Back to Journals » Journal of Inflammation Research » Volume 15
Leptin Deficiency May Influence the Divergence of Cell-Mediated Immunity Between Lepromatous and Tuberculoid Leprosy Patients
Authors Degechisa ST ![]() , Dabi YT
, Dabi YT ![]()
Received 20 September 2022
Accepted for publication 29 November 2022
Published 13 December 2022 Volume 2022:15 Pages 6719—6728
DOI https://doi.org/10.2147/JIR.S389845
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Sisay Teka Degechisa,1,2 Yosef Tsegaye Dabi1,3
1Medical Biochemistry Department, College of Health Sciences, Addis Ababa University, Addis Abeba, Ethiopia; 2Medical Laboratory Science Department, College of Medicine and Health Sciences, Arba Minch University, Arba Minch, Ethiopia; 3Medical Laboratory Science Department, Wollega University, Nekemte, Ethiopia
Correspondence: Sisay Teka Degechisa, Addis Ababa, Ethiopia, Email [email protected]
Abstract: Leprosy is a disease caused by an intracellular bacillus bacterium called Mycobacterium leprae which lives and multiplies in the hosts’ macrophages and Schwann cells. Depending on the degree of the host’s cell-mediated immunity (CMI) response to the bacilli, the disease manifests itself in five clinical spectra ranging from polar tuberculoid (TT) to polar lepromatous leprosy (LL). A very high level of T helper 1 (Th1) driven bacilli-specific CMI is seen in the TT form, whereas this response is essentially nonexistent in the LL form. As a result, there is very low or absent bacillary load and localized nodular lesions in TT patients. On the contrary, LL patients presented with high bacillary load and generalized lesions due to low CMI response. The mechanism underlying this divergence of CMI response is not clearly elucidated yet. However, mounting evidence links it to an elevated number of Th1 and Th17 suppressing CD4+ CD25+ FOXP3+ T regulatory cells (Treg cells) which are abundantly found in LL than in TT patients. The predominance of these cells in LL patients is partly attributed to a deficiency of leptin, the cytokine-like peptide hormone, in these patients. Becausea normal level of leptin promotes the proliferation and preferential differentiation of effector T cells (Th1 and Th17) while inhibiting the growth and functional responsiveness of the Treg cells. In contrast, leptin deficiency or neutralization was reported to exert the opposite effect on Treg cells and effector T cells. Other smaller subsets of lymphocytes such as gamma delta (γδ) T cells and B regulatory cells are also modulated by leptin level in the pathogenesis of leprosy. Leptin may therefore regulate the divergence of CMI between TT and LL patients by regulating the homeostasis of effector T cells and Treg cells, and this review will examine the underlying mechanism for this.
Keywords: lepromatous leprosy, tuberculosis leprosy, Th1 cell anergy, cell mediated immunity, leptin, CD4+CD25+FoxP3+ T regulatory cells, mTORC1
Introduction
Mycobacterium leprae (M. leprae), an obligate intracellular bacterium, is the primary cause of leprosy, a chronic infectious disease.1 Primarily; the disease affects the skin and peripheral nervous system. Susceptibility to infection and its clinical symptoms are linked to the degree of the host’s immune response.1–3
Using clinical, histological, and immunological criteria, Ridley and Jopling divided leprosy into two polar kinds, namely tuberculoid leprosy (TT) and lepromatous leprosy (LL). Other clinical forms between these polar forms include borderline tuberculoid (BT), mid-borderline (BB), and borderline lepromatous (BL).4 Patients with BB, BL, and LL having a bacterial index (BI) >2 at any site were classified as multibacillary (MB), whereas those with a BI < 2 at any site were classified as paucibacillary (PB).5
Inflammatory infiltrates with well-formed granulomas, differentiated macrophages, epithelioid and giant cells, a predominance of CD4+ T cells, and PB state were seen upon histopathological evaluation of samples taken from hypopigmented skin patches of TT patients4,6 TT patients are also characterized by positive lepromin skin test and enhanced T helper 1 (Th1) induced M. leprae pecific cell-mediated immunity (CMI). An increased ratio of interferon-gamma to interleukin-4 (IFN-γ: IL-4) seen in a Th1 polarized immune response activates macrophages, which will ultimately result in a decreased bacterial load and the PB state.6 Additionally, another subset of pro-inflammatory T cells, T helper 17 (Th17) cells, which mainly produce interleukin 17 (IL-17) is also abundantly found in these groups of patients.7 However, LL patients, on the other hand, are characterized by negative lepromin skin test, high T helper 2 (Th2) mediated humoral responses with high antibody titer, weak CMI due to low IFN-γIL-4 ratio, extensive skin lesions with a predominant CD8+ T cells, absence of granuloma formation, and high bacterial load. The inability to mountTh1-mediated mediated M.leprae-specific CMI response against the bacillus in LL patients allows its multiplication in large numbers and favors the transmission of the disease.8–11 The range of clinical leprosy from TT to LL thus seems to reflect a large CMI response at one extreme to a complete absence of CMI at the other.
Although the mechanism mediating the divergence of CMI in these two polar clinical forms of leprosy is not well understood, mounting evidence points to this phenomenon being linked to patients’ plasma leptin levels.12–15 Leptin, a 16 KD cytokine-like peptide hormone primarily produced by adipocytes, has the potential to modulate the dichotomy of CMI response between LL and TT patients by linking nutritional status with neuroendocrine and immune functions.12,13 Supporting this hypothesis, leptin has been shown to modulate CMI in both human and murine models. For example, it was reported that impaired CMI, leptin-deficient (ob/ob), or leptin receptor (ObR)-deficient (db/db) mice and humans had more recurring infections than their normal counterparts.14
Leptin deficiency has been correlated with nutritional deprivation/malnutrition, which in turn induces a state of immunodeficiency.15 Leprosy was connected to food deprivation and malnutrition at any age, according to case-control studies on socioeconomic factors carried out in Brazil, Bangladesh, and Ethiopia.16–18 It was also established that nutritional imbalance increases susceptibility to the disease and reduces CMI, which is the primary defense against M. leprae (reviewed in reference).19 These findings suggest that malnutrition-induced leptin deficiency may influence the CMI disparity between LL and TT patients.
