Back to Journals » International Journal of Nanomedicine » Volume 14
Lipo-PEG-PEI complex as an intracellular transporter for protein therapeutics
Authors Lin YL ![]() , Chen CH, Liu YK, Huang TH
, Chen CH, Liu YK, Huang TH ![]() , Tsai NM, Tzou SC
, Tsai NM, Tzou SC ![]() , Liao KW
, Liao KW
Received 27 September 2018
Accepted for publication 17 January 2019
Published 13 February 2019 Volume 2019:14 Pages 1119—1130
DOI https://doi.org/10.2147/IJN.S188970
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Yu-Ling Lin,1,* Chia-Hung Chen,2,* Yen-Ku Liu,2 Tse-Hung Huang,3–6,* Nu-Man Tsai,7,8 Shey-Cherng Tzou,2,9 Kuang-Wen Liao2,9–11
1Agricultural Biotechnology Research Center, Academia Sinica, Taipei, Taiwan, ROC; 2Department of Biological Science and Technology, National Chiao Tung University, Hsinchu, Taiwan, ROC; 3Department of Traditional Chinese Medicine, Chang Gung Memorial Hospital, Keelung, Taiwan, ROC; 4School of Traditional Chinese Medicine, Chang Gung University, Taoyuan, Taiwan, ROC; 5Graduate Institute of Health Industry Technology, Chang Gung University of Science and Technology, Taoyuan, Taiwan, ROC; 6School of Nursing, National Taipei University of Nursing and Health Sciences, Taipei, Taiwan, ROC; 7School of Medical Laboratory and Biotechnology, Chung Shan Medical University; 8Department of Pathology and Clinical Laboratory, Chung Shan Medical University Hospital, Taichung, Taiwan, ROC; 9Institute of Molecular Medicine and Bioengineering, National Chiao Tung University, Hsinchu, Taiwan, ROC; 10Graduate Institute of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan, ROC; 11Center for Intelligent Drug Systems and Smart Bio-devices, National Chiao Tung University, Hsinchu, Taiwan, ROC
*These authors contributed equally to this work
Background: Protein or peptide drugs are emerging therapeutics for treating human diseases. However, current protein drugs are typically limited to acting on extracellular/cell membrane components associated with the diseases, while intracellular delivery of recombinant proteins replaces or replenishes faulty/missing proteins and remains inadequate. In this study, we developed a convenient and efficient intracellular protein delivery vehicle.
Materials and methods: A cationic liposomal polyethylenimine and polyethylene glycol complex (LPPC) was developed to noncovalently capture proteins for protein transfer into cells via endocytosis. β-glucuronidase (βG) was used in vitro and in vivo as a model enzyme to demonstrate the enzymatic activity of the intracellular transport of a protein.
Results: The endocytosed protein/LPPC complexes escaped from lysosomes, and the bound protein dissociated from LPPC in the cytosol. The enzymatic activity of βG was well preserved after intracellular delivery in vitro and in vivo.
Conclusion: Using LPPC as an intracellular protein transporter for protein therapeutics, we illustrated that LPPC may be an effective and convenient tool for studying diseases and developing therapeutics.
Keywords: liposomal polyethylenimine and polyethylene glycol complex, LPPC, intracellular protein delivery, endocytosis, enhanced green fluorescent protein, EGFP, β-glucuronidase
Introduction
Therapeutic proteins, such as monoclonal antibodies and other recombinant proteins, treat diseases either by replenishing the proteins that have become defective and/or missing or by suppressing an excessive and/or dysregulated pathway in patients. Protein drugs have been popular in the treatment of cancer, diabetes, and cerebrovascular diseases, hemostasis, pulmonary and metabolic disorders.1 These protein drugs are designed to act on specific components in respective diseases; thus, conceptually, these protein drugs are not expected to interfere with normal biological processes.1 Therefore, recombinant proteins have become a hot area of drug development. As of now, there are >239 protein or peptide drugs with enzymatic activity, regulatory activity, and specific targeting that have been developed and/or approved for clinical use by the US Food and Drug Administration (FDA),2 with a total sale of US$174.7 billion in 2015 and expected to reach $248.7 billion in 2020.3
The development of protein therapeutics is confronted with a number of challenges. Protein solubility, route of administration, distribution, and stability are major factors that limit the application of protein drugs.1,4,5 Additionally, therapeutic proteins may elicit immune responses in patients.6 These immune responses can neutralize the protein and even cause a harmful reaction in the patient.7,8 These issues are partly addressed by modifying a protein with polyethylene glycol (PEG), which decreases renal clearance, retards enzymatic degradation, and increases elimination half-life of an injected protein.9–11 Second, intracellular delivery of protein drugs into diseased cells remains technically challenging.12 Currently, most protein drugs act on extracellular or cell membrane components (such as receptors). Only a few therapeutic proteins have been developed as intracellular drugs to repair damaging cellular pathways inside diseased cells.2,13 Thus far, low permeability across cell membranes, inefficient passage from the lysosome to cytosol, and degradation of the protein are major obstacles for intracellular delivery of protein drugs.
Over the last few decades, several protein delivery systems, including liposome- and polymer-based nanoparticles, bioconjugates, and hydrogels, have been attempted.14–17 Our group previously developed a lipo-PEG-polyethylenimine (PEI) complex (LPPC), which is a cationic lipocomplex that can bind four-fold amounts of active proteins on its surface.18 The LPPC was capable of capturing a variety of proteins, including β-glucuronidase (βG), antibodies, and protein immunogens.18,19 Because LPPC can capture proteins by noncovalent binding, it is easily and rapidly assembled into LPPC/protein complexes. After the captured protein is saturated, a given protein cannot be replaced by other additional proteins and still retains its biological activity.18 Furthermore, LPPC was easily complexed with a cytotoxic drug to increase the anti-tumor activity of the drug.20 Therefore, LPPC seemed a versatile vector for drug delivery.
In this study, we tested whether LPPC may be used for intracellular delivery of proteins. We began the tests by using fluorescein isothiocyanate isomer I (FITC)-modified BSA (BSA-FITC) and enhanced green fluorescent protein (EGFP) as model proteins to examine the transport process of LPPC/BSA-FITC. The LPPC-bound proteins were rapidly transported into the cells and disassociated in the lysosome. The integrity of transported proteins was tested in vitro by examining the enzymatic activity of βG after LPPC-mediated delivery into the cells (Figure 1). To test in vivo protein delivery, we intravenously injected LPPC-βG s into BALB/c mice and examined the enzyme activity of βG s in the liver tissue. These data indicate that LPPC may serve as a useful intracellular protein transporter for studying diseases in vitro and in vivo and developing protein drugs.
