Back to Journals » International Journal of Nanomedicine » Volume 18
Light-Assisted “Nano-Neutrophils” with High Drug Loading for Targeted Cancer Therapy
Authors Fan D, Wang S, Huang R, Liu X, He H ![]() , Zhang G
, Zhang G
Received 28 July 2023
Accepted for publication 31 October 2023
Published 9 November 2023 Volume 2023:18 Pages 6487—6502
DOI https://doi.org/10.2147/IJN.S432854
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Dongwoo Khang
Daopeng Fan,1,* Shuqi Wang,1,* Ran Huang,1,* Xiaoning Liu,1 Hua He,1 Gaiping Zhang1– 3
1College of Veterinary Medicine, International Joint Research Center of National Animal Immunology, Henan Agricultural University, Zhengzhou, 450046, People’s Republic of China; 2Longhu Laboratory, Zhengzhou, 450046, People’s Republic of China; 3School of Advanced Agriculture Sciences, Peking University, Beijing, 100871, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Hua He, College of Veterinary Medicine, International Joint Research Center of National Animal Immunology, Henan Agricultural University, Zhengzhou, 450046, People’s Republic of China, Email [email protected]
Background: Nanomedicine presents a promising alternative for cancer treatment owing to its outstanding features. However, the therapeutic outcome is still severely compromised by low tumor targeting, loading efficiency, and non-specific drug release.
Methods: Light-assisted “nano-neutrophils (NMPC-NPs)”, featuring high drug loading, self-amplified tumor targeting, and light-triggered specific drug release, were developed. NMPC-NPs were composed of neutrophil membrane-camouflaged PLGA nanoparticles (NPs) loaded with a hypoxia-responsive, quinone-modified PTX dimeric prodrug (hQ-PTX2) and photosensitizer (Ce6).
Results: hQ-PTX2 significantly enhanced the drug loading of NPs by preventing intermolecular π–π interactions, and neutrophil membrane coating imparted the biological characteristics of neutrophils to NMPC-NPs, thus improving the stability and inflammation-targeting ability of NMPC-NPs. Under light irradiation, extensive NMPC-NPs were recruited to tumor sites based on photodynamic therapy (PDT)-amplified intratumoral inflammatory signals for targeted drug delivery to inflammatory tumors. Besides, PDT could effectively eliminate tumor cells via reactive oxygen species (ROS) generation, while the PDT-aggravated hypoxic environment accelerated hQ-PTX2 degradation to realize the specific release of PTX, thus synergistically combining chemotherapy and PDT to suppress tumor growth and metastasis with minimal adverse effects.
Conclusion: This nanoplatform provides a prospective and effective avenue toward enhanced tumor-targeted delivery and synergistic cancer therapy.
Keywords: neutrophil, self-amplified tumor targeting, high drug loading, hypoxia-responsive, chemo-photodynamic therapy
Introduction
Breast cancer is the most common malignant tumor in women worldwide, where systemic metastasis, such as bone or lung metastasis, is the main cause of mortality.1,2 Although chemotherapy and surgical resection have advanced in cancer treatment, side effects and unsatisfactory therapeutic efficacy still remain in clinical settings.3,4 In recent years, nanomedicine has emerged as a promising alternative for cancer theranostics based on its remarkable characteristics, including safety, controlled drug release, and targeted drug delivery.5,6 Nanomedicine can provide targeted delivery of drugs to tumors through the enhanced permeability and retention (EPR) effect and ligand-mediated active targeting.7 However, the outcomes are far from satisfactory due to tumor heterogeneity, complex physiological microenvironment, and limited differences in receptors between tumor and normal cells. The ability to broadcast precise signals in biological systems provides new opportunities for effective drug delivery.8,9 Chronic inflammation serves as a persistent signal in tumors that provokes an immune response for the recruitment of immune cells to the tumor site.10,11 Among the recruited immune cells, neutrophils (NEs) are the first leukocytes to be recruited to the inflammatory tumor sites,12–14 so they can be used as natural carriers for targeted delivery of cargo to tumor sites.15 However, as terminal-differentiated cells, the short lifespan of neutrophils makes them difficult to culture in vitro, thus limiting the direct application of neutrophils as drug delivery vehicles.16–19 Extensive evidences have revealed that the majority of cells demonstrate targeting ability derived from their cell membrane,20,21 which provides an alternative strategy for targeted drug delivery via cell membrane-coated biomimetic carriers.22,23 Consequently, biomimetic drug delivery systems with neutrophil membrane coating are available for targeted drug delivery to inflammatory tumor sites.24–27
Improving drug loading of nanoparticles (NPs) and controlling specific drug release in tumor cells are critical issues that need to be addressed for nanomedicine to achieve efficient antitumor treatment.28–30 Chemo-drugs, especially hydrophobic anticancer drugs, prefer to aggregate and precipitate during encapsulation owing to π–π interactions between drugs,31 so that the majority of NPs suffer from low drug loading (< 10%) and premature drug leakage during circulation.32 Numerous studies have evidenced that prodrugs, particularly dimeric prodrugs, can substantially elevate the drug loading of NPs, since the linker in prodrugs has the ability to attenuate strong intermolecular drug interactions.33 In addition, dimeric prodrugs responsive to internal stimuli (ie, hypoxia,34 reactive oxygen species (ROS),35 pH,36,37 and glutathione (GSH))38 have been exploited to modulate the specific release of drugs at tumor sites, thus potentiating safety and antitumor efficacy.39 Especially, severe intratumoral hypoxic environment is the consequence of insufficient oxygen supply by abnormal vessels and massive oxygen consumption in tumor cells. Based on these key features of tumors, extensive hypoxia-responsive bonds have been studied, such as quinone,40 azobenzene41 and 2-nitroimidazole.42 However, heterogeneous and inadequate hypoxic environments are often prevalent in the vascular periphery and early tumor stage, thus failing to effectively trigger hypoxia-responsive bonds for drug release.43
Photodynamic therapy (PDT) has gained substantial interest in the field of anticancer treatment,44 because it features minimal invasiveness, high spatiotemporal selectivity, and enables efficient killing of tumor cells by light irradiation of photosensitizers to generate reactive oxygen species (ROS).45,46 In addition to causing tumor cell death, PDT-generated ROS can provoke acute inflammation in tumors,47–49 which dramatically enhances the targeted inflammatory signal for recruitment of more neutrophil membrane-camouflaged NPs to inflammatory tumor sites, thereby achieving targeted tumor delivery of the drug.50,51 On the other hand, a serious hypoxic environment is formed during PDT as a result of the conversion of oxygen to ROS.52 As such, a PDT-aggravated hypoxic environment can be applied to accelerate the specific release of drugs from hypoxia-responsive prodrug systems, thus boosting anticancer efficacy while reducing toxicity to normal tissues.53
Herein, we constructed light-assisted, neutrophil membrane-camouflaged poly (lactic-co-glycolic acid) (PLGA) “nano-neutrophils” that exhibited high drug loading, light-facilitated drug release, and self-amplified tumor accumulation for the targeted treatment of cancer and metastasis (Scheme 1). A hypoxia-responsive, quinone-modified paclitaxel (PTX) dimeric prodrug (hQ-PTX2) was synthesized and further mixed with a photosensitizer (chlorin e6, Ce6). Followed by PLGA NPs encapsulation, where the hypoxia-responsive linker in hQ-PTX2 could effectively attenuate strong intermolecular π–π interactions between PTX, thus enabling high drug loading of PLGA NPs (16.9%). Subsequently, the neutrophil membrane was coated on the surface of PLGA NPs to form NMPC-NPs, which endowed NPs with the biological characteristics of neutrophils, such as stability and inflammation-targeting ability. Interestingly, PDT could be utilized to amplify inflammatory signals in tumors to recruit more NMPC-NPs, yielding a superior targeted delivery of drugs to inflammatory tumor sites. Moreover, PDT not only eradicated tumor cells via ROS generation, but also PDT-aggravated hypoxic environment-induced hQ-PTX2 degradation to accelerate the specific release of PTX, thus achieving synergistic chemo-photodynamic therapy to attenuate tumor growth and metastasis. This study provides a promising approach for tumor-targeted drug delivery and synergistic cancer therapy.
