")
Back to Journals » Infection and Drug Resistance » Volume 8
Letermovir and inhibitors of the terminase complex: a promising new class of investigational antiviral drugs against human cytomegalovirus
Authors Melendez D, Razonable R
Received 30 May 2015
Accepted for publication 14 July 2015
Published 5 August 2015 Volume 2015:8 Pages 269—277
DOI https://doi.org/10.2147/IDR.S79131
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Suresh Antony
Dante P Melendez,1,2 Raymund R Razonable1,2
1Division of Infectious Diseases, 2William J von Liebig Center for Transplantation and Clinical Regeneration, Mayo Clinic, Rochester, MN, USA
Abstract: Infection with cytomegalovirus is prevalent in immunosuppressed patients. In solid organ transplant and hematopoietic stem cell transplant recipients, cytomegalovirus infection is associated with high morbidity and preventable mortality. Prevention and treatment of cytomegalovirus with currently approved antiviral drugs is often associated with side effects that sometimes preclude their use. Moreover, cytomegalovirus has developed mutations that confer resistance to standard antiviral drugs. During the last decade, there have been calls to develop novel antiviral drugs that could provide better options for prevention and treatment of cytomegalovirus. Letermovir (AIC246) is a highly specific antiviral drug that is currently undergoing clinical development for the management of cytomegalovirus infection. It acts by inhibiting the viral terminase complex. Letermovir is highly potent in vitro and in vivo against cytomegalovirus. Because of a distinct mechanism of action, it does not exhibit cross-resistance with other antiviral drugs. It is predicted to be active against strains that are resistant to ganciclovir, foscarnet, and cidofovir. To date, early-phase clinical trials suggest a very low incidence of adverse effects. Herein, we present a comprehensive review on letermovir, from its postulated novel mechanism of action to the results of most recent clinical studies.
Keywords: cytomegalovirus, letermovir, AIC246, terminase, antivirals, transplantation
Introduction
Cytomegalovirus (CMV) is a prevalent infection that causes significant morbidity and mortality in immunosuppressed patients. The management of CMV infection and disease in immunocompromised patients consists of the use of antiviral drugs, together with efforts to minimize immunosuppression. The current antiviral drugs against CMV, ie, ganciclovir (GCV), foscarnet (PFA), and cidofovir (CDV), are associated with significant adverse effects. Moreover, there is emergence of CMV strains that have developed resistance to these antiviral drugs. These circumstances limit our therapeutic options, and highlight the need to develop novel compounds with potent clinical activity against CMV.
Impeding vital stages in the life cycle of CMV has been explored as novel avenues for antiviral therapy. The disruption of the viral termination process was recently pursued as one such target for viral inhibition. Such a process relies on disrupting essential protein molecules at a step in the life cycle beyond CMV DNA synthesis. The most promising compound in this novel drug class is letermovir (previously known as AIC246). In this article, we review the development of letermovir, its mechanism of action and pharmacokinetic profile, and the results of studies that have evaluated its safety and efficacy in humans.
Cytomegalovirus, its disease, and its treatment
CMV is the fifth member of the family Herpesviridae1 and is the prototype member of the subfamily β-Herpesvirinae, which also includes human herpesviruses 6 and 7. CMV is transmitted through contact with infected body fluid and secretions such as saliva. CMV infection is very common worldwide, with seroprevalence rates that range from 50% in the developed world, such as in the USA,2 to almost 100% in the developing world.3
CMV infection in healthy individuals is often asymptomatic and generally behaves as a benign self-limiting illness. However, a deficiency (partial or complete) of a functioning immune response, whether of the humoral or the cellular type, can result in uncontrolled CMV replication that leads to clinical disease. Transplant recipients belong to the group at highest risk of CMV disease due to the necessary use of drugs that suppress the immune system in order to maintain the transplanted organs (for solid organ transplant recipients) and to treat or prevent graft-versus-host disease (for allogeneic hematopoietic stem cell transplant [HSCT] recipients). In these immunocompromised patients, clinical disease due to CMV is commonly manifested as fever, bone marrow suppression, and a variety of end-organ diseases, such as pneumonitis, enteritis, and rarely, retinitis. In addition, CMV infection is associated with damaging indirect effects, such as increased risk of other infections, graft failure, and death.4
Prevention and treatment of CMV infection and disease after solid organ transplant and HSCT is accomplished with use of antiviral drugs. Antiviral treatment is often complemented by reduction, if feasible, of drug-induced immunosuppression. The three antiviral drugs that are approved by the US Food and Drug Administration for CMV treatment are the nucleoside analog GCV, and its valine-ester prodrug valganciclovir (VGCV), the nucleotide analog CDV, and the pyrophosphate analog PFA. These three antiviral drugs are highly active against CMV, but their use is often associated with adverse effects.5–9 For example, treatment with GCV (and VGCV), which are the workhorses for management of CMV after transplantation, is commonly complicated by bone marrow suppression that limits its use particularly as prophylaxis in HSCT recipients due to concerns for delayed (or impaired) bone marrow engraftment. Such effects of GCV (and VGCV) are also a common concern among solid organ transplant recipients who are simultaneously receiving other myelosuppressive drugs (eg, trimethoprim-sulfamethoxazole and mycophenolate mofetil).
