")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Lentivirus-mediated knockdown of NLK inhibits small-cell lung cancer growth and metastasis
Authors Lv MT, Li YM, Tian X, Dai SD, Sun J, Jin GJ, Jiang SY
Received 27 April 2015
Accepted for publication 8 March 2016
Published 15 November 2016 Volume 2016:10 Pages 3737—3746
DOI https://doi.org/10.2147/DDDT.S87435
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Mutian Lv,1 Yaming Li,1 Xin Tian,2 Shundong Dai,3,4 Jing Sun,5 Guojiang Jin,6 Shenyi Jiang7
1Department of Nuclear Medicine, 2Molecular Oncology Laboratory of Cancer Research Institute, The First Affiliated Hospital of China Medical University, 3Department of Pathology, The First Affiliated Hospital, College of Basic Medical Sciences of China Medical University, 4Department of Pathology, Institute of Pathology and Pathophysiology, 5Department of Immunology and Biotherapy, Liaoning Cancer Hospital and Institute, 6Department of Laboratory Medicine, 7Department of Rheumatology, The First Affiliated Hospital of China Medical University, Shenyang, People’s Republic of China
Abstract: Nemo-like kinase (NLK), an evolutionarily conserved serine/threonine kinase, has been recognized as a critical regulator of various cancers. In this study, we investigated the role of NLK in human small-cell lung cancer (SCLC), which is the most aggressive form of lung cancer. NLK expression was evaluated by quantitative real-time polymerase chain reaction in 20 paired fresh SCLC tissue samples and found to be noticeably elevated in tumor tissues. Lentivirus-mediated RNAi efficiently suppressed NLK expression in NCI-H446 cells, resulting in a significant reduction in cell viability and proliferation in vitro. Moreover, knockdown of NLK led to cell cycle arrest at the S-phase via suppression of Cyclin A, CDK2, and CDC25A, which could contribute to cell growth inhibition. Furthermore, knockdown of NLK decreased the migration of NCI-H446 cells and downregulated matrix metalloproteinase 9. Treatment with NLK short hairpin RNA significantly reduced SCLC tumor growth in vivo. In conclusion, this study suggests that NLK plays an important role in the growth and metastasis of SCLC and may serve as a potential therapeutic target for the treatment of SCLC.
Keywords: NLK, SCLC, RNAi, proliferation, migration
Introduction
Lung cancer is the leading cause of cancer-related deaths among men in both developed and developing countries and has surpassed breast cancer as the leading cause of cancer-related deaths among women in many developed countries.1 Lung cancer can be classified into small-cell lung cancer (SCLC) and non-small-cell lung cancer (NSCLC) based on the histopathological features of the disease. SCLC accounts for ~15% of all new cases of lung cancer, with >150,000 cases every year worldwide.2 Clinically, SCLC is distinguished from NSCLC by aggressive growth and widespread metastasis.3 Most patients with SCLC already show clinically detectable metastases at diagnosis and have extremely poor prognosis even when treated with combined modalities.4 In fact, the 5-year survival rate for SCLC patients is rather low (3%–8%) compared to that for all forms of lung cancer (<15%).5 Chemotherapy is the primary treatment for SCLC; however, the drugs that are currently used for SCLC are less effective than those used for NSCLC.6 Therefore, gaining a deeper understanding of the molecular pathogenesis of human SCLC is critical for identifying novel potential targets for therapy.
Nemo-like kinase (NLK), an evolutionarily conserved serine/threonine protein kinase, plays an important role in diverse signaling pathways by phosphorylating a variety of transcription factors.7 Alteration of NLK has been shown to be associated with increased risk of cancer. NLK is known to be a tumor suppressor in various types of cancer as a negative regulator of the canonical Wnt/β-catenin signaling pathway, which phosphorylates T-cell factor and lymphoid enhancer factor, promoting the dissociation of the β-catenin/T-cell factor complex.8 It has been shown that NLK expression was suppressed in human ovarian cancer, and low NLK expression could be related to poor outcome.9 NLK also blocks the development of NSCLC by negatively modulating the Wnt signaling pathway. Knockdown of NLK resulted in a significant promotion of proliferation of A549 cells via enhancing the transcriptional activity of β-catenin/T-cell factor.10
Intriguingly, noncanonical Wnt signals inhibit canonical Wnt signaling using the MAP3K7-NLK signaling cascade.11 NLK has also been reported to be positively associated with the progression of multiple human cancers, including hepatocellular carcinoma (HCC), gallbladder cancer (GBC),12 nasopharyngeal carcinoma,13 and oral adenosquamous carcinoma.14 Overexpression of NLK is closely related to the progression of GBC, and NLK could be used as a potential prognostic marker for GBC patients.12 Moreover, NLK positivity was found to be an unfavorable prognostic indicator of overall survival in nasopharyngeal carcinoma.13 Furthermore, knockdown of NLK made laryngeal cancer cells more sensitive to Taxol treatment.15 A previous study showed that NLK overexpression inhibited growth and survival of DLD-1 colon cancer cells.16 Recently, an immunohistochemical analysis of 406 cases of primary colorectal cancer tissue and paired noncancerous tissue samples indicated that NLK expression was significantly higher in colorectal cancer tissues as well as associated with tumor invasion and metastasis.17 This discrepancy can be possibly explained by the fact that NLK can exert differing effects depending on the cellular context. It is therefore important to study the effects of altered expression of NLK in the different contexts to fully understand its biological role. To date, the functional role of NLK in human SCLC has not yet been elucidated.
In the current study, we detected NLK expression in 20 SCLC cases and found that its expression was elevated significantly in human SCLC. Targeted disruption of NLK was achieved by the lentivirus-mediated short hairpin (sh) RNA method. The effects of NLK on cell growth, cell cycle regulation, and cell migration were evaluated, and expressions of downstream molecules were observed.
