")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 16
Liraglutide Attenuates Aβ42 Generation in APPswe/SH-SY5Y Cells Through the Regulation of Autophagy
Authors Kong J, Wan L, Wang Y, Zhang H, Zhang W
Received 27 April 2020
Accepted for publication 28 June 2020
Published 27 July 2020 Volume 2020:16 Pages 1817—1825
DOI https://doi.org/10.2147/NDT.S260160
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yuping Ning
Jingjing Kong,1,* Liping Wan,2,* Yanfu Wang,1 Hua Zhang,1 Wei Zhang1
1Department of Geriatrics, The First Affiliated Hospital of Dalian Medical University, Dalian, Liaoning Province, People’s Republic of China; 2Department of Clinical Laboratory, The First Affiliated Hospital of Dalian Medical University, Dalian, Liaoning Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jingjing Kong Tel +86-18098875886
Email [email protected]
Objective: This study aimed to clarify whether liraglutide, a GLP-1 analogue, can ameliorate Aβ pathology through the regulation of autophagy in Alzheimer’s disease (AD) and to explore the related mechanisms thereof.
Methods: We used SH-SY5Y cells transiently transfected with APP695swe plasmid as an AD cellular model. Transfected cells were treated with liraglutide for 24 h in the presence or absence of 3-MA. Autophagy markers and the Aβ level were then evaluated by Western blot and ELISA. We also investigated the potential involvement of mTOR and JNK pathway in liraglutide-mediated autophagy.
Results: Our results showed that liraglutide reduced Aβ 42 generation and enhanced autophagy in APPswe/SH-SY5Y cells; however, these effects could be counteracted with 3-MA. Furthermore, our data showed that liraglutide-induced autophagy does not follow the mTOR pathway. Liraglutide might promote autophagy in APPswe/SH-SY5Y cells by activating the JNK pathway and inhibiting the beclin-1/bcl-2 complex.
Conclusion: Here, we report a novel mechanism underlying liraglutide-attenuated Aβ 42 generation through the activation of autophagy in AD cellular model.
Keywords: Alzheimer’s disease, glucagonlike peptide 1, autophagy, Aβ, JNK
Introduction
Alzheimer’s disease (AD) is the most cause of dementia among the elderly population and causes typical neuropathological changes such as the accumulation of extracellular β-amyloid (Aβ) and intracellular hyper-phosphorylated tau protein. Currently, there is no effective treatment for AD. The central role of Aβ in the onset and progression of AD has been well documented.1 On the other hand, autophagy is an evolutionarily conserved catabolic process of self-degradation of aggregated proteins and dysfunctional organelles. It has been reported that dysfunction of autophagy plays a critical role in the pathogenesis of the senile plaque.2 Therefore, autophagy is becoming an attractive target for treating neurodegenerative diseases through the selective degradation of abnormally folded proteins.
Furthermore, glucagon-like peptide 1 (GLP-1) is an intestinal hormone which regulates glycemia by stimulating glucose-dependent insulin release. In recent years, mounting evidence has shown that GLP-1 analogues have remarkable neuroprotective effects. These effects are associated with the inhibition of neuronal insulin resistance induced by Aβ.3,4 It has also been reported that the treatment of GLP-1 analogues in mouse models of AD, including aged animals can decrease Aβ plaque loads, and reduce Aβ-induced inflammatory responses, and improve neurogenesis, neuronal survival, and synaptic integrity, restore long-term potentiation and reduce cognitive decline.4–8 GLP-1 activation of GLP-1R participates in the regulation of insulin signaling pathways to improve insulin resistance, mainly including the PI3K and MAPK pathways.9
However, whether GLP-1 analogues have an effect on autophagy regulation in AD models is currently unclear. Previous studies have found that nutrient fluctuations can promote the secretion of hormones and neurotransmitters to regulate autophagy through G-protein coupled receptors (GRCRs).10 Binding of GLP-1 to its corresponding Gs-coupled receptor (GLP-1R) has been found to occur not only in pancreatic β cells but also in the brain and other tissues, and is known to lead to the activation of GLP-1R which is involved in the regulation of autophagy. The downstream targets of GPCRs are the important molecules involved in the PI3K/AKT/mTOR, MAPK, and AMPK pathways which are related to regulation of autophagy.10 Therefore, there may be common signaling pathways of GLP-1 for reducing insulin resistance and GLP-1R for mediating autophagy. Therefore, we hypothesize that GLP-1 analogues play a key role in the regulation of autophagy.
In the present study, we investigated whether the novel GLP-1 analogue liraglutide, a drug for T2DM treatment, can regulate autophagy in the APPswe/SH-SY5Y cells, an AD cellular model. We also investigated whether autophagy is required for liraglutide-mediated reduction of Aβ generation and explored the potential mechanisms of mTOR and JNK signaling.