The nephrotoxicity of CDV and PFA and the common electrolyte disturbances caused by PFA are the major barriers that restrict their use as first-line agents for prevention and treatment of CMV-related disease. Often, transplant patients are receiving other drugs (eg, tacrolimus and calcineurin inhibitors) that augment the risk for renal insufficiency.
CMV biology as targets for antiviral therapy
CMV genome and DNA synthesis
The human CMV genome is large, consisting of a 230 kbp linear double-stranded DNA. It has unique long (UL) and unique short (US) sequences that have numerous open reading frames encoding structural and functional proteins. Two of them, UL97 and UL54, are relevant in the context of antiviral therapeutics, as discussed later.
GCV and its prodrug VGCV are nucleosides that require phosphorylation in order to become active. The first-step of this triphosphorylation is catalyzed by a viral kinase encoded by UL97. Mutations in UL97 that impair viral kinase synthesis account for the most common forms of GCV resistance by CMV. Subsequent diphosphorylation and triphosphorylation are catalyzed by host cellular guanylate and phosphoglycerate kinases, resulting in active GCV triphosphate that serves as a competitive substrate for CMV DNA polymerase during viral DNA synthesis.
Similar to GCV and VGCV, the other two approved antiviral drugs (CDV and PFA) inhibit CMV replication by interfering with the function of CMV DNA polymerase. Their incorporation by CMV DNA polymerase results in the termination of viral DNA synthesis. CMV DNA polymerase is encoded by UL54. Mutations in UL54 have been reported (at a rate lower than UL97 mutation) resulting in varying degrees of cross-resistance among GCV, CDV, and PFA.10,11
DNA maturation, packaging, and the terminase complex
During CMV replication, viral DNA for each emerging virion is not produced separately. Instead, it is synthesized as a long DNA chain containing multiple repeated gene sequences (known as concatemers). Each gene sequence repetition (monomer) constitutes the genetic material for one virion. The concatemeric DNA will therefore need to undergo cleavage into multiple unit-length monomers that will be subsequently packaged into a viral capsid prior to its release as an infective virion. This maturation, packaging, and termination process is performed by a group of proteins collectively known as the “terminase complex”.12 Inhibition of the termination process is the proposed mechanism of antiviral activity by the novel drug letermovir.13,14
The CMV terminase complex is comprised of two proteins, pUL89 and pUL56, that interact synergistically.15,16 As noted earlier, the main function of the terminase complex is to cleave CMV concatemers into single units of functional CMV monomers.17,18 The terminase complex further interacts with portal (viral capsid) proteins to facilitate DNA translocation into the capsid. The structure of the terminase complex is conserved across members of herpesviruses, but it is not shared with human cells. Consequently, at least in theory, drugs that interact with this complex are virus-selective and specific, and spare human cells.
Specifically, the pUL56, which is encoded by UL56,19 participates in the DNA packaging process in three ways: it recognizes the specific sites where viral DNA will be cleaved; it produces adenosine triphosphate (ATP) necessary for DNA translocation; and it combines with capsid proteins to allow translocation of DNA into the viral capsid.12 The subunit pUL89, which is encoded by UL89,20 contains an N-terminal moiety with ATPase function that provides energy for DNA cleavage and translocation20 and a C-terminal side with nuclease activity responsible for DNA cleavage. Its C-terminal edge also serves as a potential site for synergistic interaction with pUL56, thereby enhancing overall nuclease activity21 that cleaves CMV DNA concatamer into monomeric unit-length pieces.22 The cleavage sites are indicated by two conserved motifs located at the end of each unit-length monomer.14,15
Both pUL56 and pUL89 terminase subunits are synthesized in the cytoplasm of CMV-infected cells. Hence, they need to be transported into the nucleus to exert their vital functions. The pUL56 translocation into the nucleus appears to be mediated by interaction of the “nuclear localization signal” (a short amino acid segment in the carboxy-terminal of pUL56) and translocation-protein importin a.23 The pUL56 and importin a form a stable complex with importin b, and the complex interacts with filaments of the nuclear pore for its translocation to the nucleus. Once inside, importin subunits dissociate, leaving the pUL56 available for packaging of CMV DNA.23–26 The association of pUL56 with pUL89 results in enhanced endonuclease activity.15,21,27–29
After the viral DNA concatamer undergoes cleavage, it is transported inside an empty viral procapsid.14,21 This process is orchestrated by the interaction of the terminase complex and “portal proteins” found on the surface of viral procapsids.14,30 The portal proteins are large macromolecules that are arranged in rings and associate with procapsid proteins, forming a “pore” that serves as a port of entry for viral DNA. The DNA translocation process requires the functional interaction among portal proteins, viral procapsid and the terminase complex subunits. In particular, the pUL56 forms a stable structure with viral procapsid and interacts with portal protein pUL104, forming part of the ring structure that allows DNA translocation.30 The ATP for DNA translocation is provided by pUL56.31 To complete the termination process, there is a second DNA cleavage that liberates the unit-length monomeric genome from the remaining concatemeric DNA chain.28 The proteins pUL51, pUL52, pUL77, and pUL93 are believed to be part of the terminase complex or to participate in the DNA packaging process.32 Other proteins that may also be important to the termination process include pUL86 (major capsid protein), pUL85 (minor capsid protein), pUL46 (minor capsid protein binding protein), pUL47/48 (smallest capsid protein), pUL80.5 (assembly protein), and pUL80a (proteinase precursor protein).13,14,23 Collectively, viral termination appears to be a highly complex process that is facilitated by numerous interacting protein molecules.