 |
Scheme 1 Schematic illustration demonstrating the preparation of “nano-neutrophils” (NMPC-NPs) from neutrophil membrane and their synergistic therapeutic efficacy based on their high drug loading (I), PDT-amplified tumor targeting (II) and -accelerated specific release of PTX (III). |
Materials and Methods
Materials, Cells and Animals
Chemicals were purchased from J&K (Beijing, China). Poly (D, L-lactide-co-glycolide) (PLGA, 50:50, Mw = 5000–15,000 Da) was purchased from Dalian Meilun Biotechnology Co. LTD (Dalian, China). Histopaque-1077 was purchased from Sigma-Aldrich (St Louis, MO, USA). Coumarin-6 and sodium cholate were purchased from Aladdin (Shanghai, China). MTT, BCA kit and TUNEL apoptosis assay kit were purchased from Beyotime® Biotechnology (Shanghai, China). Anti-CXCR4 and anti-Integrin beta 1 were purchased from Abcam (Cambridge Science Park, UK). Anti ATP1A1 was purchased from BOSTER (Wuhan, China). PTX was purchased from Macklin (Shanghai, China). All solvents were purchased from Sinopharm Chemical Reagent Co. Ltd (Shanghai, China).
4T1 (mouse mammary carcinoma) and RAW 264.7 cells were obtained from ATCC (Rockville, MD) and cultured in RPMI-1640 medium containing 10% fetal bovine serum (FBS). Female BALB/c mice (6–8 weeks) were purchased from Animal experimental center of Huaxing (Zhengzhou, China) and housed in clean room. All animal studies were approved by the Institutional Animal Care and Use Committee, Henan Agricultural University.
Characterization and Degradation of hQ-PTX2
The synthesis of hQ-PTX2 were shown in the Supporting Information (Scheme S1). The hQ-PTX2 was dissolved in a mixed solution (acetonitrile/PBS, v/v = 1/1) containing 10 mM Na2S2O4. Following incubation at different times and centrifugation (10,000 rpm, 5 min), the supernatant was collected and diluted with acetonitrile before high-performance liquid chromatography (HPLC) or liquid chromatography-mass spectrometry (LC-MS) analysis.
Preparation and Characterization of NMPC-NPs
A mixture of PLGA (10 mg), hQ-PTX2 (0.3 mg), and Ce6 (0.2 mg) was dissolved in DCM (2 mL) and mixed with 1% sodium cholate solution (4 mL). After sonication for 2 min, the obtained emulsion was added to a 0.5% sodium cholate solution (10 mL) and stirred for 4 h to evaporate DCM. Finally, the hQ-PTX2- and Ce6- co-loaded PLGA nanoparticles (PC-NPs) were collected by centrifugation (14,500 g, 50 min) and re-suspended in DI water for further use. The hQ-PTX2 loaded PLGA NPs (P-NPs), Ce6 loaded PLGA NPs (C-NPs), Cou6 loaded PLGA NPs (Cou6-NPs) were formulated using similar methods. Alternatively, 1 mL of the neutrophil membrane solution (1 mg/mL) in water was mixed with PC-NPs (1 mg/mL, 1mL), and further extruded using Avanti polar lipids to obtain neutrophil membrane coated NPs (NMPC-NPs) (Table S1). C-NPs, NMC-NPs, PC-NPs, and NMPC-NPs were irradiated at 660 nm (2 or 10 mW/cm2) for 30 min in both the in vitro and in vivo assays.
The particle size, zeta potential, and morphology of the NPs were determined using dynamic light scattering instrument (DLS) and transmission electron microscopy (TEM) measurement, respectively. To assess the stability of the NPs, PC-NPs and NMPC-NPs were incubated with PBS or 1640 containing 10% FBS at RT before measurement of particle size at different time points. To assess drug-loading content (DLC) and drug-loading efficiency (DLE), NMPC-NPs (100 μL) were diluted with acetonitrile (1.9 mL) and incubated overnight. The DLC and DLE of hQ-PTX2 were determined by HPLC using an acetonitrile/water mixture as the mobile phase (0.1 mL/min, λabs = 254 nm; Figure S1).
PDT-Facilitated PTX Release in vitro and in vivo
4T1 cells were seeded in 24-well plates (1 × 105 cells/well) and incubated for 24 h. Cells were treated with NMPC-NPs (60 μg PTX/mL) for 24 h, irradiated, and then continued to incubate for 2, 4 and 6 h, respectively. 200 μL of RIPA buffer was added, and the protein content in the lysate was quantified by BCA kit. The lysate was diluted with mixture solution (methanol/DCM = 1/1, v/v) overnight, centrifuged (10,000 rpm, 4 °C) for 30 min, and the PTX content in the supernatant was examined by HPLC analysis (λabs = 254 nm).
To further quantify in vivo PTX release under light irradiation, 4T1 tumor-bearing mice were intratumorally administered NMPC-NPs (10 mg PTX/kg) and irradiated at 1 h post-injection. At 3, 6, and 9 h post-irradiation, the tumors were collected, washed with PBS, weighed, and homogenized in RIPA lysis buffer. The lysate was subjected to the same treatment as described above to determine PTX content using HPLC analysis.
Detection of PDT-Induced Inflammatory Level in vitro
To evaluate PDT-induced inflammatory levels, 4T1 and RAW 264.7, cells were seeded into 24-well plates (1 × 105 cells/well) and cultured overnight, respectively. 4T1 cells were co-incubated with NMC-NPs (20 μg Ce6/mL) for 12 h and then irradiated. At 12 h post-irradiation, the supernatant of 4T1 cells was added to the medium of RAW 264.7 cells, and further cultured overnight. The mixed supernatant was collected and used to determine IL-6 and TNF-α levels using ELISA kits. The supernatant of RAW 264.7 cells without NPs treatment served as the control.
Cellular Uptake
To further evaluate cellular uptake under PDT-induced inflammatory conditions, RAW 264.7, cells were treated as described above and the inflammatory supernatant was added to 4T1 cells. Following incubation with NMC-NPs (20 μg Ce6/mL) for 4 h, cells were washed with PBS, fixed with 4% paraformaldehyde, and stained with DAPI before CLSM observation. NP-treated cells were collected and analyzed by flow cytometry. Alternatively, after treatment with NPs for different time, 4T1 cells were lysed at RT for 15 min. The content of Ce6 and protein in the mixture was measured by spectrophotometry (λex = 466 nm, λem = 504 nm) and BCA kit, respectively.
In vitro Anticancer Efficacy
To evaluate the anticancer efficacy of NPs, 4T1 cells were cultured in 96-well plates at a density of 2×104 cells/well and incubated overnight. After incubation with NMC-NPs, NMP-NPs, PC-NPs, and NMPC-NPs for 24 h, cells were irradiated and incubated for another 48 h following viability assessment by MTT assay.
