. (E) AA release was quantified in the supernatant of NFAT1-knockdown cells after HIV-1 infection. (F) The mRNA levels of inflammatory factors (IL-1β, TNF-α) were detected by RT-qPCR. (G and H) The protein level of HIV-1 p24 was determined by Elisa (G), and the mRNA levels of HIV-1 Pol and Vif were detected by RT-qPCR (H). TLR8-AS1 suppresses NFAT1 expression, which in turn regulates the prostaglandin branch of AA metabolism, inflammatory responses, and HIV-1 replication. Data are presented as mean ± SD (n ≥ 3 independent biological cell cultures). *P < 0.05; **P < 0.01; ***P < 0.001; ns, P > 0.05. |
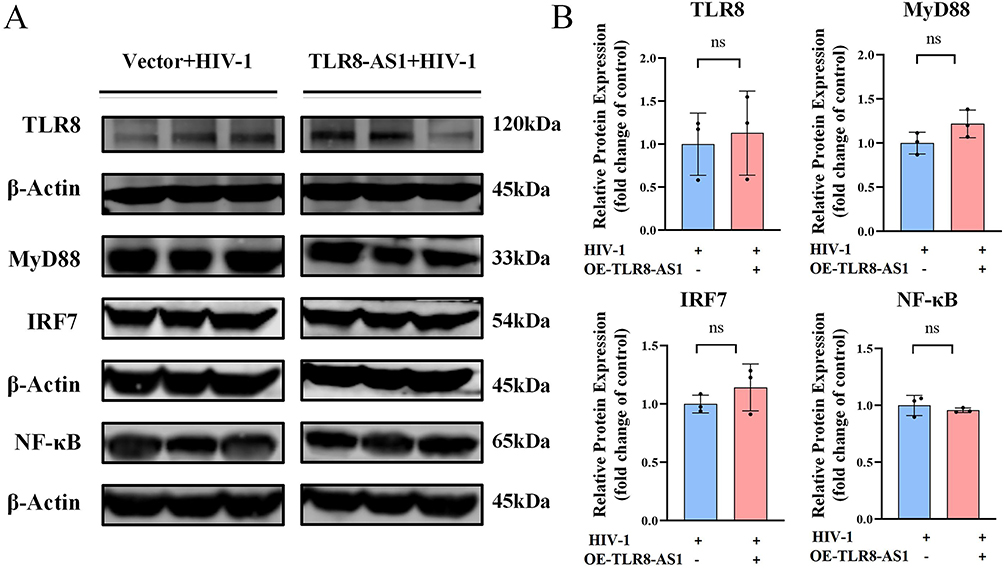 |
Figure 6 TLR8-AS1 does not regulate the TLR8 signaling pathway in HIV-1-infected macrophages. (A and B) The protein levels of TLR8 and its downstream signaling molecules (MyD88, IRF7 and NF-κB) were assessed by Western blot analysis. No significant differences were observed (P > 0.05, Student’s t-test; ns, not significant). Bar graph depicting the quantification of proteins expression levels, and the relative expression levels were normalized to β-Actin protein. Data are presented as mean ± SD (n ≥ 3 independent biological cell cultures). |
TLR8-AS1 Functions Independently of TLR8-Mediated Antiviral Signaling
Given that TLR8-AS1 is a natural antisense long non-coding RNA of the TLR8 gene, we next examined TLR8 together with the downstream signaling molecules MyD88 and IRF7, which are central mediators of TLR8-driven antiviral and inflammatory responses. However, no significant differences were observed in the protein levels of these markers following TLR8-AS1 overexpression during HIV-1 infection (Figure 6A and B, P > 0.05). These findings indicate that, in HIV-1-infected macrophages, TLR8-AS1 may exert its regulatory effects independently of TLR8 and its downstream antiviral signaling pathway.
Discussion
Growing evidence suggests that lncRNAs hold promise as diagnostic biomarkers and therapeutic targets in various infectious diseases, including HIV-1.8,30 In this study, we found that TLR8-AS1 expression in HIV-1/AIDS patients was positively correlated with CD4⁺T-cell counts. Functionally, elevated TLR8-AS1 expression suppressed inflammatory cytokine production and HIV-1 replication in macrophages. Mechanistically, TLR8-AS1 appears to function primarily through NFAT1 and the AA pathway rather than the classical TLR8 signaling axis. Collectively, these findings not only suggest that TLR8-AS1 may serve as a novel immunoregulatory factor in HIV-1 infection but also point to its potential as a therapeutic target for modulating inflammation and restricting viral persistence.
