")
Back to Journals » International Journal of Nanomedicine » Volume 16
Liposome-Coated Arsenic–Manganese Complex for Magnetic Resonance Imaging-Guided Synergistic Therapy Against Carcinoma
Authors Jin Z, Yi X, Yang J, Zhou M, Wu P, Yan G
Received 2 April 2021
Accepted for publication 13 May 2021
Published 1 June 2021 Volume 2021:16 Pages 3775—3788
DOI https://doi.org/10.2147/IJN.S313962
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Israel (Rudi) Rubinstein
Zhexiu Jin,1 Xue Yi,2 Jingjing Yang,1 Meili Zhou,1 Peifu Wu,1 Gen Yan3
1Department of Cardiovascular Medicine, The Second Affiliated Hospital of Xiamen Medical College, Xiamen, Fujian Province, 361023, People’s Republic of China; 2Department of Basic Medicine, Xiamen Medical College, Xiamen, Fujian Province, 361023, People’s Republic of China; 3Department of Radiology, The Second Affiliated Hospital of Xiamen Medical College, Xiamen, Fujian Province, 361023, People’s Republic of China
Correspondence: Gen Yan
Department of Radiology, The Second Affiliated Hospital of Xiamen Medical College, Xiamen, Fujian Province, 361023, People’s Republic of China
Tel +86 592 627 6910
Email [email protected]
Purpose: A liposome-coated arsenic–manganese complex, denoted as LP@MnAsx was constructed for the targeted delivery of arsenic trioxide (ATO) against carcinoma.
Methods: Arsenite, the prodrug of ATO, was encapsulated within a liposome via electrostatic interaction with the manganese ions. The as-prepared material was characterized with dynamic light scattering and transmission electron microscopy. The entrapment efficiency and drug loading of arsenic in the carrier were measured using inductively coupled plasma spectrometry. The in vitro release of arsenic was evaluated by using the dialysis bag method. Furthermore, the Fenton-like activity and in vitro cytodynamics research of LP@MnAsx were monitored in this work. And the cellular uptake study was used to investigate the in vitro entry mechanism. Furthermore, the cytotoxicity, cell apoptosis and cell cycle study were performed to evaluate the tumor-killing efficiency. Also, the pharmacokinetic and antitumor studies were investigated in HepG2 tumor-bearing nude mice.
Results: The as-prepared LP@MnAsx possessed a spherical morphology, uniformly distributed hydrodynamic diameter, and excellent drug-loading efficiency. LP@MnAsx displayed robust stability and sustained-release profile under physiological environments. LP@MnAsx could degrade with high sensitivity to the pH variation in the tumor microenvironment. As such, this could lead to a burst release profile of Mn2+ and arsenite to achieve a synergistic therapy of chemodynamic therapy and chemotherapy. When compared to the carrier-free arsenate, in vitro experiments revealed that LP@MnAsx exhibited enhanced cellular uptake and tumor-killing efficiency. LP@MnAsx also demonstrated significantly enhanced tumor-specific in vivo distribution of arsenic, prolonged systemic circulation lifetime, and increased accumulation at the tumor site.
Conclusion: Based on the experimental results, LP@MnAsx is an ideal arsenic-based nanodelivery system, whereby it can improve the non-specific distribution of NaAsO2 in vivo. Thus, this work can expand the research and application of arsenic trioxide against solid tumors.
Keywords: arsenic–manganese complex liposome, arsenic trioxide, pH-sensitive, hepatocellular carcinoma
Introduction
Primary carcinoma of the liver (PCL) is a type of cancer in the liver cells or intrahepatic bile duct cells, PCL is ranked as the fourth leading cause of cancer-associated deaths worldwide. Among them, hepatocellular carcinoma (HCC) accounts for the vast majority with a high incidence and mortality rate.1,2 Even though various clinically adopted HCC treatment methods have shown to be significant to ameliorate the disease, the use of chemotherapy is often associated with adverse side effects and limited therapeutic efficacy.3 Thus, due to the limitation of chemotherapy, it is necessary to explore alternative strategies for the treatment of HCC.
Arsenic trioxide (ATO) is an active ingredient in traditional Chinese medicine and it has been approved by the United States Food and Drug Administration (FDA) as a first-line antitumor drug against acute promyelocytic leukemia.4 Furthermore, ATO has also demonstrated excellent antitumor effects on various solid tumors, eg, liver cancer, glioma, and breast cancer, by inhibiting their proliferation and inducing cell apoptosis.5–10 Clinically, ATO is administered in the form of the arsenite ion (AsO2−). Even though ATO can exhibit excellent antitumor effects, the narrow therapeutic window, the lack of specificity in the body distribution, liver toxicity, neurotoxicity, and skin and nail pigmentation have significantly hindered the mass adoption of ATO in the clinical setting. Thus, to utilize ATO as high efficacy antitumor drug, it is necessary to realize targeted ATO delivery to the tumors so that it can achieve high bioavailability and reduced side effects.11–15
Manganese (Mn) is an essential element in the human body and it has been widely applied as a T1 contrast agent for cancer diagnosis due to its excellent biocompatibility.16 Moreover, Mn2+ can form insoluble complexes with multiple anionic groups, and their subsequent biodegradation can be triggered by the acidic environment of the tumor tissue.17 The released Mn2+ can then serve as a T1-weighted magnetic resonance imaging (MRI) contrast agent for real-time detection of the release and in vivo distribution of AsO2−.18 Also, Mn2+ can catalyze the tumor-abundant hydrogen peroxide (H2O2) to generate toxic hydroxyl radicals (∙OH) via Fenton-like reactions (chemodynamic therapy, CDT) to kill the cancer cells.19–22 Despite these advantages, arsenic–manganese complex exhibits limited circulation half-life and aggregation tendency, which can tremendously hinder its future application in the clinical setting.
