")
Back to Journals » International Journal of Nanomedicine » Volume 14
Liposomes targeted to MHC-restricted antigen improve drug delivery and antimelanoma response
Authors Saeed M , Zalba S , Seynhaeve ALB, Debets R , ten Hagen TLM
Received 12 October 2018
Accepted for publication 13 December 2018
Published 26 March 2019 Volume 2019:14 Pages 2069—2089
DOI https://doi.org/10.2147/IJN.S190736
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Mesha Saeed,1 Sara Zalba,1 Ann LB Seynhaeve,1 Reno Debets,2 Timo LM ten Hagen1
1Laboratory of Experimental Surgical Oncology, Section Surgical Oncology, Department of Surgery, Erasmus MC, Rotterdam, The Netherlands; 2Laboratory of Tumor Immunology, Department of Medical Oncology, Erasmus MC Cancer Institute, Rotterdam, The Netherlands
Purpose: Melanoma is the most aggressive form of skin cancer. Chemotherapy at a late stage fails due to low accumulation in tumors, indicating the need for targeted therapy.
Materials and methods: To increase drug uptake by tumor cells, we have targeted doxorubicin-containing liposomes using a T-cell receptor (TCR)-like antibody (scFv G8 and Hyb3) directed against melanoma antigen A1 (MAGE-A1) presented by human leukocyte antigen A1 (M1/A1). With the use of flow cytometry and confocal microscopy, we have tested our formulation in vitro. In vivo pharmacokinetics was done in tumor-free nu/nu mice, while biodistribution and efficacy study was done in nu/nu mice xenograft.
Results: We demonstrated two to five times higher binding and internalization of these immunoliposomes by M1+/A1+ melanoma cells in vitro in comparison with nontargeted liposomes. Cytotoxicity assay showed significant tumor cell kill at 10 µM doxorubicin (DXR) for targeted vs nontargeted liposomes. In vivo pharmacokinetics of nontargeted and targeted liposomes were similar, while accumulation of targeted liposomes was 2- to 2.5-fold and 6.6-fold enhanced when compared with nontargeted liposomes and free drug, respectively. Notably, we showed a superior antitumor activity of MAGE-A1-targeted DXR liposomes toward M1+/A1+ expressing tumors in mice compared with the treatment of M1-/A1+ tumors. Our results indicate that targeted liposomes showed better cytotoxicity in vitro and pharmacokinetics in vivo.
Conclusion: Liposomes decorated with TCR-mimicking scFv antibodies effectively and selectively target antigen-positive melanoma. We showed that DXR-loaded liposomes coupled to anti-M1/-A1 scFv inflict a significant antitumor response. Targeting tumor cells specifically promotes internalization of drug-containing nanoparticles and may improve drug delivery and ultimately antitumor efficacy. Our data argue that targeting MAGE in A1 context, by nanosized carriers decorated with TCR-like antibodies mimicking scFv, can be used as a theragnostic platform for drug delivery, immunotherapy, and potentially imaging, and diagnosis of melanoma.
Keywords: immunotherapy, targeted liposomes, TCR-mimicking scFv, melanoma, antigen A1, MAGE-A1, chemotherapy
Introduction
Melanoma is the deadliest and most aggressive among all skin cancers with increasing incidence rates.1 According to estimations of the American Cancer Society, there will be 91,270 cases of melanoma in 20182 in the USA and 9,320 deaths will occur from melanoma alone this year.2 If diagnosed at an early stage and excised by surgery, the 5-year survival rate of melanoma is 98%,2,3 whereas the survival rate decreases to 62% if melanoma is metastasized to lymph nodes and to 18% if it spreads to other body parts.2,3 Currently, the treatment for recurrent or metastatic melanoma comprises chemotherapy, radiotherapy, and immunotherapy with enhanced clinical responses in melanoma (immune checkpoint inhibitors: nivolumab and pembrolizumab);4,5 yet, a large fraction of patients still does not respond. In some cases of patients with BRAF V600 wild-type unresectable or metastatic melanoma, checkpoint inhibitors are the first choice of treatment and are approved by Food and Drug Administration. Chemotherapy is the first choice of treatment in case of primary and contained melanoma and is met with severe, dose-limiting side effects, while accumulation in tumors is low.
Drug pharmacokinetics, as well as side effects, can be changed by encapsulation into nanoparticles like liposomes. Liposomes are hollow lipid-based vesicles, with a watery core. Hydrophilic drugs can be encapsulated within the vesicle and hydrophobic drug in the bilayer. Liposomes are sterically stabilized by attaching polyethyleneglycol (PEG) at the outer membrane and shielded from opsonization, liver uptake,6,7 and sequestration by the reticuloendothelial system (RES).8 By escaping the RES, liposomes circulate longer in blood, and eventually tumor-specific accumulation of liposomes is increased.9 These PEGylated liposomes can be further functionalized by decorating the surface with antibodies7,10 specific to tumor cells. For example, targeted liposomes show increased accumulation in tumor cells overexpressing the corresponding target antigen in previous studies.11–14 However, in order to increase the number of cellular target antigens (including intracellular antigens) and selectivity (with antigens being absent in healthy tissues), liposomes could be covered by antibodies that recognize peptide-MHC (pMHC) class I molecules. MHC class I molecules are encoded by classical human leukocyte antigen A (HLA) A, B, and C genes and are highly polymorphic surface glycoproteins with a key function in antigen presentation.15 All nucleated cells express MHC class I molecules, which display endogenous peptides, which in case of alterations due to infections of cancer can be recognized by CD8+ T-cells via their T-cell receptors. Among MHC class I presented peptides, there are mainly four groups: peptides derived from differentiation antigens (ie, melanoma antigen recognized by T cells [MART-1], glycoprotein 100 [gp100]); developmental antigens (carcinoembryonic antigen [CEA]); cancer germline antigens (CGA, such as melanoma antigen A1 [MAGE-A1], MAGE-C2, and New York esophageal squamous cell carcinoma 1 [NY-ESO1]); and neoantigens (mutated protein p53 and B-raf kinase).16 In the current study, we focused on CGAs, in particular MAGE-A1, due to their tumor-selective expression and absence from mature healthy tissue, and developed dynamic drug-loaded liposomes that are specifically targeted to an M1/A1 epitope, which constitutes a natural target for T-cells and is uniquely expressed on melanoma cells.
Barrow et al17 show that MAGE-A1 has an expression of 20% in primary tumors, whereas the expression increases to 51% in distant metastases. Likewise, Brasseur et al18 reported that 48% of the metastatic melanoma has MAGE-A1 expression in comparison with 16% in primary tumors.19 It is also known that higher expression of CGA is correlated with worst outcome.19 Additionally, other known MAGEs can be used for targeted therapy in melanoma.
In previous studies, whole monoclonal antibodies have been used to target liposomes, with the Fc part of the molecule often being recognized by macrophages and other immune cells, resulting in rapid clearance7,9 from the blood stream. To address this issue, we used scFv fragments without Fc parts. These scFv fragments are the smallest fragments of antibodies (25–30 kDa) that retain complete antibody binding, but with potentially reduced immunogenicity, as a consequence of their small size, lack of Fc-domain, and complement-activating region.7,20 We have derived M1/A1-specific scFvs (G8 and Hyb3) from Fab fragments that were originally selected from a phage display library21 and have successfully converted these scFvs into chimeric antigen receptors and used to retarget T-cells.22 Additional library engineering and selections yielded antibodies with different M1/A1 affinities,23 which enabled studies into the relevance of antibody affinity for the binding and antitumor efficacy of multivalent nanoparticles. We investigated scFv G8 and Hyb3 because they are specific to pMHC complexes and have previously24 shown promising results in vitro.
Targeted nanoparticles were loaded with doxorubicin (DXR) as a proof of principle. DXR is a widely used chemotherapeutic, with a broad spectrum of activity, which makes it a good candidate for cancer therapy.25 It is a widely studied and established anthracycline, cytotoxic in nature, and optimized for encapsulation in liposomes. The amphiphilic nature of DXR enables relatively easy passage through lipid bilayers of cells.26 Moreover, DXR can be encapsulated by a so-called remote loading mechanism resulting in a near 100% efficiency.27 For these reasons, DXR is the most studied chemotherapeutic in lipid-based carriers. In fact, Doxil is a commercially available liposomal formulation encapsulating DXR stably.28 We hypothesized that by decorating DXR-encapsulating liposomes with scFvs we can target M1+/A1+ melanoma cells specifically and eventually increase the drug accumulation in tumors. To test this, we used melanoma cells with antigen expression on the cell surface in vitro and in a xenograft mouse model. By targeting specific receptors on cells, we investigated if the accumulation of nanoparticles on the cells increases and in turn will promote the delivery of cargo to the cells, thereby improving antitumor efficacy.9 Adding chemotherapy as the cargo of targeted liposomes as a proof of principle, we want to show if the delivery of cargo and its antitumor effect after liposomes have accumulated in the tumor area based on specific targeting. In this research, we show specific melanoma targeting using a novel liposomal formulation using T-cell receptor (TCR)-like antibodies G8 and Hyb3 targeting MAGE-A1/HLA-A1 (M1/A1) antigen. The novelty concerns the recognition, by scFv-decorated liposomes, of a target consisting of a specific tumor antigen together with the presenting MHC molecule, mimicking as such the T-cell receptor.
