Back to Journals » Nanotechnology, Science and Applications » Volume 14
Lipid Nanosystems and Serum Protein as Biomimetic Interfaces: Predicting the Biodistribution of a Caffeic Acid-Based Antioxidant
Authors Fernandes E ![]() , Benfeito S
, Benfeito S ![]() , Cagide F
, Cagide F ![]() , Gonçalves H
, Gonçalves H ![]() , Bernstorff S
, Bernstorff S ![]() , Nieder JB
, Nieder JB ![]() , CD Real Oliveira ME, Borges F
, CD Real Oliveira ME, Borges F ![]() , Lúcio M
, Lúcio M ![]()
Received 29 October 2020
Accepted for publication 16 January 2021
Published 9 February 2021 Volume 2021:14 Pages 7—27
DOI https://doi.org/10.2147/NSA.S289355
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Israel Rubinstein
Eduarda Fernandes,1,2 Sofia Benfeito,3 Fernando Cagide,3 Hugo Gonçalves,4 Sigrid Bernstorff,5 Jana B Nieder,2 M Elisabete CD Real Oliveira,1 Fernanda Borges,3 Marlene Lúcio1,6
1Departamento de Física da Universidade do Minho, CF-UM-UP, Centro de Física das Universidades do Minho e Porto, Campus de Gualtar, Braga, 4710-057, Portugal; 2 Ultrafast Bio- and Nanophotonics Group, INL – International Iberian Nanotechnology Laboratory, Braga, Portugal; 3CIQUP/Department of Chemistry and Biochemistry, Faculty of Sciences, University of Porto, Porto, Portugal; 4Paralab, SA, Valbom, 4420-392, Portugal; 5Elettra-Sincrotrone Trieste S. C.p.A.,, Basovizza, Trieste, I-34149, Italy; 6CBMA, Centro de Biologia Molecular e Ambiental, Departamento de Biologia, Universidade do Minho, Campus de Gualtar, Braga 4710-057, Portugal
Correspondence: Marlene Lúcio
Universidade do Minho, Campus de Gualtar, Braga 4710-057, Portugal
Email [email protected]
Fernanda Borges
CIQUP/Department of Chemistry and Biochemistry, Faculty of Sciences, University of Porto, Porto, Portugal
Email [email protected]
Purpose: AntiOxCIN3 is a novel mitochondriotropic antioxidant developed to minimize the effects of oxidative stress on neurodegenerative diseases. Prior to an investment in pre-clinical in vivo studies, it is important to apply in silico and biophysical cell-free in vitro studies to predict AntiOxCIN3 biodistribution profile, respecting the need to preserve animal health in accordance with the EU principles (Directive 2010/63/EU). Accordingly, we propose an innovative toolbox of biophysical studies and mimetic models of biological interfaces, such as nanosystems with different compositions mimicking distinct membrane barriers and human serum albumin (HSA).
Methods: Intestinal and cell membrane permeation of AntiOxCIN3 was predicted using derivative spectrophotometry. AntiOxCIN3 –HSA binding was evaluated by intrinsic fluorescence quenching, synchronous fluorescence, and dynamic/electrophoretic light scattering. Steady-state and time-resolved fluorescence quenching was used to predict AntiOxCIN3-membrane orientation. Fluorescence anisotropy, synchrotron small- and wide-angle X-ray scattering were used to predict lipid membrane biophysical impairment caused by AntiOxCIN3 distribution.
Results and Discussion: We found that AntiOxCIN3 has the potential to permeate the gastrointestinal tract. However, its biodistribution and elimination from the body might be affected by its affinity to HSA (> 90%) and by its steady-state volume of distribution (VDSS=1.89± 0.48 L∙Kg− 1). AntiOxCIN3 is expected to locate parallel to the membrane phospholipids, causing a bilayer stiffness effect. AntiOxCIN3 is also predicted to permeate through blood-brain barrier and reach its therapeutic target – the brain.
Conclusion: Drug interactions with biological interfaces may be evaluated using membrane model systems and serum proteins. This knowledge is important for the characterization of drug partitioning, positioning and orientation of drugs in membranes, their effect on membrane biophysical properties and the study of serum protein binding. The analysis of these interactions makes it possible to collect valuable knowledge on the transport, distribution, accumulation and, eventually, therapeutic impact of drugs which may aid the drug development process.
Keywords: biomimetic models, biophysical profiling, drug–membrane interaction, blood–brain barrier permeability, drug distribution, small and wide-angle X-ray diffraction
Introduction
Research over the past few decades has revealed the involvement of oxidative stress in several disease states, in particular those presenting an increased incidence with age, including cardiovascular events, cancer, diabetes mellitus and neurodegenerative diseases.1,2 The concept of an imbalance between oxidants and antioxidants has evolved in recent years embracing all the biological responses involving the disruption of redox signaling pathways. In normal circumstances, cells attempt to counteract the oxidant effects and restore the redox balance through a diversity of processes, namely the activation or silencing of genes encoding defensive enzymes and structural proteins.3 However, an overproduction of reactive species (RS) resultant from oxidation can damage the integrity of various biomolecules, such as lipids, proteins, and DNA/RNA, leading to cell death.4,5 The use of antioxidants capable to prevent and/or minimize RS levels inside the cell is considered a therapeutic solution to slow-down the progression of the radical cascade reactions. Notwithstanding, the problems found with bioavailability and druggability of antioxidants, among others, limit their success in clinical trials. This limitation is related mainly to biodistribution issues and to the fact that frequently antioxidants do not reach relevant sites of free radical generation. So, the development of new effective treatments for neurodegenerative diseases is still a challenge due to the restrictive passage across biological membranes, namely the blood brain barrier (BBB) in order to reach the Central Nervous System (CNS).6
The covalent link of a lipophilic cation, namely alkyl triphenylphosphonium (TPP), to an antioxidant has been proposed as an effective strategy to deliver this type of compounds specifically to mitochondria, the main source of RS that are generated as by-products of the electron transport chain.7–9 In this context, a rational drug discovery program based on a dietary antioxidant (caffeic acid, Figure 1A) was accomplished by our group, giving rise to the first mitochondriotropic antioxidants, commonly named AntiOxCINs, based on a phenolic acid.10 The results obtained so far indicated that AntiOxCIN3 (Figure 1B) is a potent antioxidant able of mitochondrial accumulation and devoid of mitochondrial toxicity.10
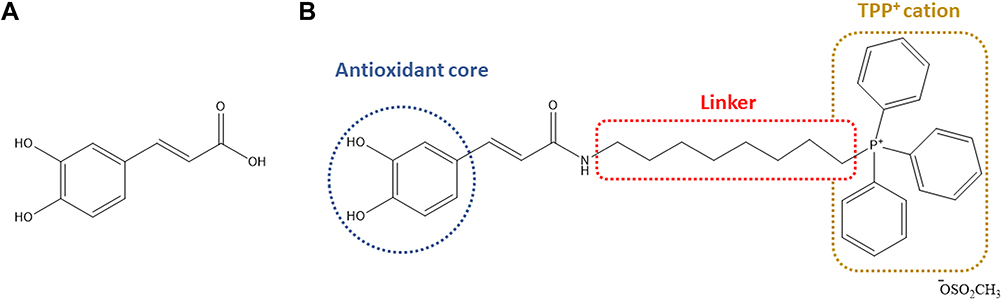 |
Figure 1 Chemical structures of caffeic acid (A) and mitochondriotropic antioxidant AntiOxCIN3 (B). |
Interestingly, TPP lipophilic cations have been described as an exception to the “rule of five” that is often used as preliminary predictor of relevant drug-like properties such as solubility, bioavailability, and ability to pass through phospholipid bilayers.11,12 This exception is due to the unusual properties of being both relatively water soluble and membrane permeant, even with a rather large molar mass and partition coefficient, an indicator of high lipophilicity.13 Among the mitochondriotropic compounds, it was found that the most lipophilic ones, till a limit on the number of methylene groups in the linker, presented the highest antioxidant potency.14 In fact, it is consensual that lipophilicity is the major determinant in drugs’ biodistribution, since it is intimately related to membrane permeability, which dictates the drug ability to pass through the different biological interfaces.15 Otherwise, increasing lipophilicity leads to a reduction in aqueous solubility and in turn, to biodistribution due to the drug’s high affinity to plasma proteins. Moreover, highly lipophilic drugs present usually poor oral absorption and are more vulnerable to CYP450 metabolism, and in consequence to a high hepatic clearance.
Considering the therapeutic interest of AntiOxCINs and assuming the oral administration of drugs as the most conventional route either by patient’s choice or due to industrial reasons,16 the oral administration of AntiOxCIN3 was hypothesized. Although oral absorption is the most popular and convenient route, it is limited by drug physicochemical properties (low aqueous solubility and/or low membrane permeability), among other factors.17 Upon oral administration of AntiOxCIN3, the drug is expected to interact promptly with a number of biological interfaces that will constitute barriers to its biodistribution.18,19 For orally administrated drugs, gastrointestinal (GI) absorption is the first physical barrier. The drug must be able to permeate through the intestinal epithelial barrier to guarantee bioavailability and biodistribution. After GI absorption, the drug will be exposed to plasma proteins in the bloodstream, the main function of which is to transport exogenous molecules through the body. However, the drug-plasma protein interaction must be reversible and balanced, since a too low or too high affinity will lead to biodistribution issues. When distributed to the body, drugs will continue to interact in a reciprocal way with other biological interfaces until they reach the therapeutic target. Also, even when the drug reaches the therapeutic target, its interaction with cell membranes can affect the rate of membrane partitioning and the subsequent penetration of the drug into the cytoplasm.18 Hence, the partitioning in cell membranes and the drug/membrane interactions should be studied and characterized.18,19
Until today, the emergence of several biomimetic model membranes based on lipid nanosystems and biophysical techniques to study and characterize drug–membrane interactions have resulted in new concerns about how a drug influences membrane properties and vice-versa.19–30 In our previous works, as well as in other studies, it is reported the correlations between the biological action and cytotoxicity of drugs and their biophysical effects in biomimetic membranes, eg by altering lipid membrane packing, decreasing lipid membrane transition temperatures or disturbing cooperativity of lipid unit cells, with consequent membrane biophysical impairment.21,26–28,31,32 Additionally, several interesting reports19,20,33–37 highlighted the importance of biomimetic models based on lipid nanosystems (liposomes, monolayers and supported lipid bilayers) to acquire molecular and functional information. The aim is using this acquired information to support the development of new drugs with better selectivity and reduced side effects34. This information can also assist to the design of new chemical entities with improved efficacy and reduced toxicity,19,34,37 or even to understand the interaction of drug delivery nanosystems with biomembranes.30,34,38,39
Therefore, in this study, the interaction of AntiOxCIN3 with the most relevant biointerfaces was studied and characterized allying in silico descriptors with different in vitro biophysical methods and biomimetic model systems to predict its biodistribution behavior. In order to mimic the journey of AntiOxCIN3 in the body after oral administration, four different biomimetic interface models were considered: (i) micelles of intestinal biliary salts; (ii) human serum albumin (HSA); (iii) lipid nanosystems (lipid vesicles and lipid bilayers) used as membrane models, which are composed by phosphatidylcholine (PC) as the main phospholipid component of cell membranes; and (iv) lipid nanosystems (lipid vesicles) composed of lipids from the endothelial membrane of BBB mimicking this important lipid biointerface. Furthermore, several features of drug/biointerfaces’ interaction were correlated with possible in vivo biodistribution issues of AntiOxCIN3, giving some outputs of the chemical modifications needed to improve its performance. Thus, biomimetic membrane model systems were used to determine: (i) membrane distribution (Kd) in biomimetic models representative of membranes found in target and off-target tissues, (ii) drug location at membrane level, (iii) plasma protein binding (PPB) to infer the biodistribution of AntiOxCIN3 and (iv) membrane biophysical characterization to predict possible toxic effects—at membrane level.
In summary, AntiOxCIN3 is a potential drug candidate with application as antioxidant for the prevention/minimization of oxidative stress ascribed to neurodegenerative diseases, for which an oral administration is envisioned. Given the therapeutic potential of AntiOxCIN310 and since there are no reports in the literature concerning the interaction of mitochondriotropic antioxidants with biomimetic interfaces, we propose the use of a membrane modelling biophysical approach to evaluate AntiOxCIN3/biointerface interactions before progressing to the in vivo studies. This study of AntiOxCIN3/biointerface interactions is a valuable step in assessing the biodistribution profile of this type of antioxidants.