The recent significant advances in nanotechnology have enabled more options in cancer diagnosis and treatment by leveraging the tumor microenvironment.23–25 Among various drug carriers, liposome, which consists of an amphiphilic lipid bilayer, is of significant importance due to its unique flexible structure and excellent biocompatibility. As such, liposomes have been used for targeted delivery of both hydrophilic and hydrophobic drugs.26,27 In particular, hybrid lipid nanoparticle with core-shell nanostructures, which consists of a functional inter-core and an outer lipid bilayer, has demonstrated numerous desirable features.28–31 Within this core-shell structure, the lipid layer can enhance the colloidal stability of the inter-core for prolonged circulation time and provide a barrier to prevent the cargo from diffusion. Thus, such a core-shell structure can achieve improved drug-loading yields and controlled release profiles, which can ultimately enhance the therapeutic efficacy of the treatment.32 Several liposomes have been approved by FDA to utilize as drug carriers, such as liposomal doxorubicin (Doxil). However, the way to directly load ATO into liposomes was limited by low encapsulation efficiency and rapid leakage of ATO, thereby causing systemic toxicity.33 Recently, metal-arsenic complex has been raised due to its high loading efficiency of ATO, but suffering from its low solubility in water. Previous attempts have shown that actively load ATO into liposome by utilizing metal ions to form a complex in the liposome was a successful strategy to improve the loading efficiency of ATO, enhance the stability of metal-arsenic complex, and prolong the in vivo circulation time in the meantime.34
Herein, liposomes were used to encapsulate the arsenic–manganese complex in this work. Based on the results, the liposome-encapsulated arsenic–manganese complex was able to exhibit significantly improved stability and distribution of arsenic in the tumor. The as-prepared liposomal nanoparticle-containing arsenic–manganese complex containing (LP@MnAsx) was able to demonstrate robust stability and sustained-release profile under physiological environments. LP@MnAsx was able to degrade with high sensitivity towards pH variation in the tumor microenvironment, and this led to a burst release profile of Mn2+ and arsenite for synergistic chemodynamic therapy and chemotherapy.18 Thus, this study on inorganic arsenic-based antitumor drugs may provide a novel approach for the treatment of HCC.
Materials and Methods
Materials
Sodium arsenite (97%) was purchased from West Asia Chemical Industry Co., Ltd. (Shanghai, China). The standard arsenic solution and standard manganese solution were purchased from Beijing Century Audiocodes Biological Technology Co., Ltd. (Beijing, China). Manganese acetate (98%) was bought from Alfa Aesar (Massachusetts, England). Hydrogenated soybean phospholipids (HSPC, 98%), cholesterol (Chol, 97%) and cholesterol-polyethylene glycol 2000 (Chol-PEG2000, 98%) were bought from Shanghai Dongsheng Biological Technology Co., Ltd. (Shanghai, China). 3,3ʹ,5,5ʹ-tetramethylbenzidine (TMB) was purchased from Solarbio Co., Ltd (Beijing, China). MTT, penicillin and streptomycin were bought from Gibco (New York, USA). PI kit and apoptosis kit were purchased from BD Biosciences (New Jersey, USA). Sodium azide (NaN3, 1%) was bought from Amresco (Washington, USA). TUNEL Assay Kit and Ki67 Cell Proliferation Kit were bought from Laval (Quebec, Canada). Nissl dye blue skies was purchased from Shanghai biological technology Co., LTD. (Shanghai, China). LO2 and HepG2 cells were obtained from Cell Bank of Chinese Academy of Sciences (Shanghai, China). Male BALB/c nude mice (6–8 weeks) and male SD rats (weighting 200 ± 15 g) were obtained from Xiamen Medical College. All animal experiments were performed under the protocols approved by the Administrative Committee of Laboratory Animals of Xiamen Medical College (Approval number: 2019008). All animal tests were approved by the Ethics Committee of Xiamen Medical College.
Preparation of LP@MnAsx
100 mg HSPC, 10 mg Chol, and 5 mg Chol-PEG2000 were dissolved in 10 mL ethanol:chloroform solution (1:1, v/v) in a round-bottom flask. The lipid film was prepared by evaporating the ethanol and chloroform, and then it was hydrated in 30 mg/mL manganese acetate solution (10 mL, pH 7.3) under sonication in a water bath using a probe-type sonicator for 15 min and 10 min, respectively. After which, the liposomes were then extruded through polycarbonate membranes, and then it was ultrafiltrated to remove the excess manganese acetate to obtain LP@Mn. LP@As was prepared using similar procedures, except that sodium arsenite solution (NaAsO2, 2 mg/mL, 10 mL) was used instead of manganese acetate solution. To prepare LP@MnAsx, NaAsO2 was firstly dissolved in deionized water, and then it was added to LP@Mn. After which, the mixture was reacted at 55°C for 30 min, and then it was poured into an ultrafiltration system. The collected concentrated solution was denoted as LP@MnAsx.
Preparation of Arsenic–Manganese Complex (AMO)
Chloroform was added to the as-obtained LP@MnAsx, and then it was sonicated with using a probe-type sonicator. The upper layer of water evaporated the chloroform, and then the deionized water was ultrasonically dissolved. Subsequently, it was repeatedly washed 10 times. The liquid was then ultrafiltered, centrifuged, and the upper solid was collected and freeze-dried to obtain AMO.
Characterization
The hydrodynamic diameter and zeta potential of liposomes were characterized via dynamic light scattering (DLS). The morphologies of the liposomes were observed under transmission electron microscopy (TEM). Inductively coupled plasma spectrometry (ICP, 6300, Thermo Electron Corporation, Waltham, MA, USA) was used to measure the entrapment efficiency (EE%) and drug loading (DL%) of the as-prepared liposomes.
Fenton-Like Activity of LP@MnAsx
TMB was used to monitor the generated ∙OH due to Mn2+ of LP@MnAs in the presence of H2O2. Briefly, 800 µL LP@MnAsx solution of different pH values were mixed with 100 µL H2O2 (10 mM) and 100 µL TMB (1 mM). After 10 min, a microplate reader was used to determine the absorbance between 400 to 800 nm.
In vitro Release Evaluation
The drug release characteristics of four formulations were investigated by dialysis bag method. The release medium was a phosphate buffer with pH values of 7.4, 6.8, and 5.5. The in vitro release rates of LP@As, LP@MnAsx, AMO, and sodium arsenite aqueous solution (NaAsO2-Sol) were investigated. The released As content was tested using ICP according to formula (1), and the release curve was obtained.