Materials and methods
Cell culture
Human SCLC NCI-H446 and human embryonic kidney 293T cell lines were purchased from the Cell Bank of Chinese Academy of Science (Shanghai, People’s Republic of China). NCI-H446 cells were maintained in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% fetal bovine serum, 100 IU/mL of penicillin, and 100 μg/mL of streptomycin (Thermo Fisher Scientific, Waltham, MA, USA). 293T cells were maintained in Dulbecco’s Modified Eagle’s Medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum. These cell lines were incubated at 37°C in a humidified atmosphere containing 5% CO2.
Quantitative real-time polymerase chain reaction
Total RNA from fresh SCLC tissues and NCI-H446 cells was isolated using TRIzol® reagent (Thermo Fisher Scientific) and synthesized into cDNA by Moloney murine leukemia virus (M-MLV) reverse transcriptase (Promega Corporation, Fitchburg, WI, USA). Real-time polymerase chain reaction (RT-PCR) was conducted using an SYBR Premix Ex Taq according to the manufacturer’s protocol (TaKaRa, Dalian, People’s Republic of China). The quantitative RT-PCR (qRT-PCR) procedure was as follows: denaturation at 95°C for 1 minute, 40 cycles of denaturation at 95°C for 5 seconds, and extension at 60°C for 20 seconds. The β-actin gene was amplified as an internal control. Relative quantitation of gene expression was calculated using the 2−ΔΔCT method. The following primers were used: NLK, 5′-ATCATCAGCACTCGCATCATC-3′ (forward) and 5′-GACCAGACAACACCAAAGGC-3′ (reverse); β-actin, 5′-GTGGACATCCGCAAAGAC-3′ (forward) and 5′-AAAGGGTGTAACGCAACTA-3′ (reverse).
Construction of NLK siRNA-containing lentivirus and transduction into SCLC cells
The NLK shRNA and control shRNA lentiviruses were obtained from GeneChem (Shanghai, People’s Republic of China). The siRNA target sequences were: 5′-GAATATCCGCTAAGGATGC-3′ and 5′-CAGATCCAAGAGATGGAAA-3′. The stem-loop-stem oligonucleotide (shRNA) corresponding to each siRNA was inserted into the pGCSIL-green fluorescence protein (GFP) vector. Recombinant lentiviruses were produced by cotransfecting 293T cells with the lentivirus expression vector and packaging plasmids pHelper 1.0 and pHelper 2.0 (GeneChem) using Lipofectamine 2000 (Thermo Fisher Scientific). The supernatant was collected at 48 hours after transfection, and lentiviral particles were purified by ultracentrifugation (4,000× g) at 4°C for 10 minutes.
For cell infection, NCI-H446 cells were seeded in six-well plates at a density of 3×104 cells/well and transduced with the constructed lentivirus-containing NLK shRNA (Lv-shNLK) or non-silencing shRNA (Lv-shCon) at a multiplicity of infection of 30. The lentiviral vectors expressed GFP, which allowed for measurement of infection efficiency in transduced cells.
Western blot analysis
After 5 days of lentivirus infection, NCI-H446 cells were lysed and the total protein content was determined by measuring its absorbance. Aliquots of 30 μg of total protein from each sample were subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis followed by transfer to polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA, USA). Nonspecific binding was blocked by incubation in Tris-Buffered Saline Tween-20 (TBST) (25 mM Tris, pH 7.4, 150 mM NaCl, and 0.1% Tween 20) containing 5% skim milk at room temperature for 2 hours. Then, the membranes were probed with primary antibodies overnight at 4°C, followed by incubation with horseradish-peroxidase-conjugated secondary antibodies at room temperature for 2 hours. The signals were visualized using the ECL Plus Western Blotting Detection System (GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA) and exposure to ECL Hyperfilm (GE Healthcare Bio-Sciences Corp.).
Rabbit anti-NLK (ab116715) antibody was obtained from Abcam (Cambridge, UK); rabbit anti-cyclin-dependent kinase 2 (CDK2; #2546), rabbit anti-matrix metalloproteinase 9 (MMP-9; #2270), and rabbit anti-β-actin (#4967) antibodies were from Cell Signaling Technology (Danvers, MA, USA); mouse anti-Cyclin A (#sc-271682), mouse anti-CDC25A (#sc-56263), horseradish-peroxidase-conjugated goat anti-rabbit (#sc-2054), and goat anti-mouse (#sc-2005) secondary antibodies were from Santa Cruz Biotechnology Inc. (Dallas, TX, USA).
In vitro cell viability assay
After lentivirus infection, NCI-H446 cells were seeded at a density of 3×103 cells/well in 96-well microplates and incubated at 37°C for 1–5 days. At each termination of culture, cell viability analysis was performed by a Cell Counting Kit-8 (CCK-8) assay according to the manufacturer’s instructions (Dojindo Laboratories, Kumamoto, Japan). After an incubation of 2 hours at 37°C, the absorbance was measured at 450 nm using a microplate reader iMark (Bio-Rad Laboratories Inc., Hercules, CA, USA).
Colony formation assay
After lentivirus infection, NCI-H446 cells (1×103 cells/well) were reseeded in six-well plates and incubated for 9 days in order to form normal colonies. The medium was replaced every 3 days. Then, the cells were washed with PBS and fixed with 4% paraformaldehyde for 30 minutes. The fixed cells were stained with freshly prepared crystal violet (Beyotime) for 20 minutes. Cell colonies were captured and counted under a microscope.
Cell cycle analysis
Cell cycle distribution was analyzed by flow cytometry using propidium iodide (PI) staining. After 4 days of lentivirus infection, NCI-H446 cells were reseeded in 6 cm dishes at a density of 1.5×105 cells/dish. After culturing overnight at 37°C, cells were harvested, fixed in 70% ethanol, and stored overnight at 4°C. The cells were then treated with NaCl/PI staining solution (50 μg/mL PI and 100 μg/mL RNase A). Following incubation for 1 hour in the dark at room temperature, cells were analyzed by flow cytometry (FACSCalibur; Becton Dickinson, San Jose, CA, USA). The fractions of the cells in G0/G1-, S-, and G2/M-phases were analyzed with dedicated software (Becton Dickinson).