To verify the antitumor effect of NPs in a hypoxic environment, 4T1 cells seeded in 96-well plates following the above method were pretreated with CoCl2 (100 μM) for 12 h, and then incubated with NMP-NPs for 72 h before viability assessment via MTT assay. To further evaluate the antitumor effect of NPs under PDT-induced hypoxic conditions, 4T1 cells in 96-well plates were pretreated with vitamin C (VC, 200 μM) for 12 h, incubated with NMPC-NPs for 24 h, irradiated, and further incubated for 48 h to assess cell viability by MTT assay.
To detect apoptosis of tumor cells, 4T1 cells were plated into 24-well plates at 1×105 cells/well and incubated for 24 h. Various NPs were added at a PTX concentration of 3.8 μg/mL and incubated with cells for 24 h. Following irradiation, cells were further incubated for 24 h, stained with the Annexin V-FITC/Propidium Iodide (PI) Apoptosis Detection Kit, and analyzed by flow cytometry. On the other hand, cells subjected to the same treatment as described above were stained with calcein-AM (1.5 μM) and PI (5 μM) for observation of dead/live cells by fluorescence microscopy.
In vivo PDT-Amplified Tumor Targeting
Mice bearing subcutaneous 4T1 tumor (~ 200 mm3) were randomized (n = 3), i.v. injected with free Ce6, C-NPs, and NMC-NPs (5 mg Ce6/kg), and irradiated tumor sites at 8 h post-injection. 96 h after the first dose, the second dose was administered to each group of mice. Fluorescence imaging was performed using in vivo imaging system at various time-points. At 104 h post-injection, mice were sacrificed, tumors and major organs were collected for ex vivo imaging and quantitative analysis. To evaluate PDT-amplified inflammation, tumors in mice were collected and the cytokine levels of TNF-α and IL-6 were measured using an ELISA kit. All animal studies were approved by the Institutional Animal Care and Use Committee of Henan Agricultural University and performed in accordance with the Guidelines on Laboratory Animal Welfare issued by the Ministry of Science and Technology of the People’s Republic of China.
Anticancer Efficacy in vivo
Mice bearing subcutaneous 4T1 tumor (~50 mm3) were randomly grouped (n = 8) and i.v. administered PBS, free PTX, NMC-NPs, NMP-NPs, PC-NPs, and NMPC-NPs (12.5 mg PTX/kg, 5 mg Ce6/kg) on day 1, 5, 9 and 13. Eight hours after dosing, the tumors in the NMC-NPs-, PC-NPs-, and NMPC-NP-treated mice were irradiated. Body weight and tumor volume were monitored every two days. Tumor volume was calculated as 0.5 × length × (width)2. The major tissues (heart, liver, spleen, lung, and kidney) and tumors were collected from one mouse in each group on day 26, fixed, embedded, sectioned, stained with H&E and TUNEL, and observed by microscopy and fluorescence microscopy, respectively. When the tumor volume was > 1000 mm3 or the mice died during the treatment period, they were identified as dead. To evaluate the in vivo safety, blood was collected at two days post the last injection, and the serum alanine aminotransferase (ALT), alanine aminotransferase (AST), glutamine aminotransferase (Cr) and urea nitrogen (BUN) level were measured via the ALT, AST, Cr detection kit (Yuanye, China) and BUN detection kit (Solarbio, China), respectively.
Anti-Metastasis Efficacy in vivo
Mice bearing orthotopic GFP/luc-4T1 tumor were randomized (n = 8) and i.v. injected with PBS, free PTX, NMC-NPs, NMP-NPs, PC-NPs, or NMPC-NPs (12.5 mg PTX/kg, 5 mg Ce6/kg) on day 14, 18, 22, 26, where NMC-NPs, PC-NPs and NMPC-NPs treated mice were irradiated at tumor sites at 8 h post-injection. To monitor tumor metastasis, d-fluorescein sodium salt (150 mg/kg) was injected intraperitoneally prior to bioluminescence imaging using an in vivo imaging system on day 13, 20, 27, and 34. The mice were sacrificed on day 36, and blood was collected. The circulating tumor cells (CTCs) in the blood were isolated using Histopaque-1077 and cultured in 24-well plates for 6 h before fluorescence microscopy observation. Besides, the lungs were collected and photographed, and the lung sections were stained with hematoxylin and eosin (H&E) following the above method before observation under a microscope.
Results and Discussion
Synthesis and Degradation of hQ-PTX2
Paclitaxel (PTX) is a common hydrophobic chemotherapeutic drug with excellent antitumor efficacy in breast and ovarian cancers.54 However, drug aggregation/precipitation during the formulation would compromise drug loading. Therefore, a hypoxia-responsive dimeric prodrug, hQ-PTX2 was designed (Scheme S1), in which the rotatable linker could weaken PTX-PTX interactions, thus improving the drug-loading capacity of NPs. To evaluate the hypoxia-responsive properties of hQ-PTX2, the hQ-PTX2 degradation under Na2S2O4 treatment was determined by HPLC analysis. After incubation with hQ-PTX2 for various times under Na2S2O4 treatment, the peak of hQ-PTX2 (13.5 min) gradually decreased and disappeared, whereas a new peak ascribed to PTX (9.6 min) increased (Figure 1A and B), suggesting that quinone reduction caused by hypoxia contributed to the degradation of hQ-PTX2. Similar results were obtained using LC-MS analysis (Figure 1C). In addition, PTX release was significantly potentiated by Na2S2O4 treatment compared with that in the absence of Na2S2O4 treatment, affording a cumulative release of 90.5% within 48 h (Figure 1D). These results collectively indicated that hypoxia-triggered hQ-PTX2 degradation could effectively facilitate PTX release.
 |
Figure 1 Hypoxia-induced hQ-PTX2 degradation and PTX release. (A) Degradation mechanism of hQ-PTX2. HPLC (B) and LC-MS (C) analysis of hQ-PTX2 degradation upon incubation with Na2S2O4 (10 mM). (D) The PTX release profiles of hQ-PTX2 with or without Na2S2O4 (n = 3). |
Preparation and Characterization of NMPC-NPs
Following the methods previously reported,55 hQ-PTX2- and Ce6- co-loaded PLGA NPs (PC-NPs) adopting the particle size of 155 nm and the zeta potential of −9.6 mV were formulated following previously reported methods (Figure 2A). Excitingly, the drug loading content (DLC) and drug loading efficiency (DLE) of hQ-PTX2 were 16.9% and 77.5%, respectively, indicating the high drug loading ability of PC-NPs. Additionally, the DLC and DLE of Ce6 were 6.9% and 66.7%, respectively. Subsequently, the cell membranes of neutrophils derived from mouse blood were decorated on the surface of the PC-NPs, thus forming NMPC-NPs. The abbreviations of the different NPs were listed in Table S1, where C-NPs, PC-NPs, NMC-NPs, and NMPC-NPs were illuminated at 660 nm (2 or 10 mW/cm2) for 30 min in all in vitro and in vivo studies unless otherwise specified. When the neutrophil membrane (NM)/PC-NPs weight ratio was > 1/1, constant expression of membrane proteins was demonstrated in NMPC-NPs, suggesting a saturated coating of NM on PC-NPs (Figure S2). Therefore, an optimized NM/PC-NP weight ratio of 1 was used in all studies, unless otherwise specified. The particle size of NMPC-NPs (165 nm) was slightly larger than that of PC-NPs, yet the zeta potential of NMPC-NPs (−12.6 mV) was similar to that of NM (−13.4 mV), implying that NM was successfully coated on the surface of PC-NPs (Figure 2A). Transmission electron microscopy (TEM) images further showed that the NM-NPs possessed a spherical morphology with a core-shell structure and a particle size of 155 nm, which was consistent with dynamic light scatter meter (DLS) results (Figure 2B). In addition, the particle size was constant when NMPC-NPs were stored in PBS or FBS for up to 15 days, indicating the excellent stability of NMPC-NPs (Figure S3).