Materials and Methods
Cell Culture and Transfection
The human neuroblastoma SH-SY5Y cell line was purchased from the Shanghai cell bank of Chinese Academy of Science. Cells were grown in DMEM (Gibco, USA) medium supplemented with 10% fetal bovine serum (Gibco, USA), 100 IU/mL penicillin and 100 ug/mL streptomycin and maintained in a humidified incubator at 37°C with 95% air and 5% CO2. Cells were grown at a density of 1 × 105 cells per well in 6 well plates. The medium was changed every 48 h. Cells at 80% confluence were subcultured every 3 days. To establish the AD cellular model, APPswe was overexpressed in SH-SY5Y cells via the transient transfection of pcDNA3.1-APP695swe using lipofectamine 3000 (Invitrogen, USA). The empty pEGFPN1 vector was used as a negative control. The high expression level of APP695 protein and increased Aβ42 secretion in APPswe/SH-SY5Y cells have been verified by Western blotting analysis and ELISA in our previous studies.11
Drug Treatment
After 24 h of transient transfection, APPswe-overexpressed cells were treated with single liraglutide (10 nM, Novo Nordisk), 3-Methyladenine (3-MA, 5 mM, MCE), or a combination of both for 24 h. The stock preparations of liraglutide and 3-MA were diluted in Gibco Water for Injection and Phosphate Buffered Saline (PBS), respectively. To investigate the involved signaling pathway(s), LY294002 (10μM, Sigma), Rapamycin (250 nM, MCE) and SP600125 (5 μM, Abcam) were separately added 1 h prior to liraglutide, followed by 24 h of co-treatment with liraglutide. The stock solutions of LY294002, Rapamycin and SP600125 were all prepared using DMSO; the final concentration of DMSO in working solution was less than 0.1% which showed no signs of cytotoxicity in preliminary experiments. The dose of liraglutide and other drugs used in this study was based on the results of preliminary experiments and values reported in literature.12,13
Measurement of Aβ42 Level by Enzyme-Linked Immunosorbent Assay (ELISA)
The cell culture medium was collected from each test group and centrifuged. The supernatants were collected and stored at −80°C before performing the ELISA test. The level of Aβ was determined using a human Aβ42 ELISA kit (Invitrogen, USA) which is a sensitive fluorescence-based sandwich ELISA. The procedures were performed according to the manufacturer’s instructions. Each group was replicated in three plates and the experiment was repeated three times.
Western Blotting
Cellular proteins were extracted using RIPA lysis buffer supplemented with 1% proteinase inhibitor. The protein concentration of cell lysates was measured using a BCA method (Beyotime, China). An equal amount of protein (20μg) in each sample was loaded into 10% SDS-PAGE gels, separated in the gels and transferred to PVDF membranes (Millipore, USA) by electrophoresis. The membranes were blocked with 5% non-fat dried milk for 2 h at room temperature and then incubated with primary antibodies overnight at 4°C. Antibodies used included anti-LC3B (1:1000; Abcam, UK), anti-Beclin-1 (1:1000; Abcam, UK), anti-p62 (1:1000; Abcam, UK), anti-AKT (1:1000; Abcam, UK), anti-p-AKT (s473) (1:1000; Abcam, UK), anti-mTOR (1:1000; Abcam, UK), anti-p-mTOR (1:1000; Abcam, UK), anti-JNK (1:1000; Abcam, UK), anti-p-JNK (1:1000; Abcam, UK), anti-bcl-2 (1:1000; PTG, USA), anti-GAPDH (1:2000, PTG, USA), and anti-α-Tubulin (1:2000; PTG, USA). The membranes were then washed and incubated with the HRP-linked secondary anti-rabbit antibody (1:5000; Beyotime, China) for 1 h at room temperature. Finally, protein bands were detected using an ECL system (Millipore, USA) and quantified using the Image J software. The blots were repeated from at least three independent experiments.
Statistical Analysis
In this study, all data were presented as the mean ± standard deviation (SD) of three independent experiments for each group. One-way ANOVA followed by Student Newman–Keuls test was used to test for statistically significant differences between groups, p<0.05 was considered statistically significant. Before analysis, the largest and the smallest variances were tested for homogeneity using the F-test. Statistical calculations were performed using SPSS18.0.