Letermovir and inhibitors of the CMV terminase complex
Benzimidazoles and sulfonamides
The first terminase inhibitors, 2,5,6-trichloro-1-β-D-ribofuranosyl benzimidazole (TCRB) and 2-bromo-5,6-dichloro-1-(β-D-ribofuranosyl)benzimidazole (BDCRB), were initially developed as anticancer drugs. The effect of BDCRB and TCRB on CMV replication did not involve the inhibition of viral DNA synthesis, but instead the drugs caused the production of virions that had left-end-truncated genomes inside their capsid.33 This finding led to the suspicion that the benzimidazole compounds inhibited CMV replication at a stage distal to viral DNA synthesis. This hypothesis was reinforced when resistance mutations to BDCRB and TCRB were mapped to UL56 (Q204R) and UL89 (D344E and A355T), which encode for proteins involved in DNA processing and packaging.20,34,35
Subsequent studies have shown that BDCRB inhibits the interaction of the C-terminal side of pUL56 and the portal protein pUL104. As mentioned previously, this is an essential step that facilitates DNA monomer translocation into viral procapsids. The Q204R mutation of UL56 is speculated as the site of action of BDCRB, and such interference with the DNA binding site by the drug would inhibit cleavage and halt the replication process.20,30 Another hypothesis is that BDCRB affects the functional structures of pUL56, which is normally a dimer of two ring-shaped structures. The point of union of these two dimers is in proximity to the zinc-finger domain affected by the Q204R mutation. Accordingly, it is possible that BDCRB inhibits the formation of pUL56 dimers and subsequently impairs the ability to recognize the DNA molecule. Notably, CMV strains resistant to TCRB and BDCRB have mutations not only in UL56 and UL89, but also in UL104 (which encodes for portal proteins).36
The sulfonamide BAY 38-4766 has been shown to inhibit CMV replication. CMV that is resistant to GCV, CDV, and PFA remained susceptible to BAY 38-4766, suggesting a unique mechanism of action other than viral DNA synthesis. Sulfonamide-resistant CMV strains were found to have mutations in UL89 and UL104,37 thereby suggesting that BAY 38-4766 interferes with the terminase complex. However, there was no cross-resistance between the sulfonamide-resistant and BCDRB-resistant strains. Thus, while these compounds interrupt the DNA maturation process, they have different resistance mutations, and are likely to act at different DNA maturation sites; these observations highlight the highly complex nature of the viral termination complex. These compounds, however, are currently not undergoing further clinical development.16,18,38,39
Letermovir
Letermovir (AIC246 or MK-8228), a 3,4-dihydro-quinazoline-4-yl-acetic acid derivative, is the prototype viral terminase complex inhibitor that is most advanced in its clinical development. The novel compound was initially developed by AiCuris after high-throughput screening, hit-to-lead optimization, structure-activity relationship, and pharmacological analyses.38 By 2009, it had undergone a Phase I clinical trial in healthy volunteers and a Phase IIa proof-of-concept trial in a small cohort of kidney transplant recipients.40 Its preclinical data and proposed mechanism of action was presented in 2010 at the International Herpesvirus Workshop (Salt Lake City, UT, USA). In October 2010, the drug was used for the compassionate treatment of a lung transplant patient with CMV disease due to multidrug-resistant CMV.41 In April 2011, the drug was granted orphan drug designation for prevention of CMV disease by the European Commission. In August 2011, the US Food and Drug Administration granted it a fast track designation. In 2012, the results of Phase IIb clinical trials using letermovir in bone marrow transplant patients were presented at various international meetings, and the data were subsequently published in 2014.42 Its continued clinical development is currently undertaken in agreement with Merck. Table 1 summarizes the information to date on letermovir. Figure 1 shows the chemical structure of letermovir, the antivirals currently approved by the US Food and Drug Administration, and other investigational drugs with anti-CMV activity.
