Back to Journals » Drug Design, Development and Therapy » Volume 17
Linarin Protects Against CCl4-Induced Acute Liver Injury via Activating Autophagy and Inhibiting the Inflammatory Response: Involving the TLR4/MAPK/Nrf2 Pathway
Authors Li L, Lan Y, Wang F, Gao T
Received 2 August 2023
Accepted for publication 9 November 2023
Published 2 December 2023 Volume 2023:17 Pages 3589—3604
DOI https://doi.org/10.2147/DDDT.S433591
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Lulu Li,1,2 Yan Lan,3 Fuqian Wang,1,2 Tiexiang Gao1
1Faculty of Pharmacy, Hubei University of Chinese Medicine, Wuhan, Hubei, People’s Republic of China; 2Department of Pharmacy, Wuhan NO.1 Hospital, Wuhan, Hubei, People’s Republic of China; 3Department of Pharmacy, Huangshi Central Hospital, Huangshi, Hubei, People’s Republic of China
Correspondence: Tiexiang Gao, Faculty of Pharmacy, Hubei University of Chinese Medicine, No. 16 Huangjiahu West Road, Hongshan District, Wuhan, Hubei, 430065, People’s Republic of China, Email [email protected]
Background: Linarin has been implicated in the inhibition of inflammatory responses and hepatoprotective effects. However, the precise mechanism by which Linarin integrates injury-induced signaling from inflammatory responses and oxidative stress remains unclear.
Methods: We evaluated the role of Linarin in a mouse model of carbon tetrachloride (CCl4)-induced acute liver injury. Mice were orally pretreated with Linarin or vehicle for seven consecutive days, followed by intraperitoneal injection with 0.2% (v/v) CCl4. To investigate the mechanism of action on oxidative stress, CCl4-stimulated HepG2 cells were utilized.
Results: Our results revealed Linarin remarkably attenuated the loss of hepatic architecture, inflammatory cell infiltration, serum transaminases, and pro-inflammatory cytokines induced by CCl4. Linarin attenuated CCl4-induced oxidative stress by increasing the expression of cytosolic Nrf2 (nuclear factor erythroid 2-related factor 2), inducing nuclear localization of Nrf2, and increasing stress-induced protein heme oxygenase-1 (HO-1). Additionally, Linarin decreased the expression of toll-like receptors (TLR)-4, and its downstream proteins, MyD88, IRAK1, and TRAF6. Furthermore, Linarin reversed CCl4-induced phosphorylation of ERK, p38, and JNK. Importantly, Linarin increased the expression of both LC3II and Beclin 1, which are hallmarks of autophagic flux. Autophagy-mediated hepatoprotective effects in Linarin-treated HepG2 cells were mitigated by the autophagy inhibitor 3-MA. However, combined treatment of Linarin with 3-MA failed to significantly reverse cell apoptosis and the production of transaminases and pro-inflammatory cytokines.
Conclusion: Linarin prevents acute liver injury, possibly by alleviating ROS-induced oxidative stress, inhibiting TLR4/MyD88 and JNK/p38/ERK-mediated inflammatory responses, and promoting Beclin 1/LC3II-mediated autophagic flux.
Keywords: Linarin, acute liver injury, autophagy, TLR4, MAPK, Nrf2
Introduction
The liver is a metabolic hub responsible for many vital functions including metabolism, detoxification and immunity.1 Acute liver injury is characterized by fulminant hepatocyte necrosis and systemic inflammatory response; without immediate treatment, substantial liver injury can progress to fibrosis and liver cirrhosis and is more likely to develop into acute liver failure and death.2,3 However, specific pharmacotherapeutic interventions for liver damage have not changed significantly and many patients fail to respond to specific treatments. Therefore, novel therapies to prevent liver injury are urgently needed.
Although the underlying mechanism has not yet been established, multiple factors are known to be involved in the pathogenesis of hepatic damage. These exists two important innate immune stimulators, pathogen-associated molecular patterns (PAMPs) caused by hepatotropic microbes and damage-associated molecular patterns (DAMPs) that are endogenous signaling derived from injured cells. Both DAMPs and PAMPs are recognized by pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs) and cytoplasmic Nod-like receptors (NLRs), to trigger the production of chemokines and other mediators, and recruit and activate innate immune cells in the liver.4–6 Many cell types participate in the innate immune response, including monocytes, macrophages, dendritic cells, neutrophils, natural killer cells (NK) or invariant natural killer T cells (iNKT cells).7 When injury is sustained, excessive reactive oxygen species (ROS) and reactive nitrogen species (RNS) are produced, which further damages hepatocytes and augments inflammatory responses.8,9 If this process is prolonged, uncontrolled inflammatory responses and activated signaling cascades further exacerbate cell and tissue death.
Chrysanthemum indicum L, one of herbal medicine recorded in “Dictionary of Chinese Materia Medica” and has been used for hundreds of years in China. Chrysanthemum indicum L has shown a relatively clear therapeutic effect in inflammatory and immune-related diseases. In terms of liver damage, Chrysanthemum indicum L has shown remarkable medicinal value. Linarin (syn. Acacetin 7-O-rhamnosyl (1’′′→6′′) glucoside, or acacetin 7-O-rutinoside) is a naturally occurring flavonol glycoside derived from Chrysanthemum indicum L.10 It is abundant in Chrysanthemum indicum L and its pharmacological activity has received increasing attention. Linarin has been reported to possess anti-inflammatory,11 antioxidative,12 anti-apoptosis activities13 in several disease models. Linarin has been shown to have liver-protective activities.14,15 Linarin treatment decreased the serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), pro-inflammatory cytokines TNF-α, IL-6, and interferon-γ in D-galactosamine (GalN)/LPS-induced liver injury.15 This cytoprotective effect is associated with enhanced STAT3 activity and increased anti-apoptotic Bcl-xL levels.15 Given the potent bioactivity of Linarin and its hepatoprotective effects, further studies are needed to clarify the underlying mechanism in liver injury.
In this study, the role of Linarin in carbon tetrachloride (CCl4)-induced mice and CCl4-stimulated HepG2 cells was investigated. We reported that Linarin administration decreased liver cell death and reduced pro-inflammatory cytokines and oxidative stress. Mechanistically, increased activation of Nrf2 signaling resulted in antioxidative effects and mitochondrial protection. Inhibition of TLR4/MyD88 and MPAK signaling was associated with anti-inflammatory activity. By inhibiting autophagy, we observed attenuated effects of Linarin in CCl4-induced HepG2 cells, suggesting that Beclin 1/LC3II-related autophagic flux was involved in Linarin-mediated hepatoprotective effects.