 |
Figure 2 NPs Characterization and PDT-elicited PTX release in vitro and in vivo. (A) Particle size and zeta potential of PC-NPs and NMPC-NPs (n = 3). (B) Representative TEM image of NMPC-NPs. (C) SDS-PAGE image of NEs, NM, NM-NPs. (D) Western blot analysis demonstrating the characteristic protein band of NEs, NM and NM-NPs. (E) The PTX level in cells after incubation with NMPC-NPs for 4 h. Cells were further irradiated and incubated for different times before HPLC analysis (n = 3). (F) Immunofluorescence staining images of tumor sections. Tumors were intratumorally injected with NMPC-NPs, irradiated at 1 h post-injection, and stained with pimonidazole. (G) The PTX level in 4T1 tumors at 3 h, 6 h, and 9 h post-irradiation. Tumors were treated with NMPC-NPs as described in (F) (n = 3). Data expressed as means ± SD. **p < 0.01. |
To confirm that membrane proteins were still present on the NM-NPs, SDS-PAGE and Western blotting were performed to evaluate the protein expression in various groups. As shown in Figure 2C and D, NM-NPs had the same protein track and representative membrane proteins (ie, β1-integrin and CXCR4) as NEs and NM. The above results revealed that the neutrophil membrane was successfully coated on the NPs, thereby rendering NPs with the biological ability of neutrophils to target inflammatory tumor sites.
PDT-Elicited Hypoxia Facilitated PTX Release in vitro and in vivo
Intracellular ROS generation and hypoxia exacerbation induced by PDT were determined by ROS/hypoxia detection kits. As demonstrated in Figures S4 and S5, NMPC-NP-treated cells exhibited enhanced green (ROS probe) and magenta (hypoxia probe) fluorescence under light irradiation, indicating that PDT could significantly generate ROS and aggravate hypoxia. Quantitative HPLC analysis was performed to detect the PDT-induced intracellular PTX release. When NMPC-NP-treated 4T1 cells were irradiated, the PTX level increased remarkably compared to that in the absence of light irradiation, particularly up to 2.5-fold at 6 h post-irradiation (Figure 2E), suggesting that the PDT-aggravated hypoxic environment could accelerate the release of PTX from NMPC-NPs.
PDT-induced intratumoral hypoxic stress was further investigated using immunofluorescence staining. After the administration of NMPC-NPs and light irradiation, serious hypoxic stress was demonstrated, as evidenced by the increased green fluorescence of the hypoxia probe (Figure 2F), suggesting severe intratumoral hypoxic stress caused by PDT. Consistent with in vitro results, HPLC analysis revealed enhanced intratumoral PTX release from NMPC-NPs under PDT treatment. As shown in Figure 2G, the PTX level in tumors treated with NMPC-NPs was significantly increased under light irradiation, with a 2.8-fold increase after 9 h of irradiation compared with that in the absence of irradiation. The in vitro and in vivo results consistently revealed that the PDT-induced severe hypoxic environment substantially accelerated the release of PTX from the NMPC-NPs.
PDT-Elicited Inflammatory Response Assisted Tumor Targeting in vitro
PDT has been reported to elicit the immunogenic death of tumor cells, resulting in the release of damage-associated molecular patterns (DAMPs). The DAMPs can serve as stimulators to induce proinflammatory cytokine production by macrophages.56 Hence, the medium of NMC-NP-treated cells with light irradiation can be used to stimulate RAW264.7 to secrete proinflammatory cytokines, yielding a PDT-boosted inflammatory environment to evaluate the uptake level of NMC-NPs (Figure 3A). The expression of proinflammatory cytokines (IL-6 and TNF-α), which are markers of inflammation in macrophages, was first determined. When cells were treated with NMC-NPs under light irradiation, the expression of IL-6 and TNF-α was significantly elevated (Figure 3B and C), suggesting that PDT contributed to the strengthening of inflammatory response.
 |
Figure 3 PDT-elicited inflammatory response assisted tumor targeting in vitro. (A) Schematic illustration of cellular uptake assay. 4T1 cells were incubated with NMC-NPs for 12 h, followed by irradiation and incubation for another 12 h. The supernatant of 4T1 cells was added to RAW 264.7 cells, and further cultured overnight to obtain inflammatory medium. 4T1 cells were treated with various NPs with or without inflammatory condition. The expression level of TNF-α (B) and IL-6 (C) from RAW 264.7 cells (n = 3). RAW 264.7 cells were treated as described in (A). (D) CLSM images of 4T1 cells following incubation with C-NPs and NMC-NPs for 4 h. 4T1 cells were pretreated with inflammatory medium for 12 h. (E) The uptake level of NPs in 4T1 cells as evaluated by flow cytometry. 4T1 cells were treated as described in (D). (F) Uptake levels of NPs in 4T1 cells. 4T1 cells were pretreated as described in (D) and further incubated with NPs for different times (n = 3). Data expressed as means ± SD. ***p < 0.001. |
The uptake levels of the various NPs were further evaluated using CLSM. As shown in Figure 3D, slight red fluorescence (Ce6) was distributed in the cytoplasm of C-NP-treated cells (w/ inflammation) and NMC-NP-treated cells (w/o inflammation). In contrast, a large number of NMC-NPs were internalized in the inflammatory environment, as evidenced by the cytoplasmic distribution of strong red fluorescence, indicating that PDT-boosted inflammation could improve the uptake level of NMC-NPs owing to the inflammatory targeting ability of the neutrophil membrane. In consistence with above results, the quantitative flow cytometric and spectrofluorimetry analysis indicated that NMC-NPs possessed the maximal uptake level, which was 4.4 and 6.8 times higher than those of NMC-NPs (w/o inflammation) and C-NPs (w/ inflammation), respectively (Figure 3E and F). Collectively, these results suggested that inflammation boosted by PDT could effectively enhance in vitro tumor targeting of neutrophil membrane-coated NPs.
Synergistic Antitumor Efficacy in vitro
The biosafety of the NPs was evaluated using the MTT assay. When 4T1 cells were co-incubated with NM-NPs for 72 h, > 90% cell viability was observed at an NPs concentration of 100 μg/mL (Figure S6), suggesting that the neutrophil membrane coating could effectively improve the biosafety of NPs. To further investigate the PDT-elicited synergistic anticancer effects of NMPC-NPs, 4T1 cells were incubated with various NPs and then irradiated. Compared with other groups, NMPC-NPs displayed remarkable synergistic antitumor efficacy, as evidenced by a combination index (CI) of < 1 between PTX and Ce6 and >1.8-fold lower IC50 of PTX in NMPC-NPs (3.5 μg/mL) than in NMP-NPs (6.4 μg/mL) (Figure 4A, Table S2). Alternatively, the cytotoxicity of the NMP-NPs under hypoxic conditions was evaluated in CoCl2 pretreated cells. As shown in Figure 4B and Table S3, the IC50 of PTX was markedly reduced under CoCl2-induced hypoxic conditions, indicating that hypoxia facilitated PTX release and improved the anticancer effect of NPs. To further investigate the ROS-assisted antitumor efficacy, cells were pretreated with VC and irradiated under NMPC-NPs treatment. As shown in Figure 4C and Table S4, attenuated anticancer effect of NMPC-NPs was demonstrated, because of PDT-generated ROS scavenged by VC. These results collectively revealed that the PDT-induced hypoxic environment contributed to PTX release to realize remarkable synergistic antitumor efficacy in vitro.