where Mn represents the cumulative release at time n, and M1 is the content in the sample.
Cellular Uptake Study
HepG2 cells were seeded in 6-well plates with a density of 5×105 cells/well, and it was allowed to incubate. The culture medium was replaced with NaAsO2-Sol, LP@As, and LP@MnAsx (all containing about 5 μg/L As) after 24 h of incubation. ICP was used to determine the intracellular drug concentrations after 2, 4, 6, 8, 12, and 24 h. Each experiment was performed in triplicate.
HepG2 cells were seeded in 6-well plates with a density of 1×106 cells/well, and then it was incubated for 24 h. It was pretreated with 5 μg/mL sodium azide (NaN3) without serum for 30 min, suctioned, and washed twice with PBS. The other experimental methods were similar to those used for the cytodynamics research.
In vitro Cytotoxicity
Firstly, approximately 8×103 HepG2 and LO2 cells were seeded into a 96-well plate. After 24 h of incubation, the medium was replaced with NaAsO2-Sol, LP@As, and LP@MnAsx contained fresh medium with different concentrations. After 24 h of incubation, the medium was replaced with 180 µL fresh medium and 20 µL MTT (0.5 mg/mL). After another 4 h of incubation, 150 µL DMSO was added and incubated for 10 min. The absorbance was subsequently determined by measuring the 490 nm absorption using a microplate reader (Synergy TM2, BIOTEK, Winooski, VT, USA). The cell viability was calculated according to equation (2).
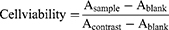
Intracellular Reactive Oxygen Species (ROS) Detection
The cell-permeable dye DCFH-DA was used to detect ROS production, whereby the ROS could oxidize the non-fluorescent dye to green fluorescent of DCF. In a typical setup, 2×105 HepG2 cells were seeded and incubated overnight. Subsequently, it was treated with LP@MnAsx for 6 h. Then, the culture medium was then replaced with DCFH-DA (10 µM), and then incubated at 37°C for 30 min. Finally, the sample was observed under a confocal laser scanning microscope (CLSM, Olympus, Tokyo, Japan). And the quantitative analysis of DCF with different treatments was assessed via flow cytometry (Guava Easycyte, Merck Millipore, Darmstadt, Germany).
Cell Apoptosis Study
HepG2 cells were seeded with a density of 5×105 cells/well. After 24 h of incubation, each plate was treated with the serum-free medium of NaAsO2-Sol, LP@As, and LP@MnAsx. Then, the cells were collected with Annexin V binding buffer for the Annexin V-FITC/PI staining and they were assessed via flow cytometry.
Cell Cycle Study
HepG2 cells were seeded with a density of 5×105 cells/well, and then it was cultured with medium of NaAsO2-Sol, LP@As and LP@MnAsx for 24 h. After which, the cells were collected and fixed with 70% ethanol for 12 h. Subsequently, it was stained with PI solution and assessed via flow cytometry.
Pharmacokinetic Study
Sprague Dawley (SD) rats were randomly divided into three groups (n = 5), and they were intravenously injected with NaAsO2-Sol, LP@As, and LP@MnAsx (As: 0.6 mg/kg). At predetermined time intervals, blood samples were obtained from the orbital vein of the SD rats. The As content in the plasma was tested using inductively coupled plasma mass spectrometer (ICP-MS), and pharmacokinetic parameters were analyzed using the PK Solver pharmacokinetic software.
Biodistribution
A total of 5×106 HepG2 cells were injected into the back of nude mice. When the tumor volume increased to 50–100 mm3, 15 tumor-bearing nude mice were randomly divided into three groups, ie, NaAsO2-Sol group, LP@As group, and LP@MnAsx group (n = 5). The mice in each group were sacrificed at 24 h after a single injection of drugs. The heart, left lobe of the liver, spleen, left lung, left kidney, brain, abdominal skin, left leg bone, and tumor tissues were collected. ICP was used to determine the drug concentration. The tissue distribution map was plotted with the percentage of tissue per gram of injection volume (% ID/g) as the ordinate.
In vitro/vivo T1-Weighted MRI Imaging Evaluation
LP@MnAsx with various Mn concentrations, ie, 0, 0.05, 0.1, 0.2, 0.4, and 0.5 mM, were dispersed in buffer solutions with various pH values for 24 h. The MRI images were then obtained via 1.5-T MRI system.
For in vivo MRI imaging, 0.5 mL LP@MnAsx was intravenously administrated at Mn concentration of 3 mg/kg. Subsequently, the MRI images were recorded at 1, 2, 4, 8, and 12 h post-injection using the 1.5-T unit MRI system.
Pharmacodynamics Evaluation
Twenty tumor-bearing nude mice were randomly divided into four groups, ie, saline, NaAsO2-Sol, LP@As, and LP@MnAsx groups (n = 5). Normal saline or medicine (approximately 0.6 As mg/kg) was injected intravenously every day. The tumor volume and body weight of nude mice were recorded every other day. The tumor volume was calculated using Formula (3). On the 15th day, the tumors were collected to weigh.

Pathological Analysis
To further prove the ROS generation in vivo, 12 tumor-bearing nude mice were intravenously injected with saline, NaAsO2-Sol, LP@As, and LP@MnAsx, respectively (n = 3). At 24 h post injection, the tumor tissues were collected and co-stained with DCFH-DA and DAPI. The generation of ROS within the tumor cells was observed via CLSM.
On Day 15, the nude mice were sacrificed, and the heart, liver, spleen, lung, kidney, hippocampus, and striatum tissue were collected and fixed with 10% formalin to prepare the histological sections. These tissues were then stained with H&E, and histological images were obtained using an optical microscope (AXIO SCOPE.A1, Zeiss, Jena, Germany).
The tumor tissues of nude mice from each group were subjected to TUNEL and Ki67 staining, and the tissue morphology was observed under a microscope.
The brain tissue of nude mice was fixed with 10% paraformaldehyde, stained with Nissl, and then observed under a microscope and imaged.