In this study, we observed that elevated TLR8-AS1 expression exerted a dual effect in HIV-1-infected macrophages by markedly suppressing the release of inflammatory cytokines (IL-1β and TNF-α) and simultaneously inhibiting the expression of the multiple key HIV-1 genes. Macrophages, as long-lived immune cells that remain persistently active throughout the course of HIV/AIDS, play a crucial role in sustaining immune activation and viral persistence.31 Importantly, excessive secretion of proinflammatory cytokines from these cells can accelerate disease progression. For instance, elevated IL-1β levels have been associated with accelerated lung function decline and enhanced atherosclerotic inflammation in HIV-infected individuals, even in those receiving effective ART treatment.32,33 Likewise, high TNF-α levels during early HIV infection correlate with increased viral loads and have been implicated in neurotoxicity.34,35 In addition to cytokine-driven inflammation, viral proteins also contribute to immune dysregulation. The pol gene encodes three essential enzymes, namely reverse transcriptase, integrase, and protease, which are critically involved in the HIV-1 replication cycle.36 Vif, a key accessory protein of HIV-1, antagonizes host antiviral defenses by promoting the ubiquitination and proteasomal degradation of APOBEC enzymes, thereby facilitating viral replication and persistence.37 The Nef protein mediates the downregulation of CD4 and MHC-I molecules on the surface of infected host cells, thereby promoting viral release and enabling immune evasion.38 The Gag protein functions as a structural component of HIV-1 and is involved in the assembly and budding of viral particles.36 LTR serves as a regulatory hub within the HIV-1 genome, containing promoter and enhancer elements that govern the transcription of viral genes.39 These findings suggested that TLR8-AS1 may contribute to antiviral activity by multiple-targeting inhibition of the expression of core genes and structural proteins of HIV-1.
Our study further revealed that TLR8-AS1 selectively suppressed AA levels and the downstream COX pathway, including PTGS2/COX-2, PTGDS, and PTGES, while leaving the ALOX pathway (ALOX5 and ALOX15) unaffected. AA is the precursor of eicosanoids, a diverse class of bioactive lipid mediators that play pivotal roles in regulating inflammatory responses.40 It is primarily metabolized by two major enzymatic pathways, COX and ALOX.41 The COX branch predominantly generates pro-inflammatory prostaglandins and thromboxanes, whereas the ALOX branch produces both pro-inflammatory leukotrienes and anti-inflammatory lipoxins.42,43 Collectively, these findings support a model in which TLR8-AS1 contributes to anti-inflammatory activity primarily through the selective downregulation of the COX-2‑dependent branch of AA metabolism, thereby modulating lipid-derived inflammatory signaling.
In this study, we also found that TLR8-AS1 is predominantly localized in the cytoplasm, and that its overexpression selectively suppressed NFAT1 expression in HIV-1-infected macrophages, accompanied by reduced AA levels, decreased pro-inflammatory cytokine production, and downregulation of key genes in the AA pathway. Cytoplasmic lncRNAs are known to exert regulatory effects through several post-transcriptional mechanisms, including functioning as competing endogenous RNAs (ceRNAs), modulating target mRNA stability, and influencing translation.44,45 Given that TLR8-AS1 reduced both NFAT1 mRNA and protein levels in our study, it is plausible that TLR8-AS1 may downregulate NFAT1 by affecting its mRNA stability or through a ceRNA-like mechanism. In addition, NFAT1 translocates to the nucleus upon activation and plays a pivotal role in immune regulation and inflammatory responses, particularly under conditions of chronic viral infection.46,47 Therefore, another possibility is that cytoplasmic TLR8-AS1 may interfere with NFAT1 nuclear translocation, thereby limiting its transcriptional activity. NFAT1 has been implicated in the development of immune reconstitution inflammatory syndrome in HIV-1/AIDS patients.48 Moreover, NFAT1 promotes COX-2 promoter-driven transcriptional activity through its binding site.49,50 Collectively, our findings identify a previously unrecognized TLR8-AS1/NFAT1/AA axis that links lncRNA-mediated regulation to HIV-1-associated inflammation and viral replication.