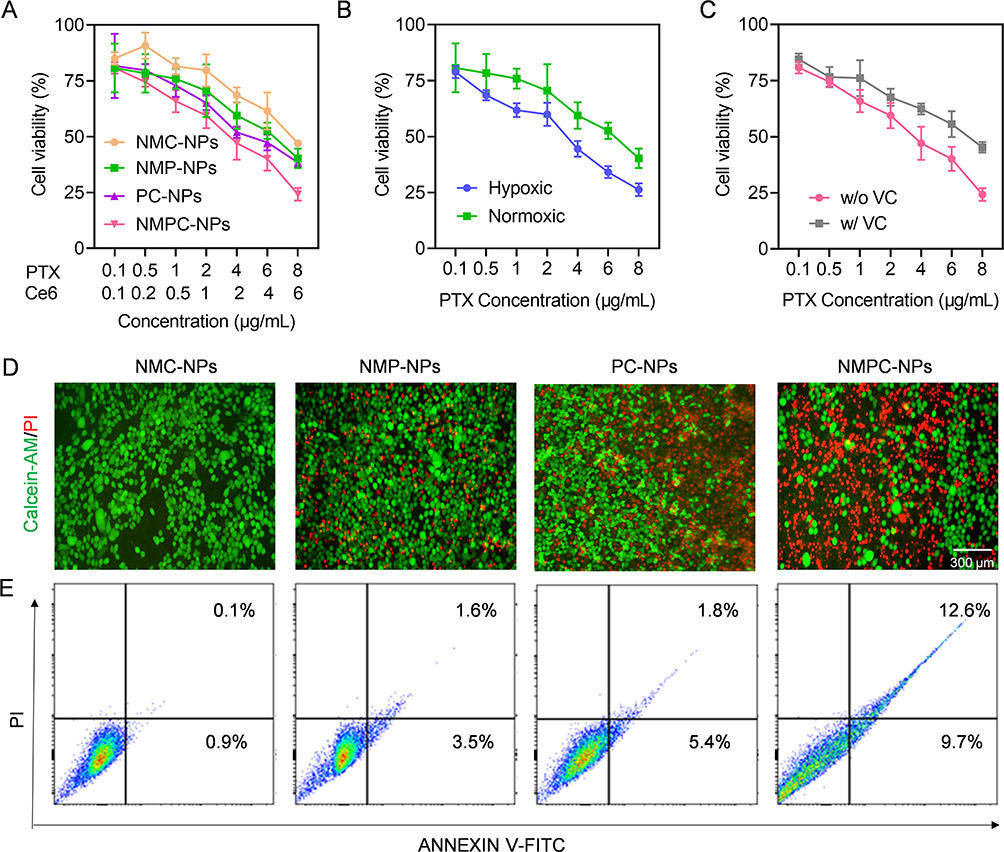 |
Figure 4 Synergistic anticancer efficacy in vitro. 4T1 cells were incubated with NMC-NPs, PC-NPs, NMPC-NPs for 24 h, followed by irradiation and incubation for 48 h. Cells incubated with NMP-NPs for 72 h served as control. (A) Cytotoxicity of various NPs in 4T1 cells (n = 3). (B) Cytotoxicity of NMP-NPs under hypoxia or normoxia (n = 3). (C) Cytotoxicity of NMPC-NPs with or without VC pretreatment (n = 3). (D) Fluorescence images of 4T1 cells treated with various NPs and stained with calcein-AM and PI. (E) Flow cytometric analysis of 4T1 cells treated with various NPs and stained with Annexin V-FITC/PI. |
To visually assess the in vitro anticancer efficacy, various NP-treated cells were stained with PI and calcein-AM to observe dead and live cells. As demonstrated in Figure 4D, strong green fluorescence (calcein-AM) was observed in cells treated with NMP-NPs and PC-NPs, whereas extensive cell death was observed in NMPC-NP-treated cells, as evidenced by the extensive red fluorescence of PI, suggesting a notable therapeutic effect of NMPC-NPs. A similar trend was noted in the apoptosis and necrosis levels of tumor cells detected using the PI/Annexin V-FITC apoptosis detection assay. As shown in Figure 4E, NMPC-NPs provoked the highest levels of apoptosis and necrosis (22.3%), significantly outperforming NMC-NPs (1.0%), NMP-NPs (5.1%), and PC-NPs (7.2%). Collectively, these results indicated that NMPC-NPs possessed remarkable anticancer efficacy ensured by the combination of chemo-drugs and PDT.
Self-Amplified Tumor Targeting in vivo
To evaluate the self-amplified tumor targeting of NMPC-NPs, 4T1 xenograft tumor-bearing mice were i.v. injected with various groups and NPs biodistribution was observed using a live animal imaging system (Figure 5A). As demonstrated in Figure 5B, due to the EPR effect-assisted tumor distribution, the fluorescence intensity of tumors treated with NPs was stronger than that of free Ce6 and reached a maximum at 8 h post-injection. Therefore, NMPC-NPs that inherited the biological characteristics of the neutrophil membrane could be effectively recruited to inflammatory tumors, enabling superior tumor accumulation. After 8 h injection in various groups, mice were irradiated and received a second injection at 96 h. Excitingly, the fluorescence intensity of NMPC-NP-treated tumors was dramatically higher under light irradiation than both without light irradiation and at the first administration, suggesting that PDT contributed to tumor accumulation of NMPC-NPs. Similar results were obtained with ex vivo fluorescence imaging. 8 hours after the second administration (96 h), the tumors were collected, imaged, sectioned and stained. As shown in Figure 5C and D, NMC-NPs with light irradiation led to 5.3- and 3.0-fold higher fluorescence intensity than C-NPs and NMC-NPs without light irradiation, respectively. Besides, numerous intratumoral distributions of red fluorescence (Ce6) was presented (Figure 5E). Alternatively, the typical inflammatory factors, including TNF-α and IL-6, have been used to investigate the PDT-induced inflammation levels. In accordance with the above results, TNF-α and IL-6 levels reached the maximum at 96 h, and were substantially elevated in NMC-NP-treated tumors with light irradiation compared to the other groups (Figure 5F and G, Figure S7), indicating increased intratumoral inflammation caused by PDT. Collectively, these results indicated that PDT-elevated inflammatory response contributed to the self-amplified recruitment of NMC-NPs to tumor sites.