Statistical Analysis
Data were presented as mean or mean ± standard deviation (SD). Statistical significance was evaluated by SPSS 22. P < 0.05 indicated a statistical difference, while P < 0.01 was indicated as a significant difference.
Results and Discussion
Characterization of LP@MnAsx
As depicted in Figure 1, the LP@MnAsx was designed to entrap arsenic by interacting with Mn2+ to realize an MRI-guided synergistic therapy of chemodynamic therapy and chemotherapy. The LP@MnAsx was prepared via a two-step method: (1) preparation of manganese acetate-containing liposome using the filming-rehydration method and (2) the formation of the arsenic–manganese complex within the interior hydrophilic space drived by ion gradient. The interactions among the arsenic–manganese complex, including the electrostatic interaction and dipole–dipole interaction, enable the sustained release of the entrapped cargo, and the liposome improves the stability of the as-prepared nanoparticles and it can also facilitate their accumulation at the tumor site for enhanced therapeutic efficacy.16
 |
Figure 1 Schematic illustration of the synthesis of LP@MnAsx and the subsequent cellular uptake and drug release. |
As shown in the TEM image (Figure 2A), LP@As, LP@Mn, and LP@MnAsx possessed spherical morphology and they were uniformly dispersed with average diameters of around 120 nm. Such a result is consistent with the results obtained by DLS (Figure 2B). When the liposome was removed from LP@MnAsx by subjecting it to chloroform and ultrasonication, the interior core of the nanoparticle was observed by TEM, suggesting the successful formation of AMO within the liposome. According to the ICP results, the As drug-loading rate and encapsulation efficiency in the LP@MnAsx were remarkably high at 4.68 ± 0.82% and 92.96 ± 5.35%, respectively. Furthermore, LP@MnAsx showed excellent colloidal stability over 30 days with negligible changes in the hydrodynamic diameter and zeta potential (Figure 2C), further illustrating the excellent stability of LP@MnAsx.
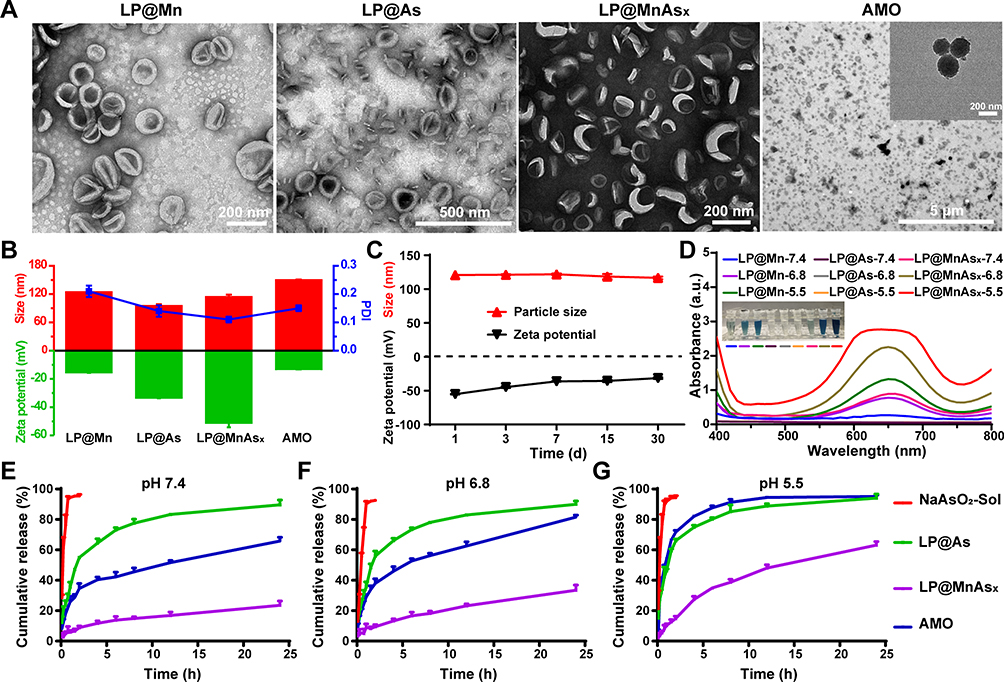 |
Figure 2 Characterization of LP@MnAsx. Notes: (A) TEM images and (B) hydrodynamic diameter, PDI, and Zeta potential of LP@Mn, LP@As, LP@MnAsx, and AMO. (C) Hydrodynamic diameter and Zeta potential stability of LP@MnAsx. (D) UV-Vis spectra and the color change of aqueous TMB of LP@MnAsx at varying pH values. Cumulative releases of NaAsO2-Sol, LP@As, LP@MnAsx and AMO at pH 7.4 (E), 6.8 (F), and 5.5 (G). |
Fenton-Like Activity of LP@MnAsx
To investigate the Fenton-like activity of Mn2+, TMB was selected as a signal for ∙OH. As shown in Figure 2D, the absorbance of TMB in LP@Mn and LP@MnAsx reaction system increased significantly as compared to that in LP@As. Such a result indicates the generation of ∙OH by the reaction between Mn2+ and H2O2. Especially, a greater extent increase of TMB was induced of LP@Mn and LP@MnAsx under pH 5.5 environment, which demonstrated that the Fenton-like reaction was more evident with the increasing acidity of PBS, displaying the pH-dependent Fenton-like reaction activity. Also, the same results could be clearly observed according to the corresponding digital images of the insert photograph in Figure 2D.