Materials and Methods
Animals and Treatment
Male BALB/c mice (20–24 g) were purchased from Hubei Experimental Animal Research Center (China). All the mice were housed under specific pathogen-free conditions. Fifty mice were provided free access to food and water. After all animals adaptability feeding a week, they were randomly divided into five groups (n=10): normal control group, CCl4 model control group, and three different concentration treatment groups (12.5 mg/kg Linarin, 25 mg/kg Linarin, and 50 mg/kg Linarin). In the Linarin-treated groups, mice were intragastrically injected with Linarin at doses of 12.5, 25, and 50 mg/kg once daily for 7 days prior to CCl4 administration. The equal volume of vehicle was intragastrically administered to mice in the control and model groups. Linarin (purity>98%) was obtained from Chengdu Purechem Standard Biotechnology Co., Ltd. (https://show.guidechem.com/purechem/). For acute liver injury, the mice were intraperitoneally injected with 0.2% (v/v) CCl4 (Tianli Chemical Reagent Co., Ltd., Tianjin, China) diluted in olive oil or vehicle (olive oil) at a dose of 10 mL/kg weight. All animals were sacrificed at 24 hours after CCl4 injection using anesthesia with 1% pentobarbital (10 mL/kg). Blood samples were collected from the orbital sinus. The serum was separated by centrifugation at 3000 rpm for 10 min, then collected and stored at −80 °C until use. In this study, all animal experiments were approved by the Ethics Committee of Hubei University of Chinese Medicine (No. HUCMS-202107011) and were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Cell Culture and Treatment
The HepG2 cells were obtained from the Key Laboratory of Traditional Chinese Medicine Resources and Chemistry of Hubei Province (Wuhan, China) and grown in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) at 37°C, 5% CO2 and humid conditions. The cells were pretreated for 1 h with Linarin at final concentrations of 10 μg/mL, 20 μg/mL, and 40 μg/mL. CCl4 (20 mM) was used to induce the cell death. To further assess the role of autophagy, 3-methyladenine (3-MA, a chemical inhibitor of autophagy) at 15 μM was added 1 h prior to incubation with Linarin. Parallel cultures were treated with equal amounts of vehicle and used as negative controls.
Histological Analysis
For histological analysis, the liver tissues were removed and fixed in 10% buffered formalin. Liver specimens were sectioned and stained with hematoxylin and eosin (H&E) for light microscopic examination. For immune histochemical analysis, liver specimens were incubated with F4/80 antibody (Abcam) or TLR4 antibody (Affinity), or LC3II antibody (Bioss). Cell death through apoptosis was detected using terminal deoxynucleotidyl transferase dUTP nick-end labelling (TUNEL) staining according to the manufacturer’s instructions (Roche) and visualized using a Nikon fluorescence microscope (Zeiss).
Determination of AST, ALT and Total Bilirubin
The enzymatic activities of ALT, AST, and total bilirubin were examined using commercial assay kits (Beyotime Biotechnology, Inc., Shanghai, China), according to the manufacturer’s instructions.
Determination of Hepatic SOD and MDA
An appropriate amount of liver tissue was collected, separated, washed, homogenized, and centrifuged with a 9-fold volume of 4°C physiological saline (4 °C, 4000 rpm, 10 min). Following centrifugation, the supernatant was collected to determine oxidative stress markers. Superoxide dismutase (SOD) and malondialdehyde (MDA) contents in the liver homogenates were determined using assay kits according to the manufacturer’s instructions (Beyotime Biotechnology, Inc., Shanghai, China).
Cytokine Measurement
The levels of IL-1β, IL-6, and TNF-α were measured using Enzyme-Linked Immunosorbent Assay (ELISA) kits (Beyotime Biotechnology, Inc., Shanghai, China) according to the manufacturer’s instructions.
Flow Cytometer Analysis
HepG2 cells were seeded in six-well plates, cultured for 24 h, and then treated as directed for 24 h. After that, cells were digested, washed in PBS, and stained using the FITC Annexin V apoptosis detection kit (Elabscience Biotechnology Co., Ltd.). The samples were analyzed by flow cytometry (Beckman Coulter) and data were analyzed with FlowJo_v10.6.2 software.
Western Blotting
Cell lysates were prepared from liver tissue and HepG2 cells using RIPA buffer containing 1% Triton X-100 lysis buffer, protease inhibitor cocktail, and PhosSTOP (Roche). The protein concentration was determined using a BCA (Bicinchoninic acid) Protein Assay Kit (Beyotime Biotechnology, Inc., Shanghai, China). Total protein from each sample was separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (PAGE). The separated samples (total protein) were transferred from the gel onto a polyvinylidene difluoride (PVDF) membrane (Absin, China). The membranes were then exposed to enhanced chemiluminescence (ECL) Western blotting substrates (Thermo Fisher). The ImageJ software was used to measure the density of the results. The following antibodies from Abcam were used: TLR4, MyD88, LC3 and Beclin 1. The following antibodies from Affinity were used: IRAK1, TRAF6, Phospho-ERK1/2 and Phospho-p38. The following antibodies from Affinity were used: Keap1. The following antibodies from Proteintech were used: phospho-JNK. Antibodies against Nrf2 and Heme oxidase-1 (HO-1) were derived from SAB and Abs, respectively.
Determination of Mitochondrial ROS
Mitochondrial probes have been used to determine mitochondrial ROS production. Briefly, HepG2 cells were treated with Linarin and incubated at 37°C with the MitoSox Green reagent and MitoTracker Red for 10 min. HepG2 cells were washed with PBS. Fluorescence microscopy (Nikon, Tokyo, Japan) was used to assess the fluorescence density.
Mitochondrial Membrane Potential Assay
The mitochondrial membrane potential (MMP) was evaluated using a 5.5′,6.6′-tetrachloro-1.1′,3.3′- tetraethylbenzimidazolylcarbocyanine iodide (JC-1) probe (Servicebio, Wuhan, China). HepG2 cells were washed with PBS and incubated in the dark with the JC-1 working solution at 37°C for 20 min. After removing the JC-1 solution, the cells were washed and resuspended in assay buffer and the sequential fluorescence of each well was measured using a fluorescence microscope (Nikon, Tokyo, Japan).
Statistical Analysis
Statistical analyses were performed using Prism 9.4.1 (GraphPad Software). All Data are presented as mean ± standard deviation (SD). Differences between multiple groups were compared using one-way analysis of variance (ANOVA). Statistical significance was set at P <0.05.