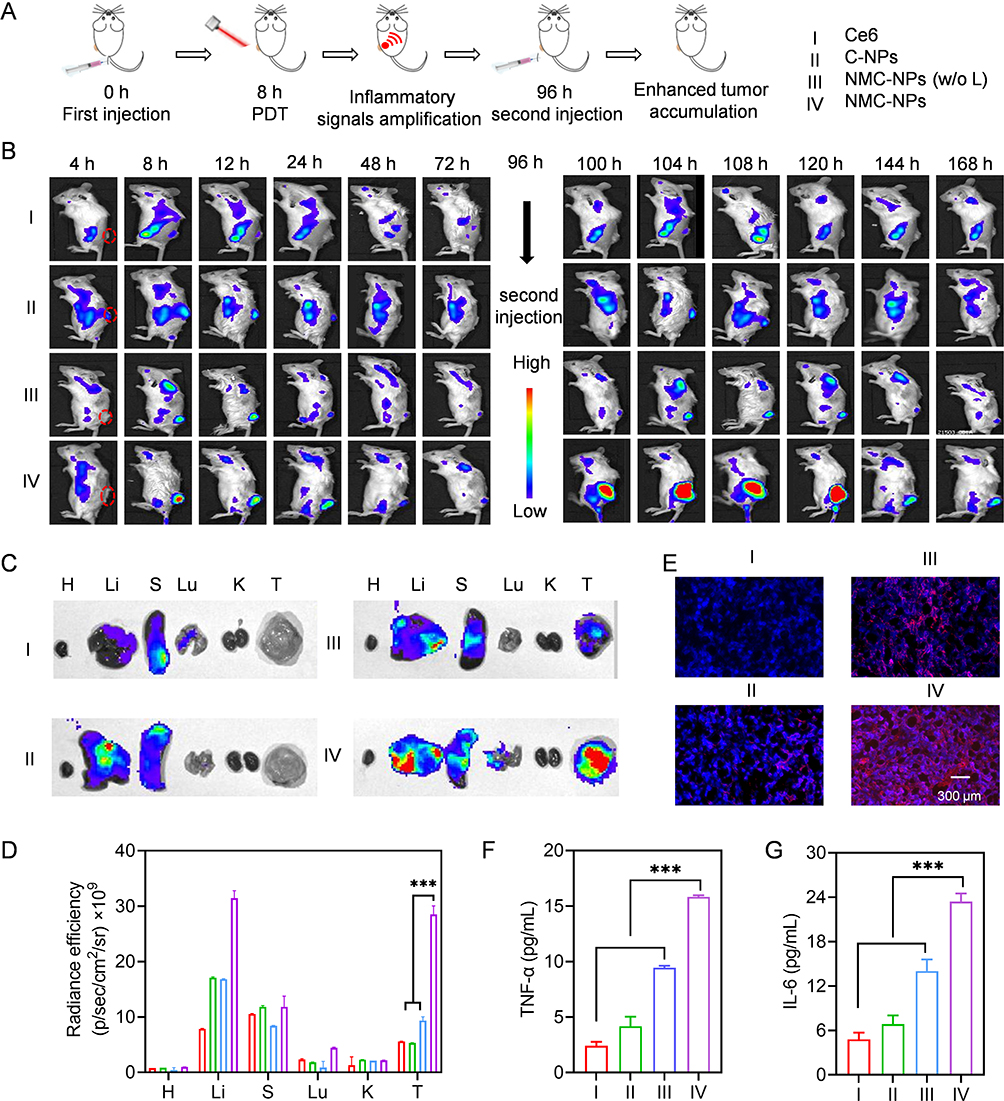 |
Figure 5 Self-amplified tumor targeting in vivo. Mice were i.v. injected with various groups, irradiated at 8 h post-injection and further received the second administration at 96 h (5 mg Ce6/kg). (A) The time line of tumor accumulation study. (B) Fluorescence images of mice at different time points. Ex vivo fluorescence images (C) and fluorescence intensity (D) of major tissues and tumors harvested at 104 h (n = 3) (H: heart, Li: liver, (S) spleen, Lu: lung, (K) kidney, (T) tumor). (E) Fluorescence images of tumor sections collected at 104 h. TNF-α (F) and IL-6 (G) level in tumors harvested at 104 h (n = 3). Data expressed as means ± SD. ***p < 0.001. |
Synergistic Antitumor and Anti-Metastasis Efficacy in vivo
4T1 xenograft tumor-bearing mice were i.v. administered various groups to evaluate in vivo therapeutic effects of NPs (Figure 6A). As shown in Figure 6B-D, NMP-NPs, NMC-NPs, and PC-NPs could suppress tumor growth to some extent, conferring the tumor growth inhibition (TGI) rates of 45.4%, 37.8%, and 62.7%, respectively. In comparison, NMPC-NPs exhibited significant tumor regression with a TGI rate of 94.3%. In particular, NMPC-NPs showed superior tumor growth inhibition compared to PC-NPs because PDT-caused intratumoral inflammation contributed to amplify the tumor accumulation of NMPC-NPs, thus achieving notable anticancer efficacy. Consistently, NMPC-NPs had the maximum survival rate during the 60d observation period and the highest level of cell death and apoptosis (Figure 6E and F). On the other hand, neither the body weight of mice nor the H&E staining of major organs were obviously affected under NPs treatment (Figures S8 and S9). Additionally, NMPC-NPs treatment did not result in abnormal ALT, AST, Cr and BUN level at two days post the last injection (Figure 7), indicating the negligible systemic toxicity of NMPC-NPs.
 |
Figure 6 PDT-assisted synergistic anticancer efficacy in vivo. (A) Time line of in vivo antitumor study. Mice were i.v. injected with various groups on day 1, 5, 9, and 13, wherein NMC-NPs, PC-NPs, NMPC-NPs treated mice were irradiated at 8 h post-injection (12.5 mg PTX/kg, 5 mg Ce6/kg). (B) Images of tumors collected on day 26. (C) Tumor volume curves of mice in different groups (n = 8). (D) The tumor growth inhibition rate of various groups (n = 8). (E) Survival rate of mice within 60-d observation period (n = 7). (F) H&E and TUNEL stained tumors harvested on day 26. Data expressed as means ± SD. ***p < 0.001. |
 |
Figure 7 In vivo safety. (A-D) The hematological assessment of mice at day 2 after i.v. injection of PBS or NMPC-NPs (12.5 mg PTX/kg) (n = 3). |
Tumor metastasis in mice that received the same administration of the antitumor treatment was further monitored using bioluminescence imaging. In contrast to the large bioluminescence distribution induced by PBS, all NPs attenuated the bioluminescence intensity of other organs, wherein NMPC-NPs provoked remarkable metastasis inhibition, as evidenced by the 9.6- and 11.1-fold lower bioluminescence intensities of NMPC-NPs than those of NMP-NPs and NMC-NPs, respectively (Figure 8A, Figure S10). Similar results were noted in the number of circulating tumor cells (CTCs) and tumor nodules in the lungs. As shown in Figure 8B-F, a minimal number of CTCs in the blood, tumor nodules, and metastatic lesions in the lungs were demonstrated in NMPC-NP-treated mice, suggesting a notable antimetastatic effect of NMPC-NPs.
 |
Figure 8 In vivo anti-metastasis efficacy. Mice were i.v. injected with various groups on day 14, 18, 22 and 26, wherein NMC-NPs, PC-NPs, NMPC-NPs treated mice were irradiated at 8 h post-injection (12.5 mg PTX/kg, 5 mg Ce6/kg). (A) Representative bioluminescence images of 4T1 orthotopic mammary tumor-bearing mice in various groups (n = 8). (B) Fluorescence images and number (C) of CTCs (GFP+ 4T1 cells) in the mouse blood isolated on day 36 (n = 3). (D) The number of metastatic nodules in lung harvested on day 36 (n = 3). (E) The inhibition rate of lung metastases in mice (n = 3). (F) The photograph and H&E staining of lung harvested on day 36. Data expressed as means ± SD. *p < 0.05; **p < 0.01; ***p < 0.001. |
Conclusion
In summary, light-assisted “nano-neutrophils (NMPC-NPs)” were developed, which consisted of neutrophil membrane-camouflaged PLGA NPs encapsulating the hypoxia-responsive PTX dimeric prodrug (hQ-PTX2) and photosensitizer (Ce6), to achieve targeted delivery of drugs toward synergistic tumor treatment and metastasis inhibition. A hypoxia-responsive hQ-PTX2 was engineered, which significantly elevated the drug loading of NPs (16.9%) due to linker-weaken intermolecular π–π interactions. In particular, NMPC-NPs were characterized by neutrophil-like biological properties, which enabled significant improvements in biosafety, serum stability, and inflammation-targeting ability. Significantly, NMPC-NP-regulated PDT could effectively amplify the tumor’s inflammatory responses to enhance NMPC-NP recruitment, resulting in prominent targeted drug delivery to inflammatory tumor sites. Moreover, NMPC-NPs could kill cancer cells via ROS generation, while the exacerbated intratumoral hypoxic environment could promote hQ-PTX2 degradation to achieve the specific release of PTX, thus optimizing their synergistic therapeutic effect and safety. The in vitro and in vivo studies consistently demonstrated that NMPC-NPs had an outstanding ability to suppress tumor growth and metastasis, as well as minimizing side effects. This study therefore sheds innovative light on targeted drug delivery and cancer treatment.