In vitro Release Evaluation
The release profiles of the four formulations, ie, NaAsO2-Sol, LP@As, LP@MnAsx, and AMO, were investigated under various pH values, ie, 7.4, 6.8, or 5.5, based on a dialysis method. As shown in Figure 2E-G, more than 95% of As was released from NaAsO2-Sol within 1 h under all pH conditions. Notably, after liposome-coating, there was a slow release of As without pH responsiveness, whereby approximately 80% As was released from LP@As after 24 h under all pH conditions. However, the LP@MnAsx demonstrated a sustained-release profile under physiological conditions (pH = 7.4) with a cumulative release of 22.69 ± 0.76% within 24 h. Such a sustained-release profile could be attributed to the synergistic effects of the interactions among the arsenic–manganese complex and the diffusional barrier provided by the liposome (Figure 2E). Upon incubation in an acidic environment, LP@MnAsx exhibited an obvious pH-dependent drug release behavior, whereby cumulative release amounts of 33.47 ± 3.43% and 63.15 ± 1.90% of As were attained at pH of 6.8 and 5.5, respectively (Figure 2F-G). The biodegradation of arsenic–manganese complex could be triggered by the acidic environment of the tumor tissue,17 which could be attributed to the dissociation of the AMO triggered by decreased pH, and the coordination between manganese and arsenic would be weakened in the meantime.
Cellular Uptake Study
HepG2 cells were incubated with NaAsO2, LP@As, or LP@MnAsx with or without NaN3 for 24 h. After which, the intracellular As concentration was determined at predetermined time intervals to investigate the internalization dynamics and pathway of various formulations. NaN3 was used as an endocytosis inhibitor to evaluate the depletion of cellular ATP.35 As shown in Figure 3A-C, NaAsO2 demonstrated a rapid fast entry into HepG2 cells, which was free from the influence of NaN3 (P > 0.05). LP@As and LP@MnAsx showed a time-dependent cellular uptake, and obviously enhanced the cellular uptake of As (P < 0.05). While with the addition of NaN3, the cellular uptake of LP@As and LP@MnAsx was significantly reduced that by more than a half. This result indicates the energy-consuming endocytosis-mediated internalization pathway.36,37 In conclusion, the cellular uptake of LP@As and LP@MnAsx would be significantly inhibited by the addition of NaN3, suggesting that their endocytosis way was ATP-dependent (P < 0.05).
 |
Figure 3 The cellular uptake, cytotoxicity, CDT activity, cell apoptosis and cell cycle of LP@MnAsx. Notes: As concentration in HepG2 cells after treatment with NaAsO2, LP@As, or LP@MnAsx in the absence (A) and presence (B) of NaN3 pre-treatment. (C) As concentration after treatment with NaAsO2-Sol, LP@As, or LP@MnAsx for 24 h for the normal group and NaN3 pre-treated groups. Cell viability of LO2 (D) and HepG2 (E) cells and IC50 value of LO2 and HepG2 (F) after treated with NaAsO2-Sol, LP@As, or LP@MnAsx. (G) ROS detection of HepG2 cells after incubating with LP@Mn, LP@As or LP@MnAsx. (H) The quantitative analysis by flow cytometry about DCF after different treatments. Cell apoptosis (I, J) and cell cycle (K, L) of HepG2 cells treated with blank medium, NaAsO2-Sol, LP@As, or LP@MnAsx. Data are expressed as means ± SD (n = 3). *P < 0.05; **P < 0.01. Abbreviations: CDT, chemodynamic therapy. |
In vitro Cytotoxicity
The cytotoxicity of these formulations on HepG2 and normal liver cells (LO2) cells was assessed via MTT method. As shown in Figure 3D-E, the viabilities of HepG2 and LO2 cells decreased in a concentration-dependent manner. Based on the IC50 value (Figure 3F), HepG2 cells were more sensitive to each of the formulation as compared to LO2 cells. Thus, the damage to LO2 could be avoided, and the as-prepared formulations could facilitate the subsequent treatment against tumors. Among all samples, LP@MnAsx exhibited the best tumor-inhibiting effect on HepG2 cells with an IC50 of 1.37 ± 0.47 μg/mL, which was remarkably lower than that of LP@As (3.15 ± 2.29 μg/mL) and NaAsO2-Sol (3.83 ± 0.85 μg/mL), indicating the synergistic therapy of chemotherapy by As and CDT by Mn2+.
Intracellular ROS Detection
DCFH-DA probes were used to detect intracellular ROS production. As shown in Figure 3G, the green fluorescence of DCF was observed for the groups with LP@Mn and LP@MnAsx due to the existence of Mn2+ when compared to the control group or LP@As. Moreover, after incubating with LP@MnAsx, HepG2 cells exhibited morphological alteration, which further illustrated the cytotoxicity of As towards tumor cells. And the results of quantitative analysis via flow cytometry was in line with CLSM (Figure 3H).
Cell Apoptosis and Cell Cycle Study
The results of the HepG2 apoptotic rates after being treated with each formulation are shown in Figure 3I-J. Compared with the control group, each treatment exerted significant effects on the apoptosis rate (P < 0.01). The group treated with LP@MnAsx exhibited the highest apoptotic rate of 42.49 ± 3.36%, which could be attributed to the combined effect of the toxic hydroxyl radicals generated by Mn2+ and the apoptosis-inducing effect mediated by ATO. According to the cell cycle analysis result (Figure 3K-L), the Control, NaAsO2-Sol, LP@As, and LP@MnAsx groups arrested 19.60 ± 1.58%, 46.43 ± 0.94%, 47.73 ± 1.31%, and 69.00 ± 0.90% of HepG2 cells at the 4N phase, respectively. Such results obtained from these groups were significantly different from that obtained from the blank group (P < 0.01). Thus, this can reasonably explain the achievement of apoptosis mainly by shortening the 2N phase and prolonging the 4N phase.