Materials and methods
Materials
pABC4 vector was a kind gift by Prof Kontermann (Stuttgart, Germany); BL21 bacteria by NEB (Leiden, the Netherlands); tryptone, yeast, NaCl, glucose, sucrose, β-mercaptoethanol, imidazole, EDTA, L-cysteine, HEPES, chloroform, methanol, and Triton X-100 were all purchased from Sigma (Zwijndrecht, the Netherlands). Immobilized metal affinity chromatography (IMAC) Ni2+ columns and AKTA FPLC system were obtained from GE Healthcare (Hoevelaken, the Netherlands), and PBS and Amicon filters MWCO 10,000 from Millipore (Amsterdam, the Netherlands). Tris carboxyethyl phosphine (TCEP) beads and powder, Pierce Spin cups cellulose acetate filters were provided by Thermo Scientific (Bleiswijk, the Netherlands). All chemicals used were HPLC grade. Horseradish peroxidase (HRP)-conjugated anti-histidine (HIS)-tag antibody was obtained from Santa Cruz (Dallas, TX, USA); mouse anti-HIS-tag antibody from both Santa Cruz, SanBio (Mountain View, CA, USA) and Abcam (Cambridge, MA, USA); goat antimouse PE from Southern Biotech (human ads-PE) (Uithoorn, the Netherlands). Mouse anti-M13 antibody and HRP-tagged anti-M13 antibody were purchased from GE Healthcare. The lipids hydrogenated soy L-α-phosphatidylcholine (HSPC), cholesterol (Ch), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-PEG2000 (DSPE-PEG2000) were purchased from Lipoid (Ludwigshafen, Germany) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-26 (ammonium salt) (DSPE-PEG2000 maleimide), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3 benzoxadiazol-4-yl) (NBD-PE), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(carboxyfluorescein) (ammonium salt) (CF-PE) were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL, USA). Nuclepore® track-etched membranes were purchased from Whatman (Maidstone, UK). Doxorubicin-HCl was purchased from Pharmachemie (Haarlem, the Netherlands).
Methods
Immunoliposome formulation
Preparation, purification, and characterization of scFv proteins
scFv G8 and Hyb3 were produced by our research group and collaborators as described previously.24 Both scFv-DNAs were cloned into the pABC4 vector (adding a HIS-tag and cysteine) and put into BL21 bacteria. Bacteria were grown at 37°C until an optical density of 0.6–0.8 at 600 nm was reached. Cultures were induced with 1 mM isopropyl β-D-1-thiogalactopyranoside and grown for an additional 4 hours at 37°C. The periplasmic fractions of BL21 bacteria were subjected to IMAC29 using an Ni2+ column to remove proteins with a HIS tag. Fractions with protein were concentrated over Amicon filters (MWCO 10 kDa), and concentrated fractions of scFv were kept at 4°C until further use. Protein content was measured by NanoDrop™. Both scFvs were characterized and evaluated for in vitro binding to APD cells before coupling to liposomes as has been shown previously.24
Preparation and characterization of liposomes
A neutral liposome composition, typically HSPC, Ch, DSPE-PEG2000, and DSPE-PEG2000 maleimide, was used in a molar ratio of 55:40:4:1, respectively.30,31 Liposomes were prepared by a film hydration method as previously described.24 Briefly, lipids were dissolved in chloroform:methanol (9:1 v/v) and solvents removed by evaporation. The film was flushed with nitrogen and hydrated with 250 mM ammonium sulfate buffer (pH 5.5), and liposomes of correct sizes were produced by extrusions through polycarbonate membranes. Size and polydispersity index (PDI) were determined with a Zetasizer (Malvern Instruments, Worcestershire, UK), and PD10 columns were used to exchange the buffer to HEPES saline buffer (pH 7.4; 10 mM HEPES, 150 mM NaCl, 5 mM EDTA). DXR was loaded with an active gradient of ammonium sulfate pH 5.5 and HEPES pH 7.432,33 for 1 hour at 60°C at a drug to lipid ratio of 0.1:1 (mol/mol). Unincorporated DXR was separated by ultracentrifugation at 29,000×g (Ti 50.2 rotor) for 2 hours at 4°C. Liposomes were resuspended at slow rotation overnight at 4°C in 10 mM HEPES buffer (pH 6.7) and preserved at 4°C until use within 2 weeks. Phospholipid content was determined before and after loading DXR by Rouser phosphate assay, and total lipid amount was calculated after Ch correction.34 DXR was quantified by fluorometry (Ex 482 nm/Em 594 nm), and loading efficiency (%) was calculated as (Da/Db)×100, where Da is the DXR to lipid ratio after loading and Db is DXR to lipid ratio before loading.35 For flow cytometry and in vitro imaging experiments, 0.3% CF-PE or 0.3% NBD-PE were used in the bilayer as fluorescent probes together with encapsulated DXR. For in vivo experiments, DXR liposomes without fluorophores were used.
Immunoliposomes formulation and characterization
Liposomes were linked to scFv proteins by the postattachment method of coupling10,36 using preformed maleimide-PEG liposomes37,38 as described previously.24 Briefly, purified scFvs were prereduced using TCEP (10 mM) for 1 hour at room temperature (RT). An amount of 340 μg of scFv was coupled to 10 μmol total lipids. Obtained immunoliposomes were subjected to phospholipid determination, and total protein coupled was calculated by Lowry–Peterson assay.39 The number of scFv molecules attached was deduced using protein and lipid determinations as follows: a: Moles of protein × Avogadro’s number = molecules of protein b: (Amount of total lipids × Avogadro’s number)/molecules of PEG = number of carriers c: Molecules of protein (a)/number of carriers (b) = number of molecules per carrier.
Finally, sizes and charges were determined and preservation of scFv was assessed by ELISA.24 Briefly, streptavidin-linked M1+/A1+ complexes were incubated with biotinylated ELISA plates for 1 hour at RT. Plates were washed and incubated with immunoliposomes at titrated concentrations for 1 hour, and plates were read according to the manufacturer’s instructions.
In vitro validation of immunoliposomes
Stability of immunoliposomes
The stability of immunoliposomes was evaluated under conditions that mimicked the in vivo setting. Thus, 50 μL of immunoliposome sample was added to a quartz cuvette and HEPES or 100% FBS were added up to 3 mL. Sample fluorescence was measured under continuous stirring every second for 1 hour in a Hitachi F4500 fluorescence spectrometer (Ex 472 nm/Em 590 nm) at 37°C, after which 10% Triton X100 was added and the total DXR signal was measured. Immunoliposome stability was calculated as DXR released:
|

where It is the fluorescence of sample at a certain time point, I0 is the blank fluorescence, and I∞ is the maximum DXR fluorescence.35 Likewise, longer time stability and DXR retention in liposomes aliquoted at 37°C were evaluated at various time points up to 24 hours.
Cellular targeting with immunoliposomes
Four human melanoma, patient-derived cell lines were used: two M1+/A1+ (MZ2Mel43 and G43) and two M1−/A1+ (Mel78 and Mel2A), with all cell lines being HLA-A1+. The melanoma cell lines (G43, Mel78, and Mel2A) were provided as a gift by P van der Bruggen. The MAGE-A1+ and HLA-A1+ melanoma cell line MZ2Mel43 were kindly provided by T Boon and P Coulie, Ludwig Cancer Research Institute, Brussels, Belgium22 (approval for using human cell lines is given by METC: Medisch Ethische Toetsings Commissie, Erasmus MC). Tumor cells were maintained in DMEM (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% FBS, 1% nonessential amino acids, 1% L-glutamine, and 1% penicillin–streptomycin solution. APD cell line,40–43 which is an Epstein–Barr virus-immortalized B-lymphoblast cell line being M1−/A1+ and used for M1 peptide (EADPTGHSY) loading, was maintained in RPMI 1640 medium supplemented with 10% FBS, 1% L-glutamine, and 1% penicillin–streptomycin solution. All cells were grown at 37°C under 5% CO2 in a humidified incubator. Cells were passaged upon reaching 80%–90% confluency. All media were phenol red free, and cells were regularly tested for mycoplasma by PCR. To assess cell binding of immunoliposomes, 500,000 melanoma cells were nonpulsed or 500,000 APD cells were pulsed with M1 peptide (1 μM) for 30 minutes at 37°C.24 Cells were then subjected to a centrifugation at 450×g for 5 minutes and resuspended in 200 μL of liposomes encapsulating DXR, for 2 hours at 37°C, in case of melanoma at 10 μM or APD cells at a concentration of 1 μM. Cells were washed and resuspended in PBS prior to data acquisition on FACSCanto™ (BD Biosciences, San Jose, CA, USA). Analysis of flow cytometry was performed with FACSDiva™ (BD Biosciences), using the FCS Express 4 Flow Research Edition (De Novo Software, Los Angeles, CA, USA).