Results
Linarin Had a Hepatoprotective Effect in CCl4-Induced Acute Injury
To determine the liver-protective activity of Linarin, a mouse model of CCl4-induced acute hepatic injury was established. The CCl4 challenge caused an increase in ALT, AST, and total bilirubin levels at 24 h. Linarin pretreatment showed significant dose-dependent protective effects, with an approximately 60% reduction in serum ALT (Figure 1A), 60% reduction in AST (Figure 1B), and 50% reduction in total bilirubin (Figure 1C). Histological evaluation revealed more severe liver injury in the model control group with more extensive cell necrosis, loss of liver structure, and features of perivascular inflammatory cell infiltration (Figure 1D). We also evaluated the protective effects of Linarin on CCl4-stimulated HepG2 cells. CCl4 induced massive hepatocyte death, which was reversed by Linarin, with cell survival rates of 79.7, 68.3, and 61.7% at doses of 40, 20, and 10 μg/mL, respectively (Figure 1E). CCl4-stimulated HepG2 cells showed elevated production of ALT (Figure 1F) and AST (Figure 1G), which was significantly down-regulated by Linarin in a dose-dependent manner. Thus, treatment with Linarin protected the hepatocytes both in vitro and in vivo.
 |
Figure 1 Linarin had a hepatoprotective effect in CCl4-induced acute injury. (A) Serum ALT, (B) AST and (C) total bilirubin levels in mice from each group. (D) Histological examination of liver sections from each group with H&E staining (original magnification, ×200). Image showed hepatocyte necrosis (black arrow), inflammatory cell infiltration (yellow arrow), cellular edema (green arrow), hepatocyte steatosis (blue arrow). (E) CCK8 assay to study cell viability. ALT (F) and AST (G) levels in the supernatant of HepG2 cells. Data are expressed as mean ± SD. *P < 0.05, ** P < 0.01, *** P < 0.001. Abbreviations: NC, normal control; MC, model control; LD, low dose of Linarin 12.5 mg/kg; MD, middle dose of Linarin 25 mg/kg; HD, high dose of Linarin 50 mg/kg; ns, non-significant. |
Linarin Inhibited Apoptosis on CCl4-Induced Hepatocytes
The results of TUNEL staining revealed that the number of TUNEL-positive cells in the liver tissues of the model control group was significantly higher than that of the normal control group. Linarin (50 mg/kg) decreased the number of TUNEL-positive cells in liver tissues of CCl4-induced mice (Figure 2A). Apoptosis of HepG2 cells was detected using flow cytometry. Our study showed that CCl4 significantly increased the proportion of early and late apoptosis to 33.6% compared with 6.0% apoptosis in the normal control group. However, Linarin alleviated CCl4-induced cell apoptosis, with the greatest reduction observed at 40 μg/mL Linarin (Figure 2B). These findings indicated that Linarin protects hepatocytes from apoptosis.
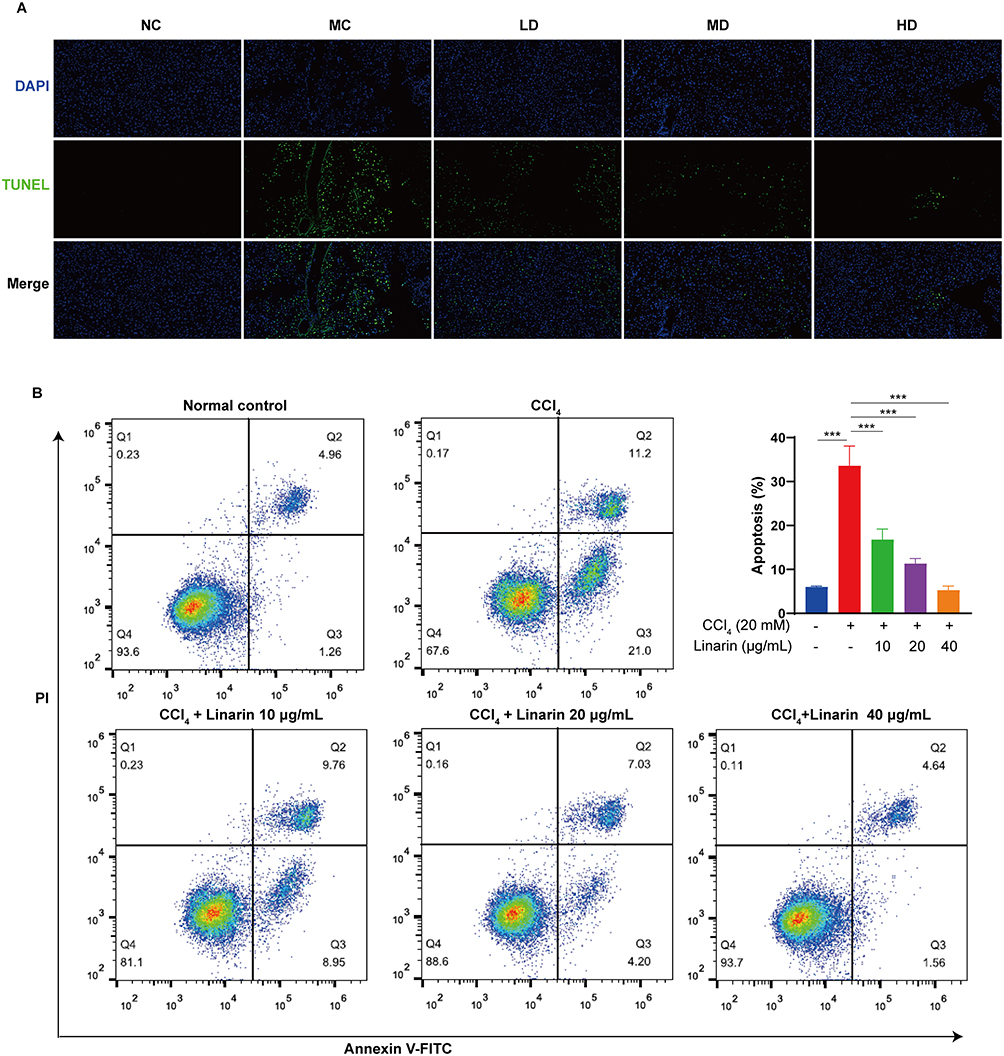 |
Figure 2 Linarin inhibited apoptosis on CCl4-induced hepatocytes. (A) TUNEL staining for apoptotic cells of the liver sections (original magnification, ×200). (B) Representative flow plots and quantification of apoptotic cells. Data are expressed as mean ± SD. *** P < 0.001. Abbreviations: NC, normal control; MC, model control; LD, low dose of Linarin 12.5 mg/kg; MD, middle dose of Linarin 25 mg/kg; HD, high dose of Linarin 50 mg/kg. |
Linarin Inhibited Excessive Inflammatory Responses
Compared to the model control group, the expression of F4/80, an activator of macrophages, was significantly decreased in Linarin-pretreated mice (Figure 3A). After exposure to CCl4, serum inflammatory cytokines significantly increased, especially IL-6 (Figure 3B), IL-1β (Figure 3C) and TNF-α (Figure 3D). However, Linarin pretreatment inhibited the production of pro-inflammatory cytokines. In particular, we observed approximate 34% decreases at the high dose for the pro-inflammatory cytokine IL-6, and 55% decreases for the cytokine IL-1β. We also examined the production of IL-6 (Figure 3E), IL-1β (Figure 3F), and TNF-α (Figure 3G) in CCl4-induced HepG2 cells. The results showed that the levels of pro-inflammatory cytokines increased in HepG2 cells after stimulation with CCl4. Linarin treatment reversed these effects in a dose-dependent manner. These results showed that the hepatoprotective effect of Linarin is associated with a reduction in inflammatory responses.
