Back to Archived Journals » Integrated Blood Pressure Control » Volume 12
Liddle’s syndrome mechanisms, diagnosis and management
Authors Enslow BT ![]() , Stockand JD, Berman JM
, Stockand JD, Berman JM ![]()
Received 25 May 2019
Accepted for publication 23 August 2019
Published 3 September 2019 Volume 2019:12 Pages 13—22
DOI https://doi.org/10.2147/IBPC.S188869
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Konstantinos Tziomalos
Video abstract presented by Jonathan M Berman.
Views: 4025
Benjamin T Enslow1, James D Stockand1, Jonathan M Berman2
1UT Health, San Antonio, TX, USA; 2New York Institute of Technology College of Osteopathic Medicine at Arkansas State University, Jonesboro, AR, USA
Corrspondence: Jonathan M Berman
New York Institute of Technology at Arkansas State University, Jonesboro, AR 72467, USA
Tel +1 870 680 8896
Email [email protected]
Abstract: Liddle’s syndrome is a genetic disorder characterized by hypertension with hypokalemic metabolic alkalosis, hyporeninemia and suppressed aldosterone secretion that often appears early in life. It results from inappropriately elevated sodium reabsorption in the distal nephron. Liddle’s syndrome is caused by mutations to subunits of the Epithelial Sodium Channel (ENaC). Among other mechanisms, such mutations typically prevent ubiquitination of these subunits, slowing the rate at which they are internalized from the membrane, resulting in an elevation of channel activity. A minority of Liddle’s syndrome mutations, though, result in a complementary effect that also elevates activity by increasing the probability that ENaC channels within the membrane are open. Potassium-sparing diuretics such as amiloride and triamterene reduce ENaC activity, and in combination with a reduced sodium diet can restore normotension and electrolyte imbalance in Liddle’s syndrome patients and animal models. Liddle’s syndrome can be diagnosed clinically by phenotype and confirmed through genetic testing. This review examines the clinical features of Liddle’s syndrome, the differential diagnosis of Liddle’s syndrome and differentiation from other genetic diseases with similar phenotype, and what is currently known about the population-level prevalence of Liddle’s syndrome. This review gives special focus to the molecular mechanisms of Liddle’s syndrome.
Keywords: ENaC, Liddle’s syndrome, hypertension, blood pressure, distal nephron
Background
Hypertension
In adults, hypertension is defined as an elevated systolic and/or diastolic blood pressure as measured on at least two separate screenings. Blood pressures greater than 120 mmHg systolic and 80 mmHg diastolic are considered elevated in adults (prehypertension), with Stage 1 Hypertension officially recognized at systolic pressures within 130–139 mmHg or diastolic pressures ranging from 80–89 mmHg.17 In children and adolescents (1–13 years of age) hypertension is officially diagnosed after two or more documented screenings where the child’s systolic and/or diastolic blood pressure are at or above the 95th percentile based upon the child’s age, sex, and height.18,19 Approximately one in three adults in the United States has hypertension, and in most patients, no identifiable cause for the hypertension can be found.17 This form of hypertension is referred to as essential or primary hypertension. Secondary hypertension refers to hypertension caused by a recognized, sometimes correctable, underlying etiology. While essential hypertension is the predominant form of elevated blood pressure in both children and adults, secondary hypertension must be excluded during the clinical evaluation of patients with elevated blood pressures, especially the very young, those with a strong family history of hypertension, and those with physical and laboratory exam findings indicative of an underlying disorder.17–21 A subset of cases of secondary hypertension are attributable to monogenic mutations that affect either the kidney or adrenal gland, consequently leading to increased sodium reabsorption and intravascular volume expansion. These disorders exhibit a Mendelian pattern of inheritance and can result in early-onset hypertension with metabolic derangements secondary to the increased activity of the epithelial sodium channel (ENaC) present within the apical membranes of the distal nephron.
ENaC
Functional ENaC is an obligate heterotrimer of an α (or δ), β, and γ subunit. Each subunit has intracellular amino and carboxy termini, two transmembrane domains, and a large extracellular loop. In the kidney ENaC is primarily expressed in principal cells of the aldosterone sensitive distal nephron, to include the late distal convoluted tubule, connecting tubule and collecting duct, where it is the hormonally controlled rate-limiting factor fine tuning sodium excretion. Increases in ENaC activity result in inappropriate sodium retention, and decreases in ENaC activity result in natriuresis and diuresis. ENaC activity is regulated by a number of factors, including aldosterone. In principal cells, aldosterone activates the mineralocorticoid receptor to upregulate positive regulators of the channel. Aldosterone also through the mineralocorticoid receptor results in a trophic increase in the transcription of α-ENaC. Regulation of ENaC activity can occur through a number of known means resulting in changes to membrane density or open probability.
Liddle’s syndrome
Liddle’s syndrome, also known as pseudohyperaldosteronism is a rare, autosomal dominant, cause of secondary hypertension. Liddle’s syndrome mimics the symptoms of mineralocorticoid excess, causing hypokalemia, hypertension, and metabolic alkalosis, but with suppressed aldosterone and renin levels. It is caused by gain of function mutations to SCNN1A, SCNN1B, and SCNN1G which encode the α, β, and γ subunits of ENaC, respectively. These mutations increase ENaC activity and cause sodium retention. The first mutations identified as causing Liddle’s syndrome increased ENaC activity, likely through multiple mechanisms. One possible mechanism is a decrease in membrane internalization by elimination of a ubiquitination site. Mutations that increase the probability that the channel is open have also been found more recently. At least 29 distinct Liddle’s syndrome polymorphisms have been described to date. A number of mutations to upstream modulators of ENaC activity can produce a Liddle’s-like phenotype, but are distinct from Liddle’s syndrome.