Importantly, leptin has been shown to reduce the proliferative potential of Th1 suppressing transforming growth factor beta (TGF-β) secreting forkhead P3 positive (FOXP3+) CD4+CD25+ T regulatory cells (Treg cells) in response to T cell receptor (TCR) stimulation.20 Palermo et al confirmed that LL patients have a higher number of these cells than TT patients.21 This negative relationship between leptin and Treg cells may be an indicator of leptin deficiency in LL patients when compared to TT patients. Additionally, Treg cells prevent Th1 cells from becoming activated and proliferating, which in turn stimulate CMI, making Th1-mediated CMI much less effective in LL patients than in TT patients.AtAt its normal level, leptin can act as a negative signal for the proliferation of Tregs.20 It controls this activity by turning on the mTORC1 signaling pathway, which increases the activity of effector T cells (Th1, Th17) while decreasing that of Treg cells.12 In this review paper, we will address the following questions: What causes the dichotomy in CMI between LL and TT patients? How does plasma leptin level modulate the divergence of CMI between the two polar clinical forms of leprosy? What other possible immune components do leptin influence? How do these factors cross-talk with leptin and affect the degree of CMI response in these patients?
How Leptin Modulates the Divergence of CMI Response in Tuberculoid and Lepromatous Leprosy?
Leptin has long been known to relate nutritional status to pro-inflammatory immunological responses, thereby directly controlling T cell proliferation and cytokine production.14 It may influence CMI against intracellular infections either directly through Th1 immune response modulation or indirectly through Treg cell suppression. Leptin, for example, appears to polarize T cells toward a Th1 phenotype by inducing the synthesis of IL-2, IL-12, and IFN while suppressing the synthesis of IL-10 and IL-4.22 Furthermore, in a mixed lymphocyte reaction, leptin has been reported to aid T-cell differentiation toward the Th1 phenotype.12,23 In its deficient state, the proliferation of Th1-suppressing Treg cells is favored and this, in turn, results in CMI suppression.21 In the following sections, we will look at two major mechanisms of how leptin deficiency may influence the divergence of CMI responses in TT and LL.
Leptin Deficiency May Induce Th1 Cell Anergy and Contribute to Variation in CMI Between Lepromatous Leprosy Patients
Despite the lack of published data indicating leptin/its receptor deficiency in LL patients, our unpublished data suggested that plasma leptin level in LL patients was significantly lower than in healthy controls.24 In line with this, a meta-analysis of 12 case-control studies25 and a recent study by Tsegaye et al26 found significantly decreased plasma levels of leptin among patients with pulmonary tuberculosis which is caused by Mycobacterium tuberculosis, an intracellular bacterium with similar characteristics to the etiology of leprosy. Additionally, as the percentage of leptin in the blood to fat mass is proportionate, hypoleptinaemia is often linked to lower body fat from nutritional insufficiency. This in turn causes secondary immunodeficiency and increased susceptibility to infection as well as infectious disease progression.11 Nutritional deficiencies, such as protein-energy malnutrition, have been linked to decreased CMI, which in the case of leprosy could result in a higher likelihood of active leprosy (90% LL form) and, consequently, an increased reservoir of a transmissible infection, according to a study conducted in Ethiopia.18 This implies that malnutrition-induced leptin deficiency may contribute to variation in CMI in the two polar forms of leprosy.
Importantly, leptin deficiency was experimentally proved to induce Th1 cell anergy. For instance, decreased plasma leptin levelsdue to starvation/malnutrition result in the suppression of Th1-mediated inflammatory responses in wild-type mice.27,28 In addition, leptin-deficient (ob/ob) mice29 and diabetic (db/db) mice30 have impaired CMI due to Th1 cell anergy. Of note, leptin-deficient mice with pulmonary tuberculosis have been shown to have anergic Th1 cells and were unable to generate IFN-γ. However, exogenous leptin supplementation restored lymphocyte trafficking, IFN-γ production, and granuloma formation in that mice.31
In humans, leptin has been shown to boost Th1 cytokine production while inhibiting Th2 cytokine secretion.27 When there is a lack of leptin, whether, from malnutrition or mutation, the opposite occurs.11,32–36 In TT patients who may have normal or higher leptin levels, leptin-induced Th1 mediated M. leprae-specific CMI responses can limit the bacilli growth in localized lesions. These responses are primarily orchestrated by IFN-γ, which is more favorable for bacilli clearance because this cytokine activates monocytes/macrophages, cytotoxic T cells, and natural killer cells for the final effector function targeting the bacilli. Unlike TT patients, LL patients areleptin-deficient and have a predominantly Th2-mediated immune response with high humoral immunity but are unable to generate Th1 responses against the bacterium, resulting in delayed clearance of the bacilli.6–8,37–39
These findings suggested that Th1 cell unresponsiveness is linked to leptin/its receptor deficiency. The observation that such unresponsiveness has been seen in LL patients but not in TT ones simply implies leptin could modulate the divergence of CMI between these clinical forms of the disease.