Interaction of immunoliposomes with cells by confocal microscopy
Immunoliposomes were formulated with 0.3% CF-PE44 enabling detection at 488 nm excitation and 500–550 nm emission. Glass cover slips were coated with 500 μL of 0.1% collagen38 or 0.1% gelatin for 30 minutes at 37°C and washed with sterile PBS. Tumor cells were harvested and grown on the coated cover slips in a concentration of 0.5×106 cells in 500 μL medium. Twenty-four hours later, cells were incubated with 10 μM of DXR in immunoliposomes for 1 hour at 37°C, carefully washed, and overlaid with medium incubated for 24 hours at 37°C. For imaging, the cell nuclei were stained with Hoechst for 10 minutes at 37°C. Nontreated cells and free DXR were used as negative and positive control, respectively. Cells were imaged with a confocal microscope (Zeiss LSM 510 META) equipped with a 405 nm, 488 nm Argon, 543 and 633 nm Helium-Neon laser. Images were acquired and analyzed with LSM AIM software (Zeiss, Oberkochen, Germany).
Toxicity
Melanoma cell lines were grown in 96-well plates for 24 hours at a seeding concentration of 10,000 cells per well, washed and exposed to free DXR, DXR-loaded liposomes (DXR-Ls), and DXR-L scFvs at 0.001, 0.01, 0.1, 1, 10, 20, 30, and 50 μM of DXR for 10, 30, 60, and 120 minutes. Liposomes were removed by washing cells twice. Cells were cultured with fresh medium for additional 72 hours, and cell survival was determined by sulforhodamine B cytotoxicity assay.45 Cell viability was calculated as percentage of growth of control or untreated cells.
In vivo validation of immunoliposomes
Pharmacokinetics and biodistribution of immunoliposomes
Animal experiments were approved by the “Instantie voor Dierenwelzijn” Erasmus MC and conducted with permissions granted by the “Nederlandse Dierexperimentencommissie.” The experiments were performed according to the European Directive 2010/63/EU on the protection of animals used for scientific purposes. NMRI nu/nu mice were housed at temperatures between 20°C and 22°C and humidity between 50% and 60%.35 Mice were fed with standard laboratory diet ad libitum (Hope Farms, Woerden, the Netherlands). Pharmacokinetics was studied in tumor-free mice. To this end, mice were injected with 4 mg/kg DXR-Ls and DXR-L scFvs, and 100 μL blood was drawn from the tail vein in heparin tubes at 5, 15, 30 minutes, 2, 4, 6, and 24 hours postinjection. Blood was centrifuged at 14,000×g for 10 minutes at RT, and plasma aliquots (50 μL) were distributed in a black plate and DXR fluorescence was quantified using a fluorescence plate reader (Wallac Victor2, PerkinElmer). The total signal of DXR was measured after adding 1% Triton X100 to the wells. One control mouse was sacrificed to subtract background signal of only plasma, without DXR. Biodistribution was studied in subcutaneous tumor-bearing mice that were subcutaneously implanted with tumor. Two cell lines were selected to induce tumors in the mice: G43 which is M1+/A1+ and Mel78 which is M1−/A1+. A bulk tumor for transplantation was generated by injecting 5×106 cells in 100 μL PBS subcutaneously in the flank region. Upon reaching a volume of 300 mm3, the tumor was excised and pieces (2×2 mm) were implanted subcutaneously into new mice. Tumors were monitored every 2 days with a caliper. Tumor volume (mm3) was calculated by the formula: 0.4 × a × b2, where “a” represents the largest diameter and “b” is perpendicular to “a.”43 When serially implanted tumor reached a volume of 300–500 mm3 size, mice were administered a dosage of 4 mg/kg DXR-Ls and DXR-L scFvs; 6 hours after administration, mice were sacrificed and tissues (tumor, liver, kidney, spleen, heart, and lungs) were harvested and snap frozen in liquid N2 until further analysis. DXR content was determined as previously described.46 Briefly, tissue was treated with acidified isopropanol (0.075 N HCl in 90% isopropanol) for 24 hours at 4°C, homogenized (Bio-Gen PRO200 homogenizer with 10 mm generator; Pro Scientific, Oxford, CT, USA), centrifuged for 20 minutes at 15,000×g, and supernatants were harvested. DXR fluorescence was determined in supernatants using a Hitachi F4500 fluorescence spectrometer (Ex 472 nm/Em 590 nm). All measurements included an internal DXR standard, and calculations were done against a standard curve of DXR in acidified isopropanol. The amount of fluorescence was calculated and presented as micrograms of DXR per gram of tissue. Organs from a nontreated mouse as well as PBS controls were used to correct for background values.
Antimelanoma efficacy
When tumors reached a volume of 100 mm3, mice were treated as follows: control (PBS), free DXR, DXR-L (nontargeted), DXR-L linked to low-affinity scFv G8 (DXR-L scFv G8), and DXR-L linked to high-affinity scFv Hyb3 (DXR-L scFv Hyb3). A minimum of six and maximum of seven mice were used for all groups except for PBS where five mice were used for G43 group and three mice were used for Mel78 group. Two different treatment protocols were used: 2 mg/kg DXR dose was injected eight times or 4 mg/kg DXR dose was injected four times every 5 days, resulting in an accumulated dose of 16 mg/kg of DXR for both groups. Weight of mice was monitored daily, and therapeutic efficacy was evaluated by measuring tumor size every other day. Classification of tumor response to the treatment was reported previously.47 Briefly, tumor size was measured every 2 days with a caliper and represented as tumor size index (TSI: the tumor volume in relation to initial tumor volume at the start of treatment). TSI of 5 was selected as a cutoff.
Survival was plotted for same number of mice as used for efficacy and was monitored for 3 weeks postinjection or until the tumors started to relapse. Mice were sacrificed due to weight loss, if exceeded >20% of the starting weight, ascites, or when the tumor volume reached the humane endpoint.
Statistical analyses
All results were processed using GraphPad Prism (version 5) and SPSS software (version 21) and analyzed by Kruskal–Wallis H tests followed by Mann–Whitney U tests for pair-wise comparisons. Survival data were analyzed by log-rank (Mantel–Cox) test.
Results
Linkage to scFv does not affect liposomal zeta potential or DXR encapsulation
DXR-Ls were produced and used as described previously and adhering to the following quality criteria: size (between 80 and 100 nm), PDI (below 0.1), zeta potential (slightly negative), and encapsulation efficiency (above 70%).24 These formulations remained stable in HEPES and at 4°C for up to 4 weeks, while only slight changes in size and PDI were observed without aggregation (Table S1). However, based on our previous results regarding scFv coupling to liposomes and its stability and functionality of scFv fragment for up to 2 weeks,24 freshly made DXR-Ls were used for all in vivo studies. DXR encapsulation was 98% for nontargeted DXR-Ls and 95% for DXR-L scFv G8 and 80% DXR-L scFv Hyb3 (Table S1), while on average >90% of the drug remained encapsulated during 4 weeks of storage at 4°C in HEPES buffer (pH 7.4). Between 30 and 50 scFv molecules were detected on DXR-L scFv G8 and Hyb3 as previously determined by size change of 10 nm immunoliposomes, SDS-PAGE (not shown but previously reported),24 and protein measurement of immunoliposomes.
Next, short-term stability of liposomal preparations was determined in HEPES buffer to mimic storage conditions and FBS to mimic in vivo conditions at 37°C. DXR remained stably encapsulated (<10% of leakage) at physiological temperature (37°C) for up to 1 hour in HEPES buffer or 100% FBS in all formulations (Figure 1). A similar result was obtained when formulations were incubated in HEPES buffer for up to 24 hours at 37°C. However, a 20% leakage was observed for both immunoliposomes when incubated in 100% FBS at 37°C for 6 hours or longer (Figure 1B), and a 10% leakage was observed at 24 hours for DXR-L, the latter being comparable with previous results.48,49 As in patient circulation times are expectedly shorter than 6 hours after injection, results indicate that our immunoliposome formulations are suitable for in vivo use.
 | Figure 1 Stability of liposomal formulation in HEPES and 100% FBS at 37°C over time. |
Immunoliposomes bind to, become internalized, and effectively kill melanoma cells expressing cognate antigen
Binding of anti-M1/-A1 scFv-coupled DXR-Ls was studied by flow cytometry using antigen-positive M1+/A1+ (MZ2Mel43 and G43) and antigen-negative M1−/A1+ (Mel78 and Mel2A) melanoma cells (Figure 2A and B). Antigen expression of these cell lines was previously established on cDNA level (Saeed et al,24 Figure S1) and rechecked by us (Figure S1) on protein level by flow cytometry assay. T-cells expressing anti-M1/-A1 TCR were incubated with melanoma cells, and they could bind only when M1 was expressed in HLA-A1 context. DXR-Ls had green carboxyfluorescein (CF-PE), a fluorochrome inserted in the lipid bilayer of liposomes and the signal showed almost equal binding/internalization when compared with DXR signal (Figure 2A). According to DXR signal, some binding and internalization at 37°C of DXR-Ls were observed, while DXR signal was quenched due to liposomal retention (Figure 2B). In the M1+/A1+ MZ2Mel43 melanoma cell line, we observed three and five times higher uptake of DXR-L scFv G8 (P=0.013) and DXR-L scFv Hyb3 (P=0.012), respectively, in comparison with nontargeted DXR-Ls (Figure 2B). Interestingly, DXR-L scFv Hyb3 was taken up 1.5–1.7 times higher in antigen-positive cells than with DXR-L scFv G8 (P=0.001). In case of M1+/A1+ cells compared with M1−/A1+ cells, DXR-L scFv G8 (P=0.013) and Hyb3 (P=0.013) uptakes were 6–15 and 8–25 times higher, respectively (Figure 2B). These liposomes were pretested on APD cells pulsed with M1 peptide and signals were compared with APD cells without peptide (Figure S2). Data showed enhanced binding of DXR-L scFv G8 and Hyb3 in comparison with DXR-Ls and APD cells without peptide.