Materials and Methods
Synthesis of AntiOxCIN3
The synthetic strategy and general procedures used to obtain the mitochondriotropic antioxidant AntiOxCIN3 have been previously described in Teixeira et al.10
Materials
The lipids 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC), 1.2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) and the brain polar lipids (BPL) were obtained from Avanti Polar Lipids Inc. (INstruchemie, Delfzijl, Netherlands). The HSA and sodium deoxycholate (NaDC) were purchased from Sigma-Aldrich (Barcelona, Spain) at a purity grade of >99% and used without further purification. The fluorescent probes (±)-n-(9-anthroyloxy) stearic acid (n=3, 6, 9 and 12, named as n-AS probes) and 1.6-diphenyl-1,3,5-hexatriene (DPH) were purchased from Invitrogen-Molecular Probes (Paisley, UK). All other chemicals were purchased either from Sigma-Aldrich or Cymit Quimica (Barcelona, Spain) and were of the highest commercially available purity. All aqueous solutions were prepared with ultrapure water produced by Millipore Milli-Q (resistivity = 18.2 MΩ·cm) or adjusted for pH with universal buffer solution.
In silico Prediction of Drug-Like Properties
Several molecular descriptors relevant to complement biophysical studies were calculated by in silico analysis using the module MarvinSketch® from ChemAxon® software (Budapest, Hungary): number of hydrogen bonds, polar (PSA) and van der Waals surface area (VWSA) and ionization state.
Preparation and Labelling of Lipid Nanosystems for Membrane Interactions
The membrane model systems were prepared by the lipid film hydration method followed by extrusion as previously reported.40 Briefly, adequate portions of DMPC, DPPC or BPL were dissolved in chloroform, and subsequently evaporated to dryness under reduced pressure in a rotary evaporator (Buchi R210 Rotavapor, VWR International, Portugal). After hydration with ultrapure water, the suspension (1.00×10−3 M) was vortexed 20 min. The hydration was performed at a temperature above the main phase transition (Tm) of the lipid or lipid mixture (37 °C for DMPC and BPL; and 60 °C for DPPC). Lipid suspensions were then submitted to several freeze/thaw cycles and sequentially extruded through 400 nm, 200 nm and 100 nm nucleopore polycarbonate filters to produce large unilamellar vesicles (LUVs). LUVs are large unilamellar vesicles with a curvature closer to that of physiological membranes. As LUVs’ preparation methods are easy and reproducible they are widely used as biomimetic systems to study the biophysical interactions between bioactives and membranes.19,33,37,41,42 Besides lipid vesicles, micelles were also used as mimetic models of the biliary salt micelles and were prepared by aqueous dispersion of NaDC at a concentration above the critical micellar concentration (6 mM).43
In order to obtain labelled membrane nanosystems, ethanolic stock solutions of fluorescent probes were added to a chloroform solution of lipids in a lipid: probe molar ratio always greater than 100:1, to prevent structural changes of the membrane model system.34 Lipid nanosystems were then prepared using a lipid film hydration method followed by extrusion as described above.
Derivative Spectrophotometry Studies to Evaluate Membrane Distribution of AntiOxCIN3
The membrane distribution of AntiOxCIN3 in NaDC (Kd(NaDC)), DMPC (Kd(PC)) and BPL (Kd(BBB)) was determined by using increasing concentrations of the lipid model systems (0–3.00×10−3 M for DMPC and BPL; 0–5.00×10−2 M for NaDC) and a fixed concentration of AntiOxCIN3 (2.00×10−4 M), which were incubated for 30 minutes at 37 °C.
The absorption spectra (200–400 nm) were acquired (Perkin-Elmer Lambda 45 UV-VIS spectrophotometer) at 37.0 ± 0.1 °C for the NaDC and BPL membrane system and at 30, 33, 37, 40 and 45.0 ± 0.1 °C for the DMPC membrane model system. Derivative spectra were obtained from absorption spectra in accordance with the procedure previously published.44 In the third derivative spectra, the spectral interferences promoted by the lipid suspensions were removed. Kd values were obtained by a non-linear regression from the graphical representation of λmax or λmin as a function of the concentration of lipid nanosystems ([MembraneModel] in mol∙L−1):
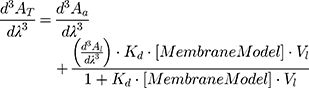
where AT stands for the total absorbance of drug (in lipid and in aqueous media) and Aa and Al are the absorbances in aqueous media and in lipid media, respectively. Vl refers to the lipid molar volume (L∙mol−1) and it is characteristic for each membrane model and can be calculated from the specific volumes of each lipid.21,45
Fluorescence Studies to Evaluate Interactions Between AntiOxCIN3 and Membrane Model Systems
Steady-State and Lifetime Fluorescence Measurements
DMPC membrane model system was labelled with n-AS probes, wherein n represents a well-defined hydrocarbon chain position of a stearic acid (carbon 2, 6, 9 or 12) where the fluorophore (anthracene group) is attached. The fluorescence of the fluorophore was monitored by lifetime and steady-state fluorescence studies to predict AntiOxCIN3 location at membrane level. Increasing concentrations of AntiOxCIN3 (0–3.0×10−2 M) and a fixed concentration of the labelled DMPC model system (5.0×10−2 M) were used. Steady-State fluorescence emission (Fluorolog-4 Spectrofluorometer, HORIBA Scientific) was obtained using λexcitation=379 nm. Fluorescence excitation and emission spectra were recorded using slits of 2 and 6 nm, respectively, and an integration time of 1 s was used. Time-resolved fluorescence measurements (ISS Chronos BH equipped with a picosecond laser C10196, Hamamatsu) were performed at λexcitation=379 nm, with a pulse duration of 80 ps, and set to a repetition rate of 10 MHz. A 435/40 nm BP filter (Brightline, Semrock) was placed in front of the photodetector (H7422P-40, Hamamatsu) whose signal was connected with the SYNC signal of the laser to the Time Correlated Single Photon Counting—TCSPC module (SPC130, Becker&Hickl). Fluorescence lifetimes were determined by fitting a bi-exponential decay model and using the ISS-VINCI analysis software. Both spectrometers were equipped with a temperature-controlled sample holder and measurements were made at 30, 37 and 45 °C to have additional data to distinguish the quenching mechanism.
The extent of fluorescence quenching induced by AntiOxCIN3 in n-AS probes (n = 3, 6, 9 or 12) was evaluated by Stern–Volmer constant (KSV) obtained by fitting data to the Stern–Volmer linear plots:
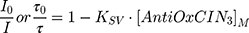
where (I, τ) and (I0, τ0) are the steady-state fluorescence emission and lifetime of the probe, respectively, in the presence or in the absence of AntiOxCIN3. [AntiOxCIN3]M is the membrane concentration of AntiOxCIN3 calculated as described elsewhere:21
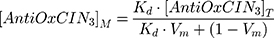
where Kd is the distribution coefficient of AntiOxCIN3 in the membrane model system (calculated as described in Equation 1), [AntiOxCIN3]T is the total concentration of AntiOxCIN3 and Vm is the membrane volume fraction.
The efficacy of AntiOxCIN3 to quench the fluorescence of each probe was evaluated by the bimolecular constant (Kq):45
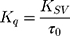
Kq is independent of intrinsic microenvironment changes sensed by each probe, which are reflected in the different values of τ0 (measured by lifetime fluorescence studies) and was used to infer the relative location of AntiOxCIN3 in the membrane model system. All fluorescence intensity data were corrected from absorption and inner filter effect.45
Fluorescence Anisotropy Measurements
The steady-state anisotropy, rSS, of DPH probe (5.0×10−6 M) incorporated in DPPC model system (5.0×10−2 M), before and after AntiOxCIN3 addition (2.0×10−2 M), was measured at 30.0 and 50.0 ± 0.1 °C (Fluorolog-4 Spectrofluorometer, HORIBA Scientific). These temperatures were chosen for the evaluation of the effect in the two lipid phases: gel phase, Lβ’, present at temperatures below Tm (ie < 41 °C), and fluid phase, Lα, at temperatures above Tm (ie > 41 °C). The probe was excited with vertically polarized light at 325 nm and the emitted fluorescence intensities were recorded between 400 and 600 nm with an integration time of 0.5 s and automatic insertion of emission polarizer parallel (IVV) or perpendicular (IVH) to the excitation polarizer allowing the determination of rSS:
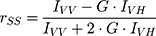
where G is the ratio between IHV and IHH and it is the internal correction factor for the sensitivity of the spectrofluorometer for vertically (IHV) and horizontally (IHH) polarized light.
Steady-State and Synchronous Fluorescence Quenching for Plasma Protein Binding Evaluation
The fluorescence excitation and emission spectra of increasing AntiOxCIN3 concentrations (0 to 3.5×10−4 M) with fixed HSA concentration (2.0×10−6 M) were acquired at 25.0, 30.0, 37.0, 40.0 and 45.0 ± 0.1 °C (Perkin-Elmer LS-50B), with λexcitation of 223 nm and λemission of 342 nm, both with slits of 5 nm. The quenching of HSA fluorescence promoted by increasing concentrations of AntiOxCIN3 can be described by the following binding isotherm equation:45
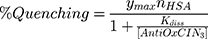
in which ymax is the maximum fluorescence quenching recorded, nHSA is the number of binding sites of HSA to AntiOxCIN3. Kdiss is the dissociation constant from which it is possible to calculate the binding constant (Kbind):
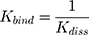
The % of plasma protein binding (PPB) can be obtained from the in vitro values of Kbind:46
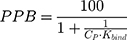
where CP is the physiological plasma protein concentration (750 μM).46
Furthermore, the next equation describes the relation between Kbind value and the Gibbs free energy (ΔG) of the binding complex by:
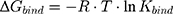
where R is the ideal gas constant and T is the temperature in Kelvin (K). This equation can also be written as the van’t Hoff equation:
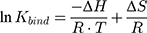
The synchronous fluorescence measurements were obtained by the simultaneous scan of the excitation and emission monochromators, taking 31 readings in the range of 200–500 nm with 5 nm of increment (Perkin Elmer LS50B). Characteristic Δλ values for tyrosine (15 nm) and tryptophan (60 nm) residues were used.47
Synchrotron Small- and Wide-Angle X-Ray Scattering Studies
For X-ray scattering experiments, the membrane model systems (DPPC and BPL) in the presence or absence of AntiOxCIN3 were transferred into X-ray transparent glass capillaries with 1.5 mm diameter (Hilgenberg, Malsfeld, Germany). Capillaries were sealed using a flame and stored at 4 °C. Small- and Wide-Angle X-ray Scattering (SAXS and WAXS) measurements were performed at the Austrian SAXS/WAXS beamline at the synchrotron light source ELETTRA (Trieste, Italy) employing monochromatic synchrotron radiation with wavelength of 1.54 Å and X-ray energy of 8 keV. WAXS and SAXS patterns were recorded using a2D Pilatus 100K and 2D Pilatus3 1M detector system, respectively, with a pixel size of 172 μm at positions that covered the typical diffraction spacing (s) range:
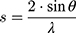
where λ is the wavelength and 2θ is the scattering angle of interest. The diffraction spacings (s) were calibrated using the lamellar peaks of silver behenate (SAXS) and p-bromo benzoic acid (WAXS) as standards. From the s values correspondent to the position of the first-order Bragg peak of each lipid phase, long distances (dL in the case of SAXS) and short distances (dS in the case of WAXS) were determined:
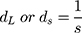
The full width at half maximum (fwhm) of the Bragg peaks were used to calculate the correlation length between the lipid bilayers (ξ):
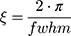
Measurements were performed in the ranges 20–56 °C and 20–90 °C, for DPPC and BPL membrane systems, respectively, both thermostated with a water bath (stability ± 0.1 °C, Unistat CC, Huber, Offenburg, Germany) to obtain the diffraction patterns of typical lipid phases (Lβ’, ripple phase Pβ’ and Lα) and the effect of AntiOxCIN3 in such phases was evaluated. The data acquired was analyzed as previously described in other reports.21
Dynamic and Electrophoretic Light Scattering Studies
The affinity of AntiOxCIN3 to HSA was confirmed by Dynamic and Electrophoretic Light Scattering (DLS and ELS). The hydrodynamic radii (Rh) and zeta-potential of free and complexed HSA were measured at 25.0, 30.0, 37.0, 40.0 and 45.0 ± 0.1 °C to follow the HSA-AntiOxCIN3 complexation.48 For each sample, at least three measurements with 100 s of stabilization time were performed.