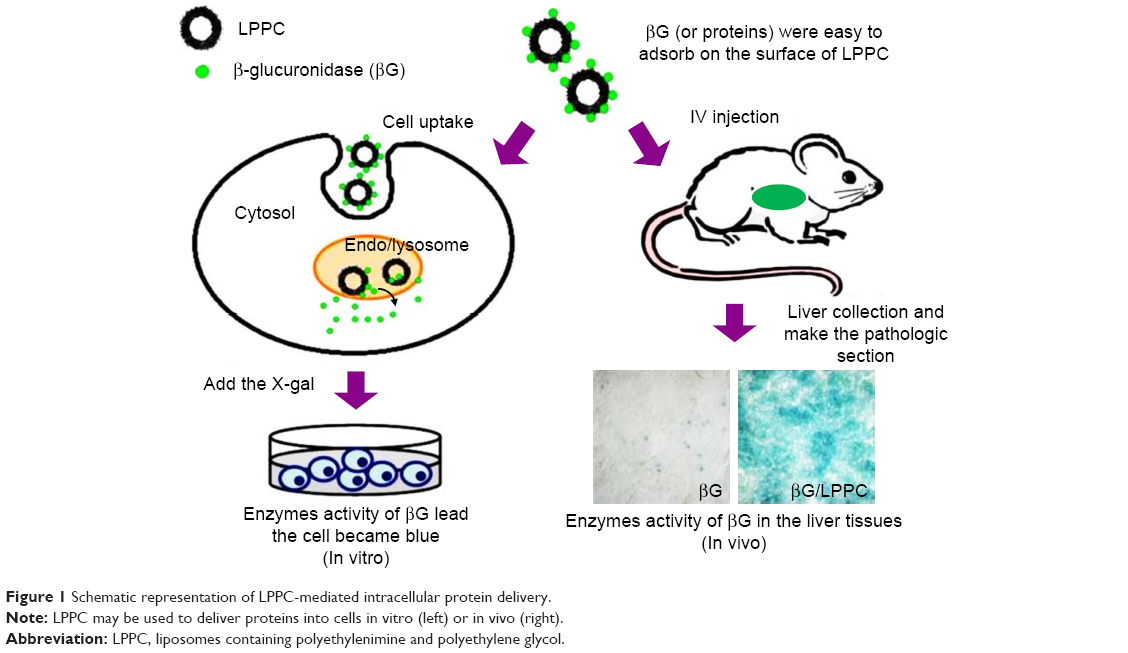 | Figure 1 Schematic representation of LPPC-mediated intracellular protein delivery. |
Materials and methods
Reagents
1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1,2-dilauroyl-sn-glycero-3-phosphocholine (DLPC) were purchased from Avanti Polar Lipids (Alabaster, AL, USA). PEG (molecular weight [MW] 1,500 and 8,000), PEI branched, (MW 25,000), p-nitrophenyl-β-d-glucuronide (PNPG), 5-bromo-4-chloro-3-indolyl-β-d-glucuronide cyclohexylamine salt (X-gluc), FITC, and paraformaldehyde, and 3,3′-dioctadecyloxacarbocyanine, p-nitrophenyl-β-d-glucuronide were obtained from Sigma-Aldrich (St Louis, MO, USA). BSA and trypan blue staining were purchased from Invitrogen (Gaithersburg, MD, USA). Triton X-100 was purchased from Amresco (Solon, OH, USA).
Preparation of LPPC
LPPC was prepared by coating an equal weight of DOPC and DLPC (total 50 mg) in 1 mL chloroform onto a round-bottom flask using a rotary evaporator (EYELA, N-1000S, Tokyo, Japan) to yield a thin lipid film. The film was hydrated with 5 mL of deionized water while dissolving an additional 675 mg PEI and 220 mg PEG-8000. At this stage, the molar ratio of phospholipids:PEI:PEG was approximately 13:5:5. The hydrated film was vigorously resuspended for 10 minutes, and the mixture was extruded nine times through a LiposoFast Extruder (Avestin Inc., Ottawa, Canada) via a 200 nm membrane. The suspension was diluted 50-fold in deionized water and centrifuged at 5,900× g for 5 minutes to remove any unincorporated materials. Finally, the pellets were resuspended in 5 mL of deionized water and stored at 4°C.
Determination of the size and zeta potential of BSA-FITC binding to LPPC
One hundred milligrams of BSA was added to 10 mL of 0.1 M bicarbonate buffer (pH 9.0). The solution was added with 584 μL of FITC (10 mg/mL) and stirred slowly at room temperature for 60–90 minutes while avoiding exposure to light. FITC/BSA solution was then added with 1 mL of 1 M glycine solution to stop the labeling reaction. The mixture was then flowed through a G-25 column (GE Healthcare Life Sciences, Pittsburgh, PA, USA) to remove unconjugated FITC from the BSA-FITC. The concentration of BSA-FITC was determined using a Coomassie Plus Bradford™ Assay Kit according to the manufacturer’s protocol.
The particle sizes of LPPC (2 mg) complexed with various amounts of BSA-FITC in 200 μL deionized water were measured before and after centrifugation by a BI-200SM dynamic laser light scattering goniometer (Brookhaven Inc., Holtsville, NY, USA). The zeta potentials were determined for 900 μL of LPPC (200 μg/mL) complexed with different amounts of BSA-FITC by Nano ZS90 (Malvern Instrument, Worcestershire, UK).
Stability of LPPC/protein complexes
BSA-FITC was incubated with LPPC for 30 minutes at 37°C, then centrifuged at 5,900× g for 5 minutes to remove any unincorporated BSA-FITC and washed. After resuspension, the LPPC/BSA-FITC complex was incubated with increasing concentrations of NaCl (100–300 mM) for 20 minutes at 37°C. Following washes and resuspension, BSA-FITC on LPPC was monitored using a spectrophotometer (Ultrospec 3100; Amersham Biosciences, Uppsala, Sweden) at 488 nm (excitation) and 515 nm (emission). A similar protocol was used to monitor whether the βG incorporated into LPPC is stable in the presence of NaCl. The activity of βG was measured by incubating the complexes with 200 μL of 0.3 mM PNPG for 20 minutes and measuring the absorbance at 450 nm.
Cell culture
HepG2 and BALB/3T3 cells (obtained from the Food Industry Research and Development Institute, Hsinchu, Taiwan) were cultured with DMEM (Invitrogen) supplemented with 10% heat inactivated FBS (Invitrogen) and 1% penicillin/streptomycin (PS; Biological Industries, Beithaemek, Israel) in a humidified atmosphere of 5% CO2 at 37°C. The cells were allowed to grow to ~70% confluence and were split at 1:3 during each passage or used for experiments.
In vitro cytotoxicity of LPPC/BSA-FITC complexes
Cells were seeded in 96-well tissue culture plates at a concentration of 1×104 cells/100 μL/well overnight. The cells were treated with serial concentrations of LPPC alone or the LPPC/BSA-FITC complexes for 48 hours. The viability of each cell line was then determined by an MTT colorimetric assay (Sigma-Aldrich).
In vitro intracellular delivery of LPPC/BSA-FITC
HepG2 cells were seeded in a 24-well plate at 1×105 cells/well in 1 mL of DMEM medium containing 10% FBS and 1% PS overnight. The medium was removed, and the cells were washed, followed by the addition of 1 mL of DMEM not containing serum. LPPC (10 μg) mixed with 40 μg of BSA-FITC in a 10 μL solution was added into each well at 37°C for varying lengths of time. The cells were trypsinized and washed with PBS. The fluorescence of cells was analyzed by flow cytometry.