Leptin May Dictate the Divergence of CMI in Tuberculoid and Lepromatous Leprosy Patients by Controlling CD4+CD25+ FOXP3+ T Regulatory Cells
The Role of CD4+CD25+ FOXP3+ Regulatory T Cells in Leprosy
The existence of an elevated number of Treg cells at infection sites and in the peripheral blood of these patients may provide a credible explanation for the pathogen’s persistence in LL patients.9,13 In order to prevent immunopathology and maintain a normal, balanced immune response, Treg cells frequently play a vital immune-regulatory role by controlling the severity of innate and adaptive immunological responses.37 However, they contribute to host disease tolerance and diminished effector function against M. leprae by releasing their cytokines such as TGF-β and interleukin-10 (IL-10).2,21,38
After the discovery of FoxP3, the lineage-specific transcription factor for this T-cell subset, their suppressor function was confirmed and this transcription factor was examined with high frequency in skin lesions from LL patients.39 It is interesting to note that Treg cells without this component lose their capacity to inhibit, and they may even change into effector T cells.10,40 Moreover, LL patients had higher gene expression of Tregs signatures (FOXP3, TGF-β, and IL-10) than TT patients.41,42 Production of TGF-β and IL-10 is regulated by FOXP3, which is associated with the anti-inflammatory nature of this cell type. These findings clearly indicated that there is a predominance of Tregs in LL patients than in TT ones and this is most probably attributed to comparably low leptin levels in LL patients.38,39
Leptin May Modulate the Proliferation of CD4+CD25+FOXP3+ T Regulatory Cells in Lepromatous Leprosy Patients
Researchers have reported that leptin can modulate the proliferation of Tregs.43–45 De Rosa et al have reported that freshly isolated Treg cells produced leptin and expressed high amounts of ObR than conventional T cells. The researchers noted that leptin produced by these cells was observed to constrain their proliferation. In vitro neutralization of these cell cultures with leptin monoclonal antibody (mAb) during anti-CD3 and anti-CD28 stimulation resulted in interleukin-2 (IL-2) dependent Treg cell proliferation20 (Figure 1). Furthermore, FoxP3 expression was elevated in leptin-neutralized Tregs while maintaining their immune-suppressive characteristics.39
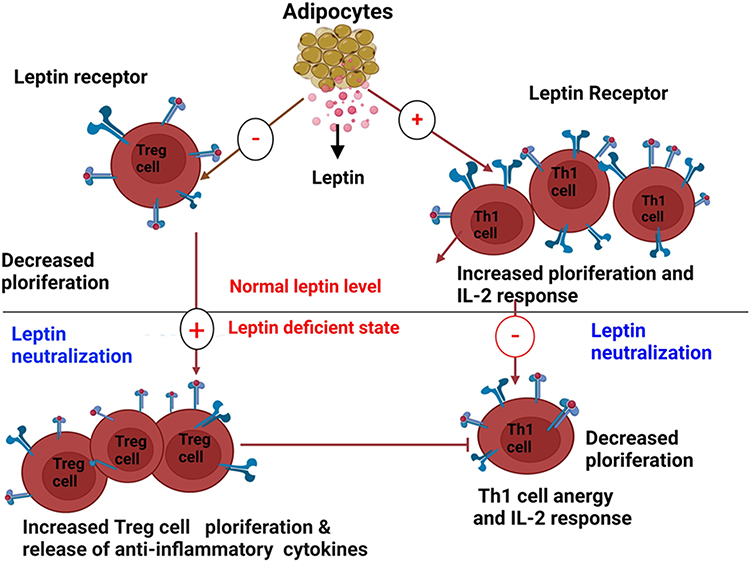 |
Figure 1 Opposite effects of leptin and its neutralization on Th1 and Treg cells (Created with https://biorender.com). Leptin generated from adipose tissue induces Th1 cells (right) to multiply and release pro-inflammatory cytokines including leptin and IL-2. On the other hand, Treg cells (left), which release leptin in an autocrine loop and regulate their hyporesponsiveness, are prevented from proliferating by leptin. These reactions are reversed by leptin neutralization, which results in a reduction in Th1 cell proliferation and cytokine release (right) and a significant expansion of Treg cells (left). |
Additionally, after treating healthy splenic Treg cells in vitro for 3 days in the presence or absence of leptin and/or leptin-neutralizing antibodies, activated Tregs had significant levels of TGF- and IL-10. However, TGF-β and IL-10 expression levels were significantly lower in leptin-treated Treg cells than in untreated Treg cells.46,47 These findings suggest that chronic leptin deficiency and leptin resistance may be associated with an increase in Tregs and a shift in immune response away from CMI in LL patients, as opposed to TT patients.
Leptin Inhibits the Proliferation of CD4+CD25+FOXP3+ T Regulatory Cells by Activating mTOR Signaling
In its physiologically normal level leptin inhibits the proliferation of Treg cells by activating the mTOR kinase, a serine/threonine kinase signaling pathway upon TCR stimulation.44 mTOR is a system that couples cell growth and homeostasis to nutrient availability and metabolic cues.46,48 In mammalian cells, there are two different multiprotein mTOR complexes called mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) having structural and functional differences.44,46,49
It has been shown that in vitro anergy of Treg cells correlated with increased leptin-mediated mTORC1 activation at baseline.44 Conversely, low leptin level as in acute starvation results in decreased mTORC1 activity and thus promotes the proliferation of Treg cells upon TCR stimulation (Reviewed).46 However, administration of leptin to starved mice activates mTORC1, hence restoring the anergic state of Treg cells.36,44 Suppression of mTORC1 with rapamycin has also been found to disrupt such an anergic state.49,50 Thus, leptin-mediated mTOR activation dampens Treg cell proliferation.