Acknowledgments
This work was supported by the Young TopNotch Talents Foundation of Henan Agricultural University (30500737), the National Natural Science Foundation of China (51903074), and the Key Technologies R&D Program of Henan Province (212102310252).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Prat A, Braso-Maristany F, Martinez-Saez O, et al. Circulating tumor DNA reveals complex biological features with clinical relevance in metastatic breast cancer. Nat Commun. 2023;14(1):1157. doi:10.1038/s41467-023-36801-9
2. Sepich-Poore GD, Zitvogel L, Straussman R, Hasty J, Wargo JA, Knight R. The microbiome and human cancer. Science. 2021;371(6536):1331. doi:10.1126/science.abc4552
3. Linares J, Sallent-Aragay A, Badia-Ramentol J, et al. Long-term platinum-based drug accumulation in cancer-associated fibroblasts promotes colorectal cancer progression and resistance to therapy. Nat Commun. 2023;14(1):746. doi:10.1038/s41467-023-36334-1
4. Rudin CM, Brambilla E, Faivre-Finn C, Sage J. Small-cell lung cancer. Nat Rev Dis Primers. 2021;7(1):3. doi:10.1038/s41572-020-00235-0
5. Baldominos P, Barbera-Mourelle A, Barreiro O, et al. Quiescent cancer cells resist T cell attack by forming an immunosuppressive niche. Cell. 2022;185(10):1694–1708. doi:10.1016/j.cell.2022.03.033
6. Altorki NK, Markowitz GJ, Gao D, et al. The lung microenvironment: an important regulator of tumour growth and metastasis. Nat Rev Cancer. 2019;19(1):9–31. doi:10.1038/s41568-018-0081-9
7. Wang Z, Li W, Park J, Gonzalez KM, Scott AJ, Lu J. Camptothesome elicits immunogenic cell death to boost colorectal cancer immune checkpoint blockade. J Controlled Release. 2022;349:929–939. doi:10.1016/j.jconrel.2022.07.042
8. Kabraji S, Ni J, Sammons S, et al. Preclinical and clinical efficacy of trastuzumab deruxtecan in breast cancer brain metastases. Clin Cancer Res. 2023;29(1):174–182. doi:10.1158/1078-0432.Ccr-22-1138
9. Fan Z, Jiang C, Wang Y, et al. Engineered extracellular vesicles as intelligent nanosystems for next-generation nanomedicine. Nanoscale Horiz. 2022;7(7):682–714. doi:10.1039/d2nh00070a
10. Zhao H, Wu L, Yan G, et al. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduction Targeted Ther. 2021;6(1):263. doi:10.1038/s41392-021-00658-5
11. Liu B, Jin Y, Yang J, et al. Extracellular vesicles from lung tissue drive bone marrow neutrophil recruitment in inflammation. J Extracell Vesicles. 2022;11(5):12223. doi:10.1002/jev2.12223
12. Pang L, Khan F, Heimberger AB, Chen P. Mechanism and therapeutic potential of tumor-immune symbiosis in glioblastoma. Trends Cancer. 2022;8(10):839–854. doi:10.1016/j.trecan.2022.04.010
13. Liang H, Du Y, Zhu C, et al. Nanoparticulate cationic poly(amino acid)s block cancer metastases by destructing neutrophil extracellular traps. ACS Nano. 2023;17(3):2868–2880. doi:10.1021/acsnano.2c11280
14. Liu Y, Tiruthani K, Wang M, et al. Tumor-targeted gene therapy with lipid nanoparticles inhibits tumor-associated adipocytes and remodels the immunosuppressive tumor microenvironment in triple-negative breast cancer. Nanoscale Horiz. 2021;6(4):319–329. doi:10.1039/d0nh00588f
15. Ding J, Lu G, Nie W, et al. Self-activatable photo-extracellular vesicle for synergistic trimodal anticancer therapy. Adv Funct Mater. 2021;33(7):2005562. doi:10.1002/adma.202005562
16. Hedrick CC, Malanchi I. Neutrophils in cancer: heterogeneous and multifaceted. Nat Rev Immunol. 2022;22(3):173–187. doi:10.1038/s41577-021-00571-6
17. Behrens LM, Berg ME. Neutrophils as immune effector cells in antibody therapy in cancer. Immunol Rev. 2022;314(1):280–301. doi:10.1111/imr.13159
18. Wang D, Wang S, Zhou Z, et al. White blood cell membrane-coated nanoparticles: recent development and medical applications. Adv Healthcare Mater. 2022;11(7):2101349. doi:10.1002/adhm.202101349
19. Buncha V, Fopiano KA, Lang L, et al. Mice with endothelial cell-selective adhesion molecule deficiency develop coronary microvascular rarefaction and left ventricle diastolic dysfunction. Physiol Rep. 2023;11(6):15643. doi:10.14814/phy2.15643
20. Oroojalian F, Beygi M, Baradaran B, Mokhtarzadeh A, Shahbazi MA. Immune cell membrane-coated biomimetic nanoparticles for targeted cancer therapy. Small. 2021;17(12):2006484. doi:10.1002/smll.202006484
21. Yi W, Xiao P, Liu X, et al. Recent advances in developing active targeting and multi-functional drug delivery systems via bioorthogonal chemistry. Signal Transduction Targeted Ther. 2022;7(1):386. doi:10.1038/s41392-022-01250-1
22. Zhang F, Zhuang J, Li Z, et al. Nanoparticle-modified microrobots for in vivo antibiotic delivery to treat acute bacterial pneumonia. Nat Mater. 2022;21(11):1324–1332. doi:10.1038/s41563-022-01360-9
23. Chen Y, Qin D, Zou J, et al. Living leukocyte-based drug delivery systems. Adv Mater. 2022;35(17):2207787. doi:10.1002/adma.202207787
24. Wang K, Lei Y, Xia D, et al. Neutrophil membranes coated, antibiotic agent loaded nanoparticles targeting to the lung inflammation. Colloids Surf, B. 2020;188:110755. doi:10.1016/j.colsurfb.2019.110755
25. Wang J, Gu X, Ouyang Y, et al. Engineering of neutrophil membrane camouflaging nanoparticles realizes targeted drug delivery for amplified antitumor therapy. Int J Nanomed. 2021;16:1175–1187. doi:10.2147/IJN.S288636
26. Cui C, Chakraborty K, Tang XA, et al. Neutrophil elastase selectively kills cancer cells and attenuates tumorigenesis. Cell. 2021;184(12):3163–3177. doi:10.1016/j.cell.2021.04.016
27. Conrad C, Looney MR. Is neutrophilic inflammation treatable in COVID-19? Lancet Respir Med. 2022;10(12):1100–1101. doi:10.1016/S2213-2600(22)00293-4
28. Guo B, Wei J, Wang J, et al. CD44-targeting hydrophobic phosphorylated gemcitabine prodrug nanotherapeutics augment lung cancer therapy. Acta Biomater. 2022;145:200–209. doi:10.1016/j.actbio.2022.04.016
29. Nirmala MJ, Kizhuveetil U, Johnson A, B G, Nagarajan R, Muthuvijayan V. Cancer nanomedicine: a review of nano-therapeutics and challenges ahead. RSC Adv. 2023;13(13):8606–8629. doi:10.1039/d2ra07863e