Transwell assay
Cell migration was examined using the Transwell chambers (8.0 μm pores, 24 wells; Corning Incorporated, Corning, NY, USA). After 4 days of lentivirus infection, NCI-H446 cells were reseeded in upper chambers at a concentration of 3×104 cells/well in 200 μL serum-free medium. The upper chambers were filled with RPMI 1640 containing 10% fetal bovine serum. After incubation for 24 hours at 37°C, the nonmigrated cells on the upper surface of the filter were gently removed using cotton swabs and the migrated cells on the lower surface were fixed with 4% paraformaldehyde, and stained with crystal violet. Migrated cells were captured and counted (five random fields per well) under a microscope.
Mouse xenograft tumor
All animal experiments were conducted in accordance with the humane treatment of animals under an animal protocol approved by the Institutional Animal Care and Use Committee of the First Affiliated Hospital of China Medical University and adhered to the animal care guidelines of the Ethics Committee of China Medical University. A total of 12 six-week-old athymic (nu/nu) mice were obtained from the Shanghai Laboratory Animal Center, Chinese Academy of Sciences (Shanghai, People’s Republic of China). Mice were kept under specific pathogen-free conditions in a temperature-controlled room (22°C±2°C) and fed with laboratory chow and water ad libitum.
NCI-H446 cells infected with Lv-shCon or Lv-shNLK were collected and suspended in serum-free medium. Then, 12 nude mice were randomly assigned to two groups: Lv-shCon- and Lv-shNLK-treated groups. Each mouse was injected subcutaneously with NCI-H446 cells (3×106) post infection into the flank region of nude mice. The diameter of the tumor in the greatest axis (a) and shortest axis (b) was measured with a Vernier caliper every 3–4 days using the following formula:
|
|
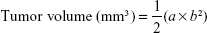
Tumor growth was followed for 3 weeks from the first injection. The mice were then sacrificed by cervical dislocation, and the tumors were excised to measure their weights.
In vivo metastasis assay
A total of 5×105 cells infected with Lv-shCon or Lv-shNLK were intravenously injected through the tail vein of 6-week-old nude mice (ten mice per group). After 8 weeks, the mice were euthanized and the number of surface metastases per lung was determined under a dissecting microscope. The lungs were excised and embedded in paraffin, whereby hematoxylin and eosin staining was performed to confirm the presence of tumors.
Statistical analysis
All data are shown as the mean ± standard deviation from three independent experiments. Statistical analysis was performed using SPSS 16.0 software package (SPSS Inc., Chicago, IL, USA). Student’s t-test was used to determine the statistical significance of the differences among the experimental groups. A P-value of <0.05 was considered statistically significant.
Results
Expression patterns of NLK in SCLC and adjacent lung tissues
To explore the role of NLK in human SCLC, we first evaluated its expression in 20 paired SCLC and adjacent lung tissue samples by qRT-PCR. As shown in Figure 1, the mRNA levels of NLK in >75% of SCLC tissues were increased by over threefold, as compared to matched normal tissues. Our results showed that NLK expression was significantly elevated in SCLC tissues and might be associated with the development of SCLC.
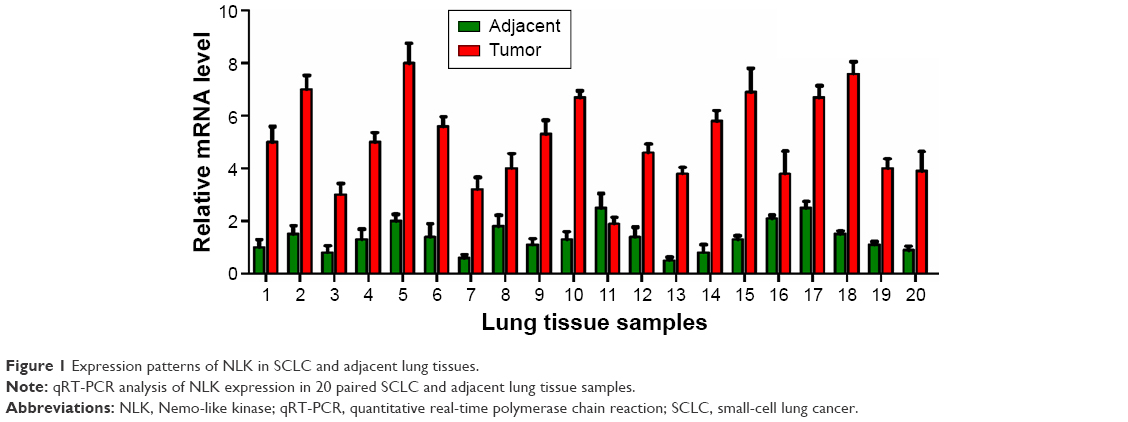 | Figure 1 Expression patterns of NLK in SCLC and adjacent lung tissues. |
Knockdown of NLK in SCLC cells by lentivirus-mediated shRNA
NCI-H446 cells were transduced with shRNA-expressing lentivirus (Lv-shCon or Lv-shNLK). GFP expression was observed by fluorescent microscopy 4 days after lentivirus infection. As shown in Figure 2A, >80% of NCI-H446 cells expressed GFP in the Lv-shCon and Lv-shNLK groups, indicating a successful infection rate. The inhibitory effect of NLK shRNA on its endogenous expression in NCI-H446 cells was examined by qRT-PCR and Western blotting. As shown in Figure 2B, the transcription level of NLK was markedly reduced in the Lv-shNLK group (P<0.01), in contrast to the uninfected (Con) and Lv-shCon groups. However, there was no significant difference between the Lv-shCon and Con groups. Immunoblotting also verified the downregulation of NLK expression in NCI-H446 cells at the translational level (Figure 2C). Therefore, lentivirus-delivered shRNA could specifically downregulate endogenous NLK expression in SCLC cells.