Pharmacokinetic Study
A plasma pharmacokinetic study was performed to investigate the pharmacokinetic behavior of various formulations. NaAsO2-Sol, LP@As, and LP@MnAsx were intravenously administered at the As dosage of 0.6 mg/kg, and the blood was collected at pre-determined time intervals for ICP-MS analyses. The As concentration–time curve and main parameters are shown in Figure 4A and Table 1, respectively. It could be observed that all the as-prepared formulations conformed to the two-compartment model. The NaAsO2-Sol was rapidly removed from the body with a short half circulation time of 14.82 ± 2.34 h, and this was shorter than those of LP@As (16.72 ± 6.59 h) and LP@MnAsx (19.54 ± 4.54 h). When compared to the NaAsO2-Sol, the area under curve (AUC) of LP@As and LP@MnAsx was increased by 1.69 and 2.06 times, and the mean residence time (MRT) increased by 23.4% and 49.0%, respectively. Such a result indicates that the liposome encapsulation could improve the plasma circulation behavior. When compared to LP@As, LP@MnAsx further improved the t1/2β and AUC0-t, and it decreased the MRT, which could be attributed to the formation of AMO for the sustained drug release. Thus, based on the collective results, LP@MnAsx displayed superior pharmacokinetic behavior of prolonged circulation time and decreased elimination from the body, benefiting from the liposome-coating.
 |
Table 1 Main Parameters of the SD Rats After Injected with of NaAsO2, LP@as, or LP@MnAsx Intravenously (n = 5) |
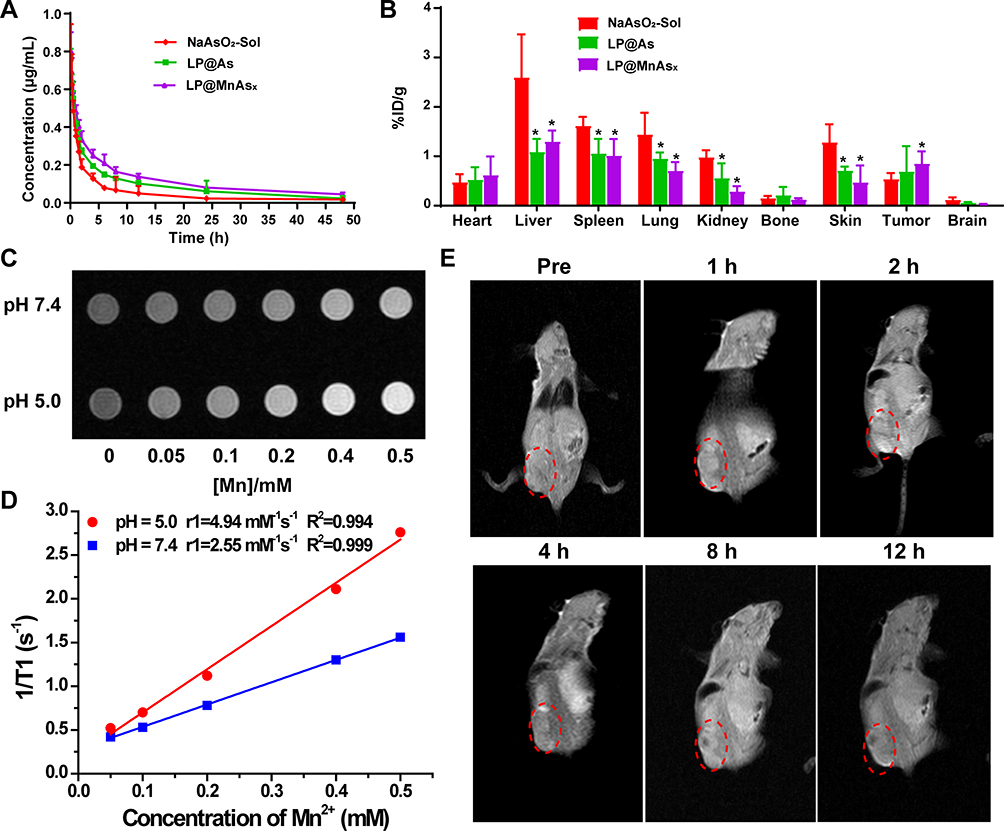 |
Figure 4 In vivo distribution and T1-weighted MR imaging of tumor-bearing nude mice. Notes: (A) The concentration-time curve and (B) tissue distribution of As after intravenous injection of NaAsO2-Sol, LP@As, or LP@MnAsx (n = 5). *P < 0.05. (C) In vitro T1-weighted MR imaging and (D) T1 relaxation rates of LP@MnAsx at various concentrations after incubating at different pH conditions. (E) In vivo T1-weighted MR imaging of tumor-bearing nude mice after being injected with LP@MnAsx intravenously. The tumor was marked by red dotted circles. |
Biodistribution
To evaluate the biodistribution of various formulations, HepG2 xenograft tumor model was constructed. NaAsO2-Sol, LP@As, and LP@MnAsx were intravenously administered, and the tissues of the heart, liver, spleen, lung, kidney, brain, abdominal skin, bone, and tumor were harvested for ICP-MS examination at 24 h post-administration. As shown in Figure 4B, NaAsO2-Sol was mainly distributed in the liver, spleen, lung, and skin, with only 0.54 ± 0.12%ID/g at the tumor site. After being encapsulated by liposomes, the liver, spleen, lung, and skin showed significantly reduced arsenic distribution, while there was increased arsenic distribution in the tumor, which indicated an improved arsenic distribution in various tissues. The amount LP@MnAsx distribution could be arranged in the following decreasing order: liver > spleen > tumor > lung > heart > skin > bone > brain. Meanwhile, when compared to other groups, the LP@MnAsx exhibited the highest accumulation at the tumor site and a decreased distribution in other major organs, which indicated superior tumor targeting ability and improved biodistribution. And the distribution in liver, spleen and lung was due to the reticuloendothelial system (RES) effect.
In vitro/vivo T1-Weighted MRI Imaging Evaluation
Due to the dissociation of LP@MnAsx triggered by an acidic environment, the released Mn2+ can serve as a contrast agent for T1-weighted MRI contrast agent. With the increased concentration, the T1-weighted MR images of LP@MnAsx at pH 5.0 revealed a greater contrasting effect as compared to those at pH 7.4 (Figure 4C). Furthermore, the r1 relaxivity of LP@MnAsx significantly increased from 2.55 mM−1s−1 due to the accelerated release of Mn2+ (Figure 4D). Subsequently, T1-weighted MR imaging was monitored at selected times after being intravenously injected with LP@MnAsx. An obvious high contrasting effect could be observed at the tumor site, whereby it continuously increased and peaked at 2 h post administration, and then gradually decreased. This result suggests that LP@MnAsx was able to respond sensitively to the tumor microenvironment (Figure 4E).