Results
Autophagy in APPswe/SH-SY5Y Cells is Induced by Liraglutide Treatment
To investigate whether liraglutide can induce autophagy in APPswe/SH-SY5Y cells, we applied 3-MA, an autophagy inhibitor. The autophagy inducer rapamycin, which is a potent mTOR inhibitor, was also used as a positive control. The activation of the autophagy process can be determined by monitoring LC3II flux.14 In addition to LC3, p62 is another potent marker that reflects the autophagy status; this is either because of the role of the p62 protein as a link between LC3 and ubiquitinated substrates or because of the negative correlation between p62 protein level and autophagy activity. Moreover, Beclin-1 also plays a key role in the initiation of autophagosome formation.15 To this end, changes in the level of LC3, p62 and beclin-1 in liraglutide-treated cells were examined by Western blots. As shown in Figure 1, liraglutide significantly increased the LC3II/LC3I ratio (1.259-fold) and the level of beclin-1 protein (1.238-fold) in APPswe/SH-SY5Y cells compared to APP control (p<0.05). Meanwhile, the level of the p62 protein in liraglutide-treated cells was significantly decreased compared to APP control (0.852-fold) (p<0.05). These results were similar to the effects of the autophagy activator rapamycin (p<0.05). Nevertheless, we still found that 3-MA significantly blocked the liraglutide-upregulated LC3II/LC3I ratio (0.675-fold) and protein level of beclin-1 (0.767-fold), as well as the liraglutide-downregulated expression of the p62 protein (1.191-fold) (Figure 2). These results suggest that liraglutide can significantly enhance autophagy in APPswe/SH-SY5Y cells.
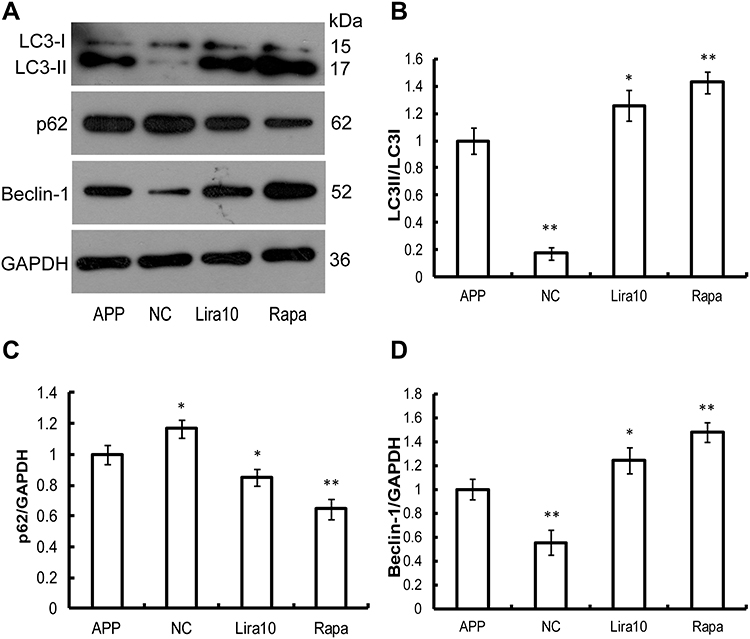 |
Figure 1 The effects of liraglutide and rapamycin on autophagy markers in APPswe/SH-SY5Y cells. (A–D) Western blotting data show that liraglutide significantly upregulates the LC3II/LC3I ratio and the level of beclin-1 protein, but down-regulates the level of p62 protein in APPswe/SH-SY5Y cells, compared to APP control. These results are similar to the effects of Rapamycin on autophagy in APPswe/SH-SY5Y cells. Data were presented as mean ± SD from three independent experiments. Data were analysed by one-way ANOVA followed by Student Newman–Keuls test. *p<0.05 vs APP control, **p<0.01 vs APP control. |
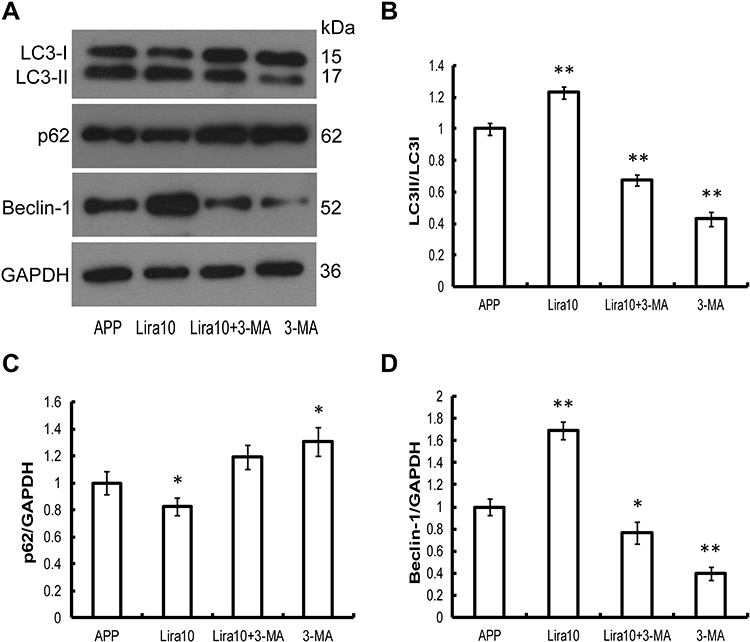 |