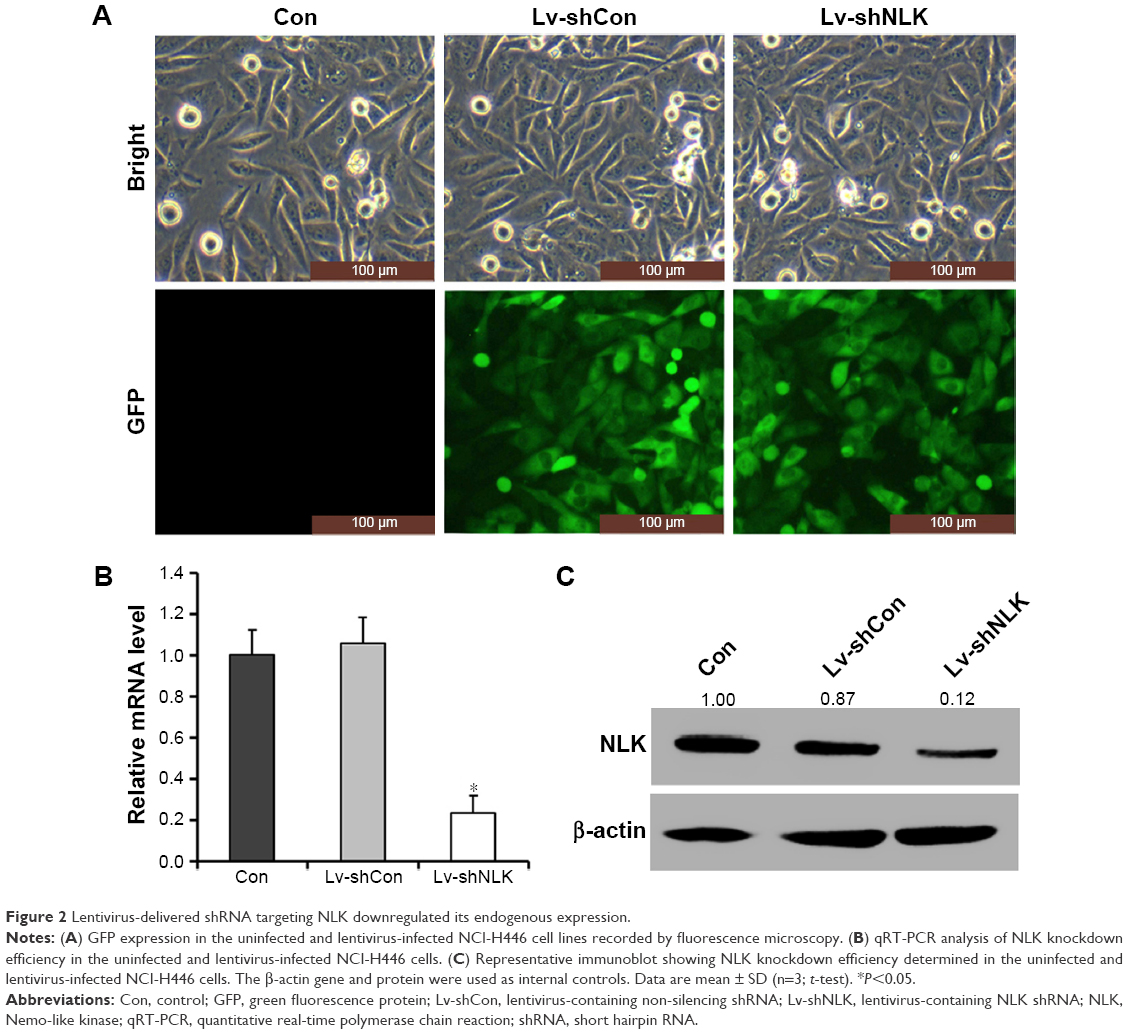 | Figure 2 Lentivirus-delivered shRNA targeting NLK downregulated its endogenous expression. |
Knockdown of NLK suppressed cell viability and proliferation in SCLC cells
To evaluate the effect of NLK knockdown on SCLC cell viability, a CCK-8 assay was performed in NCI-H446 cells post-infection. As shown in Figure 3A, the number of viable NCI-H446 cells was significantly reduced in the Lv-shNLK group on day 5 (P<0.01), in contrast to that in Con and Lv-shCon groups. This result indicated that knockdown of NLK could strongly decrease the viability of SCLC cells.
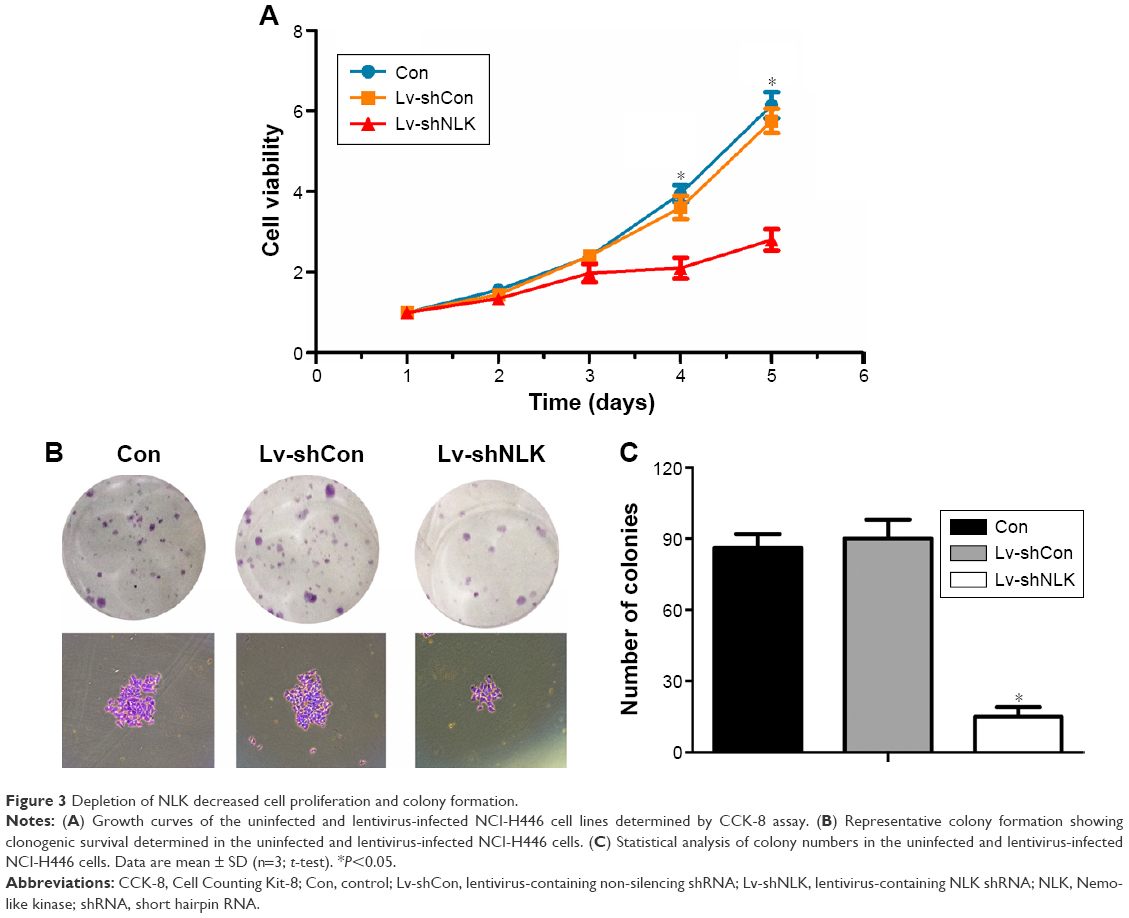 | Figure 3 Depletion of NLK decreased cell proliferation and colony formation. |
Moreover, a colony formation assay was performed in NCI-H446 cells post-infection to evaluate the effect of NLK on long-term cell proliferation. Representative microscope images of the colonies per well and the cells per colony with crystal violet staining are presented in Figure 3B. The average number of total colonies was ~90 in both the Con and Lv-shCon groups, while there was <20 colonies in the Lv-shNLK group (P<0.01; Figure 3C). Our results showed that knockdown of NLK could potently disrupt tumorigenic (colony formation) ability of SCLC cells in vitro.