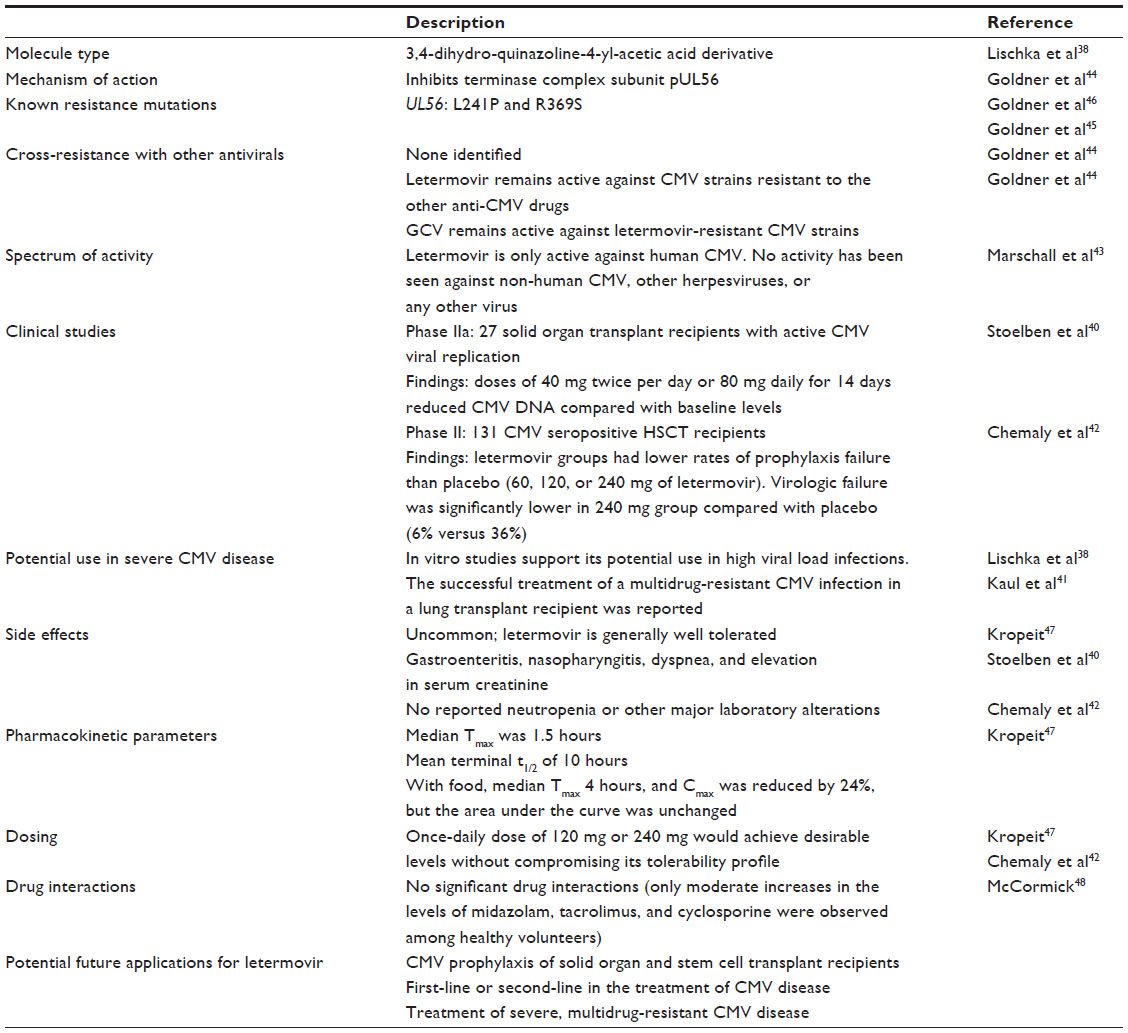 | Table 1 Properties of the terminase inhibitor letermovir (AIC246) |
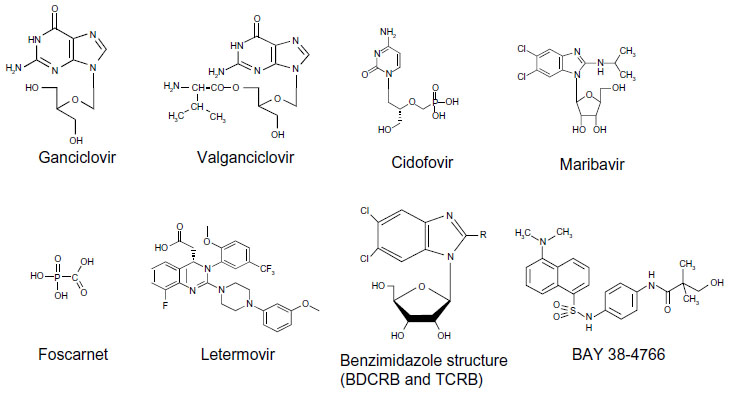 | Figure 1 Representation of the different anti-CMV antivirals currently in use as well as letermovir, benzimidazole structure (BDCRB and TCRB), and the sulfonamide BAY 38-4766. |
Mechanism of action
Letermovir is highly specific for human CMV, as it has no activity against other herpesviruses or any other virus.43 Mutational analyses indicated that letermovir resistance mapped to UL56. If this finding is sustained, inhibition of the terminase complex by letermovir can be attributed to its specific activity on pUL56.44,45 A sequence variation between amino acids 230 and 370 in pUL56 was identified as conferring letermovir resistance. These are two naturally occurring polymorphisms (L241P and R369S) that alter the inhibitory effect of letermovir on viral replication. The 50% effective concentration (EC50) was increased 160-fold with L241P and 38-fold with R396S.46 These naturally occurring mutations are the only letermovir resistance genotypes identified to date. Neither L241P nor R369S mutation has been selected during the use of letermovir in human studies, suggesting that there may be a high barrier for development of resistance.46
Because it acts through a novel mechanism, letermovir-resistant CMV does not exhibit cross-resistance with currently approved anti-CMV drugs, and remains susceptible to GCV, PFA, and CDF. Interestingly, letermovir-resistant CMV strains also do not exhibit resistance to the other terminase inhibitors (benzimidazoles and sulfonamides). This finding suggests that benzimidazoles, sulfonamides, and letermovir exhibit distinct inhibitory effects on this highly complex terminase complex.44
Preclinical studies
In vitro studies that assessed the activity of letermovir on different CMV strains using the classical cytopathic effect reduction assay and green fluorescent protein-based fluorescent reduction assay showed a very potent antiviral drug, which was more potent than GCV by 400-fold in EC50 and 2,000-fold in EC90 values. Its potency declined only slightly after an increase in virus inoculum, suggesting that it could potentially be useful even in the setting of high viral load.38 When tested against GCV-resistant CMV with UL97 mutations (M460I and C603W), letermovir retained its antiviral activity.43 Testing the drug in culture of CMV-infected fibroblasts showed that letermovir has a high efficiency in inhibiting focal expansion of the virus, and thus it can prevent cell-to-cell viral spread. Time-to-drug-addition studies showed that letermovir was effective when added as late as up to 57 hours after infection (compared with only 33 hours for GCV); this is presumed to be likely due to its effect during the later stages of viral replication (compared with GCV).