Figure 2 3-MA blocks the effect of liraglutide on autophagy in APPswe/SH-SY5Y cells. (A–D) Western blotting data show that the autophagy inhibitor 3-MA significantly blocks the liraglutide-upregulated LC3II/LC3I ratio and protein level of beclin-1, as well as the liraglutide-downregulated expression of the p62 protein in APPswe/SH-SY5Y cells. Data were presented as mean ± SD from three independent experiments. Data were analysed by one-way ANOVA followed by Student Newman–Keuls test. *p<0.05 vs APP control,**p<0.01 vs APP control. |
The Generation of Aβ42 and Activation of Autophagy in Liraglutide-Treated Cells
In this study, we also investigated the relationship between Aβ42 generation and autophagy in liraglutide-treated cells. Our results (Figure 3) showed that liraglutide significantly reduced the level of Aβ42 in the culture medium of APPswe/SH-SY5Y cells (35.294±5.000 pg/mL) compared to APP control (51.034±6.066 pg/mL) (p<0.05). However, the liraglutide-mediated decrease of extracellular Aβ42 can be blocked by 3-MA (45.374±5.348 pg/mL), suggesting that autophagy activation by liraglutide is required for the liraglutide-mediated reduction in the generation of Aβ42 in APPswe/SH-SY5Y cells.
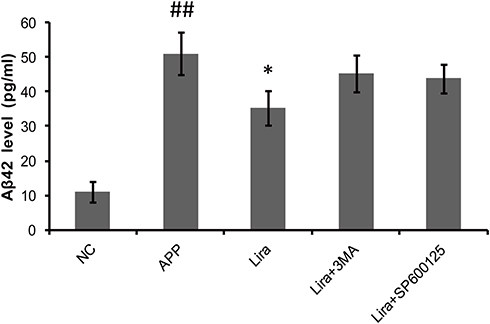 |
Figure 3 The effect of liraglutide on the generation of Aβ42 in APPswe/SH-SY5Y cells. ELISA results show that liraglutide significantly reduces the level of Aβ42 in the culture medium of APPswe/SH-SY5Y cells, compared to APP control. The decrease of extracellular Aβ42 induced by liraglutide can be blocked by 3-MA and SP600125. Data were presented as mean ± SD from three independent experiments. Data were analysed by one-way ANOVA followed by Student Newman–Keuls test. ##p<0.01 vs NC group, *p<0.05 vs APP control. |
The Role of the mTOR Signaling Pathway in Liraglutide-Mediated Autophagy
The serine/threonine protein kinase (mTOR) signaling pathway acts as the most important regulator of autophagy by suppressing the process in mammalian cells.16 Since the PI3K/AKT/mTOR signaling pathway plays a significant role in the regulation of the autophagy process,16 in this study we investigated the involvement of the PI3K/AKT/mTOR axis in liraglutide-induced autophagy pathway activation. As shown in Figure 4, liraglutide increased the ratio of both p-AKT/AKT (1.544-fold) and p-mTOR/mTOR (1.426-fold) in APPswe/SH-SY5Y cells, as compared to APP control (p<0.05). On the other hand, the PI3K inhibitor LY294002 and the mTOR inhibitor rapamycin attenuated the phosphorylation of AKT and mTOR that is induced by liraglutide. These results suggest that liraglutide-induced autophagy enhancement may be in mTOR-independent manner, although liraglutide activated PI3K/AKT/mTOR pathway.