Knockdown of NLK blocked cell cycle progression in SCLC cells
To explore the mechanism by which NLK modulates cell proliferation, postinfection NCI-H446 cells were applied to flow cytometry analysis with PI staining (Figure 4A). As shown in Figure 4B, the percentage of NCI-H446 cells in the S-phase was increased by ~30% (P<0.01), compared with the Con or Lv-shCon groups. In contrast, the populations of NCI-H446 cells in both G0/G1- and G2/M-phases were decreased in the Lv-shNLK group. Our results suggest that knockdown of NLK could inhibit SCLC cell proliferation by inducing S-phase cell cycle arrest.
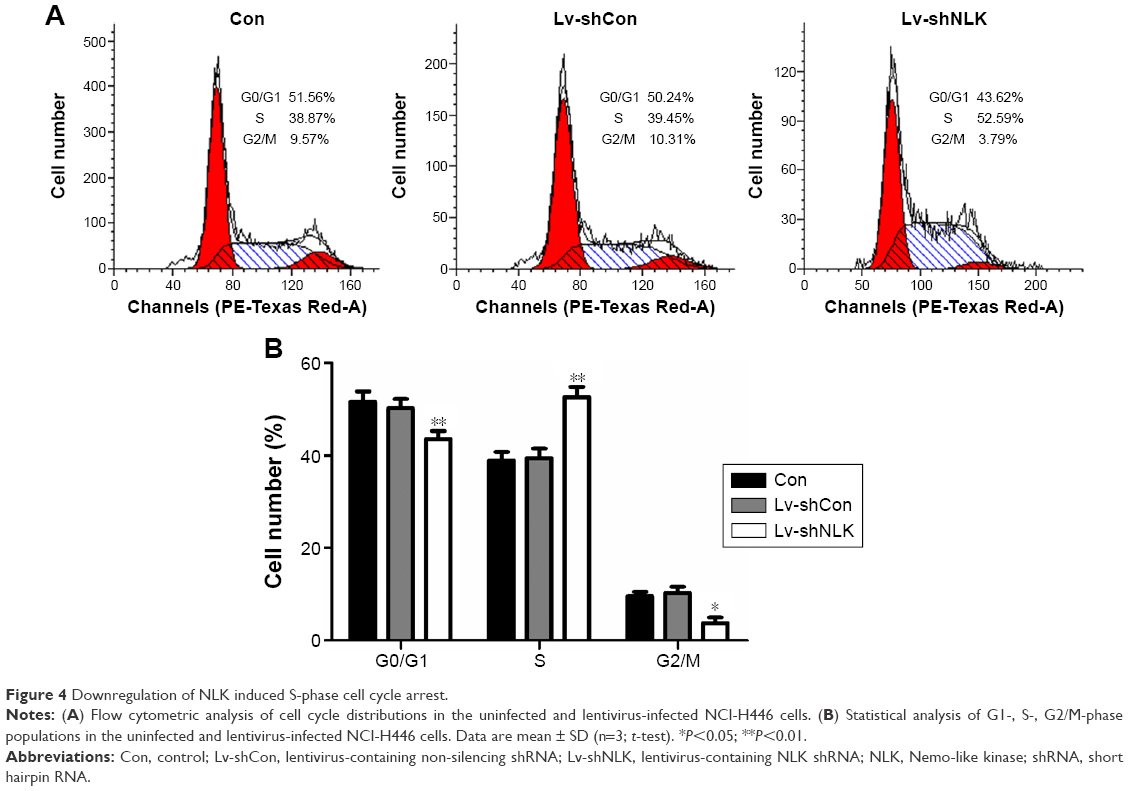 | Figure 4 Downregulation of NLK induced S-phase cell cycle arrest. |
Knockdown of NLK alleviated cell migration in SCLC cells
Furthermore, we determined the effect of NLK on SCLC cell migration using Transwell chambers. Representative microscope images of crystal-violet-stained NCI-H446 cells that migrated to the lower surface are presented in Figure 5A. The number of migrated NCI-H446 cells was significantly reduced in response to NLK knockdown (P<0.01, Figure 5B), indicating that knockdown of NLK could alleviate the migration of SCLC cells.