Letermovir has not shown any in vitro activity against herpesviruses other than human CMV, and the drug is also not active against murine or rat CMV.43 In vivo studies using a mouse xenotransplant model (transplanted with a Gelfoam sponge with infected human CMV cells) demonstrated that letermovir produced a statistically significant dose-dependent reduction of CMV titer in the transplanted cells (which was superior to VGCV).38
Human clinical studies
In a dose-finding Phase II study, the efficacy and safety of letermovir prophylaxis was compared with placebo in a cohort of 131 CMV seropositive recipients of HSCT from matched related or unrelated donors from 19 transplant centers in Germany and the USA. In this trial, different doses (60, 120, or 240 mg) of letermovir were compared with placebo for 12 weeks after stem cell engraftment. In a modified intention-to-treat analysis, the all-cause prophylaxis failure (defined as CMV infection or study drug discontinuation) was lower in all three groups that received any of the three doses of letermovir compared with 64% with placebo. However, only the groups that received letermovir 120 mg and 240 mg daily achieved a statistically significant difference when compared with placebo, with a combined failure rate of 29% (P=0.002).42 Virological failure, defined as detection of CMV DNA during study drug administration, was significantly lower in the 240 mg letermovir group when compared with placebo (6% versus 36%).42
In another Phase IIa randomized, controlled, open-label study performed in multiple centers in Germany, letermovir was given to 27 kidney transplant recipients as pre-emptive treatment for asymptomatic CMV viral replication that were detected by surveillance. In this study, letermovir at doses of 40 mg twice per day or 80 mg daily were given for 14 days, and this was compared with the local standard of care (which is VGCV). By day 15, all the three groups demonstrated significant declines in the number of copies of CMV DNA when compared with baseline. This clinical trial was designed as a proof-of-concept study with a small sample size, and many variables such as immunosuppression were not equally distributed given its open-label nature. However, none of the patients in this study developed CMV disease and there was a log10 drop in CMV load at the end of the 14 days of letermovir treatment (which is comparable with VGCV). Interestingly, there was a slower decline in CMV load in the letermovir group compared with VGCV (11 versus 4 days). This may be in part due to the fact that letermovir does not prevent CMV DNA production, but instead, it only inhibits its packaging into virions. A potential consequence of this finding is that viral load measurement may not be truly reflective of its antiviral efficacy during the first few weeks of letermovir treatment. Three patients in this pilot study had mutations in UL97 or UL54 (which conferred resistance to standard CMV drugs) and all of them responded appropriately to letermovir.40
Letermovir was used in a compassionate program for treatment of severe CMV disease in a lung transplant patient with prolonged disseminated disease due to multidrug-resistant CMV (A594T, C603W UL97 mutations; V715M and A987G UL54 mutations). The patient had failed to respond to standard treatment with GCV, VGCV, PFA, and CDV, off-label treatment with leflunomide, and compassionate use of CMX-001 (brincidofovir). The patient was eventually treated with letermovir, initially using a 120 mg daily dose but later escalated to 240 mg daily when there was no apparent response. At 4 weeks, the CMV load declined to a level below the limit of quantification and with symptom resolution.41 Reduction in the degree of immune suppression may also have facilitated the virological and clinical response in this case.41
Currently, letermovir is being evaluated in a placebo-controlled Phase III clinical trial for the prevention of CMV infection and disease in allogeneic HSCT recipients. The primary outcome of this study is the proportion of participants with clinically significant CMV infection through week 24 after transplantation. The study is expected to complete in January 2017 for primary outcome review.