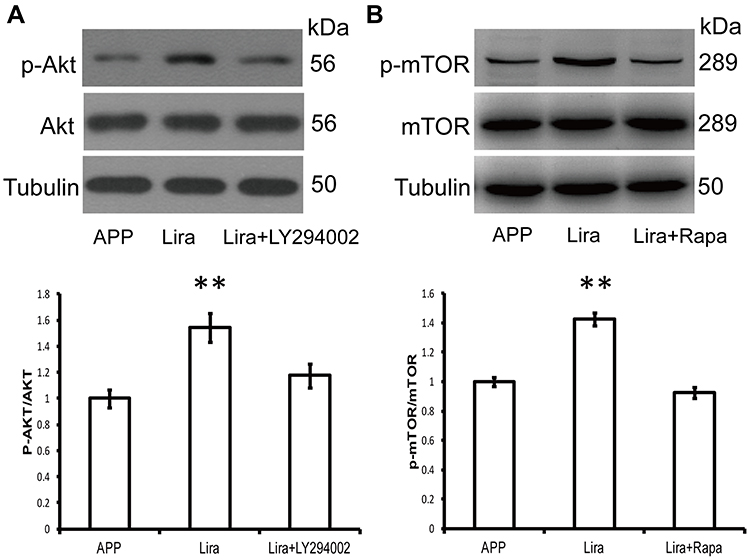 |
Figure 4 The PI3K/AKT/mTOR signaling pathway is dispensable for liraglutide-induced autophagy. (A) Western blotting data show that liraglutide significantly upregulates p-AKT/AKT ratio in APPswe/SH-SY5Y cells, compared to APP control. LY294002 blocks the liraglutide-induced phosphorylation of AKT. (B) Western blotting data show that liraglutide significantly upregulates p-mTOR/mTOR ratio in APPswe/SH-SY5Y cells, as compared to APP control. Rapamycin blocks the liraglutide-induced phosphorylation of mTOR. Data were presented as mean ± SD from three independent experiments. Data were analysed by one-way ANOVA followed by Student Newman–Keuls test. **p<0.01 vs APP control. |
The Role of the JNK Signaling Pathway in the Liraglutide-Mediated Autophagy Process
C-Jun N-terminal kinase (JNK) is a major GPCR-activated and stress-activated protein kinase (SAPK) and it belongs to the MAPK family. A variety of stress signals including autophagy can trigger JNK activation.17 To test whether JNK was involved in liraglutide-induced autophagy activation, the JNK inhibitor SP600125 was utilized in this study. As shown in Figure 5, the ratio of p-JNK/JNK was increased (1.209-fold) in liraglutide-treated cells, compared to APP control (p<0.05), but the liraglutide-induced phosphorylation of JNK could be blocked by SP600125 (0.700-fold). SP600125 also blocked both liraglutide-upregulated LC3II/LC3I (1.272-fold) and the liraglutide-mediated decrease of extracellular Aβ42 (43.818±4.045 pg/mL) (Figure 3). These results suggest that the JNK signaling pathway is involved in the liraglutide-induced autophagy process. To better understand the liraglutide-mediated regulation of autophagy, the effect of JNK on the phosphorylation of Bcl-2 and the disassociation of beclin-1 from Bcl-2 in liraglutide-treated cells was also evaluated. It has been reported that Bcl-2 inhibits the formation of autophagosomes through its binding to beclin-1,15,18 while dissociation of Beclin-1 and Bcl-2 may induce autophagy. Our results showed that liraglutide significantly decreased the expression of bcl-2 (0.561-fold) and increased the expression of beclin-1 (2.010-fold) in APPswe/SH-SY5Y cells (p<0.05). Simultaneously, SP600125 blocked the effect of liraglutide on the expression of bcl-2 (0.858-fold) and beclin-1 (1.219-fold) in APPswe/SH-SY5Y cells. These results indicated that liraglutide may promote autophagy in APPswe/SH-SY5Y cells by activating the JNK signaling pathway and inhibiting the beclin-1/bcl-2 complex.
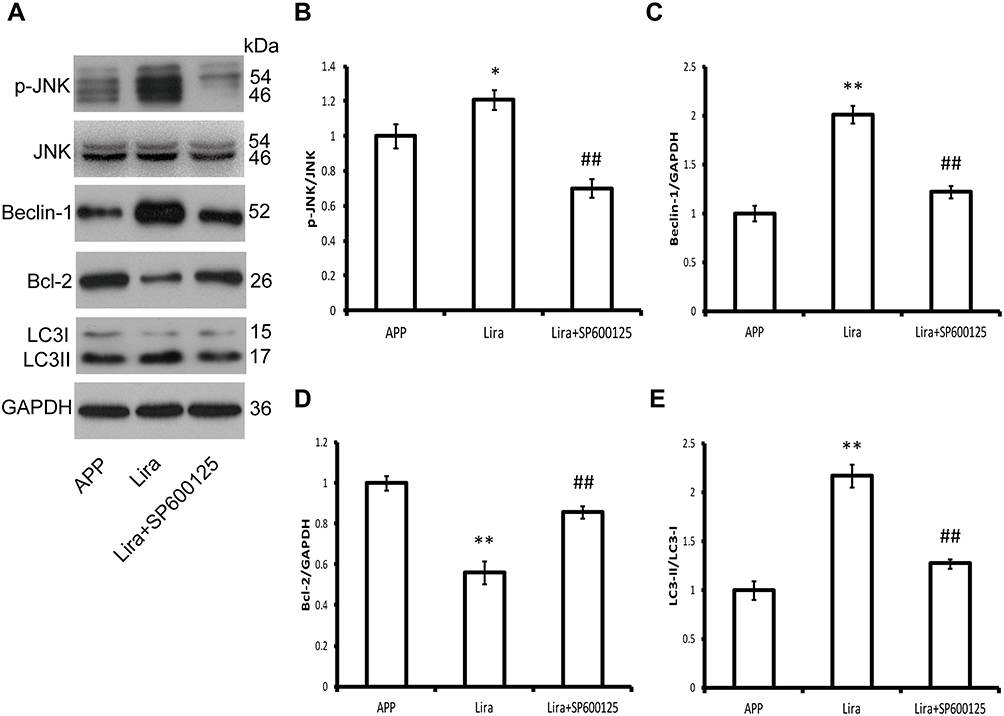 |
Figure 5 Induction of autophagy by liraglutide through activating the JNK signaling pathway. (A–E) Western blotting results show that liraglutide significantly increases the ratio of p-JNK/JNK in APPswe/SH-SY5Y cells compared to APP control. The liraglutide-induced phosphorylation of JNK and the increased LC3II/LC3I ratio can be blocked by SP600125. Furthermore, liraglutide significantly reduces bcl-2 expression, but increases beclin-1 expression, the effect of which can be blocked by SP600125. Data were presented as mean ± SD from three independent experiments. Data were analysed by one-way ANOVA followed by Student Newman–Keuls test. *p<0.05 vs APP control, **p<0.01 vs APP control, ##p<0.01 vs liraglutide group. |
Discussion
GLP-1-reduced insulin resistance and GLP-1R-enhanced autophagy may share common signaling pathways. Therefore, we speculated that GLP-1 analogues might ameliorate Aβ pathology through the regulation of autophagy in APPswe/SH-SY5Y cells, an in-vitro model of AD. Since there are few reports regarding GLP-1 analogue-mediated regulation of autophagy on Aβ pathology using AD models, in the current study we investigated the effect of liraglutide on Aβ42 generation and autophagy in APPswe/SH-SY5Y cells. Our study showed that liraglutide can reduce Aβ42 generation and simultaneously enhance autophagy in APPswe/SH-SY5Y cells. These effects, however, could be counteracted by the autophagy inhibitor 3-MA. Based on our findings, we hypothesize that the reduction of Aβ42 generation by liraglutide might through enhancing autophagy in APPswe/SH-SY5Y cells.