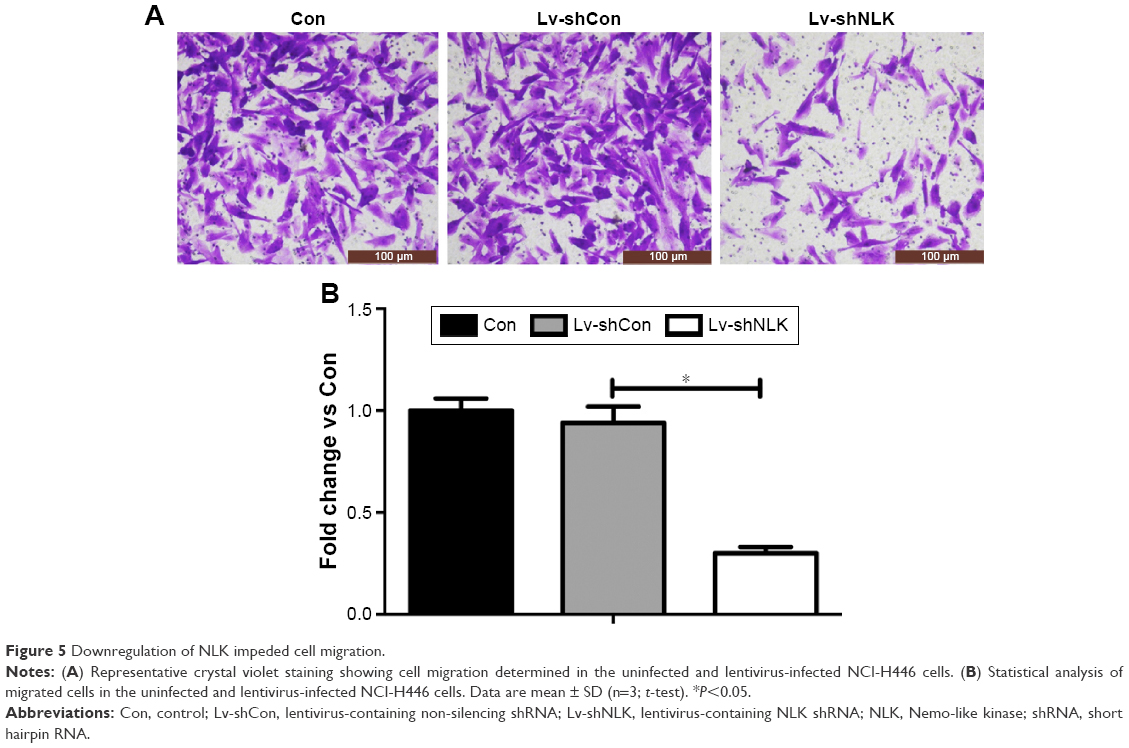 | Figure 5 Downregulation of NLK impeded cell migration. |
Knockdown of NLK decreased cell cycle and migration-related regulators
To further elucidate the molecular mechanism underlying NLK-mediated cell growth and migration, the alteration in the expression of some regulators associated with cell cycle and motility was determined in NCI-H446 cells by Western blotting. It is well known that the Cyclin A/CDK2 complex plays an essential role in DNA synthesis and S-phase progression.18 As shown in Figure 6, compared with the Con and Lv-shCon groups, the expression of Cyclin A and CDK2 as well as CDC25A, a tyrosine phosphatase that contributes to activation of CDK2,19 was noticeably downregulated in the Lv-shNLK group. In addition, NLK silencing suppressed the expression of MMP-9, which has been shown to correlate with tumor invasion.20 Our results indicate that NLK might play an essential role in SCLC growth and metastasis via suppression of the cell cycle and invasion regulatory proteins.
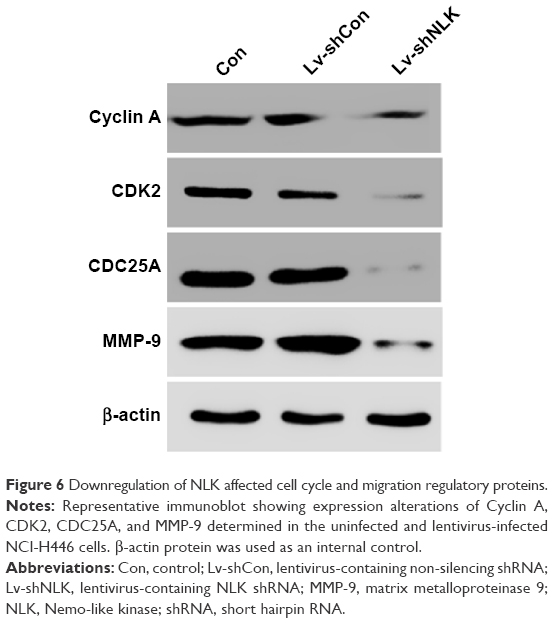 | Figure 6 Downregulation of NLK affected cell cycle and migration regulatory proteins. |
Knockdown of NLK inhibited SCLC tumor growth and lung metastasis in vivo
Since in vitro data showed that downregulation of NLK inhibited the growth of SCLC cells, we investigated the effect of NLK shRNA treatment on tumor growth in vivo. The human xenograft nude mouse models of SCLC were successfully developed using lentivirus-infected NCI-H446 cells. As the time of implantation increased, the tumor volume in each group showed a progressive increase, but the growth rate in the Lv-shNLK group was significantly slower than in the Lv-shCon group (Figure 7A). Compared with the Lv-shNLK group, the tumor weight in the Lv-shNLK group was also reduced in NCI-H446-cells-implanted mice (P<0.01, Figure 7B). Representative photographs of the solid tumors are shown in Figure 7C and D. Furthermore, in comparison with tail vein injection with Lv-shCon cells, injection with Lv-shNLK cells resulted in a lower lung metastasis frequency (10% for Lv-shNLK cells and 60% for Lv-shCon cells; Figure 7E). Representative photographs of the lung metastatic tumor nodes are shown in Figure 7F. Our results verify that knockdown of NLK could potently suppress the growth and metastasis of SCLC in vitro and in vivo.