For the localization of the LPPC–protein complex, cells were fixed in 4% paraformaldehyde at room temperature for 15 minutes, washed again in PBS, and permeabilized with 0.1% Triton X-100 (2 minutes at room temperature). Permeabilized cells were stained with a Hoechst 33342 nuclear stain (Invitrogen) and a LysoTracker Red DND-99 lysosomal stain (Invitrogen), washed twice in PBS, and examined by confocal microscopy (TCS SP2; Leica, Nussloch, Germany) at 630× magnification.
Fluorescent expression of the EGFP/LPPC complex
To address the question of whether the fluorescence of EGFP could be quenched by LPPC, a dot blot was utilized. First, LPPC was prepared with or without PEG blocking, and then EGFP was added to 1 mL of deionized water. The final samples (EGFP, PEG/EGFP, LPPC, LPPC–PEG, LPPC–EGFP and LPPC–PEG/EGFP) were spotted onto a nitrocellulose membrane. Finally, the membrane was dried and photographed under ultraviolet illumination.
In vitro intracellular delivery of the EGFP/LPPC complex
HepG2 cells were seeded in a 24-well plate at 1×105 cells/well in 1 mL of DMEM containing 10% FBS and 1% PS and incubated overnight. The medium was removed, and the cells were washed. The addition of 1 mL of DMEM not containing serum was added to the cells. EGFP (1 μg) premixed with LPPC (40 μg) was centrifuged, and the cells were resuspended in 10 μL of PBS buffer for various lengths of time. The images were analyzed by confocal microscopy as previously described.
Enzymatic activity after in vitro delivery of βG
HepG2 cells were seeded in a 24-well plate at 1×105 cells/well in 1 mL of DMEM containing 10% FBS and 1% PS and were incubated overnight. The medium was removed and the cells were washed. The addition of 1 mL of DMEM not containing serum was added to the cells. LPPC (10 μg) was premixed with βG (37.5 μg, kindly provided by Dr TL Cheng of Kaohsiung Medical University, Kaohsiung, Taiwan) in a 10 μL final volume at 25°C for 30 minutes. Following mixing, 10 μL of the complex was added to the cells at 37°C for 4 hours. The cells were perforated with 0.25% Triton X-100 (v/v) in PBS and then washed three times to remove excess detergents. X-gluc (20 μL; 5 mg/mL) in 1 mL of PBS was added to the cells at 37°C for 12 hours. The cells were then examined and photographed by a light field microscope (Olympus, Tokyo, Japan) at 400× magnification.
Biodistribution and enzymatic activity of βG delivery of in vivo
Female BALB/c mice were purchased from the National Laboratory Animal Center (Taipei, Taiwan) and maintained on a 12:12 hour light:dark cycle in an animal environmental control chamber (Micro-VENT IVC Systems, Allentown, NJ, USA). All animal studies were approved by the Institutional Animal Care and Use Committee at the National Chiao Tung University (NCTU-IACUC-105007).
Female BALB/c mice were treated intravenously with 5 mg/kg of βG, LPPC, or LPPC/βG. After 48 hours of treatment, mice were sacrificed, and major organs were collected. The organs, including heart, lung, spleen, liver, and kidney were harvested, washed with saline, weighed, and stored at −80°C until further analysis of biodistribution. To measure the enzymatic activity, liver sections were stained with X-gluc. The stained liver sections were examined and photographed by a light field microscope at 400× magnification.
Measurement of βG
Ten milligrams of tissue samples were homogenized with 100 μL of tissue lysis buffer (50 mM Tris–HCl (pH 7.4), 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM EDTA, and 10 mM NaF) on ice using a Dounce homogenizer. After centrifugation at 10,000× g for 5 minutes at 4°C, the supernatant was collected. Twenty microliters of supernatant was added into each well of a black 96-well plate. Serial dilutions of βG from 50 μg/mL to 0 μg/mL were added as the standard. Then, the concentration of βG was measured by incubating samples with 200 μL of 0.3 mM PNPG for 20 minutes and measuring the absorbance at 450 nm.
Statistical analysis
Numeric data were expressed as the mean ± SD. Data were analyzed using the SAS statistical package (SAS Institute, Inc., Cary, NC, USA). A Student’s t-test was used when comparing two independent samples, and ANOVA was used for comparing multiple samples. P<0.05 was considered statistically significant.
Results
Characterization of LPPC/BSA-FITC complexes
We use isothermal titration calorimetry (ITC) to detect whether LPPC could indeed interact with BSA-FITC. ITC measurements indicated that LPPC interacted with higher protein amounts of proteins than Lipofectamine (Figure 2A and B). In contrast, cationic 1,2-dioleoyl-3-trimethylammonium-propane liposomes did not react with BSA-FITC (Figure 2C). In these experiments, the addition of BSA could easily induce aggregation of Lipofectamine, but not the LPPC or DOTAP liposomes. Lipofectamine particles quickly reacted with the added BSA protein (Figure 2B). Figure 1A also shows that LPPC particles did not react with BSA below 40 μg.
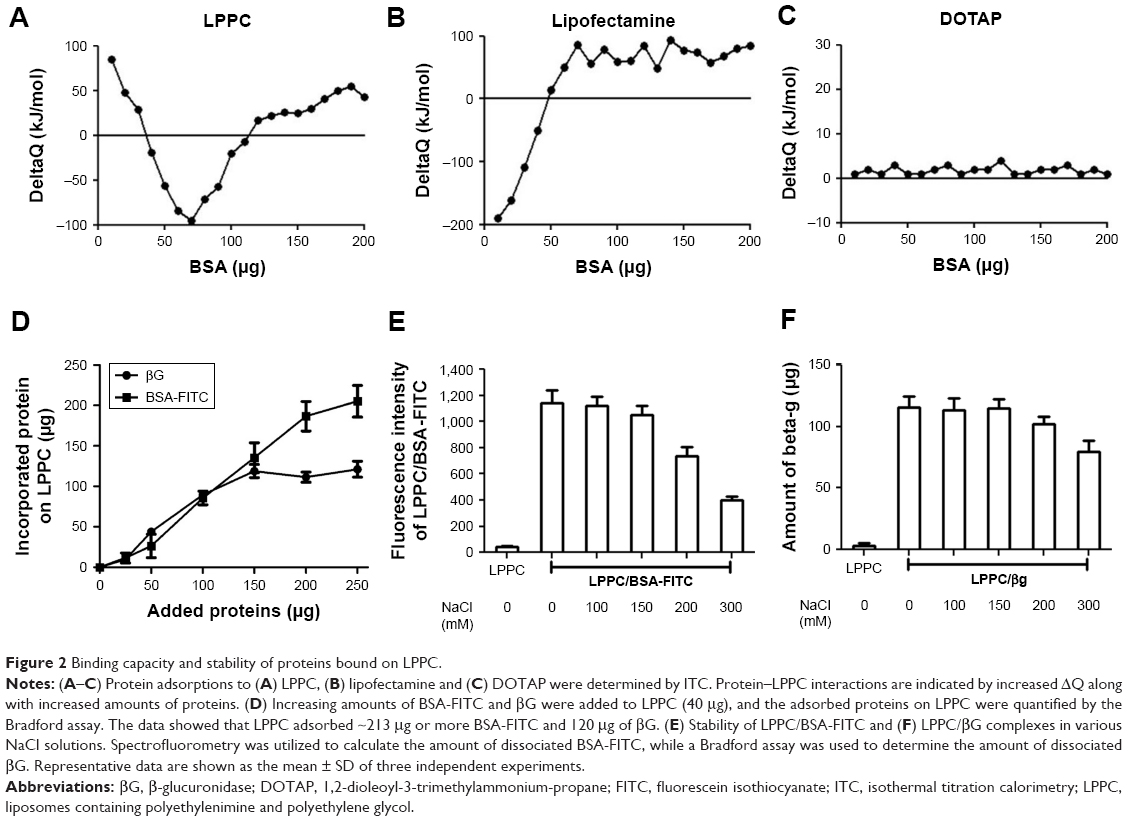 | Figure 2 Binding capacity and stability of proteins bound on LPPC. |
Next, we examined the binding capacity of LPPC and the stability of the bound proteins. Forty micrograms of LPPC adsorbed ~213 μg or more BSA-FITC and ~120 μg of βG (Figure 2D). The difference in binding capacities of LPPC for BSA-FITC and βG could be attributed to the differences in protein size. βG is a tetramer; thus, a single LPPC liposome may accommodate fewer bulky βG on the surface than the monomeric BSA-FITC. BSA-FITC and βG were relatively stable at 150 mM of NaCl, yet the protein/protein complex dissociated from LPPC when NaCl concentrations exceeded 200 mM (Figure 2E and F). BSA-FITC was also stably bound on the surface of LPPC in growth medium containing 10% FBS (Figure S1). The data suggested that the LPPC/protein complexes were stable in physiological solutions.