Clinical features of Liddle’s syndrome
The clinical and biochemical features characteristic of Liddle’s syndrome are early-onset hypertension and hypokalemic metabolic alkalosis in the setting of suppressed plasma renin activity and low plasma aldosterone concentration.1–3 The pathophysiology of Liddle’s syndrome stems from the overactivity of ENaC. Inappropriately increased renal sodium reabsorption, secondary to a gain-of-function mutation in ENaC, leads to an expanded intravascular volume, resulting in arterial hypertension. Increased sodium flux across the apical membranes of distal nephron principal cells also favors the secretion of potassium and acid into the collecting duct, resulting in hypokalemia and metabolic alkalosis. Elevated blood pressure and low serum potassium concentrations suppress the renin-angiotensin-aldosterone system, resulting in hyporeninemia. These features were first reported together in a 16-year-old female patient by Liddle et al in 1963.4 In the index case, the proband presented with severely elevated blood pressure and hypokalemia. The disorder was distinguished from the provisional diagnosis of primary hyperaldosteronism by the proband’s lack of response to mineralocorticoid synthesis inhibitors and receptor blockers and response to the ENaC blockers triamterene and amiloride.4–8 The same findings were documented in other patients and their family members by various groups, with an autosomal dominant pattern of inheritance revealed from analysis of the pedigrees.5–8 As more Liddle’s syndrome kindreds have been studied, it has become clear that the age of onset and severity of symptoms can vary greatly between and within family groups.5–10
Early-onset hypertension is a typical finding in the Liddle’s syndrome patient. For example, in one large kindred, the youngest member with Liddle’s syndrome was diagnosed with hypertension at 2 years-of-age, however Liddle’s phenotype has been observed in older patients.11,12 The largest-sampled retrospective analysis to date of patients diagnosed with Liddle’s syndrome found the average age of onset of hypertension to be 15.5±3.3 years, and mean peak blood pressure was found to be 196 (±22)/121 (±16).2 In the index case, both the proband and her brother presented with markedly elevated blood pressures in their teenage years. Similarly, another group reported a case in which significant hypertension was noted in the patients at 11 and 13 years of age.13 A systematic review of reported cases of Liddle’s syndrome conducted in 2018 revealed that hypertension is present in 92.3% of patients with Liddle’s mutation.3
Among Liddle’s syndrome patients, the severity of hypertension and the clinical symptoms associated are not uniform in all cases. Patients with more severe hypertension may only develop mild symptoms, including intermittent headache, fatigue, dizziness, and changes in vision.1–4 Some patients with Liddle’s mutations may not develop elevated blood pressure at all.1–3,10 Still others can present with frank end-organ damage such as left ventricular hypertrophy, hypertensive retinopathy and nephrosclerosis.1–3,10 The severity and type of end-organ damage likely depends on variety of factors such as degree of RAAS activation and inflammatory factors.
Complications of long-standing hypertension documented in Liddle’s syndrome patients include renal insufficiency, cardiovascular disease, and cerebrovascular accidents.1–3 Tamura et al (1996), for instance, studied two Japanese brothers with Liddle’s syndrome wherein one brother developed nephrosclerosis resulting in chronic renal failure requiring hemodialysis by age 37.14 Similarly, Liddle’s index case developed renal failure and underwent renal transplantation at the age of 49. In at least one case, a patient exhibiting signs of hypertensive encephalopathy has been reported.15 Early death, especially due to heart failure, myocardial infarction, and cerebrovascular accident (CVA), is commonly reported in Liddle’s syndrome family histories.1–3,16 In one kindred with Liddle’s syndrome, five of the twelve patients had a positive family history of at least one relative with early, sudden death. CVAs below the age of 40 were also apparent in the family histories.2
Hypokalemia and metabolic alkalosis are both highly variable findings in Liddle’s syndrome patients.1–3,10,16,17 Hypokalemia, in particular, is present in 71.8% of cases of Liddle’s syndrome.3 In the index case, both the proband and her brother exhibited a severe hypokalemia and metabolic alkalosis.4 Fasting serum chemistries for the proband and her brother showed potassium levels were as low as 2.6 and 2.7 mEq/L, and serum CO2 combining capacities of 30 and 29 mEq/L, respectively.4 Both patients also exhibited clinical signs indicative of hypokalemia and metabolic alkalosis, such as muscle weakness, tetany, paresthesia, and abdominal upset.4 EKG findings characteristic of hypokalemia were also present in the index cases, including S-T segment depression, low or inverted T waves, prominent U waves, and frequent ventricular complexes.4 Botero-Velez et al (1994) revisited the index case by Liddle et al (1963), nearly two decades later, approximately 20 months after her renal transplantation.5 The group expanded upon the original pedigree of the proband case, studying 43 additional members of the proband’s family. Overall, 18 family members were found to have hypertension, and, as a group, exhibited lower serum potassium concentrations than the normotensive family members.5 However, six of those family members with hypertension had serum potassium concentrations greater than or equal to normal. On the basis of this evidence, it was concluded that hypokalemia is not a universal finding among Liddle’s syndrome patients.5 Tamura et al (1996) reached a similar conclusion in their study of Japanese kindred with Liddle’s syndrome as confirmed through genetic sequencing, in which 2 sisters with Liddle’s syndrome had hypertension with low plasma aldosterone concentrations, but exhibited a normokalemia.14
Diagnosis
The diagnosis of Liddle’s syndrome can be difficult, as the differential for secondary sources of hypertension, especially in the child or adolescent, is broad. Furthermore, many forms of secondary hypertension can often overlap in their clinical presentation and laboratory findings. However, the end-organ damage that can arise from long-standing hypertension in the Liddle’s syndrome patient can be severe, which makes the quick recognition and appropriate treatment of the condition important.18–21