Recently, more attention has been paid to the role of autophagy in neurodegenerative diseases, including AD. Chronic dysfunction of autophagy-lysosome system has been reported to be perhaps the most likely cause of Aβ generation, aggregation and clearance. Previous studies have found that the level of Beclin-1 mRNA and protein were dramatically decreased in the brain tissue of AD cases.19,20 Both LC3-II/I, a biomarker of autophagosome formation, and markers of autophagic flux are also reduced in the AD brain and its early stages.21 In the APP transgenic mouse model, depletion of Beclin-1 significantly promotes the accumulation of both intraneuronal and extracellular Aβ deposition.19 Rapamycin, an autophagy enhancer, was reported to be effective for improving cognitive function in mouse models of AD through a mechanism related to the autophagy-mediated reduction in Aβ and tau pathology.22–24 Our results are in agreement with the above-mentioned studies. We speculate that many of the decreased Aβ42 generation is probably related to the elevation of autophagy induced by liraglutide with increased protein degradation.
GLP-1 analogues act as a PI3K activator to positively regulate the classical PI3K/Akt/mTOR signaling pathway, thus inhibiting autophagy. However, our results clearly demonstrated that liraglutide can reduce Aβ42 generation by promoting autophagy in APPswe/SH-SY5Y cells. Therefore, we consider that liraglutide-induced autophagy enhancement might be independent of the PI3K/Akt/mTOR pathway. Actually, we confirmed this idea by using PI3K/Akt/mTOR pathway inhibitors. In addition to the PI3K/AKT/mTOR signaling pathway, MAPKs such as ERK, p38, and JNK may also play an important role in regulating the autophagy process.25 JNK is an important contributor for GPCR-mediated activation of autophagy in response to stress conditions, and activation of autophagy by JNK is associated with Bcl-2/Bcl-xL phosphorylation. JNK induces Bcl-2/Bcl-xL phosphorylation, leading to the dissociation of the Beclin 1-Bcl-2/Bcl-xL complex. The released Beclin 1 as an essential inducer of autophagy can then trigger the autophagy process.26 Our study also demonstrated that JNK pathway is critical for the liraglutide-induced autophagy process as it was shown to disrupt the interaction of beclin-1/bcl-2 in our AD cellular model. Therefore, the activation of autophagy by JNK through the disassociation of beclin-1 from Bcl-2 is a possible molecular mechanism underlying the liraglutide-induced autophagy enhancement in our AD cellular model. This finding revealed that liraglutide-reduced Aβ generation may be mediated through JNK-dependent autophagy enhancement, which is consistent with previous reports. For example, Lei et al reported that proteasome inhibition upregulated BAG3 levels and improved tau clearance are dependent of JNK-activated autophagy because the inhibition of JNK or the depletion of BAG3 can attenuate the proteasome inhibition-mediated decreases of tau.27 Zhang et al reported that caffeic acid reduces A53T α-synuclein by activating JNK/Bcl-2-mediated autophagy in vitro and improved behavior and reduced α-synuclein in the substantia nigra in a mouse model of Parkinson’s disease.13
The role of autophagy in the development of AD is still highly debated. In other studies, the inhibition of autophagy has shown to have a beneficial role in AD.28 In contrary to our results, one study found that the JNK inhibitor SP600125 could improve spatial memory in Aβ-injected rats with memory deficit because the reduced Aβ-induced autophagy process.29 Although autophagy has a protective function in AD through the degradation of Aβ, autophagic activity must be limited at the proper level, otherwise excessive autophagy may cause cell death due to neurotoxicity.30 Previous studies have also found that enhanced autophagy, along with a lack of adequate lysosome-mediated clearance may lead to Aβ aggregation.31 On the other hand, enhanced autophagy also promotes Aβ generation due to autophagic vacuoles enriched by APP and proteases. The effects of autophagy activation may vary significantly depending on the pathophysiological stage of AD. Some studies suggest that rapamycin treatment may be harmful in ageing conditions with pre-existing pathology.32 Therefore, enhancing autophagy earlier in disease course before lysosomal deficits may be crucial to autophagy-based therapies.