DLS technique was also used to evaluate the effect of AntiOxCIN3 addition in the Tm and cooperativity (B) of the DPPC membrane model system.21,48 Thus, the light scattered (Mean Count Rate, MCR) by membrane system (5.0×10−2 M) in the absence and in the presence of AntiOxCIN3 (2.0×10−2 M) was monitored as function of temperature (T) between 30 and 55°C (with intervals of 1 °C and 2 min of equilibration time). The results were fitted with a sigmoidal function based on a modified Boltzmann equation:21,48
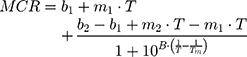
where m1 and m2 are the slopes resultant from the linear fitting to the data before and after Tm, respectively, and b1 and b2 are the corresponding y-intercepts.
All DLS and ELS studies were performed in Zetasizer Nano ZS (Malvern Panalytical, Paralab, Portugal), using disposable polystyrene cells with the addition of a dip cell (ZEN1002) when ELS studies were performed.
Modelling Biodistribution from in vitro Parameters
The LogKd value, which is dependent on the membrane model systems, can be used along with in silico descriptors to predict several biodistribution parameters using mathematical models. Thus, the value of LogKd(PC) was used to predict AntiOxCIN3 bioaccumulation (represented by the bioaccumulation constant, Kbioaccumulation), which is related to tissue blood flow (Q) and its volume (V):16
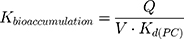
As an assessment of whether the drug is accumulated in adipose tissue, the adipose store index (ASI) of AntiOxCIN3 was calculated using the following equation:49
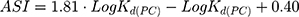
Moreover, according to Waterbeem and Kansy,50 the obtained distribution coefficient in the BPL model system – LogKd(BBB) – can be related to LogBB. LogBB can be defined as the logarithm of the ratio of drug concentrations in brain and in blood and reflects the relative affinity differences of the drug between the plasma proteins and brain tissue:51
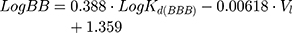
where Vl is the molar volume of the membrane model system used calculated from the specific lipid volumes.21,45
Additionally, it is possible to calculate the BBB permeability-surface area product (PS, quantified as LogPS):51
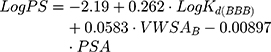
where VWSAB is the van der Walls surface area of the basic atoms that indicates the basicity of a compound.
One of the most useful pharmacokinetic parameters to describe the biodistribution of a drug is the Steady-State Volume Distribution (VDSS).52 This predictor translates the influence of the PPB, permeability, partitioning and active transport in the physiological distribution of the drug.52 The first physiological mathematical model of VDSS for predicting the biodistribution of the neutral and basic drugs was proposed with basis on human clinical pharmacokinetic data of a large set of drugs (670 drugs):53
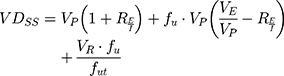
where VP and VE are the plasma and extracellular fluid volumes with corresponding value in human of 0.0436 and 0.0151 L∙Kg−1.53 RE/I represents the ratio of extravascular and intravascular proteins, and is strictly referred to the distribution of albumin, assuming an approximate value of 1.4.53 VR is defined as the physical volume into which the drug distributes minus the extracellular space (0.380 L∙Kg−1).53 fu and fut are, respectively, the unbound drug fraction in plasma and the unbound fraction in tissues.53
The value of fu can be obtained by:46
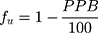
Finally, fut can be obtained by the equation proposed by Lombardo et al:53
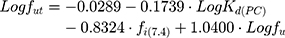
where fi(7.4) is the fraction of ionized drug at pH 7.4 which can be determined from its pKa.
Results and Discussion
Interaction Studies with Micelles Modelling Intestinal Absorption Enhancers
The NaDC is a well-known bile salt surfactant present in intestinal fluids of GI tract, which tends to form micellar structures aiding the passive absorption of compounds.54 The benefits of using NaDC to predict human intestinal absorption were described by Waters et al.54 The distribution of AntiOxCIN3 in the NaDC micellar system, expressed as LogKd(NaDC), was directly determined from the absorption spectra as the spectral interference of these systems only presents residual scattering (Figure S1).21 Plotting the absorbance values at the maximum absorbance (337 nm) as a function of NaDC micelles concentration and applying a non-linear fitting (Equation 1), a LogKd(NaDC) of 1.79 ± 0.56 was obtained. This value indicates a good solubilization of AntiOxCIN3 at small intestine level that is facilitated by the presence of the intestinal micelles. Additionally, in silico calculations showed that AntiOxCIN3 has 4 hydrogen bond acceptors and donors, a PSA value of 72.47 Å2 and a molecular weight of 619.25 Da. Following a relation described by Waters and co-workers,54 it is possible to estimate that 99% of AntiOxCIN3 will be absorbed at human intestinal level. Understandably, the pH variability of GI can influence the absorption and bioavailability of the drug.16 However, it is expected a good absorption along all the GI tract, since AntiOxCIN3 distribution in mimetic membrane/aqueous system was not dependent on pH changes (Table S1). The data is reinforced by the PSA value of AntiOxCIN3, as PSA < 140 Å2 has been described as a prerequisite for drug absorption.55 The intestinal absorption process involves the transport of the drug through the tight junctions of the intestinal epithelial and can occur by paracellular or transcellular processes, in which physicochemical properties are determinant.56 The molecular weight limit for paracellular pathway is approximately 400–500 Da,56 whereby compounds with higher molecular weights are described to permeate trough intestinal epithelium by transcellular processes.57 Accordingly, the results obtained suggest that AntiOxCIN3 has the physicochemical properties required for an effective passage through the GI tract and into the systemic circulation by transcellular pathway. As expected from a lipophilic compound, and as predicted from the biliary micelle mimetic systems, the absorption of AntiOxCIN3 might be enhanced in the presence of food due to micellar solubilization by the bile salts, uptake by the chylomicrons and lymphatic absorption.58,59
Plasma Protein Binding Studies
In systemic circulation, AntiOxCIN3 will be exposed to plasma proteins, like HSA. HSA is a globular protein, and its main physiological function is to transport endogenous and exogenous substances.60 Drug binding to HSA results in a complex that cannot be transported through epithelium tissue of blood capillaries, limiting the drug distribution to the therapeutic targets.60,61 Therefore, the drug biodistribution in target tissues is largely affected by its binding to serum proteins and the evaluation of drug-HSA binding is paramount to understand if the drugs’ biodistribution is effective.
HSA has three intrinsic fluorophores: tryptophan (Trp), tyrosine (Tyr) and phenylalanine (Phe) and changes in HSA fluorescence are associated with its interaction or binding to a variety of quenchers.62–65 As observed in Figure 2A, HSA fluorescence emission decreases with increasing concentrations of AntiOxCIN3, with a shift to higher wavelengths (from 334 to 352 nm). Emission spectral shifts can be used to estimate the surrounding hydrophobicity of the chromophore, since the fluorescence emission is strongly dependent on the local microenvironment.45
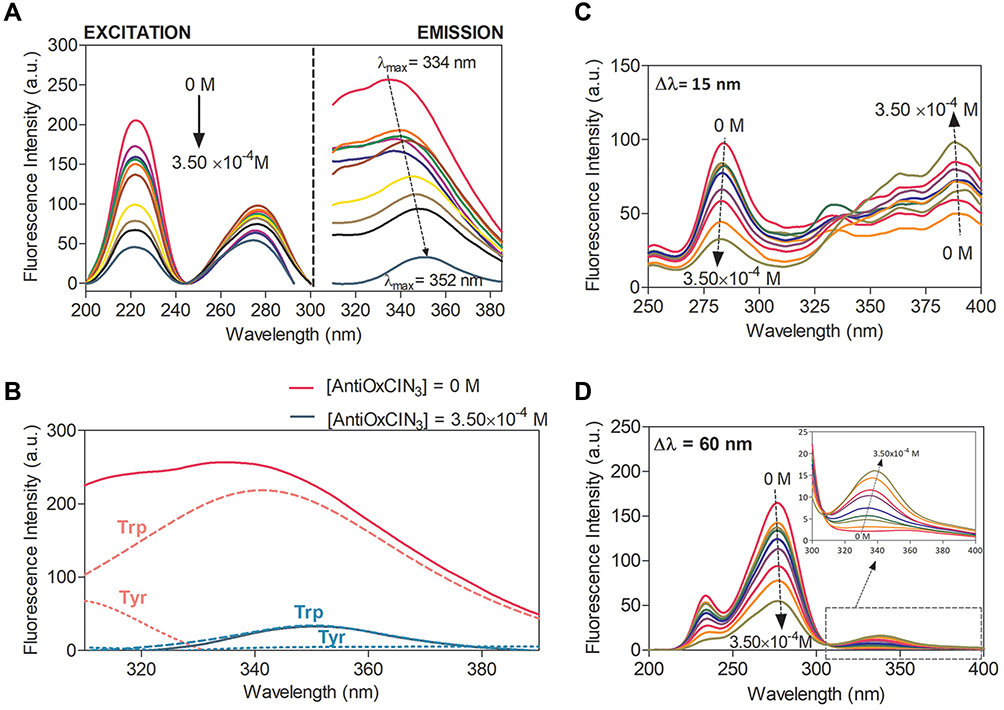 |
Figure 2 (A) Fluorescence spectra of HSA (2.0×10−6 M) in the presence of increasing AntiOxCIN3 concentrations (0 to 3.5×10−4 M) at 37 °C. (B) Deconvolution of HSA Trp (long dashes) and Tyr (short dashes) residues emission bands in the absence (pink) and in the presence (blue) of 3.5×10−4 M of AntiOxCIN3. (C) Synchronous fluorescence spectra of HSA (2.0×10−2 M) in absence and in presence of increasing concentrations of Anti-OxCIN3 (0 to 3.50×10−4 M) with Δλ = 15 nm corresponding to Tyr residues and (D) Δλ = 60 nm corresponding to Trp residues. |
Moreover, the stronger is the polarity the larger will be the solvent relaxation effect resulting ultimately in a longer emission wavelength.45 So, the observed shift is indicative of an increased polarity surrounding AntiOxCIN3 that can result from protein conformational changes or from the establishment of hydrogen bonds between AntiOxCIN3 and HSA. Similarly, to caffeic acid the hydrogen bonding may take place between the hydroxyl groups of AntiOxCIN3 and the polypeptide chain of albumin.66 Additionally, by the deconvolution of HSA emission spectra (Figure 2B), it was possible to observe that increasing AntiOxCIN3 concentrations promoted the deactivation of Tyr emission and a bathochromic shift of Trp emission band from 341 nm to 352 nm.
The conformational changes of HSA upon AntiOxCIN3 addition were evaluated by monitoring the synchronous fluorescence intensity of protein residues. By setting a Δλ value between excitation and emission wavelengths at 15 or 60 nm, it is possible to obtain the characteristic information about Tyr and Trp residues, respectively.47
In Figure 2 it is possible to observe that both Tyr (Figure 2C) and Trp residues (Figure 2D) were quenched with increasing AntiOxCIN3 concentrations, indicating that the HSA conformation has been changed. The AntiOxCIN3 addition promoted a small shift of maximum emission band from 275 nm to 277 nm in the Trp spectra (Figure 2D), indicating that the microenvironment around Trp was perturbed and the hydrophobicity decreased. On the other hand, the blue shift on the maximum emission of Tyr (Figure 2C) from 284 nm to 282 nm, indicates that the residues were exposed to a more hydrophobic environment.67
Next, similarly to other reported studies,68,69 a fluorescence quenching study was performed at different temperatures to gain insight into the molecular mechanism of AntiOxCIN3-HSA complexation and to explain the observed microenvironmental changes. The percentage of fluorescence quenching was represented as a function of AntiOxCIN3 concentration for each temperature (Figure S2A). Upon a non-linear fitting (Equation 6) of the fluorescence quenching data, Kdiss values in the magnitude of 10−5 M and Kbind values in the magnitude of 104 (Equation 7) were obtained for all the temperatures. From Kbind values it was determined a PPB of 94.76% (Equation 8) meaning that a major part of AntiOxCIN3 in solution is bound to HSA leaving only 5.24% of the free AntiOxCIN3 available to be distributed to the tissues.46 The magnitude of these values is indicative of a strong interaction between AntiOxCIN3 and HSA.45 Additionally, from the application of a linear fit of data according to van’t Hoff equation (Figure S2B), the thermodynamic parameters of the interaction were determined (Equations 9 and 10). The negative value of ΔG (−26.02 KJ·mol−1) suggests a spontaneous HSA-AntiOxCIN3 complexation.70 Furthermore, the positive values of ΔH (4.35 KJ·mol−1) and TΔS (0.03 KJ·mol−1·K−1) clearly indicate that the binding is entropically driven.71 According to Ross’s thermodynamic laws for the binding between small molecules and biomacromolecules, this type of entropic control of the binding process indicates that the hydrophobic interactions may play a predominant role in HSA-AntiOxCIN3 association.70
Gathering all the outcomes provided by fluorescence methods it was possible to predict a hydrophobic interaction between AntiOxCIN3 and the Tyr residue of HSA with a consequent protein conformational change that promotes the exposure of Trp residues to aqueous solution. As for caffeic acid, the aromaticity of the AntiOxCIN3 molecule might play an important role in hydrophobic interactions established between the phenyl group of AntiOxCIN3 and hydrophobic aminoacid residues present within the protein binding site.66
Additionally, to confirm the interaction, Rh and zeta-potential of free and complexed HSA with AntiOxCIN3 were measured by DLS and ELS, respectively, and the results obtained are summarized in Figure 3.