 |
Figure 3 Linarin inhibited excessive inflammatory responses. (A) Immunohistochemical detection of F4/80-positive cells in liver sections from each group (original magnification: ×200). Serum concentration of pro-inflammatory cytokines (B) IL-6, (C) IL-1β and (D) TNF-α in mice. The levels of (E) IL-6, (F) IL-1β and (G) TNF-α in the supernatants of HepG2 cells. Data are expressed as mean ± SD. ns, non-significant; *P < 0.05, ** P < 0.01, *** P < 0.001. Abbreviations: NC, normal control; MC, model control; LD, low dose of Linarin 12.5 mg/kg; MD, middle dose of Linarin 25 mg/kg; HD, high dose of Linarin 50 mg/kg. |
Linarin Alleviated Oxidative Stress in CCl4-Induced Hepatocytes
To analyze whether antioxidant responses are involved in Linarin-induced hepatoprotective effects, we evaluated the effects of Linarin on SOD and MDA levels in the liver. The levels of the antioxidant enzyme SOD in the model control group significantly decreased, and the levels of MDA markedly increased compared with the normal control group (Figure 4A and B). Linarin significantly increased SOD levels, but decreased MDA levels. Likewise, CCl4-induced the dysregulation of SOD and MDA was attenuated by Linarin in HepG2 cells (Figure 4C and D).
 |
Figure 4 Linarin alleviated oxidative stress in CCl4-induced hepatocytes. (A) Hepatic SOD and (B) MDA activity in mice from each group. (C) SOD and (D) MDA activity in HepG2 cells treated with Linarin. (E) Mitochondrial superoxide detected through immunofluorescence using MitoSox Green and MitoTracker Red staining. (F) Mitochondrial membrane potential was detected by JC-1 staining. Data are expressed as mean ± SD. ns, non-significant; ** P < 0.01, *** P < 0.001. Abbreviations: NC, normal control; MC, model control; LD, low dose of Linarin 12.5 mg/kg; MD, middle dose of Linarin 25 mg/kg; HD, high dose of Linarin 50 mg/kg. |
To investigate the effect of Linarin on ROS generation in vitro, CCl4-induced HepG2 cells were utilized in this experiment. The results showed that ROS were highly induced in CCl4-stimulated HepG2 cells, whereas Linarin treatment resulted in a significant reduction in intracellular ROS release (Figure 4E). We monitored the mitochondrial depolarization using the JC-1 mitochondrial membrane potential detection kit. As shown in Figure 4F, the results showed a reduction in the percentage of red-to-green fluorescence when cells were treated with CCl4 compared with untreated cells. However, in Linarin-treated cells, the reduction in the percentage of red-to-green fluorescence was reversed in a dose-dependent manner. These results suggest that Linarin effectively attenuated the decline of mitochondrial membrane potential in CCl4 treated cells.
Keap1-Nrf2 Signaling Involved in the Linarin-Alleviated Oxidative Stress
Nrf2 is a transcription factor that regulates cellular resistance to oxidants through a variety of mechanisms, including the induction of catabolism of superoxide and peroxides and the regeneration of oxidized cofactors and proteins.16 As an Nrf2 binding protein, Keap1 inhibits Nrf2 by interacting with Nrf2 and directing Nrf2 ubiquitination. The Keap1-Nrf2 pathway is a major regulator of oxidative stress.17 As shown in Figure 5A-C, CCl4 stimulation significantly decreased the expression levels of nuclear and cytosolic Nrf2, whereas Linarin increased the expression of cytosolic Nrf2 and induced nuclear localization of Nrf2. Keap1 expression was significantly increased by CCl4 exposure, whereas Linarin treatment reversed this effect in a dose-dependent manner (Figure 5D and E). Compared with the control group, the expression of HO-1 in the CCl4 stimulation group was significantly decreased, while the expression of HO-1 after Linarin treatment was significantly increased (Figure 5D and F).
 |
Figure 5 Keap1-Nrf2 signal involved in the Linarin-alleviated oxidative stress. (A) Western blot analysis of nuclear Nrf2 and cytosol Nrf2 protein in liver lysates from each group. (B) Nuclear Nrf2 amount normalized relative to expression of Lamin B1. (C) Cytosol Nrf2 amount normalized relative to expression of GAPDH. (D) Western blot analysis of HO-1 and Keap1 protein in liver lysates from each group. (E) Keap1 and (F) HO-1 amount normalized relative to expression of GAPDH. Data are expressed as mean ± SD (n = 3); *P < 0.05, ** P < 0.01, *** P < 0.001. Abbreviation: ns, non-significant. |
Effect of Linarin on TLR4 Expression and TLR4/MyD88 Signaling in CCl4-Induced Liver Injury
Based on the fact that TLR4 amplifies the inflammatory response in liver injury, we investigated whether Linarin induces hepatoprotective effects by inhibiting TLR4 signaling. Immunohistochemistry demonstrated that the liver sections from model control mice showed increased expression of TLR4 in sinusoidal endothelial cells, vascular cells, and hepatocytes after CCl4 injection, and overall TLR4 immunoreactivity was decreased by Linarin pretreatment (Figure 6A). Consistent with the above results, Western blotting showed that pretreatment with Linarin reduced the expression protein levels of TLR4 (Figure 6B and C). Western blot analysis of MyD88, IRAK1, and TRAF6, indicated that CCl4 exposure increased the expression of MyD88 (Figure 6D), IRAK1 (Figure 6E), and TRAF6 (Figure 6F), whereas pretreatment with Linarin significantly reduced the levels of these proteins.