The particle sizes and zeta potentials of LPPCs complexed with different amounts of BSA-FITC were measured. The LPPC particle size gradually increased from 215 to 441 nm as increasing amounts of BSA-FITC were added, while the zeta potentials of LPPC decreased with increasing amounts of BSA-FITC (Table 1). These results confirm that LPPC could physically bind BSA-FITC on the surface.
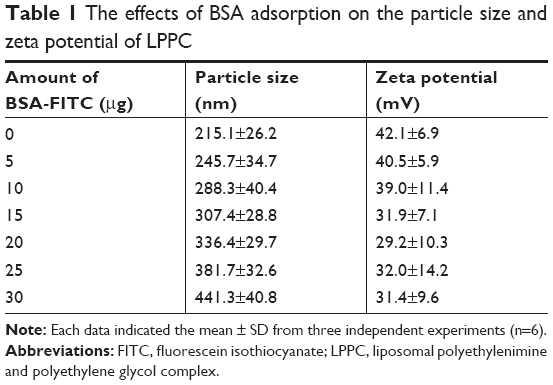 | Table 1 The effects of BSA adsorption on the particle size and zeta potential of LPPC |
The cytotoxicities of the LPPC/BSA-FITC complexes were examined sequentially. A minor inhibition of cell growth was noted after the addition of increasing amounts of LPPC or LPPC/BSA-FITC complexes. Balb/3Tc cells were slightly more sensitive to LPPC or LPPC/BSA-FITC treatment, while HepG2 cells were resistant up to 20 μg/mL of LPPC or LPPC/BSA-FITC complexes (Figure 3). Furthermore, the addition of BSA-FITC did not seem to increase the toxicity in the cells. Thus, protein-loaded LPPC complexes should be relatively safe to cells, especially HepG2 cells, in our studies.
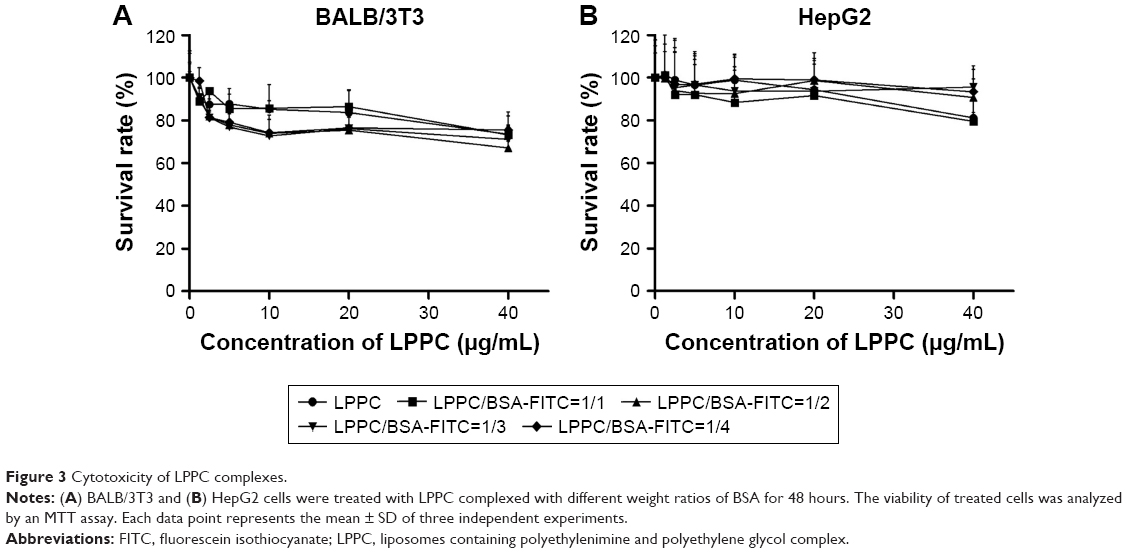 | Figure 3 Cytotoxicity of LPPC complexes. |
Efficacy of in vitro intracellular protein delivery by LPPC complexes
We used flow cytometry to test the delivery efficacy of BSA-FITC by LPPC to cells in vitro. To specifically examine the fluorescent signals emitted from intracellularly delivered BSA-FITC, cell surface fluorescent signals were quenched by incubation with trypan blue after BSA-FITC or LPPC/BSA-FITC treatments to HepG2 cells. These experiments demonstrated that LPPC/BSA-FITC efficiently entered the cells, but free BSA-FITC was unable to do so. Entry of LPPC/BSA-FITC was detected as early as 30 minutes following incubation, whereas free BSA-FITC was unable to enter the cells even after prolonged (4 hours) incubation with the cells (Figure 4). These results suggest that LPPC can efficiently deliver BSA-FITC into the cells.