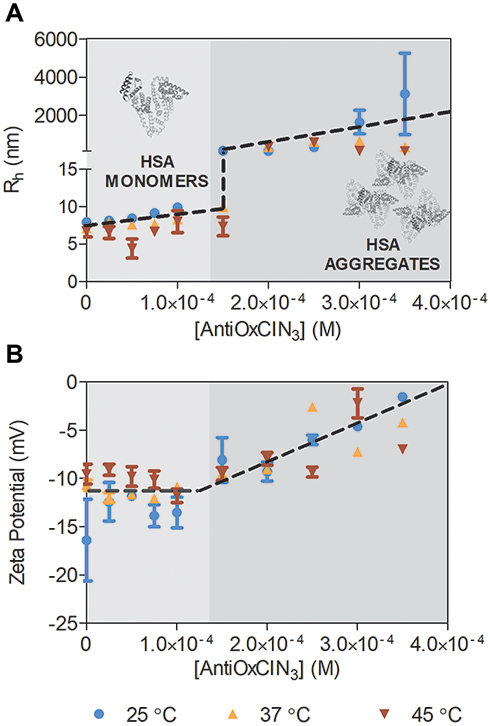 |
Figure 3 Graphic representation of Rh obtained from DLS (A) and Zeta Potential values (B) for HSA and HSA-AntiOxCIN3 ([AntiOxCIN3] from 0 to 3.50×10−4 M), at three different temperatures represented by different colors and signs. The black dashed line shows the tendency for neutralization upon AntiOxCIN3 addition observed by increase values of Rh and zeta potential. |
In the absence of AntiOxCIN3, the Rh of HSA was determined to be 7.97 nm, 7.13 nm and 6.75 nm, at 25.0, 37.0 and 45.0 °C, respectively. The data is coherent with the size range of HSA described in the literature.72 The progressive increasing of AntiOxCIN3 concentration provoked an increase in Rh values, suggesting the formation of the complex AntiOxCIN3–HSA (Figure 3A). Remarkably, at 2.0×10−4 M of AntiOxCIN3 a significant increase in the Rh value was observed, coinciding with the concentration at which the maximum fluorescence quenching was reached (Figure S2A). The large increase in Rh values suggests that HSA has reached the saturation binding point, promoting protein aggregation.
The ELS studies showed negative surface charge for free HSA, however, a charge neutralization tendency occurred upon AntiOxCIN3 addition (Figure 3B). This tendency resulted in reduced electrostatic repulsions which explain the formation of larger aggregates. These results corroborate the exposure of neutral Trp residues to the solvent upon AntiOxCIN3 interaction with Tyr residue, as predicted by fluorescence studies. Indeed, the progressive surface exposure of neutral residues will decrease the negative charge density of the protein, which justifies the observation of less negative zeta potential values, and ultimately less repulsion and higher tendency for aggregation.
As stated before, the evaluation of the binding parameters between plasma protein and drug candidates is an important factor to understand their distribution from the human plasma to body tissues and organs. Therefore, although intestinal absorption of AntiOxCIN3 is feasible, its distribution to the target tissues can be limited, by the strong binding to HSA (AntiOxCIN3 has a PPB of 94.76%). However, the strong binding to HSA cannot by itself be considered a final decisive factor for a poor biodistribution profile. Changes in protein binding may occur in vivo, for example, by competitive displacement between co-administration of drugs that share the same binding site.73 In addition, the binding of many drugs to HSA in patients can be changed due to several disease conditions (eg, kidney and liver diseases).73 Finally, it has been suggested that compounds with logD >3 and high PPB have an increase absorptive GI permeability, as the binding to albumin improves circulation and decreases the cellular accumulation, which may also lead to limited efficiency of drug’s intestinal metabolism.74 Therefore, AntiOxCIN3 may benefit from these effects being well absorbed orally, similarly to what has been predicted for related compounds, like MitoQ.74
Interaction with Lipid Nanosystems Used as Cell Membrane Mimetic Models
The ability of a drug to permeate the cell membranes through the phospholipid polar head region, diffusing through the lipophilic hydrocarbon chains and emerging into the inner region of phospholipid polar headgroups is determinant for its body distribution.18,75 The ability of AntiOxCIN3 to interact with membranes depends on an appropriate balance between liposolubility and hydrosolubility, which in turn is determined by its lipophilicity.16 Although lipophilicity is usually expressed as a partition of the drug in the octanol/water system, lipid membrane/water partition models, such as the PC model, are receiving increased attention as they can better predict drug biodistribution.15,34,48,76 In this regard, the concentration of the drug distributed in each phase of the binary lipid/water system can be determined by UV-VIS spectrophotometry.44 Increasing concentrations of PC membrane model system promoted light scattering in the absorbance spectra of AntiOxCIN3, which results in increased baseline values. Derivative method was applied to eliminate the background signals’ effect and to improve the resolution of overlapping bands (Figure 4A).21,34,44
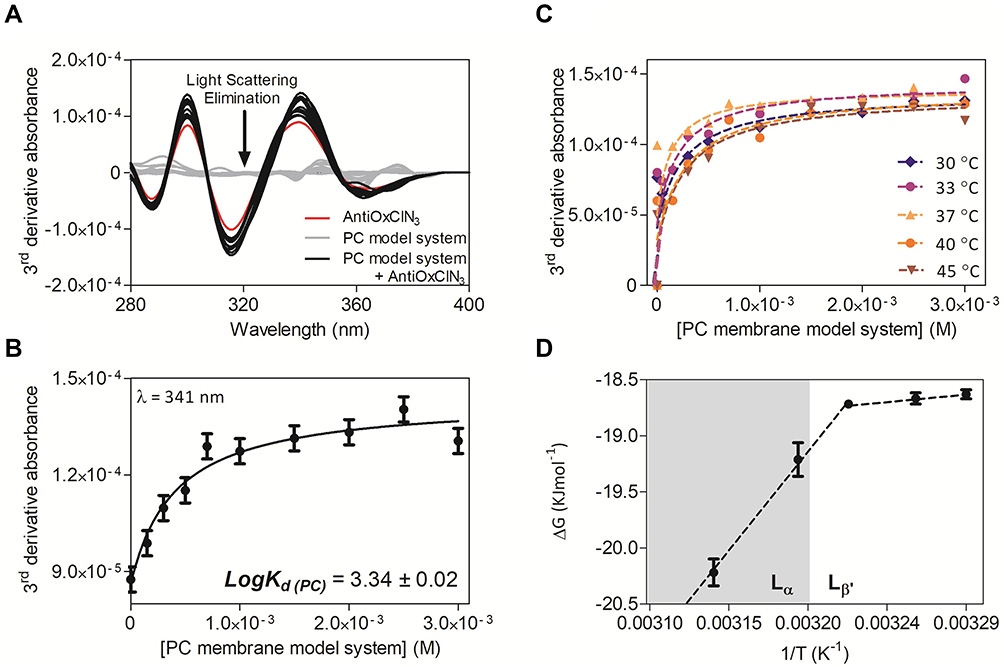 |
Figure 4 PC membrane model system partition coefficient of AntiOxCIN3 by derivative spectrophotometry: (A) Third derivative of AntiOxCIN3 (2.0×10−4 M) absorption spectra in aqueous buffered phase pH=7.4 (red), after incubation with PC membrane model system (0–3.0×10−3 M) (black) and PC membrane model system in the absence of AntiOxCIN3 (grey); (B) Non-linear fitting by Equation 1 of derivative absorbance at λ=341 nm as a function of PC membrane model system concentration. (C) Third derivative of the absorbance spectra and respective non-linear fitting by Equation 1 (dashed line) at several temperatures; (D) van’t Hoff plot representing the Gibbs free energy values as function of the inverse of temperature, in K−1, with the transition temperature between gel (Lβ’) and fluid (Lα) lipid phases highlighted. |
Thus, fitting the experimental data with Equation 1 (Figure 4B) at wavelengths where the scattering is eliminated (Figure 4A), a mean LogKd(PC) = 3.34 ± 0.02 (pH 7.4) was obtained. This value indicates a moderate to high lipophilicity (0 < LogKd < 5) and is characteristic of a drug with a good balance between solubility and permeability, which can be translated as optimal for cell membrane permeation and low metabolic liability.16,77
Additionally, AntiOxCIN3 has shown similar values of LogKd(PC) in a wide pH range (Table S1). The lipophilicity properties of AntiOxCIN3 influence its biodistribution and bioaccumulation.16 Considering the LogKd(PC) obtained by derivative spectroscopy as representative of the distribution in cell membranes of different organ/tissue, it was possible to estimate AntiOxCIN3 bioaccumulation sites (Kbioaccumulation determined using Equation 15). In general, AntiOxCIN3 is likely to bioaccumulate at more peripheral tissues: adrenal glands (61.44%), thyroid (30.72%) and kidneys (3.07%).16
Besides the estimated bioaccumulation of AntiOxCIN3 in off-target tissues, its ASI, that accounts for the partitioning of the drug in the adipose tissue, was determined to be 3.105 ± 0.016 (Equation 16), suggesting a high distribution in these tissues comparable to other neutral drugs such as clobazam.49
Additional biodistribution parameters were also determined using the values of PPB to HSA and LogKd(PC) obtained. The unbound AntiOxCIN3 fraction in plasma and tissues were respectively determined as fu=0.052 (Equation 20) and fut=0.011 (Equation 21). These parameters were used to obtain VDSS=1.89±0.43 L·Kg−1 (Equation 19), which agrees with the ASI value obtained. Indeed, drugs with VDSS values between 1 and 5 L·Kg−1 are characterized as having large volumes of distribution due to affinity for adipose tissues which hampers their elimination from the body.78 In fact the tendency to accumulate in adipose tissues and the large VDSS prolong the half-life of the compound, reducing its elimination rate.79 A large VDSS also means that the compound is more difficult to eliminate from the body by extracorporeal removal therapies (eg, dialysis).80
Lipophilicity is the net result of all intermolecular forces (hydrophobic, polar and/or ionic interactions) involved in the drug partition between two phases of different polarity.15 For the thermodynamic characterization of the intermolecular mechanisms involved in the distribution of AntiOxCIN3 in membranes, the LogKd(PC) was determined at five different temperatures (Figure 4C). The values are summarized in Table S2. The graphic representation of ΔG values as function of 1/T (K−1) (Figure 4D where data was fitted using Equations 9 and 10) evidenced a spontaneous process (ΔG < 0) with a biphasic behavior. The variation of enthalpy and entropy are related with the energetic requirements and the molecular randomness (increase or decrease in the molecular disorder), respectively, implied in AntiOxCIN3 transfer from the aqueous phase to the lipid media. When the lipid membrane model is in the fluid phase, both variations of enthalpy and entropy have positive values (ΔH= 43.9 KJ·mol−1 and TΔS= 0.200 KJ·mol−1), suggesting the presence of hydrophobic interactions between AntiOxCIN3 and PC membrane model system.70 However, when the lipid membrane is in the gel phase, the negative variation of enthalpy and positive entropy variation (ΔH = −14.9 KJ·mol−1 and TΔS = 0.012 KJ·mol−1) were indicative of the presence of strong van der Walls interactions. The biphasic behavior presented in Figure 4D suggests that with the temperature increase, the microviscosity and order of membrane decreased, which led to a better membrane penetration of AntiOxCIN3. This is also corroborated by the higher negative values of free binding energies, which indicate greater affinity of the ligand to the membrane lipid network.81 Additionally, from the gel phase to the fluid phase, a significant increase in enthalpy variation occurred indicating that intermolecular forces are stronger in the fluid phase and suggesting an entropically driven distribution of AntiOxCIN3 in the membrane model system.70 Moreover, considering that at physiological temperature (37 °C) cell membranes are mostly in the fluid phase (with more ordered domains confined to the lipid rafts),82 it can be concluded that AntiOxCIN3 tends to establish hydrophobic interactions with cell membranes in an entropic driven and spontaneous process.