As an antisense lncRNA, TLR8-AS1 would conventionally be expected to regulate its cognate sense gene TLR8 or its related signaling pathways. This expectation is further reinforced by previous studies showing that TLR8-AS1 can activate the TLR8/STAT1 signaling axis to induce preeclampsia,9 as well as promote ovarian cancer metastasis and chemotherapy resistance by enhancing the stability of TLR8 mRNA.51 Unexpectedly, our findings suggest a different mode of action. In the context of HIV-1 infection, TLR8-AS1 exerted anti-inflammatory and antiviral effects through a TLR8-independent mechanism rather than through canonical regulation of its adjacent sense gene. Increasing evidence indicates, that some antisense lncRNAs can regulate distal targets and modulate cellular functions independently of their cognate sense transcripts. For example, the immune regulation of USP30-AS1 is independent of its proximal protein-coding gene USP30.52 Similarly, Artificial LEF1-AS1 overexpression inhibited HL60 proliferation, upregulated p21/p27, and reduced ERK1/2 activation, independently of LEF1.53 Together, these observations support the possibility that TLR8-AS1 may also operate through a distinct non-canonical regulatory pathway independent of TLR8 to suppress HIV-1-induced inflammation. Further elucidation of this mechanism will be important for defining the specific role of TLR8-AS1 in HIV-1 infection.
Several limitations of the present study should be acknowledged. First, while the lack of an in vivo model is a limitation, the use of THP-1-derived macrophages and the consistent correlations observed in primary patient PBMCs provide strong translational relevance for our findings. Future studies using humanized mice or SIV-infected macaque models may be required to fully validate these metabolic and viral regulatory mechanisms in an in vivo system. Second, although our results strongly support the TLR8-AS1/NFAT1 axis, the direct physical interaction between TLR8-AS1 and NFAT1 remains to be explicitly characterized via RNA-pull down or RNA immunoprecipitation (RIP) assays in future investigations. Third, because of the well-recognized technical challenges associated with achieving high transfection efficiency and stable genetic manipulation, such as TLR8-AS1 overexpression or NFAT1 knockdown, in primary monocyte-derived macrophages (MDMs), we did not perform these functional experiments in the primary-cell system. Nevertheless, we confirmed TLR8-AS1 expression in primary MDMs to provide a proper physiological context. Collectively, the results provide a robust mechanistic foundation for future in vivo studies.
Conclusion
In summary, our findings demonstrate that TLR8-AS1 modulates immune responses in HIV-1-infected macrophages by downregulating NFAT1, which subsequently suppresses the AA signaling pathway, inhibits pro-inflammatory cytokine production, and restricts viral replication. Targeting the TLR8-AS1/NFAT1/AA axis may represent a potential therapeutic strategy to alleviate chronic immune activation and limit viral persistence in HIV-1 infection.
Data sharing statement
Data are available from the corresponding author on reasonable request.
Acknowledgment
Thanks to the diligent efforts of all participants in this study.
Author Contributions
All authors made a significant contribution to the work reported in this study, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work. Specifically, the individual contributions of each author are as follows: F.Y.W: Formal analysis, Investigation, Methodology, Data Curation, Visualization, Validation, Writing-original draft, and Writing-review and editing; S.R.L: Formal analysis, Validation, Investigation, Writing-original draft, and Writing-review and editing; J.H.Z: Validation and Data Curation, Writing-review and editing; Y.K.Z: Software, Writing-review and editing; H.L.W: Investigation, Writing-review and editing; J.L: Data Curation and Resources, Writing-review and editing; C.Y.M: Formal analysis and Validation, Writing-review and editing; J.J.J: Conceptualization, Methodology, Funding acquisition, and Resources, Writing-review and editing; L.Y: Conceptualization, Methodology, Funding acquisition, Resources, Supervision, Project administration, and Writing-review and editing; Z.X.Y: Conceptualization, Methodology, Investigation, Funding acquisition, Resources, and Writing-review and editing.
Funding
This study was supported by General Program (2025GXNSFAA069074, Special Project on Regional High-Incidence Diseases, Guangxi Medical University, to ZXY); the National Natural Science Foundation project (82560399, 82360392) and the Specific Research Project of Guangxi for Research Bases and Talents (GuikeAD23026283, to LY); and the Guangxi Medical University Training Program for Distinguished Young Scholars (DC2300001767, to JJJ).
Disclosure
The authors declare that they have no competing interests.