Tolerability profile
The target of letermovir, the viral terminase complex, is unique to herpesviruses and the drug is not known to bind to human cells, so direct toxicity in humans is anticipated to be low. The few early phase clinical trials of letermovir so far have reported a low incidence of adverse effects. Data from over 260 patients who have received letermovir in Phase I and II clinical trials showed no major changes in vital signs, electrocardiographic findings, or laboratory markers. In the Phase II trial that evaluated three different doses of letermovir compared with placebo in HSCT recipients, the drug was well tolerated, and the reported drug-related side effects were more common in the placebo group (33% versus 17% in the letermovir group).42 The most common adverse effects were gastrointestinal symptoms (but were also as common in the placebo group). The rate of neutropenia was very low (when indirectly compared with the neutropenia associated with the use of VGCV, 6% versus 58%, respectively) and similar to placebo.42 In the study of kidney transplant recipients who received 80 mg of letermovir daily, the overall rate of adverse events was high (74.1%), but the majority of these events were not believed not to be due to letermovir. Gastroenteritis, nasopharyngitis, dyspnea, and elevation in serum creatinine were reported as potential side effects of letermovir, while other serum chemistries and hematological profile were no altered.40 The lung transplant patient who received letermovir 120 mg daily for 16 days tolerated the drug well, and there were no major adverse effects when the dose was doubled to 240 mg daily.41
Pharmacokinetics and pharmacodynamics
Systemic exposure to letermovir was measured in a Phase I single dose-escalation trial in healthy volunteers, where participants received letermovir at doses from as low as 5 mg up to as high as 320 mg daily. There was an increase in measured systemic exposure to letermovir for doses up to 240 mg daily, but there were no further increases observed for doses higher than 240 mg daily. The median time to maximum serum concentration (Tmax) was 1.5 hours, with a mean terminal half-life t1/2 of 10 hours. When taken with food, the median Tmax was delayed to 4 hours, and peak serum concentration was reduced by 24%, although the area under the curve remained unchanged. Once-daily dosing did not differ from twice-daily dosing in terms of Tmax and t1/2 parameters.47
Transplant patients with subclinical CMV viremia who received letermovir 40 mg twice per day and 80 mg daily achieved stable letermovir trough levels at day 4 of treatment, and the trough levels at this doses were consistently above the 90% effective concentration (EC90) of the drug. There was minimal intra-individual variability,40 and the drug levels were not affected by hemodialysis.41 When letermovir was tested in healthy volunteers, there were moderate increases in the levels of midazolam, tacrolimus, and cyclosporine. However, in the Phase IIa trial, no significant drug interactions were observed in patients on multiple medications. The subject treated for severe multidrug-resistant CMV disease with 240 mg daily of letermovir was on tacrolimus as part of his immunosuppression regimen, and no dose adjustment was required.48
Based on the pharmacokinetic data and safety profile of letermovir obtained from early phase clinical trials, it appears that a once-daily dose of 120 mg or 240 mg would achieve desirable levels without compromising the tolerability profile. Although no definitive clinical data are yet available regarding its efficacy, it appears from the Phase II studies in kidney and allogeneic HSCT recipients that a 240 mg daily dose of letermovir should be tested further in clinical trials.41,42
Potential clinical applications of letermovir
Letermovir can potentially be used for prevention and treatment of CMV infection in transplant recipients. Given the lack of hematological side effects with letermovir, it has a promising potential role in CMV prophylaxis in allogeneic HSCT recipients. This is currently being evaluated as part of a Phase II trial (to be completed in 2 years). Another potential role of letermovir is in the treatment of active CMV replication, as suggested by the early-phase trials and a proof-of-concept trial in kidney recipients. The treatment of clinically significant CMV disease, in particular of multidrug-resistant CMV, represents another promising treatment indication of letermovir. Furthermore, given its lower risk for adverse effects compared with the currently approved drugs, letermovir may potentially replace the nucleoside analogs as a first-line drug for prevention and treatment of treatment-naïve CMV disease. One downside for clinical use is the lack of activity against other herpesviruses, which also commonly reactivate in transplant recipients.
Conclusion
Letermovir is a novel terminase complex inhibitor that appears to be a promising agent for the prevention and treatment of CMV in transplant recipients. It is highly specific against human CMV, and its potency in vitro appears to be greater than GCV. Initial studies indicate its potential for prophylaxis and treatment of CMV. Its low adverse effect profile makes it ideal as a first-line drug, if it is eventually proven to be efficacious against CMV infection. Its unique mechanism of action makes its effective even against CMV with resistance to GCV, CDV, and PFA. These characteristics open many possibilities for letermovir in the future, including its potential to be used as primary prophylaxis in allogeneic HSCT recipients. Its unique property of CMV specificity, however, would require patients to be on other antiviral drugs for the prevention of other herpesviruses such as herpes simplex. Phase III trials are currently underway, and there is cautious anticipation that this novel drug will emerge from these clinical studies as a “new kid on the block” for the management of CMV infection in the vulnerable transplant population.
Disclosure
The authors report no conflicts of interest in this work.