Importantly, FoxP3 and other immune-suppressing Treg cells’ hallmark molecules are inhibited from being expressed when mTORC1 is activated by the PI3K-Akt (protein kinase B) axis, but FoxP3 is stimulated and Treg cell differentiation is induced when mTORC1 is inhibited by the drug rapamycin.44,49,50 This implies that the proliferation of Treg cells is closely linked to dynamic changes in mTORC1 activity (Figure 2). Interestingly, this change has been confirmed to be influenced by the composition of nutrients in the extracellular environment and leptin signaling.43,46,50 Intriguingly, rapamycin has been shown to enhance Treg cell generation both in vitro and in vivo in the presence of supra-physiologic IL-2 concentrations.51–55 Considering that IL-2 activates mTORC1, the conundrum is how a concomitant inactivation of mTORC1 by rapamycin and IL-2-mediated activation of mTORC1 can result in Treg cell proliferation.44
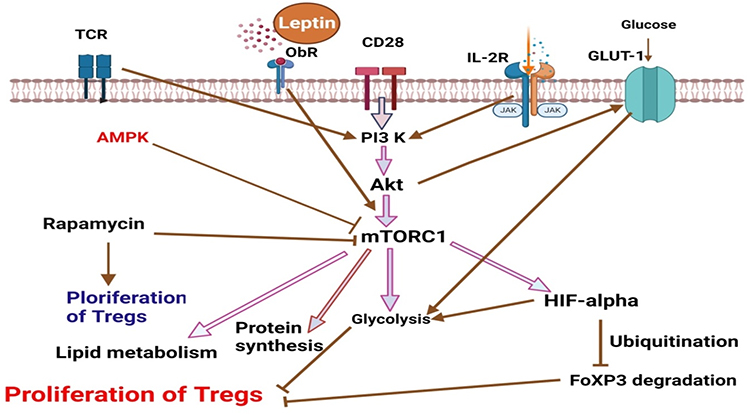 |
Figure 2 mTORC1 is a key regulator of the proliferation of T regulatory cells (Created with https://biorender.com. Leptin, IL-2, and CD28 co-stimulation, as well as cytokine signals, all contribute to TCR signaling. Together, these signals increase glycolysis by causing PI3K-dependent Akt activation, which boosts glucose transport by promoting Glut1 overexpression on the surface of T cells. Activated Akt increases the rate of protein synthesis via stimulating mTORC1. A stimulated mTORC1 increases HIF1, which in turn stimulates glycolysis and results in the degradation of FoxP3, which prevents the proliferation of Treg cells. Rapamycin, on the other hand, promotes the growth of Treg cells by inhibiting glycolysis. |
Notably, leptin neutralization reduces mTORC1 activity in Treg cells, indicating a link between autocrine leptin production by Treg cells and mTOR activation.56 Treg cells’ anergic state is maintained by increased mTORC1 activity which is mediated by leptin signaling because short mTORC1 inhibition or leptin neutralization reverses Treg cells’ hypo-responsiveness to TCR stimulation.38
Multiple researchers confirmed that reduced mTORC1 activity via leptin neutralization in newly isolated human Treg cells before TCR activation corrected their anergic status in vitro and led them to enter the cell cycle and proliferate.38,44,57–63 These findings suggested that leptin deficiency lets Treg cells proliferate abundantly. Thus, if a normal leptin level is maintained, leptin-mediated mTORC1 activation down-regulates Treg cell proliferation. However, leptin deficiency as in LL patients up regulates their proliferation and consequently induces anergy in Th1 cell response, which in turn diminishes CMI among these patients.
Other Smaller Subsets of Lymphocytes Which Play Critical Roles in Shaping Immunity in Pathogenesis of Leprosy
The Role of T Helper 17 in Pathogenesis of Leprosy
In addition to Th1 and Th2, a third subset of effector cells that generate IL-17, known as Th17, has been discovered in the last ten years. Th17’s main function is to clear pathogens that Th1 and Th2 have not been able to manage.64 According to studies, Th17 cells are more prevalent in BT and TT patients than in BL and LL patients, and they can increase IFN production while decreasing IL-10 production by Treg cells. This implies that Th17 cells also operate as a preventative measure against M. leprae infection.43,64,65 Additionally, a significant negative association between IL-10 produced by Treg cells and IL-17+ T cells in BL/LL was discovered. In BL/LL patients, inhibiting IL-10/TGF-β restored the IL-17+ T cells. TGF-β, IL-6, IL-17, and IL-23, which are Th17-related cytokines, simultaneously lowered the proportion of FoxP3+Treg cells while increasing the number of IL-17-producing CD4+ cells in LL.43 Th17 cells with signature IL-17A and its isoforms IL-17C, D, F, E and a transcription factor retinoic acid receptor related orphan receptor C (RORC) are more associated with TT form in both skin lesions and M. leprae induced PBMC cultures suggesting, its differential role in TT and LL patients.66
Studies showed that leptin increases Th17 cell proliferation while decreasing Treg cell proliferation through mTOR activation.67–69 This contributes to the divergence of the number of these cells in TT and LL patients. Leptin also metabolically licenses T effector cells including Th17 for activation to link nutrition and immunity.70,71 Leptin or its receptor deficiency in CD4+ T cells resulted in a selective defect in both autoimmune and protective Th17 responses. Reduced capacity for differentiation towards a Th17 phenotype by ObR-deficient T cells was attributed to reduced activation of the signal transducer and activator of transcription 3 (STAT3) and its downstream targets.68 Although STAT3’s expression levels were comparable in both types of leprosy patients, only TT patients had phosphorylated STAT3, even though STAT3 was expressed in equal numbers in both types of leprosy cases.68,72
The Role of Regulatory B Cells (Bregs) in the Pathogenesis of Leprosy
Because M. leprae could survive and multiply in humoral immune-dominated LL despite higher antibody responses, it was suggested that humoral immunity in leprosy was ineffective in pathogen elimination. As a result, despite being a major immune cell population with antibody secretory and antigen presentation functions, B cells are the least studied cell population in leprosy pathogenesis studies.73 Recent B cell research has revealed that three subsets of B regulatory cells (Bregs) with immunosuppressive functions may play important roles in leprosy pathogenesis. Bregs that only produce Interleukin-10 (IL-10) or Interleukin-35 (IL-35) or both are examples of these.74 Although they enhance vulnerability to infections, IL-10 and IL-35 produced by Bregs can protect against autoimmune illness. This suppression of immune responses correlates well with the bacteriological index and is linked to the development of the disease.73,74 An observation that increases in Bregs in leprosy patients in LL patients that in TT ones substantiated immune response dichotomy in these groups of patients.73
The Role of Gamma Delta (γδ) T Cells in Pathogenesis of Leprosy
Gamma delta (γδ) T cells are T-cell subsets found in both the dermis and the epidermis of humans. These cells are primarily involved in the production of IFN and IL-17 in circulation.75,76 One of the three lymphocyte lineages, γδ T cells, makes up a very small amount of total lymphocytes compared to traditional T and B cells.74 Liu et al identified skin resident γδ T cells in patients with leprosy. In skin lesions of TT patients, their results revealed that γδ T cells had dramatically increased. Dermal T cells are particularly susceptible to IL-23 stimulation, which can result in the production of IL-17A, a cytokine that may help defend against illness. They also found that the main source of IL-17A production in the skin was IL-23 sensitive dermal γδ T cells, which could be a potential target for the therapy of leprosy.77
A study reported high a level of γδ T cells in LL patients.78,79 The proportion of immunosuppressive CD4+TCR+FoxP3+ cells was shown to be the highest in LL patients when compared to that of TT patients and healthy controls.80 So, this implies that these are important in mediating pathogenesis of leprosy.