 |
Figure 6 Effect of Linarin on TLR4 expression, TLR4/MyD88 and MAPK signaling in CCl4-induced liver injury. (A) Expression of TLR4 in the liver determined by immunohistochemistry (original magnification, ×200). (B) Western blot analysis of TLR4, MyD88, IRAK1 and TRAF6 protein in liver lysates from each group. (C) TLR4, (D) MyD88, (E) IRAK1 and (F) TRAF6 amount normalized relative to expression of GAPDH. (G) Western blot analysis of p-ERK, p-JNK and p-p38 protein in liver lysates from each group. (H) p-ERK, (I) p-JNK and (J) p-p38 amount normalized relative to expression of GAPDH. Data are expressed as mean ± SD (n = 3). ns, non-significant; *P < 0.05, ** P < 0.01, *** P < 0.001. Abbreviations: NC, normal control; MC, model control; LD, low dose of Linarin 12.5 mg/kg; MD, middle dose of Linarin 25 mg/kg; HD, high dose of Linarin 50 mg/kg. |
Effect of Linarin on MAPK Signaling in CCl4-Induced Liver Injury
Mitogen-activated protein kinases (MAPKs) signaling include ERK, p38, and JNK pathways.18 Extracellular stimuli such as cytokines, TLRs, and oxidative stress can activate ERK, p38, and JNK kinase activation.19 We further investigated whether Linarin affects MAPK signaling in acute liver injury. CCl4 treatment increased hepatic expression of p-JNK, p-p38, and p-ERK. Linarin pretreatment produced the opposite effect, with a decreased expression of p-JNK, p-p38, and p-ERK (Figure 6G-J).
Linarin Promoted Autophagy in CCl4-Induced Acute Liver Injury
Given the established protective role of autophagy in acute liver injury, we investigated whether Linarin pretreatment could induce autophagy and thus promote the protective effects of Linarin against liver injury. The expression of the autophagy-specific marker LC3II was detected by immunohistochemical staining. 24 h after the CCl4 challenge, LC3II expression in the liver of CCl4 treatment group decreased, whereas LC3II expression in the liver of the Linarin pretreatment group increased (Figure 7A). Consistent with the immunostaining results, Western blot analysis showed significant increases in both Beclin 1 (Figure 7B and C) and LC3II (Figure 7B and D) in mice treated with Linarin compared to the model control group. In summary, our results suggest that Linarin promotes the protective effects of autophagy against CCl4-induced hepatic injury.
 |
Figure 7 Effect of Linarin on Beclin 1 and LC3II/I expression in CCl4-induced liver injury. (A) Expression of LC3II in the liver determined by immunohistochemistry (original magnification, ×200). (B) Western blot analysis of LC3 and Beclin 1 protein in liver lysates from each group. (C) Beclin 1 and (D) LC3II/I amount normalized relative to expression of GAPDH. Data are expressed as mean ± SD (n = 3). ns, non-significant; *P < 0.05, ** P < 0.01, *** P < 0.001. Abbreviations: NC, normal control; MC, model control; LD, low dose of Linarin 12.5 mg/kg; MD, middle dose of Linarin 25 mg/kg; HD, high dose of Linarin 50 mg/kg. |
Inhibition of Autophagy Mitigated Linarin-Induced Hepatoprotective Effect
To study the mechanisms underlying the autophagy-mediated hepatoprotective effects of Linarin, CCl4-stimulated HepG2 cells were treated with Linarin and the autophagy inhibitor, 3-MA. As expected, co-treatment with an autophagy inhibitor further increased apoptosis, which was not reversed by Linarin (Figure 8A and B). After the CCl4 challenge, co-treatment with an autophagy inhibitor further elevated the production of AST (Figure 8C) and ALT (Figure 8D) and the pro-inflammatory cytokines TNF-α (Figure 8E) and IL-6 (Figure 8F), whereas Linarin treatment failed to significantly reverse this effect. CCl4 significantly down-regulated the autophagy markers Beclin 1 and LC3II protein expressions, which were further down-regulated when co-treated with autophagy inhibitor 3-MA (Figure 8G-I). Co-treatment with Linarin did not enhance the expression of Beclin 1 and LC3II, suggesting that Linarin-mediated autophagy plays a beneficial role in protecting against liver injury triggered by CCl4 (Figure 8G-I).
 |
Figure 8 Linarin promoted autophagy in CCl4-induced acute liver injury. To further assess the role of autophagy, 3-methyladenine (3-MA, a chemical inhibitor of autophagy) at 15 μM were added 1 h prior to Linarin incubation in CCl4-stimulated HepG2 cells. (A) Representative flow plots and (B) quantification of apoptotic cells. (C) AST and (D) ALT levels in the supernatant of HepG2 cells. The levels of pro-inflammatory cytokines (E) TNF-α and (F) IL-6. (G) Western blot analysis of LC3 and Beclin 1 protein in HepG2 cells. (H) Beclin 1 and (I) LC3II/I amount normalized relative to expression of GAPDH. Data are expressed as mean ± SD (n = 3). ns, non-significant; ** P < 0.01, *** P < 0.001. |
Discussion
Liver damage is an inflammatory disease with a rising incidence each year. Unfortunately, the overall treatment of liver damage is limited. Particularly worth mentioning is that immunosuppressive agents, which have been commonly used for liver damage often have significant adverse effects. As a result, treatment of liver injury is extremely difficult. Our study focused on the traditional medicinal efficacy of Chrysanthemum indicum L, investigating whether Linarin has a certain liver-protective activity and provides a new strategy for the treatment of liver damage. In this study, further evidence was provided to support the hypothesis that Linarin alleviates liver inflammation and damage in CCl4-induced mice. Our study showed that the hepatoprotective effects of Linarin are associated with Keap1-Nrf2-dependent antioxidative stress. We also confirmed that the downregulation of TLR4 and MAKP signaling is involved in Linarin-mediated hepatoprotective effects. More importantly, our findings suggested that Linarin plays an important role in promoting autophagy to prevent normal liver cells from CCl4 induced cell damage.