References
Reddehase MJ. Antigens and immunoevasins: opponents in cytomegalovirus immune surveillance. Nat Rev Immunol. 2002;2(11):831–844. | |
Bate SL, Dollard SC, Cannon MJ. Cytomegalovirus seroprevalence in the United States: the national health and nutrition examination surveys, 1988–2004. Clin Infect Dis. 2010;50(11):1439–1447. | |
Beam E, Razonable RR. Cytomegalovirus in solid organ transplantation: epidemiology, prevention, and treatment. Curr Infect Dis Rep. 2012; 14(6):633–641. | |
Torre-Cisneros J. Toward the individualization of cytomegalovirus control after solid-organ transplantation: the importance of the “individual pathogenic balance”. Clin Infect Dis. 2009;49(8):1167–1168. | |
Humar A, Lebranchu Y, Vincenti F, et al. The efficacy and safety of 200 days valganciclovir cytomegalovirus prophylaxis in high-risk kidney transplant recipients. Am J Transplant. 2010;10(5):1228–1237. | |
Paya C, Humar A, Dominguez E, et al. Efficacy and safety of valganciclovir vs oral ganciclovir for prevention of cytomegalovirus disease in solid organ transplant recipients. Am J Transplant. 2004;4(4):611–620. | |
Einsele H, Hebart H, Kauffmann-Schneider C, et al. Risk factors for treatment failures in patients receiving PCR-based preemptive therapy for CMV infection. Bone Marrow Transplant. 2000;25(7):757–763. | |
Manuel O, Pang XL, Humar A, Kumar D, Doucette K, Preiksaitis JK. An assessment of donor-to-recipient transmission patterns of human cytomegalovirus by analysis of viral genomic variants. J Infect Dis. 2009;199(11):1621–1628. | |
Razonable RR. Epidemiology of cytomegalovirus disease in solid organ and hematopoietic stem cell transplant recipients. Am J Health Syst Pharm. 2005;62(8 Suppl 1):S7–S13. | |
Le Page AK, Jager MM, Iwasenko JM, Scott GM, Alain S, Rawlinson WD. Clinical aspects of cytomegalovirus antiviral resistance in solid organ transplant recipients. Clin Infect Dis. 2013;56(7):1018–1029. | |
Lurain NS, Chou S. Antiviral drug resistance of human cytomegalovirus. Clin Microbiol Rev. 2010;23(4):689–712. | |
Bogner E. Human cytomegalovirus terminase as a target for antiviral chemotherapy. Rev Med Virol. 2002;12(2):115–127. | |
Buerger I, Reefschlaeger J, Bender W, et al. A novel nonnucleoside inhibitor specifically targets cytomegalovirus DNA maturation via the UL89 and UL56 gene products. J Virol. 2001;75(19):9077–9086. | |
Bogner E, Radsak K, Stinski MF. The gene product of human cytomegalovirus open reading frame UL56 binds the pac motif and has specific nuclease activity. J Virol. 1998;72(3):2259–2264. | |
Hwang JS, Bogner E. ATPase activity of the terminase subunit pUL56 of human cytomegalovirus. J Biol Chem. 2002;277(9):6943–6948. | |
Pilorge L, Burrel S, Ait-Arkoub Z, Agut H, Boutolleau D. Human cytomegalovirus (CMV) susceptibility to currently approved antiviral drugs does not impact on CMV terminase complex polymorphism. Antiviral Res. 2014;111:8–12. | |
Griffiths PD, Emery VC. Taming the transplantation troll by targeting terminase. N Engl J Med. 2014;370(19):1844–1846. | |
Gable JE, Acker TM, Craik CS. Current and potential treatments for ubiquitous but neglected herpesvirus infections. Chem Rev. October 2, 2014. [Epub ahead of print.] | |
Bogner E, Reschke M, Reis B, Mockenhaupt T, Radsak K. Identification of the gene product encoded by ORF UL56 of the human cytomegalovirus genome. Virology. 1993;196(1):290–293. | |
Champier G, Couvreux A, Hantz S, et al. Putative functional domains of human cytomegalovirus pUL56 involved in dimerization and benzimidazole D-ribonucleoside activity. Antiviral Ther. 2008;13(5):643–654. | |
Couvreux A, Hantz S, Marquant R, et al. Insight into the structure of the pUL89 C-terminal domain of the human cytomegalovirus terminase complex. Proteins. 2010;78(6):1520–1530. | |
Scheffczik H, Savva CG, Holzenburg A, Kolesnikova L, Bogner E. The terminase subunits pUL56 and pUL89 of human cytomegalovirus are DNA-metabolizing proteins with toroidal structure. Nucleic Acids Res. 2002;30(7):1695–1703. | |
Giesen K, Radsak K, Bogner E. The potential terminase subunit of human cytomegalovirus, pUL56, is translocated into the nucleus by its own nuclear localization signal and interacts with importin alpha. J Gen Virol. 2000;81 Pt 9:2231–2244. | |
Adam SA, Gerace L. Cytosolic proteins that specifically bind nuclear location signals are receptors for nuclear import. Cell. 1991;66(5):837–847. | |
Weis K, Dingwall C, Lamond AI. Characterization of the nuclear protein import mechanism using Ran mutants with altered nucleotide binding specificities. EMBO J. 1996;15(24):7120–7128. | |
Melchior F, Paschal B, Evans J, Gerace L. Inhibition of nuclear protein import by nonhydrolyzable analogues of GTP and identification of the small GTPase Ran/TC4 as an essential transport factor. J Cell Biol. 1993;123(6 Pt 2):1649–1659. | |