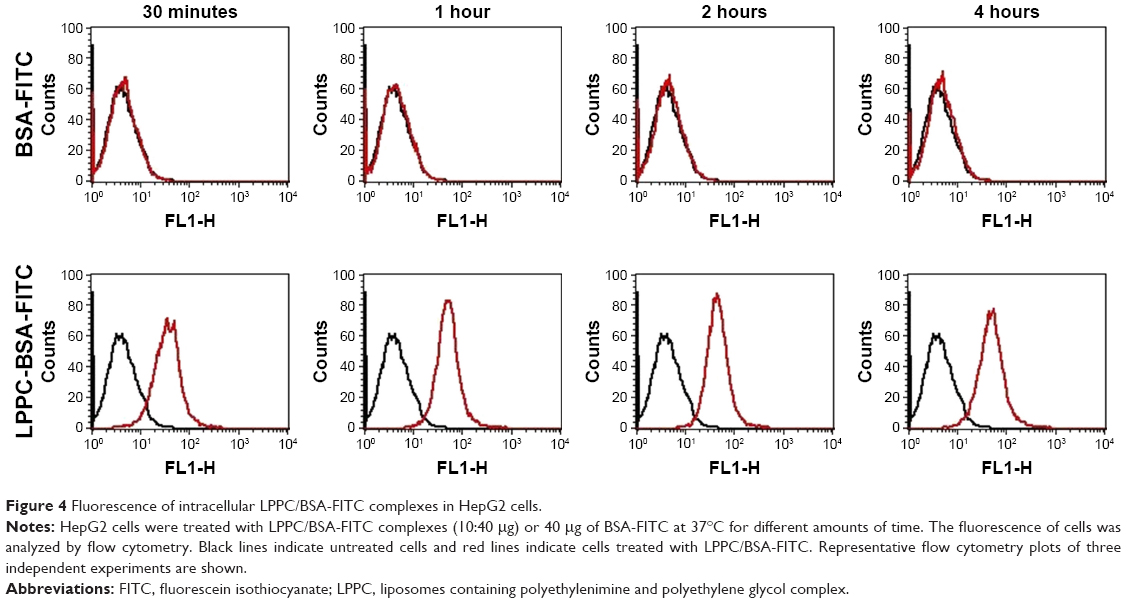 | Figure 4 Fluorescence of intracellular LPPC/BSA-FITC complexes in HepG2 cells. |
We then investigated the route by which LPPC delivered BSA-FITC into the cells. In these experiments, we used LysoTracker Red (a red fluorescent dye) to stain the lysosome for confocal microscopy. We found LPPC/BSA-FITC in the lysosome 30 minutes after LPPC/BSA-FITC treatments to the cells (Figure 5), suggesting that the LPPC/BSA-FITC was endocytosed and fused with lysosome soon after LPPC/BSA-FITC treatments. Consistent with our flow cytometry studies, no green fluorescence was recorded in the cells treated with free BSA-FITC, thus reassuring that the BSA-FITC could only be delivered by LPPC in our studies. Interestingly, green fluorescence (LPPC/BSA-FITC) was found to dissociate red fluorescence (lysosome) 2 hours after BSA-FITC/LPPC treatments to the cells (lower left panel, Figure 5), indicating that LPPC/BSA-FITC escaped from the endosome to the cytosol. However, the dissociation mechanism of LPPC/BSA-FITC is unknown.
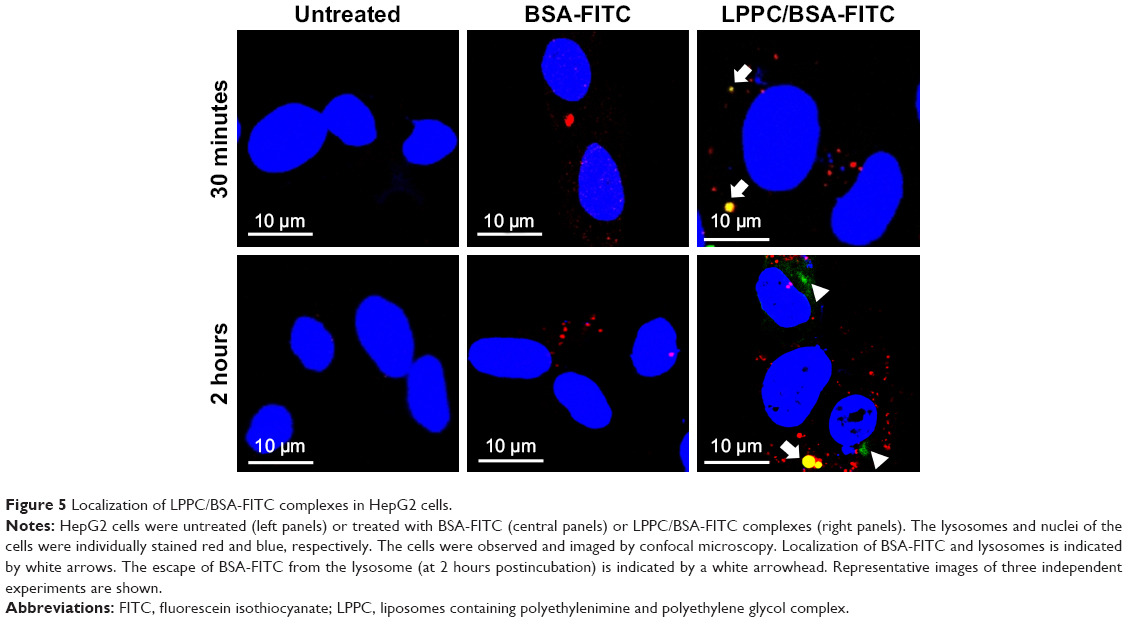 | Figure 5 Localization of LPPC/BSA-FITC complexes in HepG2 cells. |
To further explore the dissociation mechanism, we replaced BSA-FITC with EGFP for our study. We previously found that EGFP was unable to emit green fluorescence once it was complexed with LPPC (Figure 6A), possibly due to the proton buffering effect of PEI on the LPPC liposome. We reasoned that if LPPC-bound EGFP could dissociate from LPPC, then EGFP would regain its ability to fluoresce upon laser activation. Therefore, HepG2 cells were treated with EGFP alone or EGFP complexed with LPPC (LPPC/EGFP), and the cells were examined by confocal microscopy. Consistent with our results above, free EGFP was not efficiently taken into HepG2 cells (Figure 6B). However, green fluorescence was recorded in LPPC/EGFP-treated cells. More specifically, green fluorescence was not noted at 30 minutes after treatment but was recorded 2 hours after the treatment (Figure 6B). These data suggest that EGFP was still bound to LPPC within the cells at 30 minutes after the treatment but escaped from the lysosome and dissociated from LPPC 2 hours after the treatment.