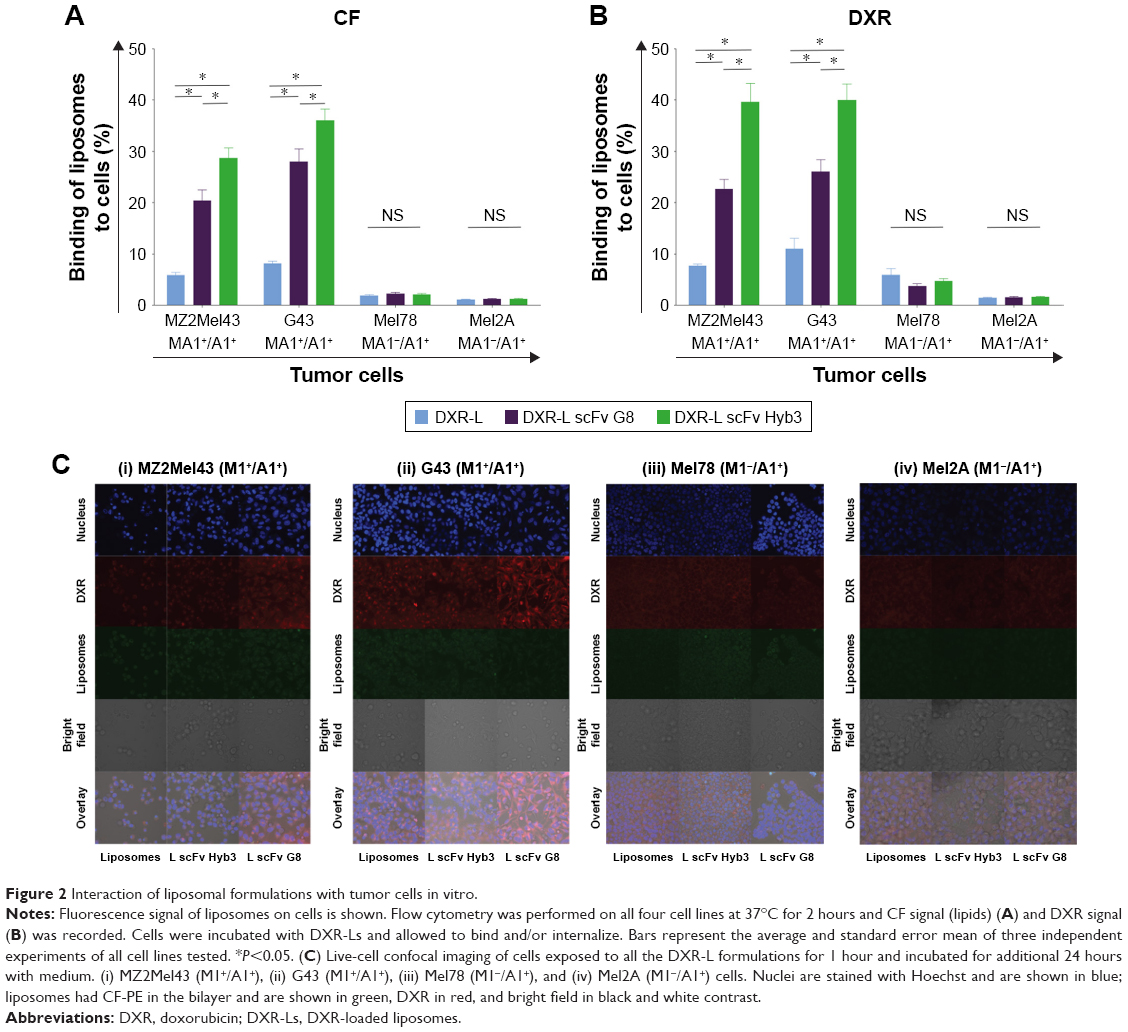 | Figure 2 Interaction of liposomal formulations with tumor cells in vitro. |
Confocal images confirmed low binding and internalization of nontargeted DXR-Ls in both antigen-positive and -negative cell lines (Figure 2C). Moreover, green (CF-PE) and red fluorescence (DXR) colocalize/overlap confirming retention of DXR in the liposome after cellular binding and internalization. We observed slightly more uptake of control DXR-Ls by antigen-positive cells vs antigen-negative cells (Figure 2C), similar to the flow cytometry data, which may be due to differences in cell membrane molecules and/or cell activity. Importantly, DXR-L-scFv G8 and Hyb3 were significantly more internalized by the M1+/A1+ G43 cells than M1−/A1+ cells (Figure 2Cii and iii). DXR-L-scFv G8 binding and/or internalization by MZ2Mel43 and G43 cells were higher compared with control DXR-Ls. Regarding Mel78 and Mel2A cells, some binding and/or internalization of DXR-Ls were observed, but no clear visible differences between liposomal formulations were observed. At 24 hours of exposure, most DXR was still present in liposomes and not released (Figure 2C), indicating intracellular entrapment of DXR. However, some leakage occurred over time resulting in DXR accumulation in the nucleus of all cell lines. Thus, M1+/A1+ cells preferentially bind to and internalize DXR-Ls decorated with anti-MAGE scFvs.
When extending our findings to in vitro toxicity toward melanoma cells, we observed that both anti-M1/-A1 scFv-coupled DXR-L formulations showed higher cytotoxicity when compared with DXR-Ls at a concentration of 10 μM liposomes or higher when incubated with M1+/A1+ melanoma cells for all exposure time points (10, 30, 60, and 120 minutes) (Figure 3Ai and ii). However, when M1−/A1+ cell lines were used, no difference in cytotoxicity was observed among all tested liposomal formulations (Figure 3Aiii and iv). As expected, free DXR shows higher toxicity due to direct availability of the drug resulting in faster uptake by cells. IC50 values extracted from the cytotoxicity curves confirm our observations, showing a two to five times lower IC50 value for scFv coupled vs noncoupled liposomal formulations toward their exposure toward antigen-positive cells (Figure 3B). In fact, Kruskal–Wallis test shows significant differences among the groups (MZ2Mel43 P=0.005, G43 P=0.005, Mel78 P=0.02, and Mel2A P=0.04) and Mann–Whitney U test shows enhanced viability for antigen-positive cell lines when free DXR or DXR-L was compared with either of the DXR-L scFv formulations (P=0.03 for both cell lines MZ2Mel43 and G43). When DXR-L scFvs were compared with each other, there were no significant differences found. For both antigen-negative cell lines, no significant differences were found among all the liposomal formulations. However, when free DXR was compared with DXR-L and DXR-L scFv formulations, it was significantly different (P=0.03 for both cell lines Mel78 and Mel2A). These data show that uptake of liposomes by antigen-positive cells was directed by scFv coupled to DXR-L, and this resulted in antigen-specific killing.
 | Figure 3 Melanoma cell survival after exposure to liposomal formulations. |
Pharmacokinetics and biodistribution of anti-M1/-A1 DXR liposomes in NMRI nu/nu mice
To evaluate pharmacokinetic profiles of our experimental liposomal formulations, NMRI nu/nu nontumor-bearing mice were treated, which revealed that DXR-Ls have a longer time of circulation compared with anti-M1/-A1 scFv-coupled DXR-L (Figure 4). These data confirmed previous findings of Allen 2002 (use of the postinsertion method for the formation of ligand-coupled liposomes) where they show that targeted formulations are cleared faster from blood stream due to possible binding to targeted cells.