Hereditary causes of hypertension are typically considered in the child or adolescent patient, though the prevalence is believed to be much lower than other causes.18,20,22–24 In children and adolescents, renal parenchymal disease and coarctation of the aorta are the cause of 70–80% of cases of secondary hypertension.18,20 In young adults, especially women less than 40 years of age, thyroid dysfunction as well as renal artery stenosis caused by fibromuscular dysplasia must be considered.20 In adults age 40–64, primary hyperaldosteronism effects up to 20% of patients with resistant hypertension, while in the elderly renal artery stenosis secondary to atherosclerotic disease should be suspected.20 It should be noted, though, that while hereditary forms of hypertension such as Liddle’s syndrome are generally diagnosed in younger patients, adulthood or advanced age does not necessarily preclude a diagnosis of Liddle’s syndrome.12
Family history may also provide clues to diagnosis of LS. Many of the monogenic causes of hypertension have known patterns of inheritance.21,22 Liddle’s syndrome is autosomal dominant, while disorders such as congenital adrenal hyperplasia (CAH) and some cases of familial glucocorticoid resistance (FGR) exhibit autosomal recessive inheritance patterns.22 Given the variable penetrance of Liddle’s syndrome, however, a clear and complete family history may be difficult to ascertain based on the symptomatology of family members alone.22
The clinical and biochemical features of Liddle’s syndrome are variably present and can overlap with the features of other causes of secondary hypertension. States of mineralocorticoid excess can mimic the electrolyte imbalances and hypertension of Liddle’s syndrome and can be renin-independent (primary hyperaldosteronism) such as with Conn’s syndrome, or renin-dependent (secondary hyperaldosteronism) such with as renovascular disease. Familial Hyperaldosteronism Type I (Glucocorticoid-remediable aldosteronism) and Type II, are caused by mutations to genes coding for enzymes involved in the mineralocorticoid synthetic pathway within the adrenal cortex, leading to the overproduction of aldosterone, and consequent hypertension and hypokalemic metabolic alkalosis.20–22 Pseudohypoaldosteronism (PHA) type I and type II are also rare, monogenic forms of hypertension resulting from improper electrolyte flux within the nephron. The patterns of inheritance and sites of mutations of PHA depend on the type and subtype. PHA-2, or Gordon syndrome, is typically autosomal dominant, and results from mutations that alter the activity of a the WNK family of serine-threonine kinases. These kinases further alter the activity and expression of other channels, such as NCC, ROMK, ENaC, and Cl transporters by mechanisms not yet fully-understood.20–22 Ultimately, these mutations result in increased sodium reabsorption and decreased potassium secretion, leading to the development of hypertension and hyperkalemia.
Other syndromes and disease can result in the development of a Liddle’s-like phenotype without a Liddle’s mutation. A rare mutation to the mineralocorticoid receptor, MRS810L, can also result in a Liddle’s-like phenotype.23 MR is normally sensitive to aldosterone, and increases ENaC activity. This mutation expanded the sensitivity of MR beyond aldosterone, so that it was activated by a number of other steroid compounds, including progesterone. As a result, patients with this mutation developed even more severe hypertension during pregnancy. This syndrome is denoted “Hypertension exacerbated by pregnancy,” and is distinct from other causes of elevated blood pressure in pregnancy, such as pre-eclampsia. Apparent Mineralocorticoid Excess also carries features that produce a Liddle’s-like phenotype.22,24 MR is sensitive to both aldosterone and cortisol, however circulating levels of cortisol are higher than that of aldosterone. To maintain low cortisol levels, principal cells produce 11-β-hydroxysteroid-dehydrogenase type II (11-β-HSD-II), an enzyme that converts cortisol to cortisone. In patients with mutations to 11-β-HSD-II, high cortisol levels result in constitutively active MR. A metabolite of licorice, glycyrrhetic acid inhibits 11-β-HSD-II. Consumption of large amounts of licorice can result in similar symptoms. Congenital adrenal hyperplasia caused by defects in 11-hydroxylase or 17-hydroxylase can also result in aberrant MR activation secondary to overproduction of 21-hydroxylated steroids. These patients, however, respond to MR blockers, and typically present with ambiguous genitalia.22 Familial Glucocorticoid Resistance also features increased cortisol, in this case due to mutations which lower the sensitivity of the glucocorticoid receptor to cortisol. Positive feedback results in chronically elevated cortisol levels, overloading the ability of 11-β-HSD-II to convert it to cortisone.25 Other forms of monogenic hypertension produce similar phenotypes, but do not have the three most important features of Liddle’s syndrome of elevated blood pressure, low plasma renin and low plasma aldosterone. Glucocorticoid remediable aldosteronism, for example, has a monogenic inheritance pattern, and increases ENaC activity through MR, but features elevated plasma aldosterone concentration.
Plasma renin activity and plasma aldosterone concentration can be useful in differentiating between possible causes of secondary hypertension.21,22 For example, increased plasma aldosterone concentration is a feature shared by causes of primary hyperaldosteronism and Gordon syndrome. A plasma aldosterone concentration/plasma renin activity ratio greater than 30 is cited as a strong indicator of primary hyperaldosteronism.21,22,26 In contrast, plasma renin activity and plasma aldosterone concentration are typically suppressed in other forms of heritable hypertension including Liddle’s syndrome, Congenital adrenal hyperplasia, and apparent mineralocorticoid excess.21,22,27,28 Genetic testing of the genes coding the three subunits of ENaC, SCNN1A, SCNN1B, and SCNN1G can provide definitive confirmation of Liddle’s syndrome.2,3 At least 29 separate alleles have been linked to LS, and nearly all mutations are frameshift, nonsense, or missense mutations that delete or alter the proline-rich PY motif of a subunit.2,3,11,14,29–33 Genetic testing should be considered in patients with early-onset treatment-resistant hypertension in the setting of suppressed plasma renin activity and plasma aldosterone concentration, despite a positive family history of the disorder.11,14,29,31,32 Genetic testing should also be recommended to all first-degree relatives of a mutation carrier, since the disease has such variable penetrance.2,3,9 However, genetic testing for known mutations cannot definitively rule out Liddle’s syndrome as not all Liddle’s syndrome causing mutations are necessarily known.