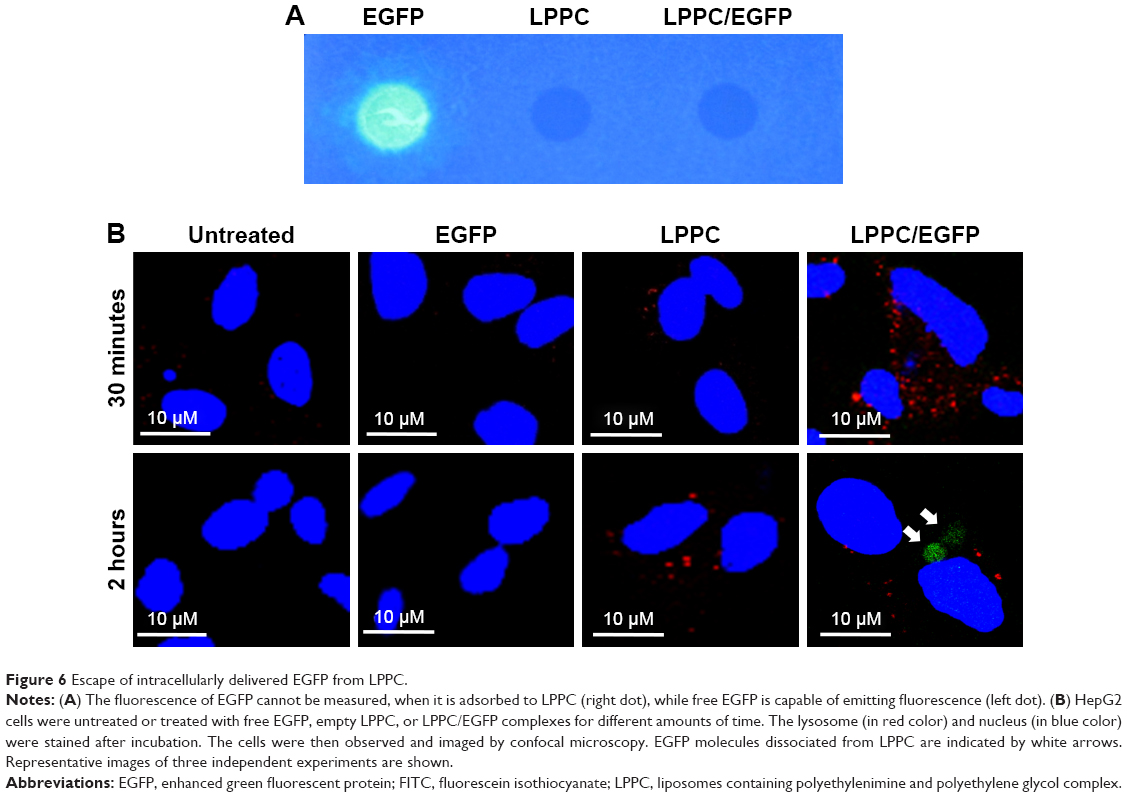 | Figure 6 Escape of intracellularly delivered EGFP from LPPC. |
Enzymatic activity of transported protein
We next tested whether the proteins retained their activities in the cytosol after LPPC-mediated intracellular delivery. To address this question, we examined the enzyme activity of βG in HepG2 cells incubated with LPPC/βG. The heightened βG activity (blue staining) was observed in the cells that had been incubated with the LPPC/βG complex (Figure 7). Thus, the enzyme retained its activity after LPPC-mediated delivery into the cell.
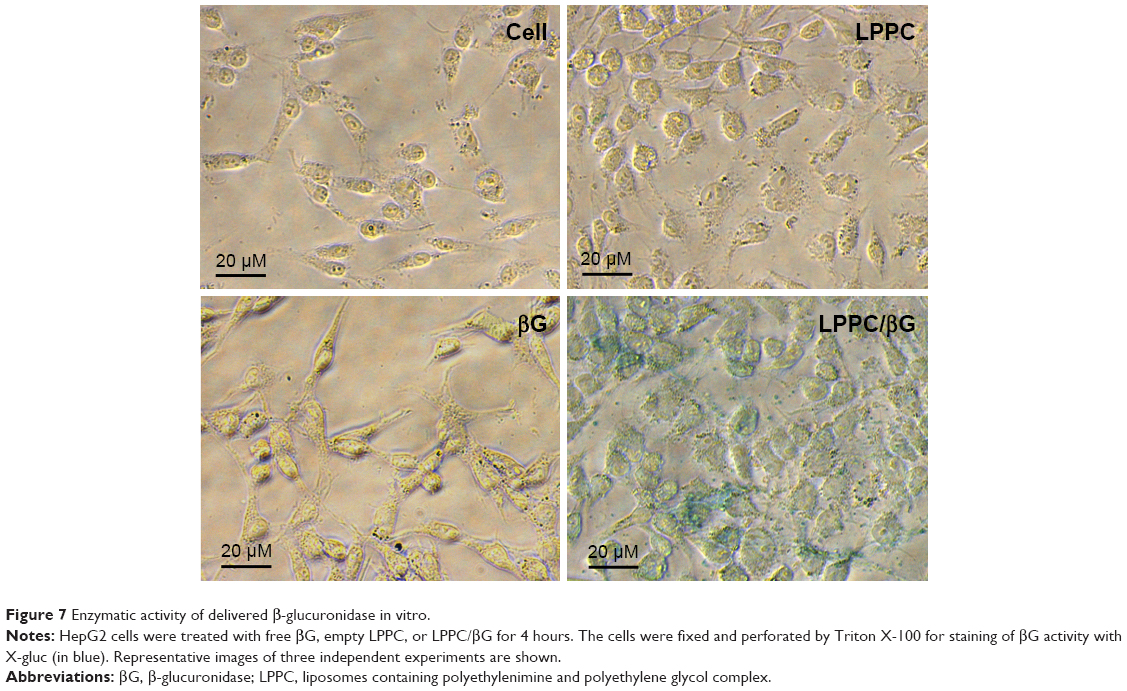 | Figure 7 Enzymatic activity of delivered β-glucuronidase in vitro. |
The activities of βG in the liver sections were examined by X-gluc staining. Consistent with the in vitro studies, livers from mice that received LPPC/βG injections expressed intense X-gluc staining (Figure 8A). In contrast, only residual, presumably endogenous, βG activities were found in the livers from mice that received LPPC or free βG injections. Then, the biodistribution of LPPC-delivered βG was analyzed after intravenous injection of LPPC/βG complexes into mice. Figure 8B shows that most βG accumulated in the liver. LPPC efficiently delivered βG to liver. These results indicated that active βG can be successfully transported to liver cells in vivo by LPPC. Thus, LPPC may be a useful tool for the in vivo delivery of functional proteins to livers.
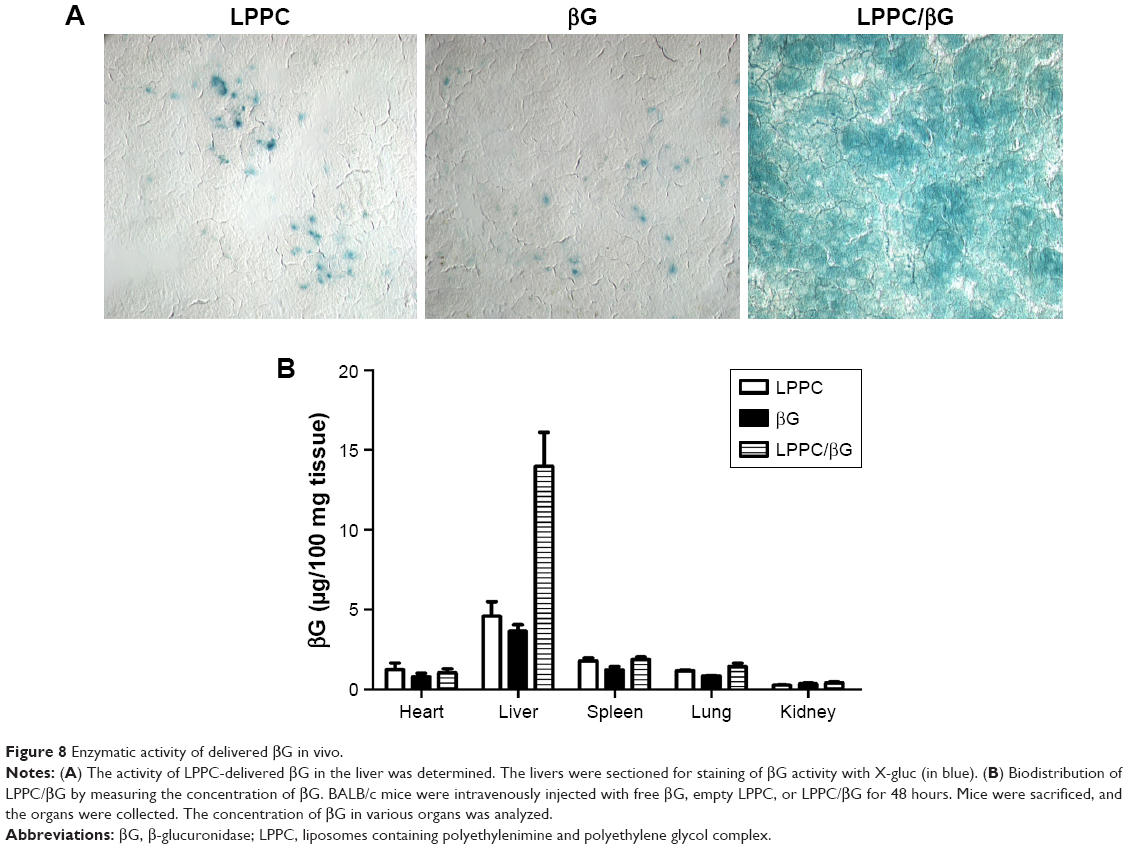 | Figure 8 Enzymatic activity of delivered βG in vivo. |
Discussion
In a previous study, we showed that LPPC is capable of binding various proteins on the surface.18 In this study, we further demonstrated that LPPC is an efficient intracellular protein delivery vehicle that assists in the delivery of active proteins or enzymes into cells. The preparation of LPPC is technically simple and does not require sophisticated instrumentation. Likewise, the adsorption of large amounts of proteins onto LPPC is straightforward and fast. Furthermore, LPPC is nontoxic to cells and animals. These features combined with the high efficacy make LPPC a useful tool for biomedical studies that require intracellular protein delivery and for the development of therapeutic proteins that correct diseases caused by faulty and/or missing proteins.