Despite these promising results, there are several limitations of our study. GLP-1 analogues such as liraglutide can freely cross the blood-brain barrier; however, whether liraglutide can promote autophagy in AD animal models is still largely unknown. To this end, it is necessary to clarify the in-vivo effect of liraglutide on autophagy using AD animal models. Furthermore, multiple signaling molecules other than JNK, such as cAMP, MAPK/ERK1/2, and AMPK may be involved in the GLP-1R-modulated autophagy process. The role of autophagy in the regulation of AD development may vary in different stages of the disease. Further studies are needed to better clarify the molecular mechanisms of liraglutide-induced autophagy using in vivo and in vitro AD models.
Conclusion
Our data strongly suggest that liraglutide, a novel GLP-1 analogue, can reduce Aβ42 generation by promoting autophagy in APPswe-overexpressed SH-SY5Y cells. Inhibition of the beclin-1/bcl-2 complex through JNK activation might be a possible molecular mechanism of liraglutide-mediated autophagy in this AD cell model. Regulation of autophagy may be considered as a potent therapeutic intervention for the treatment of neurodegenerative diseases. Our results indicate a novel neuroprotective effect of liraglutide on an AD cellular model. This result suggests that autophagy regulation is required for the neuroprotection of liraglutide which can be developed as a very promising drug for AD treatment.
Disclosure
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
References
1. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi:10.1126/science.1072994
2. Uddin MS, Stachowiak A, Mamun AA, et al. Autophagy and Alzheimer’s disease: from molecular mechanisms to therapeutic implications. Front Aging Neurosci. 2018;10:04. doi:10.3389/fnagi.2018.00004
3. Jantrapirom S, Nimlamool W, Chattipakorn N, et al. Liraglutide suppresses tau hyperphosphorylation, amyloid beta accumulation through regulating neuronal insulin signaling and BACE-1 activity. Int J Mol Sci. 2020;21(5):1725. doi:10.3390/ijms21051725
4. Bomfim TR, Forny-Germano L, Sathler LB, et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease- associated Abeta oligomers. J Clin Invest. 2012;122(4):1339–1353. doi:10.1172/JCI57256
5. McClean PL, Jalewa J, Holscher C. Prophylactic liraglutide treatment prevents amyloid plaque deposition, chronic inflammation and memory impairment in APP/PS1 mice. Behav Brain Res. 2015;293:96–106. doi:10.1016/j.bbr.2015.07.024
6. McClean PL, Holscher C. Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer’s disease. Neuropharmacology. 2014;76(Pt):
7. Chen S, Sun J, Zhao G, et al. Liraglutide improves water maze learning and memory performance while reduces hyperphosphorylation of tau and neurofilaments in APP/PS1/tau triple transgenic mice. Neurochem Res. 2017;42(8):2326–2335. doi:10.1007/s11064-017-2250-8
8. Hansen HH, Fabricius K, Barkholt P, et al. The GLP-1 receptor agonist liraglutide improves memory function and increases hippocampal CA1 neuronal numbers in a senescence-accelerated mouse model of Alzheimer’s disease. J Alzheimers Dis. 2015;46(4):877–888. doi:10.3233/JAD-143090
9. Holscher C. The incretin hormones glucagonlike peptide 1 and glucose-dependent insulinotropic polypeptide are neuroprotective in mouse models of Alzheimer’s disease. Alzheimers Dement. 2014;10(1 Suppl):S47–S54. doi:10.1016/j.jalz.2013.12.009
10. Wauson EM, Dbouk HA, Ghosh AB, Cobb MH. G protein-coupled receptors and the regulation of autophagy. Trends Endocrinol Metab. 2014;25(5):274–282. doi:10.1016/j.tem.2014.03.006