Thoma C, Borst E, Messerle M, Rieger M, Hwang JS, Bogner E. Identification of the interaction domain of the small terminase subunit pUL89 with the large subunit pUL56 of human cytomegalovirus. Biochemistry. 2006;45(29):8855–8863. | |
Nadal M, Mas PJ, Blanco AG, et al. Structure and inhibition of herpesvirus DNA packaging terminase nuclease domain. Proc Natl Acad Sci U S A. 2010;107(37):16078–16083. | |
Champier G, Hantz S, Couvreux A, et al. New functional domains of human cytomegalovirus pUL89 predicted by sequence analysis and three-dimensional modelling of the catalytic site DEXDc. Antiviral Ther. 2007;12(2):217–232. | |
Dittmer A, Drach JC, Townsend LB, Fischer A, Bogner E. Interaction of the putative human cytomegalovirus portal protein pUL104 with the large terminase subunit pUL56 and its inhibition by benzimidazole-D-ribonucleosides. J Virol. 2005;79(23):14660–14667. | |
Scholz B, Rechter S, Drach JC, Townsend LB, Bogner E. Identification of the ATP-binding site in the terminase subunit pUL56 of human cytomegalovirus. Nucleic Acids Res. 2003;31(5):1426–1433. | |
Borst EM, Kleine-Albers J, Gabaev I, et al. The human cytomegalovirus UL51 protein is essential for viral genome cleavage-packaging and interacts with the terminase subunits pUL56 and pUL89. J Virol. 2013;87(3):1720–1732. | |
McVoy MA, Nixon DE. Impact of 2-bromo-5,6-dichloro-1-beta-D-ribofuranosyl benzimidazole riboside and inhibitors of DNA, RNA, and protein synthesis on human cytomegalovirus genome maturation. J Virol. 2005;79(17):11115–11127. | |
Krosky PM, Underwood MR, Turk SR, et al. Resistance of human cytomegalovirus to benzimidazole ribonucleosides maps to two open reading frames: UL89 and UL56. J Virol. 1998;72(6):4721–4728. | |
Underwood MR, Harvey RJ, Stanat SC, et al. Inhibition of human cytomegalovirus DNA maturation by a benzimidazole ribonucleoside is mediated through the UL89 gene product. J Virol. 1998; 72(1):717–725. | |
Komazin G, Townsend LB, Drach JC. Role of a mutation in human cytomegalovirus gene UL104 in resistance to benzimidazole ribonucleosides. J Virol. 2004;78(2):710–715. | |
Reefschlaeger J, Bender W, Hallenberger S, et al. Novel non-nucleoside inhibitors of cytomegaloviruses (BAY 38-4766): in vitro and in vivo antiviral activity and mechanism of action. J Antimicrob Chemother. 2001;48(6):757–767. | |
Lischka P, Hewlett G, Wunberg T, et al. In vitro and in vivo activities of the novel anticytomegalovirus compound AIC246. Antimicrob Agents Chemother. 2010;54(3):1290–1297. | |
Lischka P, Zimmermann H. Antiviral strategies to combat cytomegalovirus infections in transplant recipients. Curr Opin Pharmacol. 2008;8(5):541–548. | |
Stoelben S, Arns W, Renders L, et al. Preemptive treatment of cytomegalovirus infection in kidney transplant recipients with letermovir: results of a Phase 2a study. Transpl Int. 2014;27(1):77–86. | |
Kaul DR, Stoelben S, Cober E, et al. First report of successful treatment of multidrug-resistant cytomegalovirus disease with the novel anti-CMV compound AIC246. Am J Transplant. 2011;11(5):1079–1084. | |
Chemaly RF, Ullmann AJ, Stoelben S, et al. Letermovir for cytomegalovirus prophylaxis in hematopoietic-cell transplantation. N Engl J Med. 2014;370(19):1781–1789. | |
Marschall M, Stamminger T, Urban A, et al. In vitro evaluation of the activities of the novel anticytomegalovirus compound AIC246 (letermovir) against herpesviruses and other human pathogenic viruses. Antimicrob Agents Chemother. 2012;56(2):1135–1137. | |
Goldner T, Hewlett G, Ettischer N, Ruebsamen-Schaeff H, Zimmermann H, Lischka P. The novel anticytomegalovirus compound AIC246 (Letermovir) inhibits human cytomegalovirus replication through a specific antiviral mechanism that involves the viral terminase. J Virol. 2011;85(20):10884–10893. | |
Goldner T, Zimmermann H, Lischka P. Phenotypic characterization of two naturally occurring human Cytomegalovirus sequence polymorphisms located in a distinct region of ORF UL56 known to be involved in in vitro resistance to letermovir. Antiviral Res. 2015;116c:48–50. | |
Goldner T, Hempel C, Ruebsamen-Schaeff H, Zimmermann H, Lischka P. Geno- and phenotypic characterization of human cytomegalovirus mutants selected in vitro after letermovir (AIC246) exposure. Antimicrob Agents Chemother. 2014;58(1):610–613. | |
Kropeit D. Phase I safety and PK data of the novel anti-HCMV terminase inhibitor AIC246. Abstract 1994 presented at the 50th Interscience Conference of Antimicrobial Agents and Chemotherapy, Boston, MA, USA, September 12–15, 2010. | |
Mccormick D. AIC246 does not exhibit clinically significant drug-drug interactions when administered with immunosuppressive drugs in transplant recipient. Paper presented at: 51th Interscience Conference of Antimicrobial Agents and Chemotherapy, Chicago, IL, USA, September 17–20, 2011. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.