References
1. UNAIDS joint united nations programme on HIV/AIDS. Available from: https://www.unaids.org/en.
2. Burnett JC, Zaia JA, Rossi JJ. Creating genetic resistance to HIV. Current Opin Immunol. 2012;24(5):625–15. doi:10.1016/j.coi.2012.08.013
3. Le Douce V, Herbein G, Rohr O, Schwartz C. Molecular mechanisms of HIV-1 persistence in the monocyte-macrophage lineage. Retrovirology. 2010;7(1):32. doi:10.1186/1742-4690-7-32
4. Kumar A, Abbas W, Herbein G. HIV-1 latency in monocytes/macrophages. Viruses. 2014;6(4):1837–1860. doi:10.3390/v6041837
5. Zhou Y, Sun W, Qin Z, et al. LncRNA regulation: new frontiers in epigenetic solutions to drug chemoresistance. Biochem Pharmacol. 2021;189:114228. doi:10.1016/j.bcp.2020.114228
6. He RZ, Luo DX, Mo YY. Emerging roles of lncRNAs in the post-transcriptional regulation in cancer. Genes Dis. 2019;6(1):6–15. doi:10.1016/j.gendis.2019.01.003
7. Imam H, Shahr Bano A, Patel P, Holla P, Jameel S. The lncRNA NRON modulates HIV-1 replication in a NFAT-dependent manner and is differentially regulated by early and late viral proteins. Sci Rep. 2015;5(1):8639. doi:10.1038/srep08639
8. Jin C, Peng X, Xie T, et al. Detection of the long noncoding RNAsnuclear‐enriched autosomal transcript 1 (
9. Peng C, Zhu J, Guo H, Zhao L, Wu F, Liu B. Long non-coding RNA TLR8-AS1 induces preeclampsia through increasing TLR8/STAT1 axis. J Hypertension. 2023;41(8):1245–1257. doi:10.1097/HJH.0000000000003410
10. Homo sapiens TLR8-AS1. Available from: https://humancyc.org/gene?orgid=HUMAN&id=G66-278562#TU.
11. Liu J. Using Transcriptome Sequencing Technology to Screen Key Genes Related to HIV-1 Unsusceptibility From HEPS Subjects. Guangxi Medical University. 2016.
12. Yuan Z, Mo C, Kang Y, et al. Integration of transcriptomics and metabolomics reveals metabolism dysregulation in HIV-1-infected macrophages. Curr Microbiol. 2025;82(5):232. doi:10.1007/s00284-025-04204-2
13. Teer E, Mukonowenzou NC, Essop MF. HIV, inflammation, and immunometabolism: a model of the inflammatory theory of disease. Viruses. 2025;17(6):839. doi:10.3390/v17060839
14. Mu W, Patankar V, Kitchen S, Zhen A. Examining chronic inflammation, immune metabolism, and T cell dysfunction in HIV infection. Viruses. 2024;16(2):219. doi:10.3390/v16020219
15. Hao Y, Li D, Xu Y, et al. Investigation of lipid metabolism dysregulation and the effects on immune microenvironments in pan-cancer using multiple omics data. BMC Bioinformatics. 2019;20(Suppl 7):195. doi:10.1186/s12859-019-2734-4
16. Bertin J, Barat C, Méthot S, Tremblay MJ. Interactions between prostaglandins, leukotrienes and HIV-1: possible implications for the central nervous system. Retrovirology. 2012;9(1):4. doi:10.1186/1742-4690-9-4
17. Lee YJ, Yeo IJ, Choi DY, et al. Amyloidogenic, neuroinflammatory and memory dysfunction effects of HIV-1 gp120. Arch Pharm Res. 2021;44(7):689–701. doi:10.1007/s12272-021-01340-8
18. Almodovar S, Wade BE, Porter KM, et al. HIV X4 variants increase arachidonate 5-lipoxygenase in the pulmonary microenvironment and are associated with pulmonary arterial hypertension. Sci Rep. 2020;10(1):11696. doi:10.1038/s41598-020-68060-9
19. Carini R, Leonarduzzi G, Camandola S, et al. Activation of human immunodeficiency virus long terminal repeat by arachidonic acid. Free Radic Biol Med. 1997;22(1–2):195–199. doi:10.1016/S0891-5849(96)00291-2
20. Liu W, Ren D, Xiong W, Jin X, Zhu L. A novel FBW7/NFAT1 axis regulates cancer immunity in sunitinib-resistant renal cancer by inducing PD-L1 expression. J Exp Clin Cancer Res. 2022;41(1):38. doi:10.1186/s13046-022-02253-0