To assess AntiOxCIN3 location in the membrane system, four probes, covering both polar and hydrophobic core of the membrane bilayer, containing the same fluorescent group located at different and well-defined depths of the bilayer (n-AS, where n= 3, 6, 9 and 12) were used.21,45 The ability of AntiOxCIN3 to quench each probe was monitored to obtain information about how deeply AntiOxCIN3 was buried within the bilayer structure.21 As the LogKd(PC) values of AntiOxCIN3 in lipid phase were previously determined, the effective concentrations of AntiOxCIN3 in the membrane model system – [AntiOxCIN3]M – are calculated using Equation 3.21 In Figure 5A, it can be observed that all quenching processes involved a static mechanism since the slope of the τ0/τ linear plot was nearly one. Static and collisional quenching can also be distinguished by their varying temperature dependence (Table S3). When Ksv values increase with the temperature, there is an indication of collisional quenching because higher temperatures result in faster diffusion. In this case, higher temperatures resulted in lower KSV values as a result of the decoupling of the complex formed between the probes and AntiOxCIN3. Therefore, from the analysis of the quenching process one can conclude that the quenching occurred by a static mechanism.
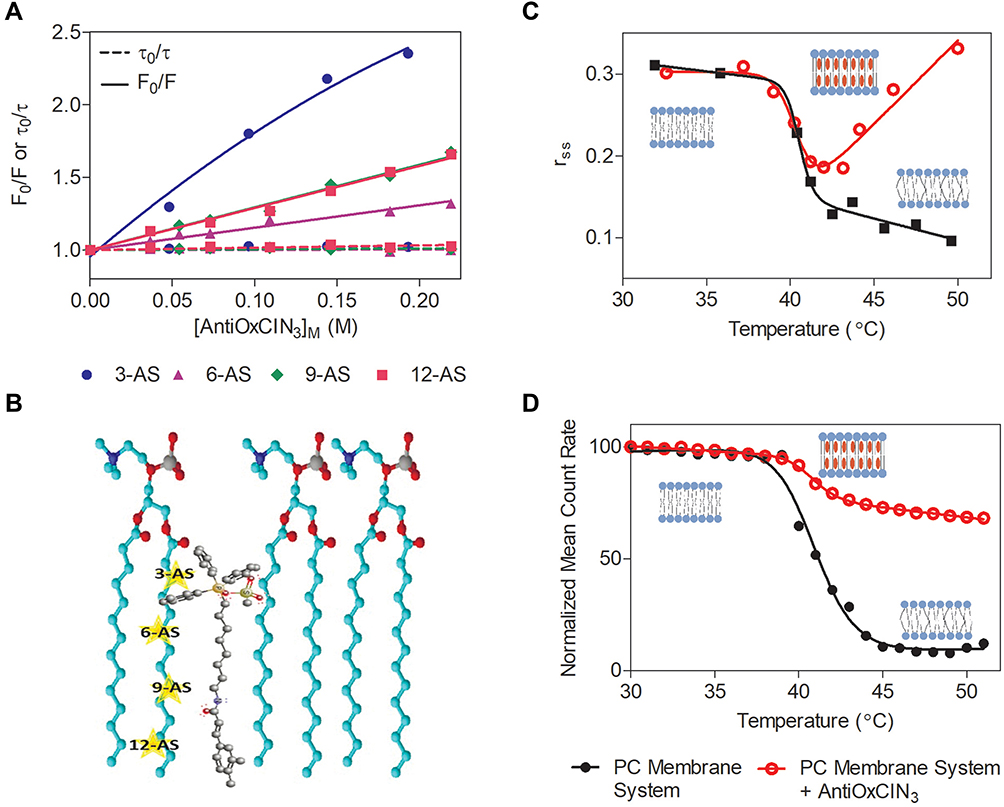 |
Figure 5 (A) Stern-Volmer plots of the probes n-AS incorporated in the PC membrane system at 37 °C as a function of AntiOxCIN3 ([AntiOxCIN3]M). Different colors and signs represent the different probes used. The full line represents the linear fitting applied to steady-state data. The dashed line represents the linear fitting applied to the data obtained from lifetime measurements. (B) Schematic representation of the possible location of the n-AS (n=3, 6, 9 and 12) fluorescent probes and AntiOxCIN3 within PC molecules of the PC membrane system. Membrane microviscosity assessment studies in the presence (red) and absence (black) of AntiOxCIN3 and respective sigmoidal fits: (C) Anisotropy, rss, profile of DPH probe in the PC membrane model system dependent on the temperature with fitting using Equation 3; (D) Sigmoidal profile of normalized mean count rate (MCR) obtained by DLS as function of temperature with sigmoidal fit using Equation 14. |
Using Equation 2, KSV values were determined from the slopes of the linear plots of the probes 6, 9 and 12-AS (Figure 5A). A deviation from linearity was observed in the Stern–Volmer representation of steady-state data related with the fluorescence quenching of 3-AS probe (Figure 5A). When the deviation is in form of an upward curvature generally it means that the extent of quenching can be explained by two types of quenching mechanisms: collisional encounters and static binding between quencher and probe.40,45 However, in our case there is no collisional mechanism of quenching because the slope of the τ0/τ linear plot was nearly one and Ksv decreases with temperature increase. Importantly, downward curvatures were observed in charged quenchers upon binding to the probe (in this case the TPP cation is closely located to the carboxylate anion from the stearic acid of 3-AS probe). Therefore, the analysis of the quenching process indicates a quenching occurring by a static mechanism, which suggests the existence of binding between 3-AS probe and a quencher group present in AntiOxCIN3. Consequently, it is possible to calculate KSV as an equilibrium binding constant (ie, the inverse of dissociation constant, Kdiss, determined by fitting data according to a nonlinear regression model described in Equation 6). From the KSV values and using Equation 4, Kq values were obtained for all the probes and summarized in Table 1.
 |
Table 1 Fluorescence Quenching Constants Obtained from Measurements of Fluorescence Quenching Studies for n-AS Fluorescence Probes in PC Membrane Model System at 37°C |
From the data acquired, it was concluded that AntiOxCIN3 was able to quench all the probes, with a more noticeable fluorescence quenching at 3-AS probe level. From the Kq values obtained, it is possible to establish a correlation with the quencher moieties present in AntiOxCIN3 and propose the most probable orientation of the drug in the lipid membrane. Considering the different chemical groups present in AntiOxCIN3 structure, aromatic rings display higher ability to quench the fluorescence emission of the probes. Hence, it is possible to predict an extended molecular orientation of AntiOxCIN3 parallel to the long axis of the phospholipid acyl chains, as represented in Figure 5B. The data agrees with the values of the high lipophilicity determined.
Once AntiOxCIN3 is deeply inserted into the membrane, it is crucial to understand how this compound can influence the physicochemical and biophysical state of the membrane bilayer. Parameters such as membrane fluidity and phospholipid order and/or packing play a key role in the maintenance of membrane dynamics that supports the most basic cellular functions21 and changes induced by drugs in these parameters can provide valuable information on their therapeutic ability and possible toxic effects at membrane level. Membrane microviscosity changes upon addition of drugs are usually monitored by steady-state fluorescence anisotropy.21 Steady-state anisotropy (rss) changes are related to the alterations in the rotational movement of the probe caused by differences in the stiffness of its surrounding matrix.21,34 Therefore, the rss of DPH probe was used to study the effect of AntiOxCIN3 on membrane’s fluidity (Equation 5). DPH locates at the hydrophobic region of the lipid bilayer, reporting the microviscosity of the acyl chains close to the center of the bilayer.83 In the gel phase (Lβ), the rss of DPH in PC membrane models was found to be between 0.32 (at 22.2 °C) and 0.23 (at 40.4 °C), and a lipid main phase transition to the fluid phase (Tm Lβ → Lα) occurred at 40.6 °C (Figure 5C black data). In the Lβ phase, the AntiOxCIN3 addition did not cause any significant change, since the rss values of the PC system with the drug were found to change between 0.33 (at 29.0 °C) and 0.24 (at 40.3 °C), presenting a Tm Lβ → Lα at 40.5 °C (Figure 5C red data). As expected, in the Lα phase, the rss of DPH in PC membrane model assumes lower values from 0.17 (at 41.2 °C) to 0.09 (at 55.7 °C). However, upon AntiOxCIN3 addition, these values increase to 0.19 (at 41.2 °C) and to 0.38 (at 54.0 °C). The surprising large increase of rss values promoted by AntiOxCIN3 reflects a higher lipid packing of the membrane model system in a deeper region where DPH is inserted. This is possibly due to AntiOxCIN3 intercalation in the hydrophobic region of the bilayer, as predicted by the location studies, and by the hydrophobic interactions that AntiOxCIN3 established with the phospholipids.
During the lipid phase transition there are distinct ordered phases,84 which are able to scatter light with different intensities. Accordingly, it is possible to monitor, within a temperature range and using DLS, the average number of photons scattered (mean count rate, MCR) by the lipid membrane system.85 This type of measurement results in a sigmoidal profile as the one presented in Figure 5D. Applying Equation 14 to the acquired data, it was possible to determine B, in the absence and presence of AntiOxCIN3, as 589.9 and 901.1, respectively. B is intimately connected with the mean size of the ordered or fluid lipid regions that can exist at different transition stages. In practical terms, a high cooperative transition is characterized as a transition where the fluid regions increase very rapidly with a simultaneous fast decrease of ordered regions, while in a less cooperative system there is a co-existence of fluid and ordered regions on an appreciable range.84 Additionally, although the Tm Lβ → Lα remained constant, there was a clear difference in the fluid phase between the PC lipid system without and with AntiOxCIN3. The AntiOxCIN3 addition provoked a clear increase in the number of photons detected in the fluid phase of PC lipid system, reflecting an increased stiffness when compared with PC membrane system without AntiOxCIN3.85 This observation corroborates the membrane stiffness effect observed by steady-state anisotropy studies. Thus, considering the gathered outcomes obtained by two different techniques, it was possible to conclude that AntiOxCIN3 addition to the PC membrane system promoted the formation of a tightly packed lipid structure.
The influence of AntiOxCIN3 on the biophysical properties of membranes, such as long-range bilayer order and hydrocarbon chain packing, was explored by SAXS and WAXS. In the Lβ phase, the SAXS pattern obtained for fully hydrated PC lipid system showed three Bragg peaks with characteristic positions of lamellar phases. Upon AntiOxCIN3 addition, the three Bragg peaks disappeared, originating a broader single peak (Figure S3). The less intense and broader Bragg peak revealed a disturbing effect of AntiOxCIN3 in the lipid structure. From the first-order Bragg peak of each lipid phase, long spacing (dL) and correlation length between bilayers (ξ) were determined (Equations 11, 12 and 13). The obtained profiles of dL values in the presence and absence of AntiOxCIN3 as function of temperature are represented in Figure S3. These profiles indicated that for all the temperatures dL increased upon AntiOxCIN3 addition. In fact, the incorporation of AntiOxCIN3 promoted an increase of the bilayer thickness up to approximately 20 Å, in the Lβ phase, and 30 Å, in the Lα phase. This substantial increase can result from changes in the hydration behavior and/or from an alteration in phospholipid headgroups orientation, with reduction of the tilt angle of the chains, due to interaction with AntiOxCIN3. According to Fernandes et al,21 a difference of 6.1 Å in the bilayer thickness is expected when the loss of tilt angle of hydrocarbon chains occurs. However, the changes observed in the tilt angle alone are not enough to explain the increased dL observed due to the incorporation of AntiOxCIN3, which means that an increase in the hydration layer is likely to occur along with the changes in the tilt angle. Additionally, the deconvolution of the first-order Bragg peak in the presence of AntiOxCIN3 (Figure S3) showed a non-influenced phase (dL similar to that of pure PC membrane system) and an influenced phase (dL higher than pure membrane system). The incorporation of AntiOxCIN3 into PC membrane systems is responsible for pronounced changes in ξ values. In PC membrane system, diffraction peaks presented high ξ values – 13,291 Å (in the Lβ phase) and 8763 Å (in the Lα phase) – which indicated a good correlation between bilayers. When AntiOxCIN3 was added, these values decreased to 574 Å (in the Lβ phase) and 895 Å (in the Lα phase), which is indicative of a disturbing effect in the global molecular organization of the multilayer stack of lipid bilayer.