Consistent with previous studies, acute CCl4 injection resulted in increased levels of serum markers of liver injury, inflammatory infiltration, and histopathological alterations.20,21 Linarin pretreatment decreased serum ALT, AST, and total bilirubin levels, protected hepatocytes, and preserved their structural integrity. Infiltration of the liver macrophages significantly increased 24 h after CCl4 injection, which was prevented by Linarin pretreatment, as determined histologically by F4/80 staining. The role of macrophages in liver injury has been well documented.22,23 Upon liver injury, inflammatory mediators are released by activated and stressed cells, which rapidly recruit bone marrow-derived monocytes to differentiate into macrophages; this inflammatory microenvironment dramatically impacts the phenotypes and function of Kupffer cells and monocyte-derived macrophages.24 Thus, it is believed that the inhibition of hepatic macrophage infiltration can relieve disease progression. In addition to macrophage expansion, hepatotoxicity leads to massive subsequent production of pro-inflammatory cytokines, especially IL-6 and TNF-α.25 After CCl4 injection, elevated levels of TNF-α, IL-1β, and IL-6 were observed, which were reversed by Linarin. Linarin pretreatment to decrease macrophage infiltration and pro-inflammatory cytokine production is important for modulating liver inflammation.
For the inflammatory pathway, TLR4/MyD88 signaling was investigated. In the liver, TLR4 is a classical immune pathway that is expressed in all parenchymal and non-parenchymal cell types. The literatures suggest that TLR4 activation induces a cascade of pro-inflammatory cytokines involved in the pathophysiology of severe liver injury.26 We found that pretreatment with Linarin decreased the levels of TLR4 protein expression in the liver, which might partly contribute to the amelioration of liver inflammation by Linarin. This is consistent with previous studies showing that Linarin can reduce TLR4 protein expression in D-galactosamine- and LPS-induced fulminant liver failure.15 In our study, Linarin attenuated the increase in MyD88, IRAK1, and TRAF6 protein expression induced by CCl4, which revealed that Linarin suppressed the TLR4-mediated MyD88-dependent pathway to alleviate the excess inflammatory response in acute injury.
Upon activation of TLR4, IRAK1 and IRAK4 are recruited and interact with TRAF6 proteins, which can eventually activate MAPK signals.27 The MAPK pathway includes p38 MAPK, ERK and JNK, which are phosphorylated to promote the production of inflammatory mediators and contribute to hepatic inflammation.28 Previous studies have demonstrated that JNK is strongly activated in response to CCl4- or acetaminophen-induced liver injury;29–31 JNK inhibition blocks liver injury.32 Phosphorylation of p38 MAPK also contributes to CCl4-induced acute liver injury, and ablation of p38 decreases inflammatory cell recruitment and antioxidative response.33 ERK signaling has been associated with liver injury, mitochondrial dysfunction, and oxidative stress.34 The role of MAPK signaling, as shown here, also illustrates protein activation via phosphorylation after CCl4 injection. Linarin decreased the levels of phosphorylated JNK, ERK, and p38 MAPK, and the levels of activation were negatively correlated with the Linarin dose. Thus, Linarin appears to attenuate CC14-induced activation of MAPK signaling.
ROS- induced oxidative stress leads to worsening of liver diseases.35 ROS overproduction affects the structure and function of cellular components. In our study, significantly reduced intracellular and mitochondrial ROS levels were observed after treatment with Linarin. In addition, the production of the hepatic oxidative marker MDA was remarkably down-regulated and the antioxidant enzyme SOD was up-regulated. ROS are primarily produced in the mitochondria and mitochondrial dysfunction is associated with increased ROS levels.36 In this study, the mitochondrial membrane potential of Linarin-treated HepG2 cells was significantly increased, suggesting that Linarin could prevent mitochondrial depolarization and restore negative membrane potential, thereby protecting mitochondrial function.
Nrf2, a redox-regulated transcription factor, plays a central role in the modulation of antioxidant defense systems to protect tissues from oxidative stress.37 During oxidative stress, Nrf2 accumulates in the cytoplasm and is then transported to the nucleus. Nrf2 then initiates the transcription of antioxidant enzymes, such as sulfiredoxin, SOD, HO-1 in the nucleus.38,39 Keap1 tightly regulates ROS-mediated Nrf2 activation by promoting proteasomal degradation of Nrf2; thus, Keap1 inhibition results in the activation of Nrf2.40 Studies have shown that Nrf2 activation prevents acute liver injury by regulating antioxidant defense-associated genes.41 Previous literatures have confirmed the hepatoprotective effect of Nrf2 in different acute liver injury models, such as LPS- and D-GalN-induced,42 CCl4-induced43,44 and acetaminophen-induced mouse acute liver injury models.45 Therefore, we explored the mechanism underlying the oxidative damage in Linarin-treated HepG2 cells. We found that CCl4 exposure obviously increased the expression of Nrf2 in both the nuclear and cytoplasm, which was reversed by Linarin treatment, suggesting that Linarin treatment significantly enhanced the nuclear translocation of Nrf2. Moreover, CCl4 exposure induced the expression of Keap1 and decreased the expression of HO-1, which were attenuated by Linarin. These results suggested that Linarin may promote Nrf2-mediated antioxidant defense to alleviate hepatic oxidative stress.
Another important finding was that Linarin pretreatment induced autophagy, which was strengthened by the results of experiments using autophagy inhibitor 3-MA-treated hepatocyte models. Autophagy, an evolutionarily conserved process in eukaryotes, preserves cellular function under normal and pathophysiological conditions.46,47 Autophagy efficiently and precisely controls the quality and quantity of cytoplasmic organelles, contributing to liver homeostasis and hepatic metabolism.48 Dysregulation of autophagy is associated with pathological symptoms and liver disease and its modulation is a potential therapeutic option.49 Autophagy exerts hepatoprotective and anti-inflammatory effects in acute liver failure. It has described that autophagy inhibits mitochondrial death pathway activation through caspase 8 in hepatocytes.50 Anti-inflammatory effects on macrophages may rely on p62-dependent mitophagy to decrease the accumulation of damaged mitochondria and generation of inflammasome-dependent IL-1β.51,52 Nevertheless, we also detected a significant reduction in LC3II and Beclin 1 expression in the liver tissues of CCl4-treated mice and CCl4-stimulated HepG2 cells. The inhibition of autophagy leads to decrease the viability of hepatocytes and increase hepatocyte apoptosis. In contrast, Linarin treatment increased the expression of LC3II and Beclin 1. These data suggest that Linarin promotes autophagy to eliminate extensively damaged cells from the liver tissue.