Concluding Remarks and Future Perspectives
It has been reported that Foxp3+ CD4+ CD25+ Tregs are abundantly found in lesions from LL patients than in TT ones. Moreover, these cells produce cytokines like Il-10 and TGF-β, which are known by suppressing responses of Th1 cells and cause their anergy/unresponsiveness. One has to ask why these cells are more abundantly found in LL patients than in TT patients and how their proliferation is controlled. Leptin has been shown to have a detrimental effect on the growth of Treg cells. Treg cells remain anergic while leptin levels are sufficient, however, it has been discovered that when leptin levels are low owing to starvation, mutation, or a lack of its receptor, Treg cell growth is induced.
Interestingly, the proliferation of Treg cells is also closely linked to dynamic changes in mTOR activity, which is influenced by the composition of nutrients in the extracellular environment and the leptin signal. Normal leptin level has been known to activate the mTORC1 pathway and inhibts its proliferation of Treg cells, but leptin deficiency diminishes mTORC1, which subsequently inhibits the proliferation of Treg cells. As a result, leptin-dependent activation of mTORC1 maintains Treg cell anergic status via an unknown mechanism. This implies that a decrease in leptin levels down-regulates mTORC1, which may result in Treg cell proliferation and activation, resulting in Th1 cell anergy and a reduced CMI response in LL patients.
Other smaller subsets of lymphocytes such as Th17 cells, gamma delta cells, and B regulatory cells which play critical roles in shaping immunity in the pathogenesis of leprosy are also involved in mediating divergence in CMI between the two polar forms of leprosy.
Despite the availability of sufficient research findings to indirectly implicate leptin’s role in modulating the divergence in CMI response between LL and TT patients, studies confirming the presence of leptin or leptin receptor deficiency or leptin resistance in LL patients are very limited. Additionally, the mechanism of activation of mTORC1 by the leptin-mTOR axis in Treg cells is not well understood and therefore needs further investigation.
Abbreviations
BB, mid-borderline; BL, borderline lepromatous; BT, borderline tuberculoid; CMI, Cell-mediated immunity; FoxP3, Fox box P3; GLUT1, glucose transporter 1; γδ T cells, gamma delta T cells; HIF1α, hypoxia-inducible factor 1 alpha; IL, interleukin; LL, lepromatous leprosy; MB, multibacillary; mTOR, mechanistic target of rapamycin; mTORC1/2, mechanistic target of rapamycin complex 1/2; ObR, leptin receptor; PB, paucibacillary; PIK3, phophosphonositide-3-kinase; RAPTOR, regulatory associated protein of mTOR of complex 1; RICTOR, RAPTOR-independent companion of mTOR of complex 2; TCR, T cell receptor; TGFβ, transforming growth factor beta; Th1/2, T helper ½; Tregs, CD4+CD25+FoxP3+ T regulatory cells; TT, tuberculoid leprosy.
Disclosure
The authors have declared that no competing interests exist.
References
1. Eichelmann K, González SG, Salas-Alanis JC, Ocampo-Candiani J. Leprosy. An update: definition, pathogenesis, classification, diagnosis, and treatment. Actas Dermo-Sifiliográficas. 2013;104(7):554–563. doi:10.1016/j.ad.2012.03.003
2. Macedo CS, Lara FA, Pinheiro RO, et al. New insights into the pathogenesis of leprosy: contribution of subversion of host cell metabolism to bacterial persistence, disease progression, and transmission. F1000Research. 2020;9:70. doi:10.12688/f1000research.21383.1
3. Pinheiro RO, de Souza Salles J, Sarno EN, Sampaio EP. Mycobacterium leprae–host-cell interactions and genetic determinants in leprosy: an overview. Future Microbiol. 2011;6(2):217–230. doi:10.2217/fmb.10.173
4. Ridley DS, Jopling WH. Classification of leprosy according to immunity: a five- group system. Int J Lepr. 1966;34:255–273.
5. World Health Organization. Chemotherapy of Leprosy for Control Programmes. Technical Report Series 675. Geneva: World Health Organization; 1983.