30. Han Z, Yuan M, Liu L, et al. pH-Responsive wound dressings: advances and prospects. Nanoscale Horiz. 2023;8(4):422–440. doi:10.1039/d2nh00574c
31. Wang F, Su H, Xu D, et al. Therapeutic supramolecular tubustecan hydrogel combined with checkpoint inhibitor elicits immunity to combat cancer. Biomaterials. 2021;279:121182. doi:10.1016/j.biomaterials.2021.121182
32. Zhou S, Hu X, Xia R, et al. A paclitaxel prodrug activatable by irradiation in a hypoxic microenvironment. Angew Chem, Int Ed Engl. 2020;59(51):23198–23205. doi:10.1002/anie.202008732
33. Cariolou M, Abar L, Aune D, et al. Postdiagnosis recreational physical activity and breast cancer prognosis: global Cancer Update Programme (CUP Global) systematic literature review and meta-analysis. Int J Cancer. 2022;152(4):600–615. doi:10.1002/ijc.34324
34. Hunter FW, Wouters BG, Wilson WR. Hypoxia-activated prodrugs: paths forward in the era of personalised medicine. Br J Cancer. 2016;114(10):1071–1077. doi:10.1038/bjc.2016.79
35. Wang K, Ye H, Zhang X, et al. An exosome-like programmable-bioactivating paclitaxel prodrug nanoplatform for enhanced breast cancer metastasis inhibition. Biomaterials. 2020;257:120224. doi:10.1016/j.biomaterials.2020.120224
36. Cai H, Dai X, Wang X, et al. A nanostrategy for efficient imaging-guided antitumor therapy through a stimuli-responsive branched polymeric prodrug. Adv Sci. 2020;7(6):1903243. doi:10.1002/advs.201903243
37. Zou C, Tang Y, Zeng P, et al. cRGD-modified nanoparticles of multi-bioactive agent conjugate with pH-sensitive linkers and PD-L1 antagonist for integrative collaborative treatment of breast cancer. Nanoscale Horiz. 2023;8(7):870–886. doi:10.1039/d2nh00590e
38. Xiao Y, Zhang T, Ma X, et al. Microenvironment-responsive prodrug-induced pyroptosis boosts cancer immunotherapy. Adv Sci. 2021;8(24):2101840. doi:10.1002/advs.202101840
39. Jiang X, Fan X, Xu W, et al. Self-assembled peptide nanoparticles responsive to multiple tumor microenvironment triggers provide highly efficient targeted delivery and release of antitumor drug. J Controlled Release. 2019;316:196–207. doi:10.1016/j.jconrel.2019.10.031
40. Reinicke KE, Bey EA, Bentle MS, et al. Development of beta-lapachone prodrugs for therapy against human cancer cells with elevated NAD(P)H:quinone oxidoreductase 1 levels. Clin Cancer Res. 2005;11(8):3055–3064. doi:10.1158/1078-0432.CCR-04-2185
41. Li S, Jiang X, Zheng R, et al. An azobenzene-based heteromeric prodrug for hypoxia-activated chemotherapy by regulating subcellular localization. Chem Commun (Camb). 2018;54(57):7983–7986. doi:10.1039/c8cc03430c
42. He X, Zhang J, Li C, et al. Enhanced bioreduction-responsive diselenide-based dimeric prodrug nanoparticles for triple negative breast cancer therapy. Theranostics. 2018;8(18):4884–4897. doi:10.7150/thno.27581
43. Li Y, Jiang M, Deng Z, Zeng S, Hao J. Low dose soft X-ray remotely triggered lanthanide nanovaccine for deep tissue co gas release and activation of systemic antitumor immunoresponse. Adv Sci. 2021;8(12):2004391. doi:10.1002/advs.202004391
44. Kuthala N, Shanmugam M, Kong X, Chiang CS, Hwang KC. Salt-mediated, plasmonic field-field/field-lattice coupling-enhanced NIR-II photodynamic therapy using core-gap-shell gold nanopeanuts. Nanoscale Horiz. 2022;7(6):589–606. doi:10.1039/d1nh00631b
45. Chen Z, Liu L, Liang R, et al. Bioinspired hybrid protein oxygen nanocarrier amplified photodynamic therapy for eliciting antitumor immunity and abscopal effect. ACS Nano. 2018;12(8):8633–8645. doi:10.1021/acsnano.8b04371
46. Zhou Z, Song J, Nie L, Chen X. Reactive oxygen species generating systems meeting challenges of photodynamic cancer therapy. Chem Soc Rev. 2016;45(23):6597–6626. doi:10.1039/c6cs00271d
47. Zhong Y, Li T, Zhu Y, et al. Targeting proinflammatory molecules using multifunctional mno nanoparticles to inhibit breast cancer recurrence and metastasis. ACS Nano. 2022;16(12):20430–20444. doi:10.1021/acsnano.2c06713
48. Pham TC, Nguyen VN, Choi Y, Lee S, Yoon J. Recent strategies to develop innovative photosensitizers for enhanced photodynamic therapy. Chem Preview. 2021;121(21):13454–13619. doi:10.1021/acs.chemrev.1c00381
49. Wei D, Qi J, Hamblin MR, Wen X, Jiang X, Yang H. Near-infrared photoimmunotherapy: design and potential applications for cancer treatment and beyond. Theranostics. 2022;12(16):7108–7131. doi:10.7150/thno.74820
50. Xu Y, Zhang X, Hu G, et al. Multistage targeted “Photoactive neutrophil” for enhancing synergistic photo-chemotherapy. Biomaterials. 2021;279:121224. doi:10.1016/j.biomaterials.2021.121224
51. Le Z, He Z, Liu H, et al. Orally administrable polyphenol-based nanoparticles achieve anti-inflammation and antitumor treatment of colon diseases. Biomater Sci. 2022;10(15):4156–4169. doi:10.1039/d2bm00540a
52. Yang J, Duan S, Ye H, et al. Pro-peptide-reinforced, mucus-penetrating pulmonary siRNA delivery mitigates cytokine storm in pneumonia. Adv Funct Mater. 2021;31(21):2008960. doi:10.1002/adfm.202008960
53. Du Z, Liu C, Song H, et al. Neutrophil-membrane-directed bioorthogonal synthesis of inflammation-targeting chiral drugs. Chem. 2020;6(8):2060–2072. doi:10.1016/j.chempr.2020.06.002
54. Yue Y-X, Zhang Z, Wang Z-H, et al. Promoting tumor accumulation of anticancer drugs by hierarchical carrying of exogenous and endogenous vehicles. Small Struct. 2022;3(10):2200067. doi:10.1002/sstr.202200067
55. Kang T, Zhu Q, Wei D, et al. Nanoparticles coated with neutrophil membranes can effectively treat cancer metastasis. ACS Nano. 2017;11(2):1397–1411. doi:10.1021/acsnano.6b06477
56. Zhang C, Gao F, Wu W, et al. Enzyme-driven membrane-targeted chimeric peptide for enhanced tumor photodynamic immunotherapy. ACS Nano. 2019;13(10):11249–11262. doi:10.1021/acsnano.9b04315
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.