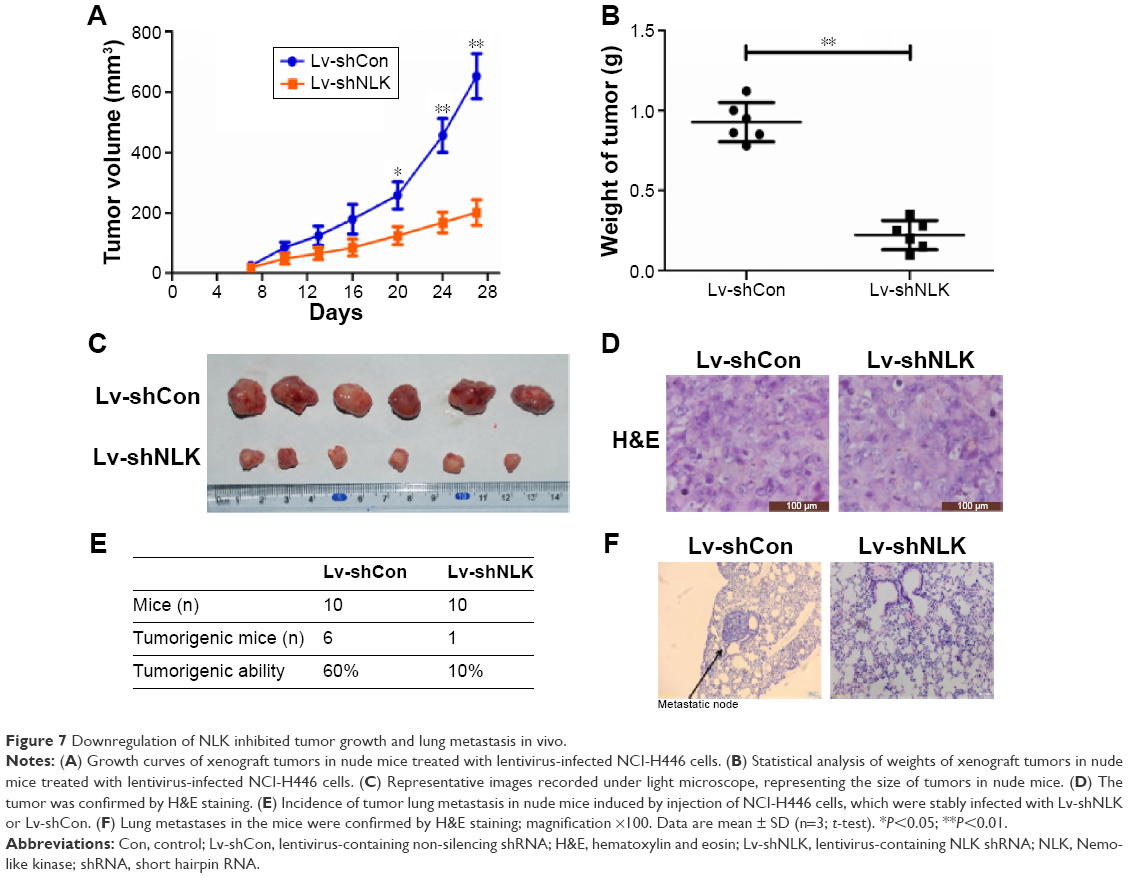 | Figure 7 Downregulation of NLK inhibited tumor growth and lung metastasis in vivo. |
Discussion
SCLC causes significant mortality worldwide, but information on the potential molecular mechanism of SCLC is limited. In the current study, we investigated the functional relationship between SCLC and a tumor-related gene NLK. We first proved that NLK expression was remarkably elevated in human SCLC tissues in contrast to normal tissues, implying the involvement of NLK in SCLC development.
Lentivirus-mediated RNAi efficiently suppressed NLK expression in NCI-H446 cells, resulting in a significant reduction in cell viability and proliferation, which suggests that NLK could be a key regulator involved in the proliferation of SCLC cells. Flow cytometry analysis of cell cycle showed that knockdown of NLK led to an obvious accumulation of NCI-H446 cells in the S-phase. Cell cycle progression is controlled by a set of Cyclin/Cdk complexes, and their abnormal expression may play an important role in the pathogenesis of human cancers.21 We further confirmed that the cell cycle arrest was accompanied by a reduction in Cyclin A, CDK2, and CDC25A expression in response to NLK knockdown. Our results were in accordance with previous reports showing that targeted disruption of NLK suppressed HCC cell growth by arresting cell cycle transition.22 Notably, ectopic miR-101 expression repressed cancer cell growth and proliferation and imitated the NLK knockdown effect on HCC cells.23 NLK, an inhibitor of Notch signaling, has also been identified as a target of miR-181 in NLK cells.24 Thus, a comprehensive microRNA (miRNA) profiling analysis on human SCLC tissues should be carried out to further identify miRNAs that may target NLK.
It has been shown that NLK is a key regulator of cell proliferation and migration. Lentivirus-mediated knockdown of NLK in GBC cell lines significantly reduced cell viability and proliferation as well as migration.25 Next, we thus evaluated the impact of NLK silencing on SCLC cell migration using Transwell chambers. Our results indicate that knockdown of NLK potently impeded the migration of NCI-H446 cells via the suppression of MMP-9.
Furthermore, we provided a deeper understanding of the role of NLK in SCLC development with the use of mouse models of SCLC. This study showed that decreased expression of NLK in SCLC cells represents an advantage for SCLC therapy resulting in reduced cell growth in vitro and in vivo, which suggests NLK as a potential therapeutic marker for the treatment of SCLC. However, Lv et al10 reported opposite results and have shown that low NLK expression is associated with NSCLC histological differentiation and clinical stage as well as the increased growth of NSCLC cells. These differing results suggest that NLK may exert different effects depending on the specific cancer type and therefore on the different signal proteins it interacts with and regulates. NLK is able to play a role in multiple processes due to its capacity to regulate a diverse array of signaling pathways, including the Wnt/β-catenin, activin, interleukin-6, and Notch signaling pathways.7 Therefore, further investigations based on the downstream signaling pathways controlled by NLK in SCLC cells are necessary.