Management
Treatment of Liddle’s syndrome is typically through the use of a potassium-sparing diuretic, such as amiloride or triamterene.1,3,6 Both diuretics work by blocking the activity of ENaC, and their efficacy in Liddle’s syndrome cases has been shown to be enhanced with dietary salt restriction (under 2 g NaCL per day).3,4 These diuretics can correct the elevated blood pressure as well as the hypokalemic metabolic alkalosis seen in Liddle’s patients. Amiloride, in particular, has been reported to be safe for Liddle’s patients who are pregnant, though the dose may need to be increased with the progression of gestational age.26 It may be valuable to closely monitor serum electrolytes while starting diuretic therapy for Liddle’s syndrome patients.
Prevalence
Overall population-prevalence of Liddle’s syndrome is not known. A cross-sectional study of 149 patients at a veteran’s clinic in Louisiana with hypertension with hypokalemia or high serum bicarbonate, found that 9 patients (6%) met diagnostic criteria for Liddle’s syndrome of low plasma renin activity and low aldosterone.34 However, these patients likely do not meet the expectation of early onset hypertension. If they exhibited the symptoms of Liddle’s syndrome at the time of enlistment, they may have been disqualified.
This raises the question of whether the early onset hypertension seen with Liddle’s syndrome is “early onset” because hypertension appears early in Liddle’s syndrome patients, or if Liddle’s syndrome is detected more often in young patients because hypertension is not unusual enough in older patients to warrant further genetic investigation.
A patient with Liddle’s syndrome mutation who does not become symptomatic until age 40 is unlikely to undergo genetic screening for the causes of her hypertension. Indeed, cases of Liddle’s syndrome have been reported in patients of advanced age.12 Therefore estimates of Liddle’s syndrome prevalence based on early-onset cases may under-represent the overall prevalence of Liddle’s syndrome. Likewise, patients who develop hypertension due to Liddle’s syndrome in pregnancy may be evaluated for preeclampsia without definitive Liddle’s syndrome diagnosis, masking some true cases of Liddle’s syndrome.
The Chinese Academy of Medical Sciences recruited 260 early onset hypertension patients and 300 normal control patients, and had a panel of nine genes including SCNN1B and SCNN1G sequenced. An additional 506 early onset patients had exon 13 of SCNN1B and SCNN1G sequenced.35 Out of 766 total investigated cases of early-onset hypertension, 7 were diagnosed with Liddle’s syndrome, and following pedigree investigations an additional 10 relatives not initially in the study were diagnosed. This suggests Liddle’s syndrome as a causative factor in about 0.9–1.5% of early onset hypertension, at least in the Chinese population. The actual prevalence may be higher considering the family members who were diagnosed due to a relative’s hypertension, and αENaC mutations being unscreened. In this population, Liddle’s syndrome was more common in patients who were first diagnosed with early-onset hypertension before the age of 30.
A question exists as to whether Liddle’s syndrome prevalence is uniformly distributed. A study of 3 putatively unrelated families with Liddle’s syndrome from the Strait of Messina used mitochondrial and Y chromosome genotyping established that these families shared a common ancestor ~13 generations ago, and that this ancestor may have left ~20 carriers alive today.10 A study of 247 severely hypertensive patients with early-onset hypertension in Japan, looked at two ENaC polymorphisms, αENaC T663A, and βT594M.36 Only the αENaC T663A polymorphism was found to be polymorphic in the Japanese population, however other ENaC polymorphisms may have existed. These studies suggest that Liddle’s syndrome may vary in regional density, and that Liddle’s syndrome causing mutations may have non-uniform distribution among populations.
Although current data on Liddle’s syndrome prevalence is incomplete, it’s likely that Liddle’s syndrome is underdiagnosed. Liddle’s syndrome symptoms appear similar to a number of other conditions, and genetic screening for Liddle’s syndrome is not widespread. Larger population-level genetic screening of all ENaC subunits would be necessary to achieve accurate estimates of Liddle’s syndrome prevalence, and screening of non-hypertensives for Liddle’s syndrome mutations would be necessary for population level estimates of Liddle’s syndrome penetrance.
Molecular mechanisms
Liddle’s syndrome is caused by mutations to the genes SCNN1A, SCNN1B, or SCNN1G, coding for the α, β, and γ subunits of ENaC. Active ENaC is an obligate heterotrimer, of at an α, β, and γ subunits. Each subunit has a large extracellular loop, two transmembrane domains, and intracellular amino and carboxy termini (Figure 1). One of the primary regulators of ENaC activity is aldosterone, which activates ENaC by a number of means, directly, and through mineralocorticoid receptor triggered expression changes in a number of upstream genes.
In the initial study of Liddle’s syndrome the paradoxical symptoms of mineralocorticoid excess while having low mineralocorticoids allowed Liddle et al to narrow the dysfunction to the distal nephron, downstream of the effects of aldosterone. When Liddle’s syndrome was revisited in the 1990s, the amiloride-sensitive sodium channel in the distal nephron had been termed ENaC. Botero-Velez et al hypothesized that constitutive activation of ENaC might be the cause of Liddle’s syndrome based on its sensitivity to the ENaC blockers amiloride and triamterene, the effectiveness of renal transplantation at restoring electrolyte balance and normotension, and the associated hypokalemia. If this hypothesis were true, the activation of ENaC in Liddle’s syndrome would have to be independent of aldosterone, due to the low circulating aldosterone levels in such patients.