6. Van Voorhis WC, Kaplan G, Sarno EN, et al. The cutaneous infiltrates of leprosy: cellular characteristics and the predominant T-cell phenotypes. N Engl J Med. 1982;307:1593–1597. doi:10.1056/NEJM198212233072601
7. Fonseca AB, Simon MD, Cazzaniga RA, et al. The influence of innate and adaptative immune responses on the differential clinical outcomes of leprosy. Infect Dis Poverty. 2017;6(1):1–8. doi:10.1186/s40249-016-0229-3
8. Modlin RL, Hofman FM, Taylor CR, Rea TH. T lymphocyte subsets in the skin lesions of patients with leprosy. J Am Acad Dermatol. 1983;8:182–189. doi:10.1016/S0190-9622(83)70021-6
9. Sridevi K, Khanna N, Chattree V, Pal PC, Haq W, Rao DN. Reversal of T cell anergy in leprosy patients: in vitro presentation with Mycobacterium leprae antigens using murabutide and Trat peptide in liposomal delivery. Int Immunopharmacol. 2003;3(12):1589–1600. doi:10.1016/S1567-5769(03)00181-4
10. Nath I. Immunopathogenesis of leprosy: a model for T cell anergy. EMJ Dermatol. 2016;4:95–101. doi:10.33590/emjdermatol/10312914
11. Maurya R, Bhattacharya P, Dey R, Nakhasi HL. Leptin functions in infectious diseases. Front Immunol. 2018;2018:2741.
12. Conde J, Scotece M, Gómez R, Gómez-Reino JJ, Lago F, Gualillo O. At the crossroad between immunity and metabolism: focus on leptin. Expert Rev Clin Immunol. 2010;6(5):801–808. doi:10.1586/eci.10.48
13. Mackey-Lawrence NM, Petri WA. Leptin and mucosal immunity. Mucosal Immunol. 2012;5(5):472–479. doi:10.1038/mi.2012.40
14. Iikuni N, Kwan Lam QL, Lu L, Matarese G, Cava AL. Leptin and inflammation. Curr Immunol Rev. 2008;4(2):70–79. doi:10.2174/157339508784325046
15. Matarese G. Leptin and the immune system: how nutritional status influences the immune response. Eur Cytokine Netw. 2000;11(1):7–14.
16. Kerr-Pontes LR, Barreto ML, Evangelista CM, Rodrigues LC, Heukelbach J, Feldmeier H. Socioeconomic, environmental, and behavioural risk factors for leprosy in North-east Brazil: results of a case–control study. Int J Epidemiol. 2006;35(4):994–1000. doi:10.1093/ije/dyl072
17. Feenstra SG, Nahar Q, Pahan D, Oskam L, Richardus JH. Recent food shortage is associated with leprosy disease in Bangladesh: a case-control study. PLoS Negl Trop Dis. 2011;5(5):e1029. doi:10.1371/journal.pntd.0001029
18. Anantharam P, Emerson LE, Bilcha KD, Fairley JK, Tesfaye AB. Undernutrition, food insecurity, and leprosy in North Gondar Zone, Ethiopia: a case-control study to identify infection risk factors associated with poverty. PLoS Negl Trop Dis. 2021;15(6):e0009456. doi:10.1371/journal.pntd.0009456
19. Dwivedi VP, Banerjee A, Das I, et al. Diet and nutrition: an important risk factor in leprosy. Microb Pathog. 2019;137:103714. doi:10.1016/j.micpath.2019.103714
20. De Rosa V, Procaccini C, Calì G, et al. A key role of leptin in the control of regulatory T cell proliferation. Immunity. 2007;26(2):241–255. doi:10.1016/j.immuni.2007.01.011
21. Palermo ML, Pagliari C, Trindade MA, et al. Increased expression of regulatory T cells and down-regulatory molecules in lepromatous leprosy. Am J Trop Med Hyg. 2012;86(5):878. doi:10.4269/ajtmh.2012.12-0088
22. Mattioli B, Straface E, Quaranta MG, Giordani L, Viora M. Leptin promotes differentiation and survival of human dendritic cells and licenses them for Th1 priming. J Immunol. 2005;174(11):6820–6828. doi:10.4049/jimmunol.174.11.6820
23. Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol. 2012;12(5):325–338. doi:10.1038/nri3198
24. Dabi YT, Degechisa ST, Bobosha K, Wassie L. Changes in plasma level of endocrine hormones in lepromatous leprosy patients. IJID Regions. 2022.
25. Ye M, Bian LF. Association of serum leptin levels and pulmonary tuberculosis: a meta-analysis. J Thorac Dis. 2018;10(2):1027. doi:10.21037/jtd.2018.01.70
26. Tsegaye Y, Admassu W, Edao A, et al. Alteration of endocrine hormones and antibody responses in different spectrum of tuberculosis disease. Front Immunol. 2022;25:731.
27. Skinsnes LK, Higa LH. The role of protein malnutrition in the pathogenesis of ulcerative “Lazarine” leprosy. Int J Lepr Other Mycobact Dis. 1976;44:346–358.
28. Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature. 1998;394(6696):897–901. doi:10.1038/29795
29. Matarese G, Di Giacomo A, Sanna V, et al. Requirement for leptin in the induction and progression of autoimmune encephalomyelitis. J Immunol. 2001;166(10):5909–5916. doi:10.4049/jimmunol.166.10.5909
30. Chandra RK. Cell-mediated immunity in genetically obese (C57BL/6J ob/ob) mice. Am J Clin Nutr. 1980;33(1):13–16. doi:10.1093/ajcn/33.1.13
31. Mandel MA, Mahmoud AA. Impairment of cell-mediated immunity in mutation diabetic mice (db/db). J Immunol. 1978;120(4):1375–1377.