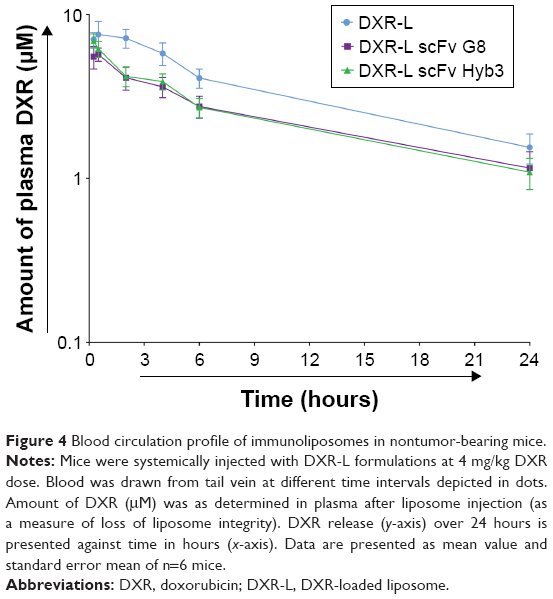 | Figure 4 Blood circulation profile of immunoliposomes in nontumor-bearing mice. |
Ratio of DXR (encapsulated) to CF (liposomal bilayer) over time suggests overall stability of DXR-L (0.5 hour=0.48) and possible drug release from DXR-L scFv G8 (0.5 hour=0.94) and Hyb3 (0.5 hour=0.95) at 4 hours (DXR-L scFv G8=3.78 and Hyb3=2.69). The correlation between liposome and DXR is considered as a more robust measure of stability (P=0.4). No significant differences among the groups were found during blood circulation, when DXR-L was compared with either DXR-L scFv G8 or DXR-L scFv Hyb3. Half-life of DXR-L was ~7.03 hours, DXR-L scFv G8 was 5.243 hours, and DXR-L scFv Hyb3 was 5.413 hours (Figure 4). Of note, DXR-L scFv G8 and Hyb3 showed similar circulation times, indicating identical clearance properties and stability after injection.
To evaluate biodistribution of our experimental liposomal formulations, NMRI nu/nu mice with tumors derived from either the G43 (M1+/A1+) or the Mel78 (M1−/A1+) cell line were treated. DXR-L scFv G8 and Hyb3 accumulated similarly in tumors; however, their accumulation was significantly higher in comparison with DXR-L and free DXR (Figure 5). In tumor tissue, DXR-L scFv Hyb3 accumulates 2.3 times more than nontargeted formulation DXR-L, even in the M1−/A1+-negative cell line, pointing to some off target binding that has been discussed earlier24 and may be due to HLA-A1 antigen. DXR-L scFv G8 tends to accumulate at higher concentrations in all healthy organs in mice bearing G43-positive tumors in comparison with DXR-L scFv Hyb3 (Figure 5). This accumulation could be due to physiology of G43 cells and how these cells interact or affect other organs. Distribution to organs and nontargeted tissues is substantial for both nontargeted50 and targeted liposomes compared with free DXR as shown previously. This however is not accompanied by side effects due to low cell uptake in nontargeted tissues.51
 | Figure 5 Tissue distribution of immunoliposomes in tumor-bearing mice. |
In mice bearing Mel78 tumors, DXR-L scFv G8 is accumulated higher than DXR-Ls but similar compared with DXR-L scFv Hyb3. Accumulation of DXR-L scFv G8 on the whole was higher in G43 tumor tissue than all organs except lungs. DXR-L scFv Hyb3 accumulation was the highest in tumor tissue in comparison with other organs. For Mel78, accumulation of DXR-L scFv Hyb3 was high in all organs. Both DXR-L scFvs accumulated higher in kidneys, spleen, lungs, and heart.
Efficacy of anti-M1/-A1 DXR liposomes and survival
Finally, mice bearing human melanoma tumors were treated with the immunoliposomes using two dosage schemes: either four injections of 4 mg/kg DXR (Figure 6A and C) or eight injections of 2 mg/kg (Figure 6B and D) in free form or liposomal encapsulated with an interval of 5 days. For the treatment of G43 and Mel78 tumors, a minimum of six and maximum of seven mice were used for all groups except for PBS where five mice were used for G43 group and three mice were used for Mel78 group. A dose of 2 mg/kg free DXR resulted in a transient inhibition of tumor growth for about 14 days (Figure 6A and B). The treatment of G43 tumor with 2 or 4 mg/kg nontargeted DXR-Ls had an intermediate effect, where three and four out of six tumors had a growth delay. In these groups no true antitumor responses, such as tumor shrinkage or disappearance, were observed. We observed tumor inhibition from the start in G43 tumors when treated with 4 mg/kg DXR-L scFvs (Figure 6A and E). Notably, we observed five out of seven antigen-positive tumors treated with 4 mg/kg DXR-L scFv G8 that went into complete remission and four remained undetectable until the end of experiment, one went back into recurrence and was palpable at day 42 (Figure 6A and E). Furthermore, six out of seven antigen-positive tumors showed clear growth inhibition following treatment with DXR-L Hyb3 4 mg/kg (Figure 6A and E), while two went into complete remission and remained undetected until the end of the experiment.
 | Figure 6 DXR-L scFvs show significant and sustained antimelanoma effect. |
Administration of 2 mg/kg dose resulted in prolonged antitumor response in G43 tumor-bearing mice for both immunoliposome formulations DXR-L scFv G8 and Hyb3 (Figure 6). When treated with 2 mg/kg DXR-L scFv G8, two out of five tumors showed profound growth inhibition (Figure 6B and E). We observed that seven out of seven antigen-positive tumors treated with DXR-L scFv Hyb3 2 mg/kg showed growth inhibition while two went into remission and remained undetectable. In G43 tumor-bearing mice, superior activity of targeted treatment (DXR-L scFv G8 and Hyb3) became apparent after fourth dose (Figure 6B and E).
Interestingly, in Mel78 tumor-bearing mice, following a treatment dose of 4 mg/kg DXR-L-scFv Hyb3, a prolonged antitumor response was observed in five out of six mice (Figure 6C and E). DXR-L-scFv G8 administration only had a transient effect on these antigen-negative tumors with two out of seven mice showing stable disease. Repeated administration of 2 mg/kg of both DXR-L-scFv formulations resulted in comparable (nonsignificant) tumor growth reduction toward the antigen-negative tumor Mel78 (Figure 6D).
An overview of normalized tumor growth values at days 6 (after single dose), 20 (4 doses), and 36 (toward the end of experiment) is given calculated against initial tumor measurement at the start of the experiment (Figure 6E). We observed that at day 6 after single dose, all mice had a TSI below 5%. At day 20, in both treatment schedules, none of the M1+/A1+ G43 bearing animals treated with both targeted formulations showed a TSI above 5. We also observed that nontargeted DXR-L already had quite an effect over G43 tumors, which could be explained by higher uptake of DXR-L by G43 tumor cells as previously observed (Figure 2Cii) in combination with a higher sensitivity of these cells to DXR as seen in in vitro (Figure 3). This in contradiction to M1–/A1+ Mel78 bearing animals showed more DXR-Ls, nontargeted as well as targeted, treated animals with a TSI exceeding 5.
Survival data, log-rank test (Mantel–Cox test), show significantly extended survival in all the groups (G43 4 mg/kg P=0.0006, G43 2 mg/kg P=0.001, Mel78 4 mg/kg P=0.02, and Mel78 2 mg/kg P=0.0006) (Figure S3). Treatment with 4 mg/kg dosage of DXR-L scFv G8 (P=0.0004) and DXR-L scFv Hyb3 (P=0.005) enhanced life span, almost doubled, when comparing mice with G43 tumors (mean survival of 93 days) vs those with Mel78 tumors (mean survival of ~41.5 days) (Figure S3A, C and E). Treatments compared within G43 tumor group 4 mg/kg dosage showed significant differences when DXR-L scFvs were compared with either control (Figure S3A and E). When nontargeted DXR-L was compared with DXR-L scFv G8 (P=0.02) or DXR-L scFv Hyb3 (P=0.04), significant differences were found (Figure S3A and E). However, when mice were treated with 2 mg/kg dosage, only DXR-L scFv Hyb3 showed a better survival for G43 tumors (mean survival of 71 days and P=0.0042 in comparison with DXR-L) (Figure S3B and E). No significant differences were found among the DXR-L scFvs in all groups. When Mel78 2 mg/kg group treatments were compared, significant differences were only found against PBS group, rest of the treatments showed no significant differences (Figure S3D and E).