A previous study showed that in LPS-induced acute injury, Linarin attenuates hepatic apoptosis by reducing pro-apoptotic Bim phosphorylation and increasing anti-apoptotic Bxl-xL.15 Our study confirmed the protective effects of Linarin on CCl4-induced acute hepatic injury in vitro and in vivo. In addition, we revealed an alternative mechanism for hepatocyte protection by Linarin. Linarin attenuated ROS-induced oxidative stress, inhibited TLR4/MyD88-mediated inflammatory responses, and promoted Beclin 1/LC3II-mediated autophagic flux (Figure 9). Further studies are required to determine the triggers of autophagy and to identify the mechanism by which Linarin inhibits TLR4 signaling.
 |
Figure 9 Mechanisms of protective effects of Linarin in CCl4-induced acute liver injury. |
Data Sharing Statement
Data that support the findings of this study will be made available on request from the corresponding author, Tiexiang Gao.
Funding
This study was supported by the Scientific Research Project of Wuhan Municipal Health Commission (WZ22C44).
Disclosure
The authors declare that there are no competing interests associated with the manuscript.
References
1. Kalra A, Yetiskul E, Wehrle CJ, Tuma F. Physiology, Liver. StatPearls. 2022.
2. Mokdad AA, Lopez AD, Shahraz S, et al. Liver cirrhosis mortality in 187 countries between 1980 and 2010: a systematic analysis. BMC Med. 2014;12:145. doi:10.1186/s12916-014-0145-y
3. Stravitz RT, Kramer DJ. Management of acute liver failure. Nat Rev Gastroenterol Hepatol. 2009;6(9):542–553. doi:10.1038/nrgastro.2009.127
4. Han H, Desert R, Das S, et al. Danger signals in liver injury and restoration of homeostasis. J Hepatol. 2020;73(4):933–951. doi:10.1016/j.jhep.2020.04.033
5. Tiegs G, Horst AK. TNF in the liver: targeting a central player in inflammation. Semin Immunopathol. 2022;44(4):445–459. doi:10.1007/s00281-022-00910-2
6. Chung RT, Stravitz RT, Fontana RJ, et al. Pathogenesis of liver injury in acute liver failure. Gastroenterology. 2012;143(3):e1–e7. doi:10.1053/j.gastro.2012.07.011
7. Zindel J, Kubes P. DAMPs, PAMPs, and LAMPs in Immunity and Sterile Inflammation. Annu Rev Pathol. 2020;15:493–518. doi:10.1146/annurev-pathmechdis-012419-032847
8. Lebeaupin C, Proics E, de Bieville CH, et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 2015;6(9):e1879. doi:10.1038/cddis.2015.248
9. Sutti S, Jindal A, Locatelli I, et al. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology. 2014;59(3):886–897. doi:10.1002/hep.26749
10. Mottaghipisheh J, Taghrir H, Boveiri Dehsheikh A, et al. Linarin, a Glycosylated Flavonoid, with Potential Therapeutic Attributes: a Comprehensive Review. Pharmaceuticals. 2021;14(11). doi:10.3390/ph14111104
11. Tian D, Yang Y, Yu M, et al. Anti-inflammatory chemical constituents of Flos Chrysanthemi Indici determined by UPLC-MS/MS integrated with network pharmacology. Food Funct. 2020;11(7):6340–6351. doi:10.1039/d0fo01000f
12. Zeng J, Hu W, Li H, et al. Purification of linarin and hesperidin from Mentha haplocalyx by aqueous two-phase flotation coupled with preparative HPLC and evaluation of the neuroprotective effect of linarin. J Sep Sci. 2021;44(12):2496–2503. doi:10.1002/jssc.202001243
13. Lou H, Fan P, Perez RG, Lou H. Neuroprotective effects of linarin through activation of the PI3K/Akt pathway in amyloid-beta-induced neuronal cell death. Bioorg Med Chem. 2011;19(13):4021–4027. doi:10.1016/j.bmc.2011.05.021
14. Ma BX, Meng XS, Tong J, Ge LL, Zhou G, Wang YW. Protective effects of Coptis chinensis inflorescence extract and linarin against carbon tetrachloride-induced damage in HepG2 cells through the MAPK/Keap1-Nrf2 pathway. Food Funct. 2018;9(4):2353–2361. doi:10.1039/c8fo00078f
15. Kim SJ, Cho HI, Kim SJ, et al. Protective effect of linarin against D-galactosamine and lipopolysaccharide-induced fulminant hepatic failure. Eur J Pharmacol. 2014;738:66–73. doi:10.1016/j.ejphar.2014.05.024
16. Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401–426. doi:10.1146/annurev-pharmtox-011112-140320
17. Tu W, Wang H, Li S, Liu Q, Sha H. The Anti-Inflammatory and Anti-Oxidant Mechanisms of the Keap1/Nrf2/ARE Signaling Pathway in Chronic Diseases. Aging Dis. 2019;10(3):637–651. doi:10.14336/AD.2018.0513
18. Yue J, Lopez JM. Understanding MAPK Signaling Pathways in Apoptosis. Int J Mol Sci. 2020;21(7). doi:10.3390/ijms21072346
19. Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev. 2012;92(2):689–737. doi:10.1152/physrev.00028.2011
20. Ullah H, Khan A, Baig MW, et al. Poncirin attenuates CCL4-induced liver injury through inhibition of oxidative stress and inflammatory cytokines in mice. BMC Complement Med Ther. 2020;20(1):115. doi:10.1186/s12906-020-02906-7
21. Dai C, Xiao X, Li D, et al. Chloroquine ameliorates carbon tetrachloride-induced acute liver injury in mice via the concomitant inhibition of inflammation and induction of apoptosis. Cell Death Dis. 2018;9(12):1164. doi:10.1038/s41419-018-1136-2
22. Shan Z, Ju C. Hepatic Macrophages in Liver Injury. Front Immunol. 2020;11:322. doi:10.3389/fimmu.2020.00322
23. Starkey Lewis P, Campana L, Aleksieva N, et al. Alternatively activated macrophages promote resolution of necrosis following acute liver injury. J Hepatol. 2020;73(2):349–360. doi:10.1016/j.jhep.2020.02.031
24. Roohani S, Tacke F. Liver Injury and the Macrophage Issue: molecular and Mechanistic Facts and Their Clinical Relevance. Int J Mol Sci. 2021;22(14). doi:10.3390/ijms22147249
25. Robinson MW, Harmon C, O’Farrelly C. Liver immunology and its role in inflammation and homeostasis. Cell Mol Immunol. 2016;13(3):267–276. doi:10.1038/cmi.2016.3
26. Guo J, Friedman SL. Toll-like receptor 4 signaling in liver injury and hepatic fibrogenesis. Fibrogenesis Tissue Repair. 2010;3:21. doi:10.1186/1755-1536-3-21
27. Liang W, Han B, Hai Y, et al. The Role of Microglia/Macrophages Activation and TLR4/NF-kappaB/MAPK Pathway in Distraction Spinal Cord Injury-Induced Inflammation. Front Cell Neurosci. 2022;16:926453. doi:10.3389/fncel.2022.926453
28. Westenberger G, Sellers J, Fernando S, et al. Function of Mitogen-Activated Protein Kinases in Hepatic Inflammation. J Cell Signal. 2021;2(3):172–180.