Conclusion
The current study suggests that NLK is a key regulator involved in the proliferation and migration of SCLC. Our findings may extend our knowledge of the molecular pathogenesis of SCLC and provide a novel therapeutic target for the treatment of SCLC.
Acknowledgment
This work was supported by grants from the National Natural Science Foundation of China (numbers 81202955, 81401881, and 81372287).
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Siege LR, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62(1):10–29. | ||
Sakamoto S, Inoue H, Ohba S, et al. A new metastatic model of human small cell lung cancer by orthotopic transplantation in mice. Cancer Sci. 2015;106(4):367–374. | ||
Ehrlich D, Wang B, Lu W, Dowling P, Yuan R. Intratumoral anti-HuD immunotoxin therapy for small cell lung cancer and neuroblastoma. J Hematol Oncol. 2014;7(1):91. | ||
Bi N, Cao J, Song Y, et al. A microRNA signature predicts survival in early stage small-cell lung cancer treated with surgery and adjuvant chemotherapy. PLoS One. 2014;9(3):e91388. | ||
Fang C, Zhang J, Qi D, et al. Evodiamine induces G2/M arrest and apoptosis via mitochondrial and endoplasmic reticulum pathways in H446 and H1688 human small-cell lung cancer cells. PLoS One. 2014;9(12):e115204. | ||
Ishitani T, Ishitani S. Nemo-like kinase, a multifaceted cell signaling regulator. Cell Signal. 2013;25(1):190–197. | ||
Ohkawara B, Shirakabe K, Hyodo-Miura J, et al. Role of the TAK1-NLK-STAT3 pathway in TGF-beta-mediated mesoderm induction. Genes Dev. 2004;18(4):381–386. | ||
Zhang Y, Peng C, Wu G, et al. Expression of NLK and its potential effect in ovarian cancer chemotherapy. Int J Gynecol Cancer. 2011;21(8):1380–1387. | ||
Lv L, Wan C, Chen B, et al. Nemo-like kinase (NLK) inhibits the progression of NSCLC via negatively modulating WNT signaling pathway. J Cell Biochem. 2014;115(1):81–92. | ||
Katoh M. Network of WNT and other regulatory signaling cascades in pluripotent stem cells and cancer stem cells. Curr Pharm Biotechnol. 2011;12(2):160–170. | ||
Li M, Zhang S, Wang Z, et al. Prognostic significance of nemo-like kinase (NLK) expression in patients with gallbladder cancer. Tumour Biol. 2013;34(6):3995–4000. | ||
Chen S, Ma Z, Chen X, Zhang J. Prognostic significance of nemo-like kinase in nasopharyngeal carcinoma. Mol Med Rep. 2014;10(1):131–136. | ||
Zhang B, Li KY, Chen HY, et al. Lentivirus-based RNA silencing of Nemo-like kinase (NLK) inhibits the CAL 27 human adenosquamos carcinoma cells proliferation and blocks G0/G1 phase to S phase. Int J Med Sci. 2013;10(10):1301–1306. | ||
Dong JR, Guo N, Zhao JP, Liu PD, Feng HH, Li Y. Inhibition of nemo-like kinase increases taxol sensitivity in laryngeal cancer. Asian Pac J Cancer Prev. 2013;14(12):7137–7141. | ||
Yasuda J, Yokoo H, Yamada T, Kitabayashi I, Sekiya T, Ichikawa H. Nemo-like kinase suppresses a wide range of transcription factors, including nuclear factor-kappaB. Cancer Sci. 2004;95(1):52–57. | ||
Chen J, Han Y, Zhao X, et al. Nemo-like kinase expression predicts poor survival in colorectal cancer. Mol Med Rep. 2015;11(2):1181–1187. | ||
Tu YS, Kang XL, Zhou JG, Lv XF, Tang YB, Guan YY. Involvement of Chk1-Cdc25A-cyclin A/CDK2 pathway in simvastatin induced S-phase cell cycle arrest and apoptosis in multiple myeloma cells. Eur J Pharmacol. 2011;670(2–3):356–364. | ||
Busino L, Chiesa M, Draetta GF, Donzelli M. Cdc25A phosphatase: combinatorial phosphorylation, ubiquitylation and proteolysis. Oncogene. 2004;23(11):2050–2056. | ||
Zhang BG, Du T, Zang MD, et al. Androgen receptor promotes gastric cancer cell migration and invasion via AKT-phosphorylation dependent upregulation of matrix metalloproteinase 9. Oncotarget. 2014;5(21):10584–10595. | ||
Grana X, Reddy EP. Cell cycle control in mammalian cells: role of cyclins, cyclin dependent kinases (CDKs), growth suppressor genes and cyclin-dependent kinase inhibitors (CKIs). Oncogene. 1995;11(2):211–219. | ||
Jung KH, Kim JK, Noh JH, et al. Targeted disruption of Nemo-like kinase inhibits tumor cell growth by simultaneous suppression of cyclin D1 and CDK2 in human hepatocellular carcinoma. J Cell Biochem. 2010;110(3):687–696. | ||
Shen Q, Bae HJ, Eun JW, et al. MiR-101 functions as a tumor suppressor by directly targeting nemo-like kinase in liver cancer. Cancer Lett. 2014;344(2):204–211. | ||
Cichocki F, Felices M, McCullar V, et al. Cutting edge: microRNA-181 promotes human NK cell development by regulating Notch signaling. J Immunol. 2011;187(12):6171–6175. | ||
Tan Z, Li M, Wu W, et al. NLK is a key regulator of proliferation and migration in gallbladder carcinoma cells. Mol Cell Biochem. 2012;369(1–2):27–33. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.