To monitor toxicity body weight and other signs of toxicity, such as diarrhea, lethargy, less appetite, or improper gait, were also recorded. Body weight measurements showed that mice treated with free DXR or DXR-L did not lose weight but became slow and lost appetite as a consequence of prolonged tumor growth (Figure S4) and were taken out of the experiment as mandatory by the regulations in animal welfare. Treatment with DXR-L scFvs 4 mg/kg dosage lowered weight after injection in both antigen-positive and -negative tumor-bearing mice, rebounced to normal levels a few days later, and stayed at normal levels till the end of the experiments (Figure S4). Animals treated with 2 mg/kg dose of DXR did not lose weight or showed other discomforts due to treatment. Two G43 tumor-holding mice treated with DXR-L Hyb3 4 mg/kg had diarrhea for 2 days after the first dose. Three mice with G43 tumors developed ascites (one in PBS group and two treated with 4 mg/kg DXR-L scFv G8). Two mice with G43 tumors became lethargic and eventually were sacrificed (one 2 mg/kg DXR-L scFv G8 and one 4 mg/kg free DXR). Five mice with Mel78 tumors developed ulcers (three from PBS group and two from 2 mg/kg DXR-L group). Two mice with Mel78 tumors had weight loss over 20% of initial weight (both 4 mg/kg dose: one DXR-L scFv G8 and one DXR-L scFv Hyb3), and therefore had to be sacrificed.
Discussion
Previously, we showed that melanoma cells can be targeted with antibodies mimicking TCRs.22,24 These antibodies recognize tumor-specific peptides expressed by MHC-I molecules on tumor cell surfaces and, compared with classical antibodies, cover a higher number of cellular target antigens. In case tumor antigens are targeted that are absent in healthy tissues, the coupling of TCR-like antibodies to nanosized carriers, such as liposomes, enhances the tumor selectivity of liposome targeting. However, the making of immunoliposomes is challenged by liposome; opsonization as well as recognition by macrophages,52,53 mainly due to the Fc part of antibodies, causes these challenges. Therefore, we and others exploit scFv antibodies to generate immunoliposomes, the smallest fragment that still retains full binding capacity.54,55 The TCR-like scFv G8 and Hyb3 coupled to liposomes mediate specific binding and internalization by M1/A1-positive melanoma cells in vitro.24 Here, we have built on these earlier data and linked M1/A1 targeting, using scFvs with different binding affinities, to DXR-based liposomal chemotherapy and included a series of appropriate tests, including those related to stability, pharmacokinetics, biodistribution, and in vitro and in vivo antitumor efficacy. Steric stabilization of liposomes with PEG renders these liposomes stealth-like, ie, due to PEG on the outside liposomes is not recognized by opsonins and circulates therefore relatively long after systemic injection.56 Here, we use a unique antibody as it recognizes a peptide/MHC-I complex.22,57 Moreover, the antigen, MAGE, is only expressed on tumor cells in this context, providing a melanoma-specific target. Thus, in short, we have shown that when anti-M1/-A1 antibodies are coupled to liposomes carrying DXR as cargo, uptake of these liposomes is enhanced, which leads to enhanced cell-specific killing. In vivo, we show that these targeted DXR-Ls show better efficacy and survival when compared with nontargeted treatments and antigen-negative tumors.
Decoration of DXR-L with TCR-like scFvs did not affect long-term DXR encapsulation, which is in line with findings for other targeted formulations.58 In addition, flow cytometry and confocal microscopy revealed higher binding of DXR-L-scFvs to M1+/A1+ cells compared with M1−/A1+ cells and DXR-Ls. It is noteworthy that liposomal binding to antigen-negative cells was lower but present for both DXR-L scFvs; however, we observed minimal binding of targeted liposomes DXR-L scFv G8 and Hyb3 to antigen-negative melanoma cells in vitro, indicating excellent specificity. We postulate that this could be the result of more rapid processing of DXR-L scFvs followed by a higher degree of degradation compared with DXR-Ls.
Confocal microscopy data revealed that slightly higher uptake of DXR-L scFv G8, and binding site barrier (BSB) effect could be responsible for this high uptake of DXR-L scFv G8 in comparison with Hyb3.59,60 This BSB effect was proposed for antibodies, and a study by Saga et al showed that BSB effect could be seen in small micrometastases of size 300 μm. They also showed that increasing the dose of antibody partially overcame the BSB effect but lost antibodies lose their specificity. We hypothesize here that DXR-L scFv Hyb3 is possible to have been saturated by antigens first encountered, and due to the high affinity of Hyb3 it is not possible to escape it. DXR-L scFv G8 having the low affinity can penetrate deeper and may be that contributes to a higher signal in confocal. Based on previous findings,24 we had the impression that scFv Hyb3 has a high affinity and binding to MHC molecule: HLA-A1. G8 and Hyb3 scFvs are both selected against M1 only when presented by HLA-A1,21 yet a higher affinity of Hyb3 scFv may render enough binding force to enable sufficient interaction with HLA-A1 alone.
Besides due to PEG on the outside liposomes are not recognized by opsonins and circulate therefore relatively long up to 6 hours after systemic injection.56 However, encapsulation of DXR in such sterically stabilized liposomes, as we use here, impairs in vitro cytotoxicity.56 Likewise, we observed a >100-fold higher cytotoxicity of free DXR vs DXR-Ls. But importantly, DXR-L-scFv G8 and Hyb3 were significantly more toxic toward M1+/A1+ melanoma cells but not antigen-negative melanoma cells compared with nontargeted DXR-Ls. This results from antigen-specific binding and internalization mediated by the attached scFv.
Generally, circulation time of PEGylated liposomes becomes compromised when moieties are attached on the outside.57 Although our cytotoxicity data argue that coupling liposomes to TCR-like scFvs enhances efficacy of liposomes, which extends data from earlier studies with liposomes coupled to classical scFvs,61,62 faster clearance, instability, and off target binding are observed as well with scFv-coupled liposomes.57 Indeed, our scFv-decorated liposomes exhibit shorter circulation time compared with nontargeted DXR-L. Especially in the first hour after injection in mice, a drop in blood levels of immunoliposomes is observed followed by a second phase of clearance, which runs parallel to that of DXR-L. Although the initial faster clearance of targeted DXR-L will affect accumulation in tumor tissue, we argue that specificity of targeting enhances safety of immunoliposomes and is able to compensate for any potential compromise toward efficacy of these liposomes (ie, enhanced delivery at tumor and less/negligible delivery at other tissues). MAGE-A1 is only expressed on tumor cells, and our biodistribution experiments indeed demonstrated that our immunoliposomes preferentially accumulated in antigen-positive tumors, while DXR-L and free DXR accumulated at lower levels and in accordance to previous observations.57 scFv G8 and Hyb3 differ in affinity toward M1/A1 (scFv G8 KD is 250 nM and scFv Hyb3 KD is 14 nM). In spite of a higher affinity, DXR-L-scFv Hyb3 localized to the same extent in antigen-positive G43 tumors when compared with DXR-L-scFv G8. However, due to yet unknown reasons, accumulation of DXR-L scFv G8 was highest in all examined healthy tissues. We could hypothesize that there is some effect of G43 tumor cells on other organs, and more experiments are needed to prove this interaction.
Although, in vivo, accumulation of targeted liposomes in antigen-negative tumors was about 3-fold lower when compared with accumulation in antigen-positive tumors, localization of DXR-Ls and free DXR in the former tumors was also lower, making interpretation of targeting specificity difficult. Possible explanations could include difference in vascular density and tumor perfusion between G43 and Mel78 tumors, sensitivity to DXR, or low expression of M1 in vivo by the otherwise antigen-negative melanoma Mel78. In vivo, however, treatment of antigen-negative melanoma with DXR-L scFv Hyb3 did result in a better tumor response when compared with DXR-L scFv G8-treated mice. These results may argue that besides antigen specificity, other tumor-related factors as those mentioned above may be responsible. When targeting self-antigens, self-recognition might occur causing side effects. Here, we show that DXR-L scFv G8 does not show off target binding, while DXR-L scFv Hyb3 shows some binding to M1− cells. We hypothesize that maturation of the antibody made it less specific to M1, while binding to HLA is preserved, which may affect the toxicity profile.
Conclusion
A dominating hurdle in cancer therapy with systemically injected compounds is the lack of local accumulation.63 Whether chemotherapeutics, immune-modulating agents, or small molecules are used, systemic distribution results in dose-limiting toxicity to vital organs. The architecture of a tumor, specifically poor perfusion resulting from low vascular density, sluggish blood flow, elevated interstitial pressure, and abundance of intra-tumoral stroma, works against drug delivery.64,65 Previous studies have already demonstrated a possible benefit for nanosized carriers as these may impair side effects and augment accumulation in tumors.63,64 Here, we clearly demonstrate stability, specificity, and effectiveness of antitumor therapy with liposomes coupled to TCR-like scFv. Therapeutic efficacy was recorded by tumor growth inhibition, weight loss, and survival percentage of mice. We showed that DXR-L scFvs inhibited tumor growth significantly compared with other groups. These liposomes are specifically targeted to tumor cells and deposit large quantities of drug in tumor proximity or within cells. In addition, we would like to develop these further for imaging purposes; when loaded with contrast agents they can assist in the detection of micrometastases. In future, these can be ultimately used to study the possibilities of targeting various compounds and co-encapsulting them together leading to image-guided drug delivery.
Disclosure
The authors report no conflicts of interest in this work.