29. Li W, Yang GL, Zhu Q, et al. TLR4 promotes liver inflammation by activating the JNK pathway. Eur Rev Med Pharmacol Sci. 2019;23(17):7655–7662. doi:10.26355/eurrev_201909_18889
30. Matsumaru K, Ji C, Kaplowitz N. Mechanisms for sensitization to TNF-induced apoptosis by acute glutathione depletion in murine hepatocytes. Hepatology. 2003;37(6):1425–1434. doi:10.1053/jhep.2003.50230
31. Kluwe J, Pradere JP, Gwak GY, et al. Modulation of hepatic fibrosis by c-Jun-N-terminal kinase inhibition. Gastroenterology. 2010;138(1):347–359. doi:10.1053/j.gastro.2009.09.015
32. Hanawa N, Shinohara M, Saberi B, Gaarde WA, Han D, Kaplowitz N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in Acetaminophen-induced liver injury. J Biol Chem. 2008;283(20):13565–13577. doi:10.1074/jbc.M708916200
33. Rius-Perez S, Tormos AM, Perez S, et al. p38alpha deficiency restrains liver regeneration after partial hepatectomy triggering oxidative stress and liver injury. Sci Rep. 2019;9(1):3775. doi:10.1038/s41598-019-39428-3
34. Lee HC, Yu HP, Liao CC, Chou AH, Liu FC. Escin protects against Acetaminophen-induced liver injury in mice via attenuating inflammatory response and inhibiting ERK signaling pathway. Am J Transl Res. 2019;11(8):5170–5182.
35. Cichoz-Lach H, Michalak A. Oxidative stress as a crucial factor in liver diseases. World J Gastroenterol. 2014;20(25):8082–8091. doi:10.3748/wjg.v20.i25.8082
36. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014;94(3):909–950. doi:10.1152/physrev.00026.2013
37. He F, Ru X, Wen T. NRF2, a Transcription Factor for Stress Response and Beyond. Int J Mol Sci. 2020;21(13). doi:10.3390/ijms21134777
38. He F, Antonucci L, Karin M. NRF2 as a regulator of cell metabolism and inflammation in cancer. Carcinogenesis. 2020;41(4):405–416. doi:10.1093/carcin/bgaa039
39. He CH, Gong P, Hu B, et al. Identification of activating transcription factor 4 (ATF4) as an Nrf2-interacting protein. Implication for heme oxygenase-1 gene regulation. J Biol Chem. 2001;276(24):20858–20865. doi:10.1074/jbc.M101198200
40. Canning P, Sorrell FJ, Bullock AN. Structural basis of Keap1 interactions with Nrf2. Free Radic Biol Med. 2015;88(Pt B):101–107. doi:10.1016/j.freeradbiomed.2015.05.034
41. Wu KC, Liu JJ, Klaassen CD. Nrf2 activation prevents cadmium-induced acute liver injury. Toxicol Appl Pharmacol. 2012;263(1):14–20. doi:10.1016/j.taap.2012.05.017
42. Pan CW, Pan ZZ, Hu JJ, et al. Mangiferin alleviates lipopolysaccharide and D-galactosamine-induced acute liver injury by activating the Nrf2 pathway and inhibiting NLRP3 inflammasome activation. Eur J Pharmacol. 2016;770:85–91. doi:10.1016/j.ejphar.2015.12.006
43. Cao M, Wang H, Guo L, et al. Dibenzoylmethane Protects Against CCl4-Induced Acute Liver Injury by Activating Nrf2 via JNK, AMPK, and Calcium Signaling. AAPS J. 2017;19(6):1703–1714. doi:10.1208/s12248-017-0133-1
44. Peng X, Dai C, Liu Q, Li J, Qiu J. Curcumin Attenuates on Carbon Tetrachloride-Induced Acute Liver Injury in Mice via Modulation of the Nrf2/HO-1 and TGF-beta1/Smad3 Pathway. Molecules. 2018;23(1). doi:10.3390/molecules23010215
45. Huang YJ, Chen P, Lee CY, et al. Protection against Acetaminophen-induced acute liver failure by omentum adipose tissue derived stem cells through the mediation of Nrf2 and cytochrome P450 expression. J Biomed Sci. 2016;23:5. doi:10.1186/s12929-016-0231-x
46. Lahiri V, Hawkins WD, Klionsky DJ. Watch What You (Self-) Eat: autophagic Mechanisms that Modulate Metabolism. Cell Metab. 2019;29(4):803–826. doi:10.1016/j.cmet.2019.03.003
47. Vijayakumar K, Cho GW. Autophagy: an evolutionarily conserved process in the maintenance of stem cells and aging. Cell Biochem Funct. 2019;37(6):452–458. doi:10.1002/cbf.3427
48. Ueno T, Komatsu M. Autophagy in the liver: functions in health and disease. Nat Rev Gastroenterol Hepatol. 2017;14(3):170–184. doi:10.1038/nrgastro.2016.185
49. Allaire M, Rautou PE, Codogno P, Lotersztajn S. Autophagy in liver diseases: time for translation? J Hepatol. 2019;70(5):985–998. doi:10.1016/j.jhep.2019.01.026
50. Amir M, Zhao E, Fontana L, et al. Inhibition of hepatocyte autophagy increases tumor necrosis factor-dependent liver injury by promoting caspase-8 activation. Cell Death Differ. 2013;20(7):878–887. doi:10.1038/cdd.2013.21
51. Zhong Z, Umemura A, Sanchez-Lopez E, et al. NF-kappaB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell. 2016;164(5):896–910. doi:10.1016/j.cell.2015.12.057
52. Ilyas G, Zhao E, Liu K, et al. Macrophage autophagy limits acute toxic liver injury in mice through down regulation of interleukin-1beta. J Hepatol. 2016;64(1):118–127. doi:10.1016/j.jhep.2015.08.019
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.