Following the cloning of the human βENaC gene, SCNN1B, a variant allele was shown to have 100% linkage to the family of the proband.37 Sequencing showed this Liddle’s syndrome causing allele to be the result of a C->T mutation at A564, resulting in a stop codon at this position. DNA from additional patients from four other families affected by Liddle’s syndrome were screened, identifying one patient with a mutation identical to the proband, and three novel mutations, Q589->stop, and two frameshift mutations at positions 592, and 595. One of these families had been previously diagnosed with Liddle’s syndrome, confirming that Liddle’s syndrome was caused by mutations to ENaC.38
The functional consequences of the βENaC A564 truncation were studied in rat ENaC expressed exogenously in Xenopus laevis oocytes.39 This system allows for measurements of sodium currents as a proxy for ENaC activity in oocytes heterologously expressing the channel. Oocytes expressing the truncated βENaC, along with wild type alpha and gamma subunits, had multiple fold greater overall currents, compared to oocytes expressing all wild type subunits, showing that this truncation resulted in an ENaC gain of function. Truncation of the γENaC subunit at a homologous position, but not αENaC also resulted in a gain of function which was additive to the βENaC truncation effect. Single channel patch clamp experiments ruled out changes to single channel conductance, open-close times, or sodium selectivity as explaining the high activity of these channels but did suggest a greater number of active channels present at the membrane. This provided direct evidence that the symptoms of Liddle’s syndrome were the result of an ENaC gain of function, possibly through an increase in membrane density, or possibly through activation of quiescent channels. Other investigators have found that the truncated βENaC subunit both increases surface expression, and increases the channel open probability.40 Investigation of a large kindred with Liddle’s syndrome that had previously been identified in Japan found a truncated γENaC subunit, but not βENaC subunit defect, confirming the experimental finding that a γENaC mutation can also cause Liddle’s syndrome.13,41 A rodent ENaC with this γENaC truncation also produced high sodium currents in Xenopus oocytes, adding evidence that ENaC gain of function due to truncation of the β or γ subunits was the cause of Liddle’s syndrome.
This left open the question of which amino acids in the carboxy termini of β and γ ENaC were critical for this gain of function. Mutagenesis studies identified a short proline rich segment (PPPXY), called a PY motif, present in all three subunits as being necessary and sufficient to cause the Liddle’s gain of function.42 A companion paper identified Nedd4, a ubiquitin ligase, as the binding partner of the ENaC PY motifs by yeast two-hybrid screening and in vitro binding assays.43 The binding interaction was narrowed to the WW domains of rNEDD4, and the key amino acids were identified as the PPXY motif. These interactions were also present in γENaC, and to a lesser degree αENaC. Although a mutation to the αENaC PY motif could potentially lead to Liddle’s syndrome, no kindred has as yet been identified with this mutation.
Co-expression of Nedd4 and ENaC RNA in Xenopus oocytes resulted in reduced ENaC activity, and mutation of the PY motif eliminated this inhibition.44 This confirmed that loss of interaction with a Nedd4 family member was a likely mechanism for Liddle’s syndrome. This inhibition likely occurred through a decrease in cell surface expression, rather than a change to single channel properties. However, mammals have two Nedd4 family member proteins, Nedd4-1 and Nedd4-2. It is Nedd4-2 that is the physiological regulator of ENaC activity. Nedd4-2 has greater homology to the Nedd4 homolog found in Xenopus oocytes, Nedd4-2 is expressed in the kidney, and Nedd4-2 interacts with ENaC, but Nedd4-1 does not.45,46
Ubiquitylation normally targets proteins for rapid degradation, usually in the 26S proteosome, but in the case of transmembrane proteins, often in the lysosomal pathway. A series of enzymes, including a ubiquitin activating enzyme, ubiquitin-conjugating enzyme, and a ubiquitin-protein ligase like Nedd4-2 are involved in covalently attaching either a single ubiquitin, or a polyubiquitin chain to lysine residues. Studies in transiently transfected cultured cells showed ubiquitination of the ENaC α, and γ subunits, but not β.47 Together these data provided a potential mechanism underlying Liddle’s syndrome that the Nedd4’s WW domains bind ENaC’s PY motifs, and ubiquitinates ENaC, resulting in decreased membrane density. Absent this ubiquitination in Liddle’s syndrome patients, ENaC accumulates at the membrane, resulting in greater sodium reabsorption from the urine, leading to intravascular volume expansion.
The Liddle’s syndrome phenotype overlaps some with that of hyperaldosteronism, despite low serum aldosterone. This left open the question of whether Liddle’s syndrome mimicked a physiological state normally induced by high aldosterone. Serum and gluococorticoid-related kinase (SGK1) was identified as being upregulated in response to aldosterone, and as stimulating ENaC activity.48,49 The effects of SGK1 on ENaC are through increased membrane density, similar to the proposed mechanism for Liddle’s syndrome.50 SGK1 was shown to phosphorylate Nedd4-2, inhibiting its ability to ubiquitinate ENaC, leading to enhanced ENaC cell surface expression and activity.51 Therefore, Liddle’s syndrome mimics aldosterone mediated SGK1 activity.
ENaC gain of function explains the symptoms of Liddle’s syndrome. Increased ENaC activity leads to increased sodium reabsorption, volume expansion, and hypertension. Elevated blood pressure is sensed by the juxtaglomerular apparatus, leading to decreased renin production, and decreased aldosterone production. However because the mutant ENaC in Liddle’s syndrome patients is unable to respond to regulation by MR through the SGK1 and NEDD4-2 pathway, mineralocorticoid receptor antagonists such as spironolactone are ineffective at lowering blood pressure. The defects in sodium absorption in the distal nephron also explain the hypokalemia associated with LIddle’s syndrome. Distal nephron potassium channels including the renal outer medullary potassium channel, and the voltage and calcium activated big K channel are responsible for potassium efflux in this segment.52 There is coupling of K+ and Na+ movement in principal cells of the distal nephron because movement of both ions across the serosal membrane occurs through active countertransport through the Na+/K+ ATPase and movement across the luminal membrane is in opposite directions following the electrochemical gradient established by the Na+/K+ ATPase. In Liddle’s syndrome, inappropriately high sodium reabsorption results in inappropriately high potassium secretion. Additionally, sodium reabsorption creates an environment favorable to H+ secretion in neighboring intercalated cells by creating a lumen negative potential.This model, showing the regulatory mechanisms associated with the dysfunction seen in Liddle’s syndrome is illustrated in Figure 2.
Dietary sodium restriction and potassium sparing diuretics are effective at treating the symptoms of Liddle’s syndrome because filtered sodium modulates ENaC activity, and triamterene’s mechanism of action is to inhibit ENaC directly.