32. Wieland CW, Florquin S, Chan ED, et al. Pulmonary Mycobacterium tuberculosis infection in leptin-deficient ob/ob mice. Int Immunol. 2005;17(11):1399–1408. doi:10.1093/intimm/dxh317
33. Hasenkrug KJ. The leptin connection: regulatory T cells and autoimmunity. Immunity. 2007;26(2):143–145. doi:10.1016/j.immuni.2007.02.002
34. Matarese G, La Cava A, Sanna V, et al. Balancing susceptibility to infection and autoimmunity: a role for leptin? Trends Immunol. 2002;23(4):182–187. doi:10.1016/S1471-4906(02)02188-9
35. Procaccini C, Jirillo E, Matarese G. Leptin as an immunomodulator. Mol Aspects Med. 2012;33(1):35–45. doi:10.1016/j.mam.2011.10.012
36. Cava AL, Matarese G. The weight of leptin in immunity. Nat Rev Immunol. 2004;4(5):371–379. doi:10.1038/nri1350
37. Alti D, Sambamurthy C, Kalangi SK. Emergence of leptin in infection and immunity: scope and challenges in vaccines formulation. Front Cell Infect Microbiol. 2018;8:147. doi:10.3389/fcimb.2018.00147
38. Bobosha K, Wilson L, van Meijgaarden KE, et al. T-cell regulation in lepromatous leprosy. PLoS Negl Trop Dis. 2014;8(4):e2773. doi:10.1371/journal.pntd.0002773
39. Cao Q, Wang L, Du F, et al. Down regulation of CD4+CD25+ regulatory T cells may underlie enhanced Th1 immunity caused by immunization with activated autologous T cells. Cell Res. 2007;17(7):627–637. doi:10.1038/cr.2007.46
40. Takahashi T, Kuniyasu Y, Toda M, et al. Immunologic self-tolerance maintained by CD25+ CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int Immunol. 1998;10(12):1969–1980. doi:10.1093/intimm/10.12.1969
41. Zhou X, Bailey-Bucktrout SL, Jeker LT, et al. Instability of the transcription factor FOXP3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol. 2009;10(9):1000–1007. doi:10.1038/ni.1774
42. Saini C, Ramesh V, Nath I. Increase in TGF-β secreting CD4+ CD25+ FOXP3+ T regulatory cells in anergic lepromatous leprosy patients. PLoS Negl Trop Dis. 2014;8(1):e2639. doi:10.1371/journal.pntd.0002639
43. Sadhu S, Khaitan BK, Joshi B, Sengupta U, Nautiyal AK, Mitra DK. Reciprocity between regulatory T cells and Th17 cells: relevance to polarized immunity in leprosy. PLoS Negl Trop Dis. 2016;10(1):e0004338. doi:10.1371/journal.pntd.0004338
44. Procaccini C, De Rosa V, Galgani M, et al. An oscillatory switch in mTOR kinase activity sets regulatory T cell responsiveness. Immunity. 2010;33(6):929–941. doi:10.1016/j.immuni.2010.11.024
45. Parente JN, Talhari C, Schettini AP, Massone C. T regulatory cells (CD4+ CD25+ FOXP3+) distribution in the different clinical forms of leprosy and reactional states. An Bras Dermatol. 2015;90:41–47. doi:10.1590/abd1806-4841.20153311
46. Zeng H, Chi H. The interplay between regulatory T cells and metabolism in immune regulation. Oncoimmunology. 2013;2(11):e26586. doi:10.4161/onci.26586
47. Wei R, Hu Y, Dong F, Xu X, Hu A, Gao G. Hepatoma cell‐derived leptin downregulates the immunosuppressive function of regulatory T‐cells to enhance the anti‐tumor activity of CD8+ T‐cells. Immunol Cell Biol. 2016;94(4):388–399. doi:10.1038/icb.2015.110
48. Maya-Monteiro C, Bozza P. Leptin and mTOR: partners in metabolism and inflammation. Cell Cycle. 2008;7(12):1713–1717. doi:10.4161/cc.7.12.6157
49. Chen Y, Colello J, Jarjour W, Zheng SG. Cellular metabolic regulation in the differentiation and function of regulatory T cells. Cells. 2019;8(2):188. doi:10.3390/cells8020188
50. Coe DJ, Kishore M, Marelli-Berg F. Metabolic regulation of regulatory T cell development and function. Front Immunol. 2014;5:590. doi:10.3389/fimmu.2014.00590
51. Battaglia M, Stabilini A, Migliavacca B, Horejs-Hoeck J, Kaupper T, Roncarolo MG. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J Immunol. 2006;177:8338–8347. doi:10.4049/jimmunol.177.12.8338
52. Valmori D, Tosello V, Souleimanian NE, et al. Rapamycin-mediated enrichment of T cells with regulatory activity in stimulated CD4+ T cell cultures is not due to the selective expansion of naturally occurring regulatory T cells but to the induction of regulatory functions in conventional CD4+ T cells. J Immunol. 2006;177:944–949. doi:10.4049/jimmunol.177.2.944
53. Turnquist HR, Raimondi G, Zahorchak AF, Fischer RT, Wang Z, Thomson AW. Rapamycin-conditioned dendritic cells are poor stimulators of allogeneic CD4+ T cells, but enrich for antigen-specific Foxp3+ T regulatory cells and promote organ transplant tolerance. J Immunol. 2007;178:7018–7031. doi:10.4049/jimmunol.178.11.7018
54. Strauss L, Czystowska M, Szajnik M, Mandapathil M, Whiteside TL. Differential responses of human regulatory T cells and effector T cells to rapamycin. PLoS One. 2009;4:e5994. doi:10.1371/journal.pone.0005994
55. Zeiser R, Leveson-Gower DB, Zambricki EA, et al. Differential impact of mammalian target of rapamycin inhibition on CD4+CD25+Foxp3+ regulatory T cells compared with conventional CD4+ T cells. Blood. 2008;111:453–462. doi:10.1182/blood-2007-06-094482
56. Chapman NM, Chi H. mTOR signaling, Tregs and immune modulation. Immunotherapy. 2014;6(12):1295–1311. doi:10.2217/imt.14.84
57. Delgoffe GM, Kole TP, Zheng Y, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832–844. doi:10.1016/j.immuni.2009.04.014
58. Haxhinasto S, Mathis D, Benoist C. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J Exp Med. 2008;205:565–574. doi:10.1084/jem.20071477
59. Galgani M, De Rosa V, La Cava A, Matarese G. Role of metabolism in the immunobiology of regulatory T cells. J Immunol. 2022;2022:2569–2575.