Cationic liposome or PEI enter cells via energy-dependent endocytosis.21–23 LPPC is a cationic lipocomplex mainly composed of lipid, PEG, and PEI; thus, it is likely that LPPC also delivers protein cargo by the same mechanism. Intracellular protein delivery by LPPC is fast and efficient since we can detect BSA-FITC in the cells within 30 minutes after incubation. We found that the LPPC/protein complex entered the cytoplasm at 37°C but did not enter at 4°C (data not shown), suggesting that delivery involves an energy-dependent process. As an endosome is often fused with a lysosome, our data showed that LPPC/BSA-FITC colocalized with lysosomes and provided additional evidence that endocytosis is the route by which LPPC/proteins are delivered into a cell.
Once an endosome is fused to a lysosome, an endocytosed protein may be degraded or denatured by lysosomal proteases or acidic environments. Escape from an endo/lysosome seems to be a critical step for intracellular protein delivery. Cationic complexes or polymers can assist proteins in escaping from an endo/lysosome by a proton sponge effect or by destabilization of lysosomal membranes.24,25 PEI, a major component in LPPC, has been shown to induce proton sponge effects and leads to endo/lysosome swelling and eventually leakage of the vesicles.24 This mechanism is consistent with our data that the LPPC/protein complex was released from endo/lysosomes, and functionally active proteins dissociated from LPPC within 2 hours.
Although >239 protein drugs have been approved for clinical use by the FDA, only a few are delivered intracellularly.1 This observation is clinically pertinent since faulty or missing proteins within cells are often the underlying mechanisms of diseases. A well-known example is mutant oncoproteins, such as Ras, which result in the transformation of normal cells.26,27 Moreover, defects of intracellular proteins or enzymes, especially those involved in metabolic pathways, may cause cellular damage and eventually lead to the loss of organ function. For example, glycogen storage diseases are diseases manifesting as developmental abnormalities, weakness, and confusion that can occur when defects occur in the enzymes involved in the metabolism of glycogen.28 These diseases point to an urgent need in the development of intracellular protein drug therapy to correct the disease within the cells.
A major consideration in intracellular protein drug therapy is the passage of the protein across the plasma membrane. Different approaches have been developed to address this issue. For example, recombinant β-glucocerebrosidase has been modified with mannose to increase its binding to endocytic carbohydrate receptors on macrophages and other cell types, allowing the enzyme to enter these cells.29 In addition, nanoparticles have also been shown to assist molecules across the plasma membrane.15,30,31 Virus-like particles (VLPs) have also been used to deliver functional proteins into certain cells by displaying targeting molecules on the surface of the VLPs.32 However, safety concerns, stability, immunogenicity (in the case of VLP or other virus-mediated delivery), and production methods and/or instrumentations may limit the development of these protein delivery platforms.15 In this report, LPPC did not decrease cell survival and did not induce noticeable pathology in the liver, suggesting LPPC is a safe vehicle for intracellular protein delivery. In addition, large-scale production of LPPC/protein complexes is straightforward and fast; hundreds of milligrams of LPPC/protein complexes can be produced in 2 days. No sophisticated/expensive instruments are needed in the production, and simple chromatography and centrifugation are used to remove impurities. Thus, LPPC is an efficient and economical tool for the intracellular delivery of proteins in vitro and in vivo.
Conclusion
Owing to its high protein-binding capacity, safety, and stability as well as its ease of preparation and efficient intracellular delivery, LPPC may be a useful tool for the development of therapeutic proteins that aim to treat diseases caused by faulty/missing proteins in the cells.
Acknowledgments
This work was supported by grants from the Ministry of Science and Technology of Taiwan (MOST 107 – 2320-B-182A-007 and MOST 105 – 2320-B-009-002) and from the Chang-Gung Memorial Hospital Research Foundation (CMRPG2F0141-2 and CMRPG2D0091-3). The authors thank the core facility of Multiphoton and Confocal Microscope System (MCMS) at the National Chiao Tung University, Hsinchu, Taiwan.