References
Maio M. Melanoma as a model tumour for immuno-oncology. Ann Oncol. 2012;23(suppl 8):viii10–viii14. | ||
Cancer.Net. Melanoma: Statistics. Available from: http://www.cancer.net/cancer-types/melanoma/statistics. Accessed June 1, 2014. | ||
Aris M, Barrio MM. Combining immunotherapy with oncogene-targeted therapy: a new road for melanoma treatment. Front Immunol. 2015;6(2):46. | ||
Fucà G, de Braud F, Di Nicola M. Immunotherapy-based combinations: an update. Curr Opin Oncol. 2018;30(5):345–351. | ||
Festino L, Vanella V, Trojaniello C, Ascierto PA. Selecting immuno-oncology-based drug combinations – what should we be considering? Expert Rev Clin Pharmacol. 2018;11(10):971–985. | ||
Maruyama K. PEG-immunoliposome. Biosci Rep. 2002;22(2):251–266. | ||
Allen TM. Ligand-targeted therapeutics in anticancer therapy. Nat Rev Cancer. 2002;2(10):750–763. | ||
Sun X, Yan X, Jacobson O, et al. Improved tumor uptake by optimizing liposome based RES blockade strategy. Theranostics. 2017;7(2):319–328. | ||
Deshpande PP, Biswas S, Torchilin VP. Current trends in the use of liposomes for tumor targeting. Nanomedicine. 2013;8(9):1509–1528. | ||
Iden DL, Allen TM. In vitro and in vivo comparison of immunoliposomes made by conventional coupling techniques with those made by a new post-insertion approach. Biochim Biophys Acta. 2001;1513(2):207–216. | ||
Shmeeda H, Tzemach D, Mak L, Gabizon A. HER2-targeted pegylated liposomal doxorubicin: retention of target-specific binding and cytotoxicity after in vivo passage. J Control Release. 2009;136(2):155–160. | ||
Deshpande P, Jhaveri A, Pattni B, Biswas S, Torchilin V. Transferrin and octaarginine modified dual-functional liposomes with improved cancer cell targeting and enhanced intracellular delivery for the treatment of ovarian cancer. Drug Deliv. 2018;25(1):517–532. | ||
Sugiyama I, Kaihatsu K, Soma Y, Kato N, Sadzuka Y. Dual-effect liposomes with increased antitumor effects against 67-kDa laminin receptor-overexpressing tumor cells. Int J Pharm. 2018;541(1–2):206–213. | ||
Lu L, Ding Y, Zhang Y, et al. Antibody-modified liposomes for tumor-targeting delivery of timosaponin AIII. Int J Nanomedicine. 2018;13:1927–1944. | ||
Kunert A, Straetemans T, Govers C, et al. TCR-Engineered T cells meet new challenges to treat solid tumors: choice of antigen, T cell fitness, and sensitization of tumor milieu. Front Immunol. 2013;4:1–16. | ||
Coulie PG, van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer. 2014;14(2):135–146. | ||
Barrow C, Browning J, Macgregor D, et al. Tumor antigen expression in melanoma varies according to antigen and stage. Clin Cancer Res. 2006;12(3 Pt 1):764–771. | ||
Brasseur F, Rimoldi D, Liénard D, et al. Expression of MAGE genes in primary and metastatic cutaneous melanoma. Int J Cancer. 1995;63(3):375–380. | ||
Caballero OL, Chen YT. Cancer/testis (CT) antigens: potential targets for immunotherapy. Cancer Sci. 2009;100(11):2014–2021. | ||
Pavlinkova G, Colcher D, Booth BJ, Goel A, Wittel UA, Batra SK. Effects of humanization and gene shuffling on immunogenicity and antigen binding of anti-TAG-72 single-chain Fvs. Int J Cancer. 2001;94(5):717–726. | ||
Chames P, Hufton SE, Coulie PG, Uchanska-Ziegler B, Hoogenboom HR. Direct selection of a human antibody fragment directed against the tumor T-cell epitope HLA-A1-MAGE-A1 from a nonimmunized phage-Fab library. Proc Natl Acad Sci. 2000;97(14):7969–7974. | ||
Willemsen RA, Debets R, Hart E, Hoogenboom HR, Bolhuis RL, Chames P. A phage display selected Fab fragment with MHC class I-restricted specificity for MAGE-A1 allows for retargeting of primary human T lymphocytes. Gene Ther. 2001;8(21):1601–1608. | ||
Chames P, Willemsen RA, Rojas G, et al. TCR-like human antibodies expressed on human CTLs mediate antibody affinity-dependent cytolytic activity. J Immunol. 2002;169(2):1110–1118. | ||
Saeed M, van Brakel M, Zalba S, et al. Targeting melanoma with immunoliposomes coupled to anti-MAGE A1 TCR-like single-chain antibody. Int J Nanomedicine. 2016;11:955–975. | ||
Amselem S, Gabizon A, Barenholz Y. Optimization and upscaling of doxorubicin-containing liposomes for clinical use. J Pharm Sci. 1990;79(12):1045–1052. | ||
Khan DR, Webb MN, Cadotte TH, Gavette MN. Use of targeted liposome-based chemotherapeutics to treat breast cancer. Breast Cancer. 2015;9(Suppl 2):1–5. | ||
Mohammadi ZA, Aghamiri SF, Zarrabi A, Talaie MR. Liposomal doxorubicin delivery systems: effects of formulation and processing parameters on drug loading and release behavior. Curr Drug Deliv. 2016;13(7):1065–1070. | ||
Barenholz Y, Barenholz Y. Doxil® – the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160(2):117–134. | ||
Hochuli E. Large-scale chromatography of recombinant proteins. J Chromatogr. 1988;444:293–302. | ||
Messerschmidt SKE, Beuttler J, Rothdiener M. Immunoliposomes. In: Kontermann R, Dubel S, editors. Antibody Engineering. Vol 2. New York: Springer; 2010:401–428. | ||
Abraham SA, Waterhouse DN, Mayer LD, Cullis PR, Madden TD, Bally MB. The liposomal formulation of doxorubicin. Methods Enzymol. 2005;391:71–97. | ||
Haran G, Cohen R, Bar LK, Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim Biophys Acta. 1993;1151(2):201–215. | ||
Li L, Ten Hagen TL, Hossann M, et al. Mild hyperthermia triggered doxorubicin release from optimized stealth thermosensitive liposomes improves intratumoral drug delivery and efficacy. J Control Release. 2013;168(2):142–150. | ||
Rouser G, Fkeischer S, Yamamoto A. Two dimensional then layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids. 1970;5(5):494–496. | ||
Dicheva BM, Ten Hagen TL, Seynhaeve AL, Amin M, Eggermont AM, Koning GA. Enhanced specificity and drug delivery in tumors by cRGD-anchoring thermosensitive liposomes. Pharm Res. 2015;32(12):3862–3876. | ||
Kontermann RE. Immunoliposomes for cancer therapy. Curr Opin Mol Ther. 2006;8(1):39–45. | ||
Rüger R, Müller D, Fahr A, Kontermann RE. Generation of immunoliposomes using recombinant single-chain Fv fragments bound to Ni-NTA-liposomes. J Drug Target. 2005;13(7):399–406. | ||