From the WAXS patterns obtained for PC membrane system (Figure 6A), a single symmetric Bragg peak was observed that allowed determining a short spacing dS value around 4.13 Å (Equations 11 and 12), characteristic of a hexagonal chain packing and a cross-sectional area of 19.65 Å2.26 The high ξ value (Equation 13) obtained (1718 Å) indicated a good correlation between bilayers. As expected, after Tm Lβ → Lα (T>45 °C) there was no Bragg peak in the WAXS region since the lipid bilayers were in the fluid phase. Upon AntiOxCIN3 addition, the symmetric Bragg peaks gave rise to a double asymmetric Bragg peak with short spacings characteristic of a pseudo-hexagonal lattice of the chains in the bilayers (d20 = 4.14 Å and d11=4.18 Å) and a cross-sectional area of 19.93 Å2 (Figure 6B).
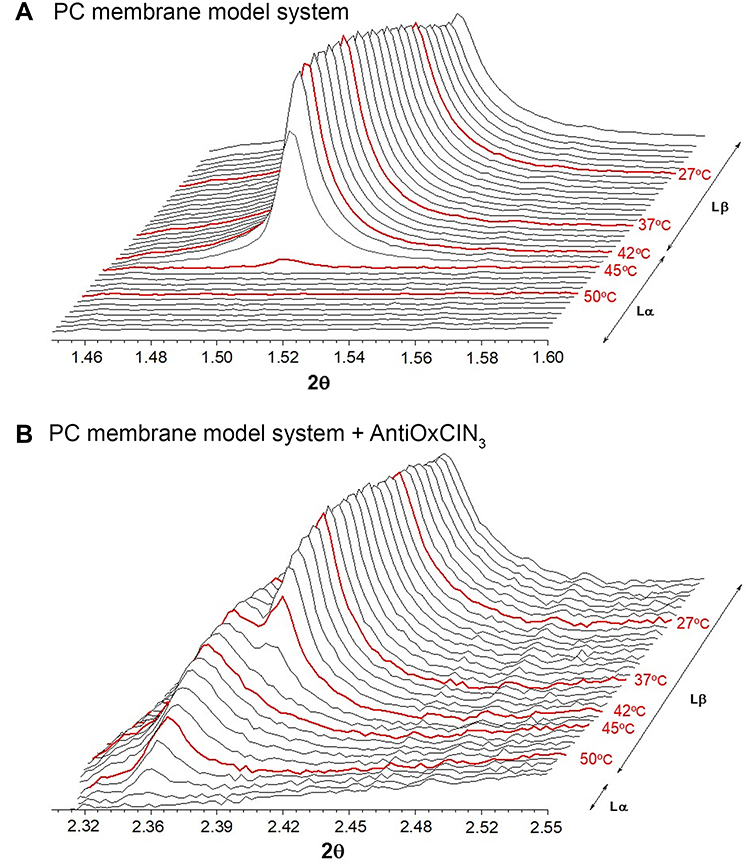 |
Figure 6 WAXS patterns of PC membrane system in the absence (A) and presence (B) of AntiOxCIN3 with some temperatures highlighted in red. |
The intercalation of AntiOxCIN3 reduced the ξ (ξ = 471 Å), suggesting a disturbing effect in membrane multilayer structure. For T>45 °C, where no Bragg peaks were expected to appear (due to the fluidization of the lipid bilayer), it was still visible an asymmetric single Bragg peak, with ds≈4.23 Å and a cross-sectional area of 20.81 Å2, which only disappeared at T>50 °C (Figure 6B). The results obtained by a third different technique corroborate the membrane stiffening effect caused by AntiOxCIN3 which was also observed by the other two techniques (DLS and steady-state anisotropy). As suggested by the thermodynamic studies, AntiOxCIN3 establishes hydrophobic interactions with membrane phospholipids. These interactions might be the cause for the membrane stiffening effect observed. Finally, the observed increase of lipid cross-sectional area upon AntiOxCIN3 addition indicates the penetration of the drug within the aliphatic core of the membrane confirming its intercalation between the hydrophobic lipid chains observed in the location studies.
BBB Membrane Interaction Studies
Drug delivery to the CNS represents a challenging task due to the presence of the BBB. It is one of the most important blood–CNS interfaces, protecting the brain from potentially harmful xenobiotics while simultaneously regulating the transport of essential molecules and maintaining a stable milieu for neuronal function.86 The BBB gatekeeper function is essentially a result of the endothelial cells lining the brain capillaries sealed together by an intricate network of tight junctions, which restrict severely the transport of the drugs. Since one of AntiOxCIN3 potential therapeutic targets is the CNS, where the new chemical entity can exert its antioxidant effect, it is important to predict its ability to permeate through BBB. Therefore, the BBB endothelial cellular membranes were mimicked by a lipid nanosystem composed of brain phospholipids (BPL) and used as model to predict how AntiOxCIN3 interacts with BBB lipid barrier. This interaction can be inferred using derivative spectroscopy, as performed in the previous studies (Figure S4). In the third derivative of absorbance spectra an evident bathochromic shift occurred, suggesting changes in polarity of microenvironment surrounding AntiOxCIN3 due to its distribution in the lipid phase. A LogKd(BBB) of 3.64 ± 0.25 (Equation 1) was obtained which is indicative of AntiOxCIN3 tendency to distribute in BBB endothelial cell membrane. Moreover, several mathematical models and/or in silico descriptors can be used to classify a drug accordingly to its BBB distribution. Thus, a drug can be classified as BBB+ or BBB- using the threshold cut-offs described in the literature.87 From the application of Equation 17, it was possible to determine a LogBB of 2.77 ± 0.10, which suggests potential BBB permeability.88 Moreover, using Equation 18 and considering VWSAB as 0.59, a LogPS of −1.88 was obtained. Bearing in mind the reported cut-off schemes, AntiOxCIN3 is classified as BBB+.87 Thus, from the interaction studies of AntiOxCIN3 with BBB membrane model system, it is predicted that AntiOxCIN3 can permeate BBB.
Considering the high distribution coefficient of AntiOxCIN3 in the BBB membrane system, SAXS studies were performed in BPL to study the possible order perturbations caused by the drug in this biological interface. The SAXS pattern of BPL in the presence of AntiOxCIN3 presents two Bragg peaks characteristic of a lamellar phase organization at 20 °C with a dL = 5.83 nm (Figure 7). A high correlation between bilayers (ξ = 8918 Å) was obtained, suggesting that the drug does not disturb the multilayer stack of the lipid bilayer. However, as previously reported,89 at 20 °C the SAXS pattern of BPL presents a Lα phase with dL = 9.86 nm that upon heating, at 40 °C, is converted into an inverted hexagonal phase (HII), with a lattice parameter a=2d/√3=7.72 nm. The 4 nm decrease of dL value associated with the high correlation between bilayers suggests that AntiOxCIN3 addition provokes a reduction of the hydration layer between the lipid bilayers. Furthermore, upon the addition of AntiOxCIN3 to BPL a HII phase with a lattice parameter a = 7.59 nm is observed already at 30 °C (Figure 6C). The ability of AntiOxCIN3 to significantly decrease the Lα-to-HII phase transition temperature (TH) is relevant from a therapeutic point of view. Actually, a similar behavior was observed for some natural bioactive fatty acids on lipid membranes.90,91 The effects of these bioactive fatty acids included the capacity to reduce the blood pressure (HII phase induction has regulatory effects on G protein-associated signaling cascades); antiproliferative effects (HII phase induction promotes the subsequent recruitment of protein kinase C to the cell membrane) and the ability to promote membrane fusion and thus interfere with related cellular processes (endo/exocytosis, cell division).90 Focusing on the effect of AntiOxCIN3 in BPL, the observed TH reduction can as well be related to membrane fusion processes92,93 and to an increase of BBB permeability,94 however this type of effects must be confirmed by cellular assays in a near future. However, at this point, it is possible to assure that the TH reduction effect observed by SAXS agrees with the BBB+ classification and supports the assumption that AntiOxCIN3 is able to permeate BBB.
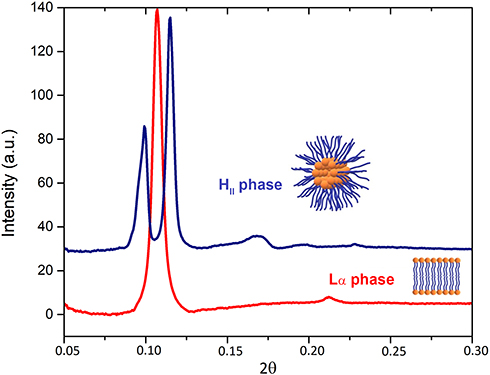 |
Figure 7 SAXS patterns of a BPL membrane model system in the presence of AntiOxCIN3 before (red line) and after (blue line) the transition between lamellar phase (Lα) to inverted hexagonal phase (HII). |
Conclusions
In this study, the drug-like properties of AntiOxCIN3, a novel mitochondriotropic antioxidant previously described by Teixeira et al,10 were characterized using lipid nanosystems and serum protein as relevant biomimetic interfaces to obtain important information correlated with its in vivo biodistribution.
Our results demonstrated that AntiOxCIN3 is expected to be effectively absorbed in GI tract through transcellular process and reach the bloodstream. At the bloodstream, the compound is predicted to strongly interact with the plasma albumin, with more than 90% of AntiOxCIN3 binding to albumin, causing changes in protein conformation. The high affinity of AntiOxCIN3 to plasma protein is in line with its determined high lipophilicity. Tendency to bioaccumulate in peripheral tissues and adipose tissues is also predicted and corroborated by the VDSS value (1.89±0.43 L·Kg−1). This value is in the range of the drugs often characterized by having a large volume of distribution and a slow rate of elimination from the body. Additionally, AntiOxCIN3 interacts with cellular membranes through hydrophobic forces and tends to be located parallel to the long axis of phospholipid acyl chains. In this interaction there are no signs of major disturbance on the membrane transition temperature and cooperativity which points to the absence of membrane biophysical impairment and thus lack of membrane toxicity. However, the preferred location of the drug at bilayer level explains the observed increase in membrane stiffness confirmed by all the complementary techniques applied (steady-state anisotropy, DLS, SAXS and WAXS). Considering the widespread involvement of oxidative stress in several diseases, including disturbance effects observed at CNS level (eg, brain homeostasis disorders that predispose to neurodegenerative conditions, harmful effects on the expression of important molecules involved in BBB integrity, and apoptosis of pericytes95) the predicted ability of AntiOxCIN3 to overcome BBB and exert its antioxidant effect in the brain, is a positive outcome when considering its potential therapeutic use.