Pharmacodynamics Evaluation
To assess the in vivo therapeutic efficacy, 20 HepG2 tumor-bearing mice were randomly divided into four groups, ie, treated with saline, NaAsO2-Sol, LP@As, and LP@MnAsx. During the 14 days of treatment, tumor volume and body weight of mice were recorded each day. Based on the results, the average tumor volumes of the groups treated with saline, NaAsO2-Sol, LP@As, and LP@MnAsx were 1192.34 ± 482.21 mm3, 648.53 ± 198.62 mm3, 499.67 ± 116.72 mm3, and 316.20 ± 110.45 mm3, respectively, showing that NaAsO2-Sol, LP@As and LP@MnAsx have a difference degrees in the inhibition of the growth of tumor (Figure 5A-C). Compared with the group of NaAsO2-Sol, LP@As and LP@MnAsx could significantly inhibit the tumor, which might be owing to the tumor-targeting ability of liposome and the synergistic therapy of chemotherapy and CDT (P < 0.01). In addition, the body weight of the mice treated with NaAsO2-Sol group was significantly lower than that of the saline group, indicating systemic cytotoxicity caused by the non-specific distribution of NaAsO2. In contrast, the body weight of the mice treated with LP@As and LP@MnAsx kept increased continuously, which suggests the attenuation of adverse side effects, showing the good biosafety of LP@As and LP@MnAsx (Figure 5D).
 |
Figure 5 In vivo anti-tumor evaluation. Notes: Tumor volume (A), tumor weight (B), digital photos of tumors (C) and body weight (D) of tumor-bearing nude mice treated with saline, NaAsO2-Sol, LP@As, or LP@MnAsx (n = 5). (E) ROS generation in tumor tissues collected from different groups at 24 h post injection and co-stained with DCFH-DA (green) and DAPI (blue). (F) Tunel and Ki67 staining of the tumor, and Nissl images of the hippocampus after saline, NaAsO2-Sol, LP@As, or LP@MnAsx treatment. (G) IOD values of Tunel and Ki67, and the number of Nissl bodies (n = 3). *P < 0.05; **P < 0.01. |
Pathological Analysis
To further display the in vivo ROS generation, the immuno-histochemical analysis of tumor tissues were investigated by using DCFH-DA as the ROS probe. As shown in Figure 5E, the obvious green fluorescence could be seen in the tumor tissues after LP@MnAsx treatment, suggesting a large amount of ROS generation in vivo.
Ki67 and TUNEL staining were used to perform the histological evaluation and statistical analysis so as to determine the tumor cell apoptosis caused by treatment, whereby saline group was used as the control (Figure 5F-G). When compared to other groups, the group treated with LP@MnAsx resulted in the largest area of apoptotic cells, and therefore it achieved the best proliferation inhibiting effect. When considering the side effects of nerve cytotoxicity caused by arsenic treatment, Nissl staining was used to observe the damage inflicted onto the nerve cells. Based on the results, the number of Nissl bodies in the hippocampus of the LP@As and LP@MnAsx groups were significantly less than that in NaAsO2-Sol groups (P < 0.05). Furthermore, morphological alterations could be detected, which were indicative of severe nerve cytotoxicity. Meanwhile, LP@MnAsx showed attenuated damage to the nerve cells with a comparable number of Nissl bodies (P < 0.01), and this result is consistent with the biodistribution results. According to the H&E staining result, administrating of NaAsO2-Sol, LP@As, and LP@MnAsx to the mice did not lead to obvious damage to the major organs relative to the control group (Figure 6).
 |
Figure 6 H&E staining image of major organs collected from nude mice after treated with NaAsO2-Sol, LP@As, and LP@MnAsx. |
Conclusion
In summary, the LP@MnAsx was successfully developed for the carcinoma-targeted delivery of the arsenic–manganese complex via magnetic resonance imaging-guided synergistic therapy of chemodynamic therapy and chemotherapy. The enhanced therapeutic efficacy exhibited by the LP@MnAsx was realized by the pH-sensitive drug release, prolonged circulation time, and improved tumor accumulation. Hence, based on the collective results, the use of the LP@MnAsx is a promising strategy for carcinoma treatment, overcoming the shortage of ATO, and propelling the clinical translation process of arsenic trioxide based formulation.
Acknowledgments
This study was supported by the Science and Technology Program of Fujian Province of China (No. 2017D017); The Joint Funds for the Health and Education of Fujian Province, China (No. 2019-WJ-31); Institute of Respiratory Diseases, Xiamen Medical College (No. HXJB-06, 15).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Fu X, Liang Q-R, Luo R-G, et al. An arsenic trioxide nanoparticle prodrug (ATONP) potentiates a therapeutic effect on an aggressive hepatocellular carcinoma model via enhancement of intratumoral arsenic accumulation and disturbance of the tumor microenvironment. J Mater Chem B. 2019;7(19):3088–3099. doi:10.1039/C9TB00349E
2. Shi ZY, Chu CC, Zhang Y, et al. Self-assembled metal-organic nanoparticles for multimodal imaging-guided photothermal therapy of hepatocellular carcinoma. J Biomed Nanotechnol. 2018;14:1934–1943.
3. Kim DW, Talati C, Kim R. Hepatocellular carcinoma (HCC): beyond sorafenib-chemotherapy. J Gastrointest Oncol. 2017;8:256–265.
4. Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood. 2008;111:2505–2515.
5. Shen L, Zhang GS, Lou ZH, et al. Cryptotanshinone enhances the effect of Arsenic trioxide in treating liver cancer cell by inducing apoptosis through downregulating phosphorylated-STAT3 in vitro and in vivo. BMC Complem Altern M. 2017;17:106.
6. Sun YY, Wang C, Wang LG, et al. Arsenic trioxide induces apoptosis and the formation of reactive oxygen species in rat glioma cells. Cell Mol Biol Lett. 2018;23:13.
7. Du J, Zhou NN, Liu HX, et al. Arsenic induces functional re-expression of estrogen receptor alpha by demethylation of DNA in estrogen receptor-negative human breast cancer. PLoS One. 2012;7:e35957.