In addition to changes to Nedd4-2 binding to ENaC, complementary mechanisms have been identified that can result in Liddle’s syndrome. These include changes to ENaC activity resulting from absence of the PY motif, such as a change in sensitivity to high intracellular [Na+],53 changes to activation of ENaC by proteolytic cleavage,54 non-NEDD4-2 mediated changes to trafficking,55 and increases to channel open probability.56 Additionally, a Liddle’s syndrome-causing mutation has been identified that does not act on the PY-motif, but increases open probability-αENaC C479R.57 The overall prevalence of this alternative Liddle’s syndrome mechanism is unknown.
Interestingly, the end-organ damage associated with severe hypertension may also depend in part on ENaC, as well. ENaC is expressed in a number of tissues and has recently been identified in dendritic cells.58 ENaC in these cells is involved in mediating sodium induced inflammation,59 suggesting an alternative means by which ENaC mutation directs the course of Liddle’s syndrome, by mediating the immune component of end-organ damage. However, β ENaC has not been detected in dendritic cells, meaning that the bulk of Liddle’s syndrome mutations would be unlikely to affect ENaC activity in dendritic cells.
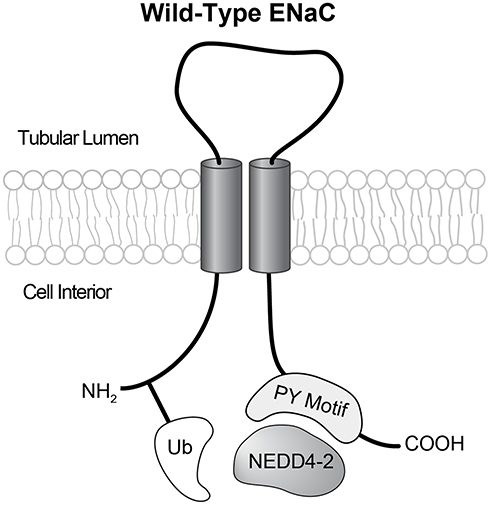 |
Figure 1 Each ENaC subunit has intracellular amino and carboxy termini, two transmembrane domains, and an extracellular loop. When an intact PY motif is present the E-3 ubiquitin ligase NEDD4-2 can bind to ENaC carboxy termini, and ubiquitinate amino termini. In typical Liddle’s syndrome this mechanism is disrupted. |
 |
Figure 2 Model of a principal cell of the distal nephron. In normal conditions aldosterone activates mineralocorticoid receptor (MR), which can increase transcription of ENaC subunits, or an inhibitor of NEDD4-2, SGK. The Na+/K+ ATPase creates the electrochemical gradient for the reabsorption of sodium, and potassium efflux in these cells. In Liddle’s syndrome, additional sodium reabsorption in these cells can result in additional potassium efflux through ROMK, and BK channels. |
Glossary of terms
Liddle’s-like Phenotype– Used here to mean the constellation of symptoms: Elevated blood pressure, low plasma aldosterone, low plasma renin, and sometimes hypokalemia, and metabolic alkalosis.
Liddle’s Mutation– Used here to mean any gain of function mutation to an Epithelial Sodium Channel subunit that results in Liddle’s syndrome, usually through changes to membrane density or open probability.
Liddle’s syndrome– Used here to mean Liddle’s Phenotype secondary to a Liddle’s Mutation.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Palmer BF, Alpern RJ. Liddle’s syndrome [Internet]. Am J Med. 1998;301–309. doi:10.1016/s0002-9343(98)00018-7
2. Cui Y, Tong A, Jiang J, Wang F, Li C. Liddle syndrome: clinical and genetic profiles [Internet]. J Clin Hypertens. 2017;524–529. doi:10.1111/jch.12949
3. Tetti M, Monticone S, Burrello J, et al. Liddle Syndrome: review of the literature and description of a new case. Int J Mol Sci. 2018;19. doi:10.3390/ijms19030812.
4. Liddle GW. A familial renal disorder simulating primary aldosteronism but with negligible aldosterone secretion. Trans Assoc Am Physicians. 1963;76:199–213.
5. Botero-Velez M, Curtis JJ, Warnock DG. Liddle’s syndrome revisited – a disorder of sodium reabsorption in the distal tubule. N Engl J Med. 1994;330:178–181.
6. Rodriguez JA, Biglieri EG, Schambelan M. Pseudohyperaldosteronism with renal tubular resistance to mineralocorticoid hormones. Trans Assoc Am Physicians. 1981;94:172–182.
7. Wang C, Chan TK, Yeung RT, Coghlan JP, Scoggins BA, Stockigt JR. The effect of triamterene and sodium intake on renin, aldosterone, and erythrocyte sodium transport in Liddle’s syndrome. J Clin Endocrinol Metab. 1981;52:1027–1032. doi:10.1210/jcem-52-5-1027
8. Nakada T, Koike H, Akiya T, et al. Liddle’s syndrome, an uncommon form of hyporeninemic hypoaldosteronism: functional and histopathological studies. J Urol. 1987;137:636–640. doi:10.1016/S0022-5347(17)44161-9
9. Büyükkaragöz B, Yilmaz AC, Karcaaltincaba D, Ozdemir O, Ludwig M. Liddle syndrome in a Turkish family with heterogeneous phenotypes. Pediatr Int. 2016;58:801–804. doi:10.1111/ped.12790
10. Pagani L, Diekmann Y, Sazzini M, et al. Three reportedly unrelated families with Liddle Syndrome inherited from a common ancestor. Hypertension. 2018;71:273–279. doi:10.1161/HYPERTENSIONAHA.117.10491
11. Findling JW, Raff H, Hansson JH, Lifton RP. Liddle’s syndrome: prospective genetic screening and suppressed aldosterone secretion in an extended kindred. J Clin Endocrinol Metab. 1997;82:1071–1074.