60. Howie D, Waldmann H, Cobbold S. Nutrient sensing via mTOR in T cells maintains a tolerogenic microenvironment. Front Immunol. 2014;5:409. doi:10.3389/fimmu.2014.00409
61. Matarese G, Procaccini C, De Rosa V, Horvath TL, La Cava A. Regulatory T cells in obesity: the leptin connection. Trends Mol Med. 2010;16(6):247–256. doi:10.1016/j.molmed.2010.04.002
62. Kempkes RW, Joosten I, Koenen HJ, He X. Metabolic pathways involved in regulatory T cell functionality. Front Immunol. 2019;10:2839. doi:10.3389/fimmu.2019.02839
63. Sun IH, Oh MH, Zhao L, et al. mTOR complex 1 signaling regulates the generation and function of central and effector Foxp3+ regulatory T cells. J Immunol. 2018;201(2):481–492. doi:10.4049/jimmunol.1701477
64. Saini C, Siddiqui A, Ramesh V, Nath I. Leprosy reactions show increased Th17 cell activity and reduced FOXP3+ tregs with concomitant decrease in TGF-beta and increase in IL-6. PLoS Negl Trop Dis. 2016;10:e0004592. doi:10.1371/journal.pntd.0004592
65. Salgame P, Yamamura M, Bloom BR, Modlin RL. Evidence for functional subsets of CD4+ and CD8+ T cells in human disease: lymphokine patterns in leprosy. Chem Immunol. 1992;54:44–59.
66. Saini C, Ramesh V, Nath I. CD4+ Th17 cells discriminate clinical types and constitute a third subset of non Th1, Non Th2 T cells in human leprosy. PLoS Negl Trop Dis. 2013;7(7):e2338. doi:10.1371/journal.pntd.0002338
67. Pérez-Pérez A, Sánchez-Jiménez F, Vilariño-García T, Sánchez-Margalet V. Role of leptin in inflammation and vice versa. Int J Mol Sci. 2020;21(16):5887. doi:10.3390/ijms21165887
68. Reis BS, Lee K, Fanok MH, et al. Leptin receptor signaling in T cells is required for Th17 differentiation. J Immunol. 2015;194:5253–5260. doi:10.4049/jimmunol.1402996
69. Yu Y, Liu Y, Shi F-D, Zou H, Matarese G, La Cava A. Cutting edge: leptin-induced RORγt expression in CD4+ T cells promotes Th17 responses in systemic lupus erythematosus. J Immunol. 2013;190:3054–3058. doi:10.4049/jimmunol.1203275
70. Saucillo DC, Gerriets VA, Sheng J, Rathmell JC, MacIver NJ. Leptin metabolically licenses T cells for activation to link nutrition and immunity. J Immunol. 2014;192(1):136–144. doi:10.4049/jimmunol.1301158
71. Gerriets VA, Danzaki K, Kishton RJ, et al. Leptin directly promotes T‐cell glycolytic metabolism to drive effector T‐cell differentiation in a mouse model of autoimmunity. Eur J Immunol. 2016;46(8):1970–1983. doi:10.1002/eji.201545861
72. Saini C, Tarique M, Rai R, Siddiqui A, Khanna N, Sharma A. T helper cells in leprosy: an update. Immunol Lett. 2017;184:61–66. doi:10.1016/j.imlet.2017.02.013
73. Tarique M, Naz H, Kurra SV, et al. Interleukin-10 producing regulatory B cells transformed CD4+ CD25− into Tregs and enhanced regulatory T cells function in human leprosy. Front Immunol. 2018;9:1636. doi:10.3389/fimmu.2018.01636
74. Mi Z, Liu H, Zhang F. Advances in the immunology and genetics of leprosy. Front Immunol. 2020;11:567. doi:10.3389/fimmu.2020.00567
75. Shen P, Roch T, Lampropoulou V, et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature. 2014;507(7492):366–370. doi:10.1038/nature12979
76. Saini C, Tarique M, Ramesh V, Khanna N, Sharma A. γδ T cells are associated with inflammation and immunopathogenesis of leprosy reactions. Immunol Lett. 2018;200:55–65. doi:10.1016/j.imlet.2018.07.005
77. Liu Y, Shi C, Ma S, et al. The protective role of tissue-resident IL-17A-producing gamma delta T cells in Mycobacterium leprae infection. Front Immunol. 2022;2022:6386.
78. Sridevi K, Neena K, Chitralekha KT, Arif AK, Tomar D, Rao DN. Expression of costimulatory molecules. (CD80, CD86, CD28, CD152), accessory molecules. (TCR alphabeta, TCR gammadelta) and T cell lineage molecules. (CD4+, CD8+) in PBMC of leprosy patients using Mycobacterium leprae antigen. (MLCWA) with murabutide and T cell peptide of Trat protein. Int Immunopharmacol. 2004;4:1–14. doi:10.1016/j.intimp.2003.09.001
79. Tarique M, Naqvi RA, Ali R, Khanna N, Rao DN. CD4 (+) TCRgammadelta(+) FoxP3(+) cells: an unidentified population of immunosuppressive cells towards disease progression leprosy patients. ExpDermatol. 2017;26:946–948.
80. Ochoa MT, Teles R, Haas BE, et al. A role for interleukin-5 in promoting increased immunoglobulin M at the site of disease in leprosy. Immunology. 2010;131:405–414. doi:10.1111/j.1365-2567.2010.03314.x
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.