Rothdiener M, Beuttler J, Messerschmidt SK, Kontermann RE. Antibody targeting of nanoparticles to tumor-specific receptors: immunoliposomes. Methods Mol Biol. 2010;624:295–308. | ||
Peterson RC. Application of Lowry protein determination to influenza vaccine. J Pharm Sci. 1969;58(1):141–142. | ||
Willemsen RA, Ronteltap C, Chames P, Debets R, Bolhuis RL. T cell retargeting with MHC class I-restricted antibodies: the CD28 costimulatory domain enhances antigen-specific cytotoxicity and cytokine production. J Immunol. 2005;174(12):7853–7858. | ||
Schaft N, Lankiewicz B, Drexhage J, et al. T cell re-targeting to EBV antigens following TCR gene transfer: CD28-containing receptors mediate enhanced antigen-specific IFNgamma production. Int Immunol. 2006;18(4):591–601. | ||
Roszik J, Sebestyén Z, Govers C, et al. T-cell synapse formation depends on antigen recognition but not CD3 interaction: studies with TCR: ζ, a candidate transgene for TCR gene therapy. Eur J Immunol. 2011;41(5):1288–1297. | ||
Sebestyén Z, Schooten E, Sals T, et al. Human TCR that incorporate CD3 induce highly preferred pairing between TCR and chains following gene transfer. J Immunol. 2008;180(11):7736–7746. | ||
Dicheva BM, Ten Hagen TL, Schipper D, et al. Targeted and heat-triggered doxorubicin delivery to tumors by dual targeted cationic thermosensitive liposomes. J Control Release. 2014;195:37–48. | ||
Skehan P, Storeng R, Scudiero D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990;82(13):1107–1112. | ||
Hoving S, Seynhaeve AL, van Tiel ST, Eggermont AM, Ten Hagen TL. Addition of low-dose tumor necrosis factor-alpha to systemic treatment with STEALTH liposomal doxorubicin (Doxil) improved anti-tumor activity in osteosarcoma-bearing rats. Anticancer Drugs. 2005;16(6):667–674. | ||
Seynhaeve ALB, Hoving S, Schipper D, et al. Tumor necrosis factor α mediates homogeneous distribution of liposomes in murine melanoma that contributes to a better tumor response. Cancer Res. 2007;67(19):9455–9462. | ||
Shi C, Guo D, Xiao K, Wang X, Wang L, Luo J. A drug-specific nanocarrier design for efficient anticancer therapy. Nat Commun. 2015;6(1):7449. | ||
Hioki A, Wakasugi A, Kawano K, Hattori Y, Maitani Y. Development of an in vitro drug release assay of PEGylated liposome using bovine serum albumin and high temperature. Biol Pharm Bull. 2010;33(9):1466–1470. | ||
Luo R, Li Y, He M, et al. Distinct biodistribution of doxorubicin and the altered dispositions mediated by different liposomal formulations. Int J Pharm. 2017;519(1–2):1–10. | ||
Seynhaeve ALB, Dicheva BM, Hoving S, Koning GA, Ten Hagen TLM. Intact Doxil is taken up intracellularly and released doxorubicin sequesters in the lysosome: evaluated by in vitro/in vivo live cell imaging. J Control Release. 2013;172(1):330–340. | ||
Cheng WW, Allen TM. The use of single chain Fv as targeting agents for immunoliposomes: an update on immunoliposomal drugs for cancer treatment. Expert Opin Drug Deliv. 2010;7(4):461–478. | ||
Ternant D, Paintaud G. Pharmacokinetics and concentration-effect relationships of therapeutic monoclonal antibodies and fusion proteins. Expert Opin Biol Ther. 2005;5(Suppl 1):S37–S47. | ||
Baum P, Müller D, Rüger R, Kontermann RE. Single-chain Fv immunoliposomes for the targeting of fibroblast activation protein-expressing tumor stromal cells. J Drug Target. 2007;15(6):399–406. | ||
Thorpe SJ, Turner C, Heath A, et al. Clonal analysis of a human antimouse antibody (HAMA) response. Scand J Immunol. 2003;57(1):85–92. | ||
Gabizon A, Shmeeda H, Horowitz AT, Zalipsky S. Tumor cell targeting of liposome-entrapped drugs with phospholipid-anchored folic acid-PEG conjugates. Adv Drug Deliv Rev. 2004;56(8):1177–1192. | ||
Cheng WW, Allen TM. Targeted delivery of anti-CD19 liposomal doxorubicin in B-cell lymphoma: a comparison of whole monoclonal antibody, Fab’ fragments and single chain Fv. J Control Release. 2008;126(1):50–58. | ||
Amin M, Badiee A, Jaafari MR. Improvement of pharmacokinetic and antitumor activity of pegylated liposomal doxorubicin by targeting with N-methylated cyclic RGD peptide in mice bearing C-26 colon carcinomas. Int J Pharm. 2013;458(2):324–333. | ||
Miao L, Newby JM, Lin CM, et al. The binding site barrier elicited by tumor-associated fibroblasts interferes disposition of nanoparticles in stroma-vessel type tumors. ACS Nano. 2016;10(10):9243–9258. | ||
Saga T, Neumann RD, Heya T, et al. Targeting cancer micrometastases with monoclonal antibodies: a binding-site barrier. Proc Natl Acad Sci U S A. 1995;92(19):8999–9003. | ||
Sapra P, Allen TM. Internalizing antibodies are necessary for improved therapeutic efficacy of antibody-targeted liposomal drugs. Cancer Res. 2002;62(24):7190–7194. | ||
Nielsen UB, Kirpotin DB, Pickering EM, et al. Therapeutic efficacy of anti-ErbB2 immunoliposomes targeted by a phage antibody selected for cellular endocytosis. Biochim Biophys Acta. 2002;1591(1–3):109–118. | ||
Manzoor AA, Lindner LH, Landon CD, et al. Overcoming limitations in nanoparticle drug delivery: triggered, intravascular release to improve drug penetration into tumors. Cancer Res. 2012;72(21):5566–5575. | ||
Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2–25. | ||
Prabhakar U, Maeda H, Jain RK, et al. Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res. 2013;73(8):2412–2417. |
Supplementary materials
 | Table S1 Linkage to scFv does not affect liposomal zeta potential or DXR encapsulation |
 | Figure S1 Anti-MAGE-A1/HLA-A1 TCR T-cells show activation after binding to MAGE-A1/HLA-A1+ melanoma cells. |
 | Figure S2 Immunoliposomes bind to cognate peptide. |
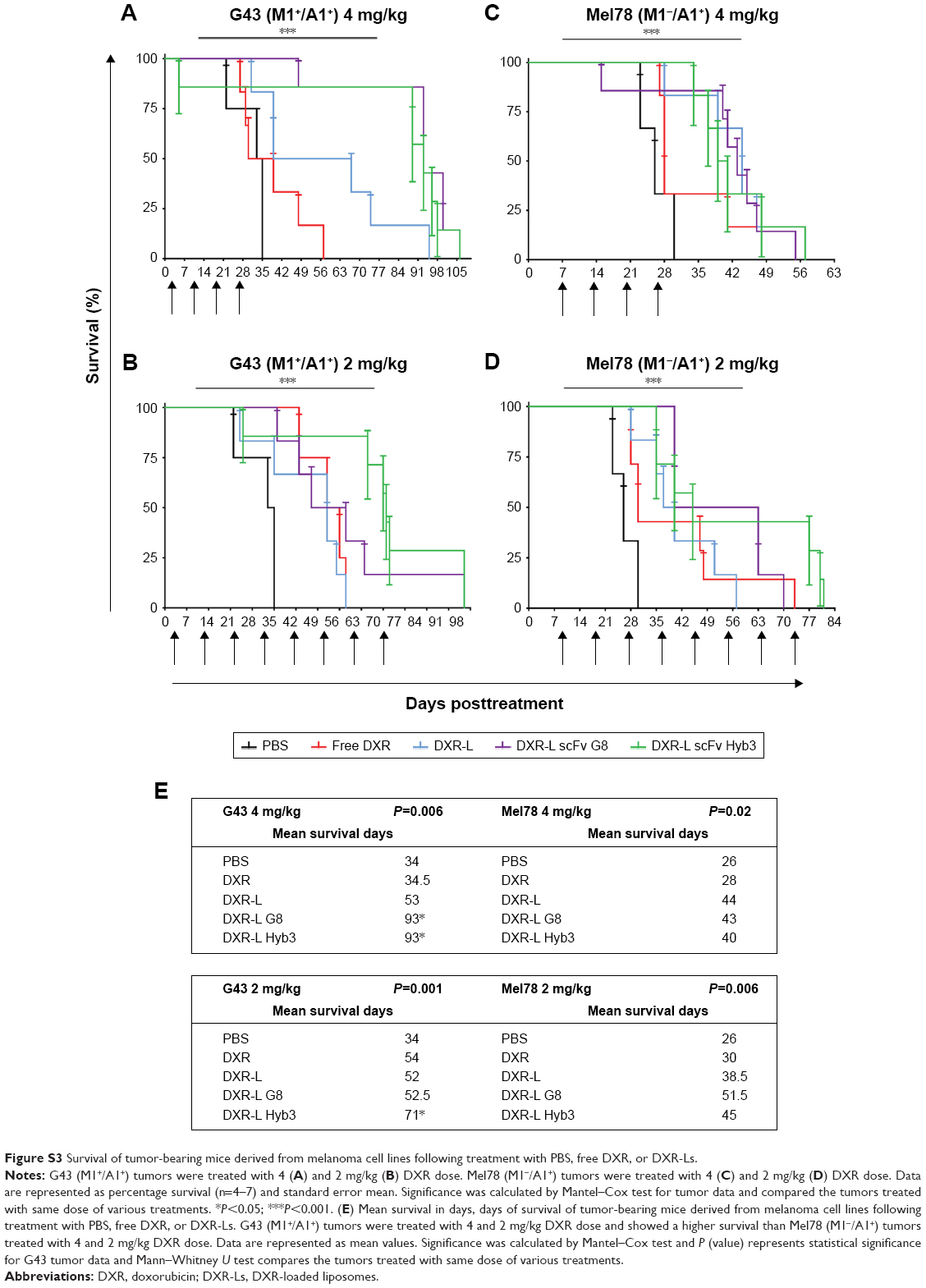 | Figure S3 Survival of tumor-bearing mice derived from melanoma cell lines following treatment with PBS, free DXR, or DXR-Ls. |
 | Figure S4 Weight loss of tumor-bearing mice derived from melanoma cell lines following treatment with PBS, free DXR, or DXR-Ls. G43 (M1+/A1+) tumors were treated with 4 (A) and 2 mg/kg (B) DXR-L dose. Mel78 (M1−/A1+) tumors were treated with 4 (C) and 2 mg/kg (D) DXR-L dose. Data are represented as mean weight index values (n=4–7) and SD. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.