12. Pepersack T, Allegre S, Jeunemaître X, Leeman M, Praet J-P. Liddle syndrome phenotype in an octogenarian. J Clin Hypertens. 2015;17:59–60. doi:10.1111/jch.2015.17.issue-1
13. Hansson JH, Nelson-Williams C, Suzuki H, et al. Hypertension caused by a truncated epithelial sodium channel γ subunit: genetic heterogeneity of Liddle syndrome. Nat Genet. 1995;11:76–82. doi:10.1038/ng0995-76
14. Tamura H, Schild L, Enomoto N, Matsui N, Marumo F, Rossier BC. Liddle disease caused by a missense mutation of beta subunit of the epithelial sodium channel gene. J Clin Invest. 1996;97:1780–1784. doi:10.1172/JCI118606
15. Kota SK, Kota SK, Panda S, Modi KD. A case of Liddle’s syndrome; unusual presentation with hypertensive encephalopathy. Saudi J Kidney Dis Transpl. 2014;25:869–871. doi:10.4103/1319-2442.135185
16. Gong L, Chen J, Shao L, Song W, Hui R, Wang Y. Phenotype–genotype analysis in two Chinese families with Liddle syndrome. Mol Biol Rep. 2014;41:1569–1575. doi:10.1007/s11033-013-3003-7
17. Rossi E, Farnetti E, Nicoli D, et al. A clinical phenotype mimicking essential hypertension in a newly discovered family with Liddle’s syndrome. Am J Hypertens. 2011;24:930–935. doi:10.1038/ajh.2011.76
18. Flynn JT, Kaelber DC, Baker-Smith CM, et al. Clinical practice guideline for screening and management of high blood pressure in children and adolescents. Pediatrics. 2017;140. doi:10.1542/peds.2017-1904.
19. Ingelfinger JR. The child or adolescent with elevated blood pressure. N Engl J Med. 2014;1075.
20. Viera AJ, Neutze DM. Diagnosis of secondary hypertension: an age-based approach. Am Fam Physician. 2010;82:1471–1478.
21. Mumford E, Unwin RJ, Walsh SB, Liquorice L. Bartter or Gitelman—how to differentiate? Nephrol Dial Transplant. 2019;34:38–39. doi:10.1093/ndt/gfy199
22. Vehaskari VM. Heritable forms of hypertension. Pediatr Nephrol. 2009;24:1929–1937. doi:10.1007/s00467-007-0537-8
23. Geller DS, Farhi A, Pinkerton N. et al. Activating mineralocorticoid receptor mutation in hypertension exacerbated by pregnancy. Science. 2000;119–123. doi:10.1126/science.289.5476.119.
24. Al-Harbi T, Al-Shaikh A. Apparent mineralocorticoid excess syndrome: report of one family with three affected children. J Pediatr Endocrinol Metab. 2012;25:1083–1088. doi:10.1515/jpem-2012-0113
25. Hurley DM, Accili D, Stratakis CA, et al. Point mutation causing a single amino acid substitution in the hormone binding domain of the glucocorticoid receptor in familial glucocorticoid resistance. J Clin Invest. 1991;87:680–686. doi:10.1172/JCI115046
26. Caretto A, Primerano L, Novara F, Zuffardi O, Genovese S, Rondinelli M. A therapeutic challenge: Liddle’s syndrome managed with amiloride during pregnancy. Case Rep Obstet Gynecol. 2014;2014:156250.
27. Terjung R, editor, Pathophysiology, Diagnosis, and Treatment of Mineralocorticoid Disorders. Comprehensive Physiology. Hoboken, NJ, USA: John Wiley & Sons, Inc.; 2011:1083–1119.
28. Funder JW, Carey RM, Mantero F, et al. the management of primary aldosteronism: case detection, diagnosis, and treatment: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2016;101:1889–1916. doi:10.1210/jc.2015-4061
29. Hansson JH, Schild L, Lu Y, et al. A de novo missense mutation of the beta subunit of the epithelial sodium channel causes hypertension and Liddle syndrome, identifying a proline-rich segment critical for regulation of channel activity [Internet]. Proc Natl Acad Sci. 1995;11495–11499. doi:10.1073/pnas.92.25.11495.
30. Yang K-Q, Lu C-X, Fan P, et al. Genetic screening of SCNN1B and SCNN1G genes in early-onset hypertensive patients helps to identify Liddle syndrome. Clin Exp Hypertens. 2018;40:107–111. doi:10.1080/10641963.2017.1334799
31. Bogdanović R, Kuburović V, Stajić N, et al. Liddle syndrome in a Serbian family and literature review of underlying mutations. Eur J Pediatr. 2012;171:471–478. doi:10.1007/s00431-011-1581-8
32. Gao PJ, Zhang KX, Zhu DL, et al. Diagnosis of Liddle syndrome by genetic analysis of β and γ subunits of epithelial sodium channel–a report of five affected family members. J Hypertens. 2001;19:885–889. doi:10.1097/00004872-200103000-00017
33. Fan P, Lu C-X, Zhang D, et al. Liddle syndrome misdiagnosed as primary aldosteronism resulting from a novel frameshift mutation of SCNN1B. Endocr Connect. 2018. doi:10.1530/EC-18-0484
34. Tapolyai M, Uysal A, Dossabhoy NR, et al. High prevalence of liddle syndrome phenotype among hypertensive US Veterans in Northwest Louisiana. J Clin Hypertens. 2010;12:856–860. doi:10.1111/j.1751-7176.2010.00359.x
35. Liu K, Qin F, Sun X, et al. Analysis of the genes involved in Mendelian forms of low-renin hypertension in Chinese early-onset hypertensive patients. J Hypertens. 2018;36:502–509. doi:10.1097/HJH.0000000000001556
36. Sugiyama T, Kato N, Ishinaga Y, Yamori Y, Yazaki Y. Evaluation of selected polymorphisms of the Mendelian hypertensive disease genes in the Japanese population. Hypertens Res. 2001;24:515–521. doi:10.1291/hypres.24.515