21. Sun Y, Tao Y, Geng Z, et al. The activation of CaN/NFAT signaling pathway in macrophages aggravated lactobacillus casei cell wall extract-induced kawasaki disease vasculitis. Cytokine. 2023;169:156304. doi:10.1016/j.cyto.2023.156304
22. Wu Y, Wang X, Huang Y, et al. Immunogenicity of an inactivated COVID-19 vaccine in people living with HIV in guangxi, China: a prospective cohort study. Viruses. 2024;16(9):1481. doi:10.3390/v16091481
23. Phuangbubpha P, Thara S, Sriboonaied P, Saetan P, Tumnoi W, Charoenpanich A. Optimizing THP-1 macrophage culture for an immune-responsive human intestinal model. Cells. 2023;12(10):1427. doi:10.3390/cells12101427
24. Zhang J, Yuan Z, Li X, et al. Activation of the JNK/COX-2/HIF-1α axis promotes M1 macrophage via glycolytic shift in HIV-1 infection. Life Sci Alliance. 2023;6(12):e202302148. doi:10.26508/lsa.202302148
25. NationalCenterforAIDS/STD Control and Prevention, ChinaCDC. Based on the 4th Edition of the China National Hand-Book of Free Antiviral Therapy for AIDS.
26. Cao Z, Pan X, Yang Y, Huang Y, Shen HB. The lncLocator: a subcellular localization predictor for long non-coding RNAs based on a stacked ensemble classifier. Bioinformatics. 2018;34(13):2185–2194. doi:10.1093/bioinformatics/bty085
27. Su ZD, Huang Y, Zhang ZY, et al. iLoc-lncRNA: predict the subcellular location of lncRNAs by incorporating octamer composition into general PseKNC. Bioinformatics. 2018;34(24):4196–4204. doi:10.1093/bioinformatics/bty508
28. Wang T, Fu X, Chen Q, et al. Arachidonic acid metabolism and kidney inflammation. IJMS. 2019;20(15):3683. doi:10.3390/ijms20153683
29. Wang B, Wu L, Chen J, et al. Metabolism pathways of arachidonic acids: mechanisms and potential therapeutic targets. Sig Transduct Target Ther. 2021;6(1):94. doi:10.1038/s41392-020-00443-w
30. Liu H, Hu PW, Couturier J, Lewis DE, Rice AP. HIV-1 replication in CD4+ T cells exploits the down-regulation of antiviral NEAT1 long non-coding RNAs following T cell activation. Virology. 2018;522:193–198. doi:10.1016/j.virol.2018.07.020
31. Woottum M, Yan S, Sayettat S, et al. Macrophages: key cellular players in HIV infection and pathogenesis. Viruses. 2024;16(2):288. doi:10.3390/v16020288
32. Hsue PY, Li D, Ma Y, et al. IL-1β inhibition reduces atherosclerotic inflammation in HIV infection. J Am Coll Cardiol. 2018;72(22):2809–2811. doi:10.1016/j.jacc.2018.09.038
33. Thudium RF, Arentoft NS, Hoel H, et al. Elevated levels of interleukin-1β and interleukin-10 are associated with faster lung function decline in people with well-treated human immunodeficiency virus. J Infect Dis. 2023;228(8):1080–1088. doi:10.1093/infdis/jiad233
34. Vaidya SA, Korner C, Sirignano MN, et al. Tumor necrosis factor α is associated with viral control and early disease progression in patients with HIV type 1 infection. J Infect Dis. 2014;210(7):1042–1046. doi:10.1093/infdis/jiu206
35. Brabers NACH, Nottet HSLM. Role of the pro‐inflammatory cytokines TNF‐α and IL‐1β in HIV‐associated dementia. Eur J Clin Invest. 2006;36(7):447–458. doi:10.1111/j.1365-2362.2006.01657.x
36. Sundquist WI, Krausslich HG. HIV-1 assembly, budding, and maturation. Cold Spring Harb Perspect Med. 2012;2(7):a006924–a006924. doi:10.1101/cshperspect.a006924
37. Wang Y, Qian G, Zhu L, et al. HIV-1 vif suppresses antiviral immunity by targeting STING. Cell Mol Immunol. 2022;19(1):108–121. doi:10.1038/s41423-021-00802-9