Author contributions
All authors contributed toward data analysis, drafting and revising the paper, gave final approval of the version to be published and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Leader B, Baca QJ, Golan DE. Protein therapeutics: a summary and pharmacological classification. Nat Rev Drug Discov. 2008;7(1):21–39. | ||
Usmani SS, Bedi G, Samuel JS, et al. THPdb: database of FDA-approved peptide and protein therapeutics. PLoS One. 2017;12(7):e0181748. | ||
Agyei D, Ahmed I, Akram Z, Iqbal HM, Danquah MK. Protein and peptide biopharmaceuticals: an overview. Protein Pept Lett. 2017;24(2):94–101. | ||
Putney SD, Burke PA. Improving protein therapeutics with sustained-release formulations. Nat Biotechnol. 1998;16(2):153–157. | ||
Mahmood I, Green MD. Pharmacokinetic and pharmacodynamic considerations in the development of therapeutic proteins. Clin Pharmacokinet. 2005;44(4):331–347. | ||
Schellekens H. Bioequivalence and the immunogenicity of biopharmaceuticals. Nat Rev Drug Discov. 2002;1(6):457–462. | ||
Scharrer I, Bray GL, Neutzling O. Incidence of inhibitors in haemophilia a patients – a review of recent studies of recombinant and plasma-derived factor VIII concentrates. Haemophilia. 1999;5(3):145–154. | ||
Gilles JG, Arnout J, Vermylen J, Saint-Remy JM. Anti-factor VIII antibodies of hemophiliac patients are frequently directed towards nonfunctional determinants and do not exhibit isotypic restriction. Blood. 1993;82(8):2452–2461. | ||
Zündorf I, Dingermann T. PEGylation – a well-proven strategy for the improvement of recombinant drugs. Pharmazie. 2014;69(5):323–326. | ||
Yang Z, Wang J, Lu Q, et al. PEGylation confers greatly extended half-life and attenuated immunogenicity to recombinant methioninase in primates. Cancer Res. 2004;64(18):6673–6678. | ||
Ekladious I, Colson YL, Grinstaff MW. Polymer–drug conjugate therapeutics: advances, insights and prospects. Nat Rev Drug Discov. Epub 2018 Dec 12. | ||
Brown LR. Commercial challenges of protein drug delivery. Expert Opin Drug Deliv. 2005;2(1):29–42. | ||
Robinson M, Fraser I, McKee E, Scheck K, Chang L, Willerth SM. Transdifferentiating astrocytes into neurons using ASCL1 functionalized with a novel intracellular protein delivery technology. Front Bioeng Biotechnol. 2018;6:173. | ||
di Marco M, Shamsuddin S, Razak KA, et al. Overview of the main methods used to combine proteins with nanosystems: absorption, bioconjugation, and encapsulation. Int J Nanomedicine. 2010;5:37–49. | ||
Rawat M, Singh D, Saraf S, Saraf S. Nanocarriers: promising vehicle for bioactive drugs. Biol Pharm Bull. 2006;29(9):1790–1798. | ||
Salmaso S, Bersani S, Semenzato A, Caliceti P. Nanotechnologies in protein delivery. J Nanosci Nanotechnol. 2006;6(9–10):2736–2753. | ||
Yuan H, Jiang S-P, du Y-Z, Miao J, Zhang X-G, Hu F-Q. Strategic approaches for improving entrapment of hydrophilic peptide drugs by lipid nanoparticles. Colloids Surf B Biointerfaces. 2009;70(2):248–253. | ||
Liu YK, Lin YL, Chen CH, et al. A unique and potent protein binding nature of liposome containing polyethylenimine and polyethylene glycol: a nondisplaceable property. Biotechnol Bioeng. 2011;108(6):1318–1327. | ||
Chen CH, Lin YL, Liu YK, et al. Liposome-based polymer complex as a novel adjuvant: enhancement of specific antibody production and isotype switch. Int J Nanomedicine. 2012;7:607–621. | ||
Lin YL, Liu YK, Tsai NM, et al. A lipo-PEG-PEI complex for encapsulating curcumin that enhances its antitumor effects on curcumin-sensitive and curcumin-resistance cells. Nanomedicine. 2012;8(3):318–327. | ||
Wrobel I, Collins D. Fusion of cationic liposomes with mammalian cells occurs after endocytosis. Biochim Biophys Acta. 1995;1235(2):296–304. | ||
Wong AW, Scales SJ, Reilly DE. DNA internalized via caveolae requires microtubule-dependent, Rab7-independent transport to the late endocytic pathway for delivery to the nucleus. J Biol Chem. 2007;282(31):22953–22963. | ||
Thomas M, Klibanov AM. Enhancing polyethylenimine’s delivery of plasmid DNA into mammalian cells. Proc Natl Acad Sci U S A. 2002;99(23):14640–14645. | ||
Benjaminsen RV, Mattebjerg MA, Henriksen JR, Moghimi SM, Andresen TL. The possible “proton sponge” effect of polyethylenimine (PEI) does not include change in lysosomal pH. Mol Ther. 2013;21(1):149–157. | ||
du L, Zhou J, Meng L, et al. The pH-triggered triblock nanocarrier enabled highly efficient siRNA delivery for cancer therapy. Theranostics. 2017;7(14):3432–3445. | ||
Charette N, Vandeputte C, Stärkel P. Ras in digestive oncology: from molecular biology to clinical implications. Curr Opin Oncol. 2014;26(4):454–461. | ||
Takahashi N, Yamada Y, Taniguchi H, et al. Clinicopathological features and prognostic roles of KRAS, BRAF, PIK3CA and NRAS mutations in advanced gastric cancer. BMC Res Notes. 2014;7:271. | ||
Hicks J, Wartchow E, Mierau G. Glycogen storage diseases: a brief review and update on clinical features, genetic abnormalities, pathologic features, and treatment. Ultrastruct Pathol. 2011;35(5):183–196. | ||
Grabowski GA, Barton NW, Pastores G, et al. Enzyme therapy in type 1 Gaucher disease: comparative efficacy of mannose-terminated glucocerebrosidase from natural and recombinant sources. Ann Intern Med. 1995;122(1):33–39. | ||
Cohen S, Coué G, Beno D, Korenstein R, Engbersen JF. Bioreducible poly(amidoamine)s as carriers for intracellular protein delivery to intestinal cells. Biomaterials. 2012;33(2):614–623. | ||
Coué G, Freese C, Unger RE, Kirkpatrick CJ, Engbersen JF. Bioresponsive poly(amidoamine)s designed for intracellular protein delivery. Acta Biomater. 2013;9(4):6062–6074. | ||
Kaczmarczyk SJ, Sitaraman K, Young HA, Hughes SH, Chatterjee DK. Protein delivery using engineered virus-like particles. Proc Natl Acad Sci U S A. 2011;108(41):16998–17003. |
Supplementary material
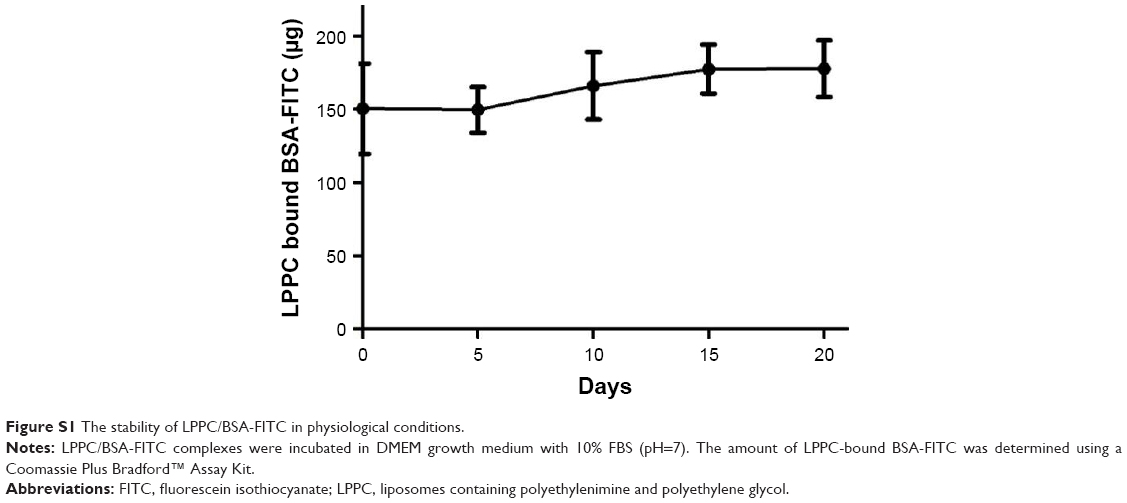 | Figure S1 The stability of LPPC/BSA-FITC in physiological conditions. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.