37. Shimkets RA, Warnock DG, Bositis CM, et al. Liddle’s syndrome: heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell. 1994;79:407–414. doi:10.1016/0092-8674(94)90250-X
38. Gardner JD, Lapey A, Simopoulos P, Bravo EL. Abnormal membrane sodium transport in Liddle’s syndrome. J Clin Invest. 1971;50:2253–2258. doi:10.1172/JCI106722
39. Schild L, Canessa CM, Shimkets RA, Gautschi I, Lifton RP, Rossier BC. A mutation in the epithelial sodium channel causing Liddle disease increases channel activity in the Xenopus laevis oocyte expression system. Proc Natl Acad Sci U S A. 1995;92:5699–5703. doi:10.1073/pnas.92.12.5699
40. Firsov D, Schild L, Gautschi I, Mérillat AM, Schneeberger E, Rossier BC. Cell surface expression of the epithelial Na channel and a mutant causing Liddle syndrome: a quantitative approach. Proc Natl Acad Sci U S A. 1996;93:15370–15375. doi:10.1073/pnas.93.26.15370
41. Fukutake N, Kawashima S, Matsumoto T, Ryo K, Mitani Y, Iwasaki T. [A case of Liddle’s syndrome with familial occurrence]. Nihon Naika Gakkai Zasshi. 1988;77:441–442. doi:10.2169/naika.77.441
42. Schild L, Lu Y, Gautschi I, Schneeberger E, Lifton RP, Rossier BC. Identification of a PY motif in the epithelial Na channel subunits as a target sequence for mutations causing channel activation found in Liddle syndrome. Embo J. 1996;15:2381–2387. doi:10.1002/j.1460-2075.1996.tb00594.x
43. Staub O, Dho S, Henry P, et al. WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle’s syndrome. Embo J. 1996;15:2371–2380. doi:10.1002/j.1460-2075.1996.tb00593.x
44. Goulet CC, Volk KA, Adams CM, Prince LS, Stokes JB, Snyder PM. Inhibition of the Epithelial Na + Channel by Interaction of Nedd4 with a PY Motif Deleted in Liddle’s syndrome. J Biol Chem. 1998;273:30012–30017. doi:10.1074/jbc.273.45.30012
45. Kamynina E, Debonneville C, Bens M, Vandewalle A, Staub O. A novel mouse Nedd4 protein suppresses the activity of the epithelial Na+ channel. Faseb J. 2001;15:204–214. doi:10.1096/fj.00-0191com
46. Kamynina E, Tauxe C, Staub O. Distinct characteristics of two human Nedd4 proteins with respect to epithelial Na(+) channel regulation. Am J Physiol Renal Physiol. 2001;281:F469–77. doi:10.1152/ajprenal.2001.281.3.F469
47. Staub O, Gautschi I, Ishikawa T, et al. Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. Embo J. 1997;16:6325–6336. doi:10.1093/emboj/16.3.659
48. Chen SY, Bhargava A, Mastroberardino L, et al. Epithelial sodium channel regulated by aldosterone-induced protein sgk. Proc Natl Acad Sci U S A. 1999;96:2514–2519. doi:10.1073/pnas.96.5.2514
49. Náray-Fejes-Tóth A, Canessa C, Cleaveland ES, Aldrich G, Fejes-Tóth G. Sgk is an aldosterone-induced kinase in the renal collecting duct. Effects on epithelial Na+ channels. J Biol Chem. 1999;274:16973–16978. doi:10.1074/jbc.274.24.16973
50. Alvarez de la Rosa D, Zhang P, Náray-Fejes-Tóth A, Fejes-Tóth G, Canessa CM. The serum and glucocorticoid kinase sgk increases the abundance of epithelial sodium channels in the plasma membrane of Xenopus oocytes. J Biol Chem. 1999;274:37834–37839. doi:10.1074/jbc.274.53.37834
51. Debonneville C. Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na channel cell surface expression [Internet]. Embo J. 2001;7052–7059. doi:10.1093/emboj/20.24.7052
52. Welling PA, Ho K. A comprehensive guide to the ROMK potassium channel: form and function in health and disease. Am J Physiol Renal Physiol. 2009;297:F849–63. doi:10.1152/ajprenal.00181.2009
53. Kellenberger S, Gautschi I, Rossier BC, Schild L. Mutations causing Liddle syndrome reduce sodium-dependent downregulation of the epithelial sodium channel in the Xenopus oocyte expression system. J Clin Invest. 1998;101:2741–2750. doi:10.1172/JCI2325
54. Knight KK, Olson DR, Zhou R, Snyder PM. Liddle’s syndrome mutations increase Na+ transport through dual effects on epithelial Na+ channel surface expression and proteolytic cleavage. Proc Natl Acad Sci U S A. 2006;103:2805–2808. doi:10.1073/pnas.0511184103
55. Snyder PM. Liddle’s syndrome mutations disrupt cAMP-mediated translocation of the epithelial Na+ channel to the cell surface. J Clin Invest. 2000;105:45–53. doi:10.1172/JCI7869
56. Anantharam A, Tian Y, Palmer LG. Open probability of the epithelial sodium channel is regulated by intracellular sodium. J Physiol. 2006;574:333–347. doi:10.1113/jphysiol.2006.109926
57. Salih M, Gautschi I, van Bemmelen MX, et al. A missense mutation in the extracellular domain of αENaC causes Liddle Syndrome. J Am Soc Nephrol. 2017;28:3291–3299. doi:10.1681/ASN.2016080886
58. Barbaro NR, Foss JD, Kryshtal DO, et al. Dendritic cell amiloride-sensitive channels mediate sodium-induced inflammation and hypertension. Cell Rep. 2017;21:1009–1020. doi:10.1016/j.celrep.2017.10.002
59. Van Beusecum JP, Barbaro NR, McDowell Z, et al. High salt activates CD11c+ antigen-presenting cells via SGK (Serum Glucocorticoid Kinase) 1 to promote renal inflammation and salt-sensitive hypertension. Hypertension. 2019;74:555–563. doi:10.1161/HYPERTENSIONAHA.119.12634
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.