8. Zhang L, Zhang ZW, Mason RP, et al. Convertible MRI contrast: sensing the delivery and release of anti-glioma nano-drugs. Sci Rep. 2015;5:09874.
9. Tao JY, Fei WD, Tang HX, et al. Angiopep-2-conjugated “core-shell” hybrid nanovehicles for targeted and pH-triggered delivery of arsenic trioxide into glioma. Mol Pharmaceut. 2019;16:786–797.
10. Lu YP, Han SP, Zheng HY, et al. A novel RGDyC/PEG co-modified PAMAM dendrimer-loaded arsenic trioxide of glioma targeting delivery system. Int J Nanomed. 2018;13:5937–5952.
11. Alamolhodaei NS, Shirani K, Karimi G. Arsenic cardiotoxicity: an overview. Environ Toxicol Phar. 2015;40:1005–1014.
12. Zhang SY, Zhang B, Wang M, et al. Calcium homeostasis and endoplasmic reticulum stress are involved in Salvianolic acid B-offered protection against cardiac toxicity of arsenic trioxide. Oncotarget. 2017;8:97384–97393.
13. Pace C, Dagda R, Angermann J. Antioxidants protect against arsenic induced mitochondrial cardio-toxicity. Toxics. 2017;5:38.
14. Mizrachi A, Shamay Y, Shah J, et al. Tumour-specific PI3K inhibition via nanoparticle-targeted delivery in head and neck squamous cell carcinoma. Nat Commun. 2017;8:14292.
15. Shi XW, Ma R, Lu YP, et al. iRGD and TGN co-modified PAMAM for multi-targeted delivery of ATO to gliomas. Biochem Bioph Res Co. 2020;527:117–123.
16. Zhao ZH, Wang XM, Zhang ZJ, et al. Real-time monitoring of arsenic trioxide release and delivery by activatable T-1 imaging. ACS Nano. 2015;9:2749–2759.
17. Xu HW, Li CQ, Wei YH, et al. Angiopep-2-modified calcium arsenite-loaded liposomes for targeted and pH-responsive delivery for anti-glioma therapy. Biochem Bioph Res Co. 2021;551:14–20.
18. Zhang K, Lin HY, Mao JJ, et al. An extracellular pH-driven targeted multifunctional manganese arsenite delivery system for tumor imaging and therapy. Biomater Sci. 2019;7:2480–2490.
19. Fu LH, Hu YR, Qi C, et al. Biodegradable manganese-doped calcium phosphate nanotheranostics for traceable cascade reaction-enhanced anti-tumor therapy. ACS Nano. 2019;13:13985–13994.
20. Wang P, Liang C, Zhu JW, et al. Manganese-based nanoplatform as metal ion-enhanced ROS generator for combined chemodynamic/photodynamic therapy. ACS Appl Mater Inter. 2019;11:41140–41147.
21. Tang ZM, Liu YY, He MY, et al. Chemodynamic therapy: tumour microenvironment-mediated Fenton and Fenton-like reactions. Angew Chem Int Edit. 2019;58:946–956.
22. Tang HX, Li CQ, Zhang Y, et al. Targeted manganese doped silica nano GSH-cleaner for treatment of liver cancer by destroying the intracellular redox homeostasis. Theranostics. 2020;10:9865–9887.
23. Chaturvedi VK, Singh A, Singh VK, et al. Cancer nanotechnology: a new revolution for cancer diagnosis and therapy. Curr Drug Metab. 2019;20:416–429.
24. Piao JG, Gao F, Yang LH. Acid-responsive therapeutic polymer for prolonging nanoparticle circulation lifetime and destroying drug-resistant tumors. ACS Appl Mater Inter. 2016;8:936–944.
25. Piao JG, Gao F, Li YN, et al. pH-sensitive zwitterionic coating of gold nanocages improves tumor targeting and photothermal treatment efficacy. Nano Res. 2018;11:3193–3204.
26. Jain A, Tiwari A, Verma A, et al. Combination cancer therapy using multifunctional liposomes. Crit Rev Ther Drug. 2020;37:105–134.
27. Khan AA, Allemailem KS, Almatroodi SA, et al. Recent strategies towards the surface modification of liposomes: an innovative approach for different clinical applications. 3 Biotech. 2020;10:163.
28. Yan W, Leung SSY, To KKW. Updates on the use of liposomes for active tumor targeting in cancer therapy. Nanomedicine. 2020;15:303–318.
29. Tan SW, Li X, Guo YJ, et al. Lipid-enveloped hybrid nanoparticles for drug delivery. Nanoscale. 2013;5:860–872.
30. Kumar R, Mondal K, Panda PK, et al. Core-shell nanostructures: perspectives towards drug delivery applications. J Mater Chem B. 2020;8:8992–9027.
31. Mandal B, Bhattacharjee H, Mittal N, et al. Core-shell-type lipid-polymer hybrid nanoparticles as a drug delivery platform. Nanomed-Nanotechnol. 2013;9:474–491.
32. Deodhar S, Dash AK. Long circulating liposomes: challenges and opportunities. Ther Deliv. 2018;9:857–872.
33. Zhang L, Zhang ZW, Mason RP, et al. Convertible MRI contrast: sensing the delivery and release of anti-glioma nano-drugs. Sci Rep. 2015;5:e09874.
34. Chen HM, MacDonald RC, Li SY, et al. Lipid Encapsulation of arsenic trioxide attenuates cytotoxicity and allows for controlled anticancer drug release. J Am Chem Soc. 2006;128:13348–13349.
35. Hu X, Yang FF, Liu CY, et al. In vitro uptake and transport studies of PEG-PLGA polymeric micelles in respiratory epithelial cells. Eur J Pharm Biopharm. 2017;114:29–37.
36. Ozawa M, Asano A. The preparation of cell fusion-inducing proteoliposomes from purified glycoproteins of HVJ (Sendai virus) and chemically defined lipids. J Biol Chem. 1981;256:5954–5956.
37. Xiang GY, Wu J, Lu YH, et al. Synthesis and evaluation of a novel ligand for folate-mediated targeting liposomes. Int J Pharmaceut. 2008;356:29–36.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.