38. Collins KL, Chen BK, Kalams SA, Walker BD, Baltimore D. HIV-1 nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature. 1998;391(6665):397–401. doi:10.1038/34929
39. Karn J, Stoltzfus CM. Transcriptional and posttranscriptional regulation of HIV-1 gene expression. Cold Spring Harb Perspect Med. 2012;2(2):a006916–a006916. doi:10.1101/cshperspect.a006916
40. Jang Y, Kim M, Hwang SW. Molecular mechanisms underlying the actions of arachidonic acid-derived prostaglandins on peripheral nociception. J Neuroinflamm. 2020;17(1):30. doi:10.1186/s12974-020-1703-1
41. Regulska M, Szuster-Głuszczak M, Trojan E, Leśkiewicz M, Basta-Kaim A. The emerging role of the double-edged impact of arachidonic acid- derived eicosanoids in the neuroinflammatory background of depression. CN. 2020;19(2):278–293. doi:10.2174/1570159X18666200807144530
42. Li XJ, Suo P, Wang YN, et al. Arachidonic acid metabolism as a therapeutic target in AKI-to-CKD transition. Front Pharmacol. 2024;15:1365802. doi:10.3389/fphar.2024.1365802
43. Kennedy BM, Harris RE. Cyclooxygenase and lipoxygenase gene expression in the inflammogenesis of colorectal cancer: correlated expression of EGFR, JAK STAT and src genes, and a natural antisense transcript, RP11-C67.2.2. Cancers. 2023;15(8):2380. doi:10.3390/cancers15082380
44. Zhang X, Shi L, Xing M, et al. Interplay between lncRNAs and the PI3K/AKT signaling pathway in the progression of digestive system neoplasms (review). Int J Mol Med. 2025;55(1):15. doi:10.3892/ijmm.2024.5456
45. Piergentili R, Sechi S, De Paola L, Zaami S, Marinelli E. Building a hand-curated ceRNET for endometrial cancer, striving for clinical as well as medicolegal soundness: a systematic review. Noncoding RNA. 2025;11(3):34. doi:10.3390/ncrna11030034
46. Luo C, Burgeon E, Carew JA, et al. Recombinant NFAT1 (NFATp) is regulated by calcineurin in T cells and mediates transcription of several cytokine genes. Mol Cell Biol. 1996;16(7):3955–3966. doi:10.1128/MCB.16.7.3955
47. Cron RQ, Bartz SR, Clausell A, Bort SJ, Klebanoff SJ, Lewis DB. NFAT1 enhances HIV-1 gene expression in primary human CD4 T cells. Clin Immunol. 2000;94(3):179–191. doi:10.1006/clim.1999.4831
48. Sun J, Chen H, Xie Y, et al. Nuclear factor of activated T cells and cytokines gene expression of the T cells in AIDS patients with immune reconstitution inflammatory syndrome during highly active antiretroviral therapy. Mediators Inflamm. 2017;2017:1–7. doi:10.1155/2017/1754741
49. Tie X, Han S, Meng L, Wang Y, Wu A. NFAT1 is highly expressed in, and regulates the invasion of, glioblastoma multiforme cells. PLoS One. 2013;8(6):e66008. doi:10.1371/journal.pone.0066008
50. Abraham F, Sacerdoti F, De León R, Gentile T, Canellada A. Angiotensin II activates the calcineurin/NFAT signaling pathway and induces cyclooxygenase-2 expression in rat endometrial stromal cells. PLoS One. 2012;7(5):e37750. doi:10.1371/journal.pone.0037750
51. Xu Q, Lin YB, Li L, Liu J. LncRNA TLR8-AS1 promotes metastasis and chemoresistance of ovarian cancer through enhancing TLR8 mRNA stability. Biochem Biophys Res Commun. 2020;526(4):857–864. doi:10.1016/j.bbrc.2020.03.087
52. C Y, Awh C, G H, et al. An interferon-stimulated long non-coding RNA USP30-AS1 as an immune modulator in influenza a virus infection. PLoS Pathogens. 2025;21(1). doi:10.1371/journal.ppat.1012854
53. Congrains-Castillo A, Niemann FS, Santos Duarte AS, Olalla-Saad ST. LEF1-AS1, long non-coding RNA, inhibits proliferation in myeloid malignancy. J Cell Mol Med. 2019;23(4):3021–3025. doi:10.1111/jcmm.14152
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.