As reviewed by Murphy et al,13 the covalent link of a TPP lipophilic cation to improve the delivery of antioxidants has proven to be an efficient strategy in vivo. In fact, the results reviewed showed that this class of mitochondriotropic antioxidants (in which AntiOxCIN3 is included) are able to pass easily through all biological membranes, including BBB, and showed a tendency to be located at the hydrophobic portion of the lipid/water interface, ie, close to the phospholipids’ fatty acyl groups level. The ability of other type mitochondriotropic antioxidants to be absorbed from gut to the bloodstream was also reported.13 Overall, our results are in accordance with these studies, which reinforces the importance to use biomimetic models as an approach for the prediction of the drug biological behavior, as well as, to understand how the molecular structure might affect such behavior. In this regard, our studies indicate that AntiOxCIN3 might have a good biodistribution profile, being able to be absorbed at GI tract and overcome membrane barriers of off-target tissues and BBB. The only hindrance to the effective biodistribution of AntiOxCIN3 may be its strong binding to HSA. However, this apparent drawback can be tuned with either a structural optimization to reduce AntiOxCIN3 lipophilicity or its encapsulation in stealth nanocarriers systems. On the other hand, the strong HSA binding of MitoQ10 (lipophilic compounds similar to AntiOxCIN3) has been reported as an advantage to avoid cellular accumulation of compounds leading to limited intestinal metabolic conversion and accelerating absorptive transport.74
Regarding the membrane toxicity effects of AntiOxCIN3, they seem to present no concern, as high molar ratios of the drug did not induce relevant membrane impairment, which can cause loss of membrane integrity. The major biophysical effects of this mitochondriotropic antioxidant was its surprising capacity to increase membrane stiffness in the fluid phase and its ability to facilitate HII phase in BBB endothelial membrane model systems. The underlying mechanisms by which AntiOxCIN3 and other mitochondriotropic antioxidants influence mitochondrial membrane structure is currently being studied by our group and will be object of a follow up study.
In conclusion, herein we provide an innovative toolbox of biophysical studies, in silico predictors and lipid nanosystems used as mimetic models to study the interaction between the drug and biological interfaces towards a comprehensive molecular-level understanding of drug’s biodistribution profile. This toolbox is a simple and useful approach for predicting future pharmacokinetic issues of novel drug candidates, in the early stages of pre-clinical studies of the drug development process. It can be used to scrutinize several optimize leads and select the ones that have potential better biodistribution profile to be tested in vivo. These in vitro assays are not surrogating in vivo assays, as they have limitations in mimicking the complexity of biological barriers but will be essential to understand the molecular-level properties of novel drugs and drug candidates. Moreover, under controlled conditions that are not feasible in vivo, the in vitro assays may help to rationalize and predict interactions with the bioenvironment. Finally, the work herein proposed is a contribution for the protection of animals used for scientific purposes (Directive 2010/63/EU) in agreement with the EU principles of Replacement (of animals for other in vitro models), Reduction (of animal tests) and Refinement (the “Three Rs”).
Acknowledgments
This work was supported by Fundação para a Ciência e Tecnologia (FCT) in the framework of the Strategic Funding Funding [UID/FIS/04650/2019], and by the project CONCERT [POCI-01-0145-FEDER-032651 and PTDC/NAN-MAT/326512017], co-financed by the European Regional Development Fund (ERDF), through COMPETE 2020, under Portugal 2020, and FCT I.P. The authors thank Elettra Sincrotone, Trieste, Italy, for beam time and support through the project 20155321. We also acknowledge UIDB/00081/2020, PTDC/MED-FAR/29391/2017, PTDC/BIA-MOL/28607/2017, PTDC/MED-QUI/29164/2017. M Lúcio thanks FCT and ERDF for doctoral position [CTTI-150/18-CF (1)] in the ambit of the project CONCERT. Jana Nieder acknowledges funding by the CCDR-N via the grant: NORTE-01-0145-FEDER-000019. Sofia Benfeito and Eduarda Fernandes (SFRH/BD/147938/2019) grants are supported by FCT, POPH and FEDER/COMPETE.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408(6809):239–247. doi:10.1038/35041687
2. Islam MT. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol Res. 2017;39(1):73–82. doi:10.1080/01616412.2016.1251711
3. Scandalios JG. Genomic responses to oxidative stress. Mol Cell Biol Mol Med. 2006;489–512.
4. Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 2003;17(10):1195–1214. doi:10.1096/fj.02-0752rev
5. Chakravarti B, Chakravarti DN. Oxidative modification of proteins: age-related changes. Gerontology. 2007;53(3):128–139. doi:10.1159/000097865
6. Ballatore C, Huryn DM, Smith AB
7. Murphy MP. Targeting lipophilic cations to mitochondria. Biochim Biophys Acta. 2008;1777(7–8):1028–1031. doi:10.1016/j.bbabio.2008.03.029
8. Smith RA, Murphy MP. Mitochondria-targeted antioxidants as therapies. Discov Med. 2011;11(57):106–114.
9. Amorim R, Benfeito S, Teixeira J, Cagide F, Oliveira PJ, Borges F. Targeting mitochondria: the road to mitochondriotropic antioxidants and beyond. Mitochondrial Biol Exp Ther. 2018;333–358.
10. Teixeira J, Cagide F, Benfeito S, et al. Development of a mitochondriotropic antioxidant based on caffeic acid: proof of concept on cellular and mitochondrial oxidative stress models. J Med Chem. 2017;60(16):7084–7098. doi:10.1021/acs.jmedchem.7b00741
11. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1–3):3–26. doi:10.1016/S0169-409X(00)00129-0
12. Benfeito S, Oliveira C, Fernandes C, et al. Fine-tuning the neuroprotective and blood-brain barrier permeability profile of multi-target agents designed to prevent progressive mitochondrial dysfunction. Eur J Med Chem. 2019;167:525–545. doi:10.1016/j.ejmech.2019.01.055
13. Murphy MP, Smith RA. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol. 2007;47(1):629–656. doi:10.1146/annurev.pharmtox.47.120505.105110
14. Teixeira J, Oliveira C, Amorim R, et al. Development of hydroxybenzoic-based platforms as a solution to deliver dietary antioxidants to mitochondria. Sci Rep. 2017;7(1):6842. doi:10.1038/s41598-017-07272-y
15. Liu X, Testa B, Fahr A. Lipophilicity and its relationship with passive drug permeation. Pharm Res. 2011;28(5):962–977. doi:10.1007/s11095-010-0303-7
16. Di L, Kerns EH. Drug-Like Properties: Concepts, Structure Design and Methods from ADME to Toxicity Optimization. Elsevier Science; 2015.
17. Seydel JK. Drug-membrane interaction and pharmacokinetics of drugs. Drug Membrane Interact. 2003;141–215.
18. Pignatello R, Musumeci T, Basile L, Carbone C, Puglisi G. Biomembrane models and drug-biomembrane interaction studies: involvement in drug design and development. J Pharm Bioallied Sci. 2011;3(1):4–14. doi:10.4103/0975-7406.76461
19. Lucio M, Lima JL, Reis S. Drug-membrane interactions: significance for medicinal chemistry. Curr Med Chem. 2010;17(17):1795–1809. doi:10.2174/092986710791111233
20. Stefaniu C, Brezesinski G, Mohwald H. Langmuir monolayers as models to study processes at membrane surfaces. Adv Colloid Interface Sci. 2014;208:197–213. doi:10.1016/j.cis.2014.02.013
21. Fernandes E, Soares TB, Goncalves H, et al. A molecular biophysical approach to diclofenac topical gastrointestinal damage. Int J Mol Sci. 2018;19(11):3411. doi:10.3390/ijms19113411
22. Lúcio M, Bringezu F, Reis S, Lima JLFC, Brezesinski G. Binding of nonsteroidal anti-inflammatory drugs to DPPC: structure and thermodynamic aspects. Langmuir. 2008;24(8):4132–4139. doi:10.1021/la703584s
23. Lucio M, Ferreira H, Lima FC, Reis S. Interactions between oxicams and membrane bilayers: an explanation for their different COX selectivity. Med Chem (Los Angeles). 2006;2(5):447–456. doi:10.2174/157340606778250199
24. Lúcio M, Ferreira H, Lima JLFC, Reis S. Use of liposomes as membrane models to evaluate the contribution of drug–membrane interactions to antioxidant properties of etodolac. Redox Rep. 2013;13(5):225–236. doi:10.1179/135100008X308939
25. Lúcio M, Ferreira H, Lima JLFC, Reis S. Use of liposomes to evaluate the role of membrane interactions on antioxidant activity. Anal Chim Acta. 2007;597(1):163–170. doi:10.1016/j.aca.2007.06.039
26. Nunes C, Brezesinski G, Lima JL, Reis S, Lucio M. Synchrotron SAXS and WAXS study of the interactions of NSAIDs with lipid membranes. J Phys Chem B. 2011;115(24):8024–8032. doi:10.1021/jp2025158
27. Nunes C, Brezesinski G, Pereira-Leite C, Lima JL, Reis S, Lucio M. NSAIDs interactions with membranes: a biophysical approach. Langmuir. 2011;27(17):10847–10858. doi:10.1021/la201600y
28. Nunes C, Brezesinski G, Lopes D, Lima JLFC, Reis S, Lúcio M. Lipid–drug interaction: biophysical effects of tolmetin on membrane mimetic systems of different dimensionality. J Phys Chem B. 2011;115(43):12615–12623. doi:10.1021/jp206013z
29. Bailey-Hytholt CM, Shen TL, Nie B, Tripathi A, Shukla A. Placental trophoblast-inspired lipid bilayers for cell-free investigation of molecular interactions. ACS Appl Mater Interfaces. 2020;12(28):31099–31111. doi:10.1021/acsami.0c06197
30. Uehara TM, Marangoni VS, Pasquale N, Miranda PB, Lee KB, Zucolotto V. A detailed investigation on the interactions between magnetic nanoparticles and cell membrane models. ACS Appl Mater Interfaces. 2013;5(24):13063–13068. doi:10.1021/am404042r
31. Khadka Nawal K, Cheng X, Ho Chian S, Katsaras J, Pan J. Interactions of the anticancer drug tamoxifen with lipid membranes. Biophys J. 2015;108(10):2492–2501. doi:10.1016/j.bpj.2015.04.010
32. Sarpietro MG, Accolla ML, Santoro N, et al. Calorimetry and Langmuir–Blodgett studies on the interaction of a lipophilic prodrug of LHRH with biomembrane models. J Colloid Interface Sci. 2014;421:122–131. doi:10.1016/j.jcis.2014.01.040
33. Peetla C, Stine A, Labhasetwar V. Biophysical interactions with model lipid membranes: applications in drug discovery and drug delivery. Mol Pharm. 2009;6(5):1264–1276. doi:10.1021/mp9000662
34. Fernandes E, Soares TB, Goncalves H, Lucio M. Spectroscopic studies as a toolbox for biophysical and chemical characterization of lipid-based nanotherapeutics. Front Chem. 2018;6:323. doi:10.3389/fchem.2018.00323
35. Seddon AM, Casey D, Law RV, Gee A, Templer RH, Ces O. Drug interactions with lipid membranes. Chem Soc Rev. 2009;38(9):2509. doi:10.1039/b813853m
36. Bunea A-I, Harloff-Helleberg S, Taboryski R, Nielsen HM. Membrane interactions in drug delivery: model cell membranes and orthogonal techniques. Adv Colloid Interface Sci. 2020;281:281. doi:10.1016/j.cis.2020.102177
37. Jelinek R, Kolusheva S. Membrane interactions of host-defense peptides studied in model systems. Curr Protein Pept Sci. 2005;6(1):103–114. doi:10.2174/1389203053027511
38. Nunes C, Sousa CT, Proença MP, et al. Synchrotron small angle X-ray scattering for the evaluation of the interaction of silica nanotubes with lipid membranes. RSC Adv. 2013;3(26):10323. doi:10.1039/c3ra22974b
39. Wang S, Guo H, Li Y, Li X. Penetration of nanoparticles across a lipid bilayer: effects of particle stiffness and surface hydrophobicity. Nanoscale. 2019;11(9):4025–4034. doi:10.1039/C8NR09381D
40. Monteiro JP, Martins AF, Lucio M, et al. Nimesulide interaction with membrane model systems: are membrane physical effects involved in nimesulide mitochondrial toxicity? Toxicol in Vitro. 2011;25(6):1215–1223. doi:10.1016/j.tiv.2011.05.014
41. Hu X, Tam K. Biomembrane mimics and their roles in anti-bacterial drug discovery. ADMET DMPK. 2017;5(1):9. doi:10.5599/admet.5.1.375
42. Sarkis J, Vie V. Biomimetic models to investigate membrane biophysics affecting lipid-protein interaction. Front Bioeng Biotechnol. 2020;8:270. doi:10.3389/fbioe.2020.00270
43. Jojart B, Posa M, Fiser B, Szori M, Farkas Z, Viskolcz B. Mixed micelles of sodium cholate and sodium dodecylsulphate 1:1 binary mixture at different temperatures–experimental and theoretical investigations. PLoS One. 2014;9(7):e102114. doi:10.1371/journal.pone.0102114
44. Magalhaes LM, Nunes C, Lucio M, Segundo MA, Reis S, Lima JL. High-throughput microplate assay for the determination of drug partition coefficients. Nat Protoc. 2010;5(11):1823–1830. doi:10.1038/nprot.2010.137
45. Lakowicz JR. Principles of Fluorescence Spectroscopy.
46. Musteata FM. Clinical utility of free drug monitoring. Ther Drug Monit. 2012;75–101.
47. Rahnama E, Mahmoodian-Moghaddam M, Khorsand-Ahmadi S, Saberi MR, Chamani J. Binding site identification of metformin to human serum albumin and glycated human serum albumin by spectroscopic and molecular modeling techniques: a comparison study. J Biomol Struct Dyn. 2015;33(3):513–533. doi:10.1080/07391102.2014.893540
48. Carvalho AM, Fernandes E, Goncalves H, et al. Prediction of paclitaxel pharmacokinetic based on in vitro studies: interaction with membrane models and human serum albumin. Int J Pharm. 2020;580:119222. doi:10.1016/j.ijpharm.2020.119222
49. Seydel JK, Wiese M. Drug-Membrane Interactions Analysis, Drug Distribution, Modeling. Vol. 5. Weinheim: Wiley-VCH; 2002.