11. Kong J, Ren G, Jia N, et al. Effects of nicorandil in neuroprotective activation of PI3K/AKT pathways in a cellular model of Alzheimer’s disease. Eur Neurol. 2013;70(3–4):233–241. doi:10.1159/000351247
12. Panagaki T, Michael M, Holscher C. Liraglutide restores chronic ER stress, autophagy impairments and apoptotic signalling in SH-SY5Y cells. Sci Rep. 2017;7(1):16158. doi:10.1038/s41598-017-16488-x
13. Zhang Y, Wu Q, Zhang L, et al. Caffeic acid reduces A53T α-synuclein by activating JNK/Bcl-2-mediated autophagy in vitro and improves behaviour and protects dopaminergic neurons in a mouse model of parkinson’s disease. Pharmacol Res. 2019;150:104538. doi:10.1016/j.phrs.2019.104538
14. Yoshii SR, Mizushima N. Monitoring and measuring autophagy. Int J Mol Sci. 2017;18(9):1865. doi:10.3390/ijms18091865
15. Liang XH, Jackson S, Seaman M, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402(6762):672–676. doi:10.1038/45257
16. Perluigi M, Di Domenico F, Butterfield DA. mTOR signaling in aging and neurodegeneration: at the crossroad between metabolism dysfunction and impairment of autophagy. Neurobiol Dis. 2015;84:39–49. doi:10.1016/j.nbd.2015.03.014
17. Kyriakis JM, Banerjee P, Nikolakaki E, et al. The stress-activated protein kinase subfamily of c-Jun kinases. Nature. 1994;369(6476):156–160. doi:10.1038/369156a0
18. Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–939. doi:10.1016/j.cell.2005.07.002
19. Pickford F, Masliah E, Britschgi M, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008;118(6):2190–2199. doi:10.1172/JCI33585
20. Rohn TT, Wirawan E, Brown RJ, Harris JR, Masliah E, Vandenabeele P. Depletion of Beclin-1 due to proteolytic cleavage by caspases in the Alzheimer’s disease brain. Neurobiol Dis. 2011;43(1):68–78. doi:10.1016/j.nbd.2010.11.003
21. Tramutola A, Triplett JC, Di Domenico F, et al. Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J Neurochem. 2015;133(5):739–749. doi:10.1111/jnc.13037
22. Bove J, Martinez-Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci. 2011;12(8):437–452. doi:10.1038/nrn3068
23. Richardson A, Galvan V, Lin AL, Oddo S. How longevity research can lead to therapies for Alzheimer’s disease: the rapamycin story. Exp Gerontol. 2015;68:51–58. doi:10.1016/j.exger.2014.12.002
24. Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. 2010;285(17):13107–13120. doi:10.1074/jbc.M110.100420
25. Belaid A, Ndiaye PD, Klionsky DJ, Hofman P, Mograbi B. Signalphagy: scheduled signal termination by macroautophagy. Autophagy. 2013;9(10):1629–1630. doi:10.4161/auto.25880
26. Zhou F, Yang Y, Xing D. Bcl-2 and Bcl-xL play important roles in the crosstalk between autophagy and apoptosis. FEBS J. 2011;278(3):403–413. doi:10.1111/j.1742-4658.2010.07965.x
27. Lei Z, Brizzee C, Johnson GV. BAG3 facilitates the clearance of endogenous tau in primary neurons. Neurobiol Aging. 2015;36(1):241–248. doi:10.1016/j.neurobiolaging.2014.08.012
28. Lipinski MM, Zheng B, Lu T, et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2010;107(32):14164–14169. doi:10.1073/pnas.1009485107
29. Mohammadi M, Guan J, Khodagholi F, et al. Reduction of autophagy markers mediated protective effects of JNK inhibitor and bucladesine on memory deficit induced by Abeta in rats. Naunyn Schmiedebergs Arch Pharmacol. 2016;389(5):501–510. doi:10.1007/s00210-016-1222-x
30. Huang J, Klionsky DJ. Autophagy and human disease. Cell Cycle. 2007;6(15):1837–1849. doi:10.4161/cc.6.15.4511
31. Tung YT, Wang BJ, Hu MK, et al. Autophagy: a double-edged sword in Alzheimer’s disease. J Biosci. 2012;37(1):157–165. doi:10.1007/s12038-011-9176-0
32. Carosi JM, Sargeant TJ. Rapamycin and Alzheimer disease: a double-edged sword? Autophagy. 2019;15(8):1460–1462. doi:10.1080/15548627.2019.1615823
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.