50. van de Waterbeemd H, Kansy M. Hydrogen-bonding capacity and brain penetration. CHIMIA Int J Chem. 1992;46(7–8):299–303.
51. Liu X, Tu M, Kelly RS, Chen C, Smith BJ. Development of a computational approach to predict blood-brain barrier permeability. Drug Metab Dispos. 2004;32(1):132–139. doi:10.1124/dmd.32.1.132
52. Korzekwa K, Nagar S. Drug distribution part 2. predicting volume of distribution from plasma protein binding and membrane partitioning. Pharm Res. 2017;34(3):544–551. doi:10.1007/s11095-016-2086-y
53. Lombardo F, Obach RS, Shalaeva MY, Gao F. Prediction of volume of distribution values in humans for neutral and basic drugs using physicochemical measurements and plasma protein binding data. J Med Chem. 2002;45(13):2867–2876. doi:10.1021/jm0200409
54. Waters LJ, Shokry DS, Parkes GM. Predicting human intestinal absorption in the presence of bile salt with micellar liquid chromatography. Biomed Chromatogr. 2016;30(10):1618–1624. doi:10.1002/bmc.3731
55. Clark DE. Rapid calculation of polar molecular surface area and its application to the prediction of transport phenomena. 1. Prediction of intestinal absorption. J Pharm Sci. 1999;88(8):807–814. doi:10.1021/js9804011
56. Kramer SD. Absorption prediction from physicochemical parameters. Pharm Sci Technolo Today. 1999;2(9):373–380. doi:10.1016/S1461-5347(99)00188-1
57. Camenisch G, Alsenz J, Waterbeemd Hvd FG, Folkers G. Estimation of permeability by passive diffusion through Caco-2 cell monolayers using the drugs’ lipophilicity and molecular weight. Eur J Pharm Sci. 1998;6(4):313–319. doi:10.1016/S0928-0987(97)10019-7
58. Katayama K, Fujita T. Studies on lymphatic absorption of 1ʹ,2ʹ-(3 H)-coenzyme Q 10 in rats. Chem Pharm Bull (Tokyo). 1972;20(12):2585–2592. doi:10.1248/cpb.20.2585
59. Zaki NM. Strategies for oral delivery and mitochondrial targeting of CoQ10. Drug Deliv. 2016;23(6):1868–1881. doi:10.3109/10717544.2014.993747
60. Lambrinidis G, Vallianatou T, Tsantili-Kakoulidou A. In vitro, in silico and integrated strategies for the estimation of plasma protein binding. A review. Adv Drug Deliv Rev. 2015;86:27–45.
61. Wan H, Holmen A. High throughput screening of physicochemical properties and in vitro ADME profiling in drug discovery. Comb Chem High Throughput Screen. 2009;12(3):315–329. doi:10.2174/138620709787581701
62. Naik KM, Kolli DB, Nandibewoor ST. Elucidation of binding mechanism of hydroxyurea on serum albumins by different spectroscopic studies. Springerplus. 2014;3(1):360. doi:10.1186/2193-1801-3-360
63. Shamsi A, Ahmed A, Bano B. Probing the interaction of anticancer drug temsirolimus with human serum albumin: molecular docking and spectroscopic insight. J Biomol Struct Dyn. 2018;36(6):1479–1489. doi:10.1080/07391102.2017.1326320
64. Shamsi A, Ahmed A, Khan MS, Husain FM, Bano B. Rosmarinic acid restrains protein glycation and aggregation in human serum albumin: multi spectroscopic and microscopic insight - possible therapeutics targeting diseases. Int J Biol Macromol. 2020;161(161):187–193. doi:10.1016/j.ijbiomac.2020.06.048
65. Shamsi A, Ahmed A, Khan MS, Al Shahwan M, Husain FM, Bano B. Understanding the binding between rosmarinic acid and serum albumin: in vitro and in silico insight. J Mol Liq. 2020;311(311):113348. doi:10.1016/j.molliq.2020.113348
66. Precupas A, Sandu R, Leonties AR, Anghel D-F, Popa VT. Complex interaction of caffeic acid with bovine serum albumin: calorimetric, spectroscopic and molecular docking evidence. New J Chem. 2017;41(24):15003–15015. doi:10.1039/C7NJ03410E
67. Hu YJ, Li W, Liu Y, Dong JX, Qu SS. Fluorometric investigation of the interaction between methylene blue and human serum albumin. J Pharm Biomed Anal. 2005;39(3–4):740–745. doi:10.1016/j.jpba.2005.04.009
68. Anwar S, Shamsi A, Kar RK, et al. Structural and biochemical investigation of MARK4 inhibitory potential of cholic acid: towards therapeutic implications in neurodegenerative diseases. Int J Biol Macromol. 2020;161:596–604. doi:10.1016/j.ijbiomac.2020.06.078
69. Anwar S, Shamsi A, Shahbaaz M, et al. Rosmarinic acid exhibits anticancer effects via MARK4 inhibition. Sci Rep. 2020;10(1). doi:10.1038/s41598-020-65648-z.
70. Ross PD, Subramanian S. Thermodynamics of protein association reactions: forces contributing to stability. Biochemistry. 1981;20(11):3096–3102. doi:10.1021/bi00514a017
71. Satish L, Rana S, Arakha M, et al. Impact of imidazolium-based ionic liquids on the structure and stability of lysozyme. Spectrosc Lett. 2016;49(6):383–390. doi:10.1080/00387010.2016.1167089
72. Suvarna M, Dyawanapelly S, Kansara B, Dandekar P, Jain R. Understanding the stability of nanoparticle–protein interactions: effect of particle size on adsorption, conformation and thermodynamic properties of serum albumin proteins. ACS Appl Nano Mater. 2018;1(10):5524–5535. doi:10.1021/acsanm.8b01019
73. Yamasaki K, Chuang VT, Maruyama T, Otagiri M. Albumin-drug interaction and its clinical implication. Biochim Biophys Acta. 2013;1830(12):5435–5443. doi:10.1016/j.bbagen.2013.05.005
74. Li Y, Fawcett JP, Zhang H, Tucker IG. Transport and metabolism of MitoQ10, a mitochondria-targeted antioxidant, in Caco-2 cell monolayers. J Pharm Pharmacol. 2007;59(4):503–511. doi:10.1211/jpp.59.4.0004
75. Bemporad D, Essex JW, Luttmann C. Permeation of small molecules through a lipid bilayer: a computer simulation study. J Phys Chem B. 2004;108(15):4875–4884. doi:10.1021/jp035260s
76. van Balen GP, Martinet C, Caron G, et al. Liposome/water lipophilicity: methods, information content, and pharmaceutical applications. Med Res Rev. 2004;24(3):299–324. doi:10.1002/med.10063
77. Di L, Kerns EH. Profiling drug-like properties in discovery research. Curr Opin Chem Biol. 2003;7(3):402–408. doi:10.1016/S1367-5931(03)00055-3
78. Ballard P, Brassil P, Bui KH, et al. Metabolism and pharmacokinetic optimization strategies in drug discovery. Drug Des Devel. 2013;135–155.
79. Greenblatt DJ, Abernethy DR, Divoll M. Is volume of distribution at steady state a meaningful kinetic variable? J Clin Pharmacol. 1983;23(8–9):391–400. doi:10.1002/j.1552-4604.1983.tb02753.x
80. Filler G. Extracorporeal therapies for poisoning. Compr Pediatr Nephrol. 2008;1045–1052.
81. Saha S, Panieri E, Suzen S, Saso L. The interaction of flavonols with membrane components: potential effect on antioxidant activity. J Membr Biol. 2020;253(1):57–71. doi:10.1007/s00232-019-00105-1
82. Simons K, Vaz WL. Model systems, lipid rafts, and cell membranes. Annu Rev Biophys Biomol Struct. 2004;33(1):269–295. doi:10.1146/annurev.biophys.32.110601.141803
83. Poojari C, Wilkosz N, Lira RB, et al. Behavior of the DPH fluorescence probe in membranes perturbed by drugs. Chem Phys Lipids. 2019;223:104784. doi:10.1016/j.chemphyslip.2019.104784
84. Marsh D, Watts A, Knowles PF. Cooperativity of the phase transition in single- and multibilayer lipid vesicles. Biochim Biophys Acta Biomembr. 1977;465(3):500–514. doi:10.1016/0005-2736(77)9028-1
85. Michel N, Fabiano AS, Polidori A, Jack R, Pucci B. Determination of phase transition temperatures of lipids by light scattering. Chem Phys Lipids. 2006;139(1):11–19. doi:10.1016/j.chemphyslip.2005.09.003
86. Domínguez A, Álvarez A, Hilario E, Suarez-Merino B, Goñi-de-Cerio F. Central nervous system diseases and the role of the blood-brain barrier in their treatment. Neurosci Discov. 2013;1(1):3. doi:10.7243/2052-6946-1-3
87. Carpenter TS, Kirshner DA, Lau EY, Wong SE, Nilmeier JP, Lightstone FC. A method to predict blood-brain barrier permeability of drug-like compounds using molecular dynamics simulations. Biophys J. 2014;107(3):630–641. doi:10.1016/j.bpj.2014.06.024
88. Oliveira C, Bagetta D, Cagide F, et al. Benzoic acid-derived nitrones: a new class of potential acetylcholinesterase inhibitors and neuroprotective agents. Eur J Med Chem. 2019;174:116–129. doi:10.1016/j.ejmech.2019.04.026
89. Koynova R, Macdonald RC. Natural lipid extracts and biomembrane-mimicking lipid compositions are disposed to form nonlamellar phases, and they release DNA from lipoplexes most efficiently. Biochim Biophys Acta. 2007;1768(10):2373–2382. doi:10.1016/j.bbamem.2007.04.026
90. Ibarguren M, Lopez DJ, Escriba PV. The effect of natural and synthetic fatty acids on membrane structure, microdomain organization, cellular functions and human health. Biochim Biophys Acta. 2014;1838(6):1518–1528. doi:10.1016/j.bbamem.2013.12.021
91. Lopez S, Bermudez B. Membrane composition and dynamics: a target of bioactive virgin olive oil constituents. Biochim Biophys Acta. 2014;1838(6):1638–1656.
92. Hoekstra D, Martin OC. Transbilayer redistribution of phosphatidylethanolamine during fusion of phospholipid vesicles. Dependence on fusion rate, lipid phase separation, and formation of nonbilayer structures. Biochemistry. 1982;21(24):6097–6103. doi:10.1021/bi00267a011
93. Rappolt M, Hickel A, Bringezu F, Lohner K. Mechanism of the lamellar/inverse hexagonal phase transition examined by high resolution x-ray diffraction. Biophys J. 2003;84(5):3111–3122. doi:10.1016/S0006-3495(03)70036-8
94. Corazzi L, Roberti R. Lipids of brain mitochondria. Handbook Neurochem Mol Neurobiol. 2009;199–221.
95. Carvalho C, Moreira PI. Oxidative stress: a major player in cerebrovascular alterations associated to neurodegenerative events. Front Physiol. 2018;9:806. doi:10.3389/fphys.2018.00806
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.