Back to Journals » Drug Design, Development and Therapy » Volume 11
Layer-by-layer nanoparticles co-loading gemcitabine and platinum (IV) prodrugs for synergistic combination therapy of lung cancer
Authors Zhang R, Ru Y, Gao Y, Li J, Mao S
Received 1 June 2017
Accepted for publication 2 August 2017
Published 5 September 2017 Volume 2017:11 Pages 2631—2642
DOI https://doi.org/10.2147/DDDT.S143047
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sukesh Voruganti
Rongrong Zhang, Yun Ru, Yiping Gao, Jinyin Li, Shilong Mao
Department of Pharmacy, Shanghai Xuhui District Central Hospital, Zhongshan Hospital Affiliated to Fudan University Xuhui Hospital, Shanghai, People’s Republic of China
Purpose: Cisplatin plus gemcitabine (GEM) is a standard regimen for the first-line treatment of advanced non-small cell lung cancer. The aim of this study was to prepare biocompatible and biodegradable polymeric prodrugs and construct nanoparticles (NPs) with layer-by-layer (LbL) technique.
Methods: Platinum (Pt) (IV) complex with a carboxyl group was conjugated to the amino group of chitosan (CH), resulting in a CH-Pt conjugation with positive charge. GEM with amino group was conjugated to the carboxyl group of hyaluronic acid (HA), resulting in a HA-GEM conjugation with negative charge. Novel LbL NPs consisting of the CH-Pt core and the HA-GEM layer, named as HA-GEM/CH-Pt NPs, were constructed. The physicochemical properties of the HA-GEM/CH-Pt NPs were investigated. In vitro cytotoxicity against human non-small lung cancer cells (NCl-H460 cells) was investigated, and in vivo antitumor efficiency was evaluated on mice bearing NCl-H460 cells xenografts.
Results: HA-GEM/CH-Pt NPs have a size of about 187 nm, a zeta potential value of -21 mV and high drug encapsulation efficiency of 90%. The drug release of HA-GEM/CH-Pt NPs exhibited a sustained behavior. HA-GEM/CH-Pt NPs could significantly enhance in vitro cytotoxicity and in vivo antitumor effect against lung cancer animal model compared to the single-drug-loaded NPs and free drug solutions.
Conclusion: The results demonstrated that the HA-GEM/CH-Pt NPs might be a promising system for the synergetic treatment of lung carcinoma.
Keywords: lung cancer, combination chemotherapy, cisplatin, gemcitabine, layer-by-layer technology
Introduction
Lung cancer remains the leading cause of cancer-associated deaths worldwide, and the non-small cell lung cancer (NSCLC) constitutes about 85% of lung cancer cases.1,2 Currently, the main clinical therapeutic regimens for NSCLC still include surgery, radiotherapy and chemotherapy. Because the majority of patients are diagnosed at later or metastatic stages, chemotherapy or palliative treatment is the main therapeutic schedule.3,4 Combination chemotherapy is effective in the treatment of cancer for its benefits, which include maximized therapeutic efficacy, reduced side effects and overcome drug resistance.5 Based on a large number of clinical trials, treatment with platinum-based combination chemotherapy is considered the first-line therapy for patients with advanced NSCLC and superior to the single-agent treatment in terms of overall survival.6,7
Cisplatin (CIS) plus gemcitabine (GEM) is a standard regimen for the first-line treatment of advanced NSCLC.8,9 CIS has been approved by the FDA for the treatment of epithelial malignancies such as lung, ovarian and bladder cancer. However, its poor hydrophilic and hydrophobic property, severe toxicity such as renal toxicity, neurotoxicity and drug resistance have hindered its clinical application.10,11 Intensive efforts have been implemented to improve its toxicity and drug resistance, which include prodrugs especially platinum (IV) (Pt) prodrugs and nanocarrier-based formulations.12,13 Compared to CIS, Pt (IV) shows better pharmacokinetics and decreased side effects. GEM is an efficacious antitumor agent, which has a wide spectrum of antitumor activity including NSCLC, pancreatic cancer, bladder cancer, breast cancer and so on. Because of its short plasma half-life and hydrophilic nature, researchers have devoted to finding novel prodrug or nanotechnology and improving its in vivo pharmacokinetics and pharmcodynamics.14 However, it is difficult to deliver both CIS and GEM with drastically different physical chemistry in the same nanoparticles (NPs).
Nanocarriers based on the conjugation of a polymer with a drug have attracted considerable attention in the field of cancer treatment due to their passive or active tumor-targeted efficiency, enhanced drug bioavailability and balanced pharmacokinetics for hydrophilic and hydrophobic drugs.15–17 Among the various polymeric nanocarriers, layer-by-layer (LbL) NPs are another research hot topic. The systems are designed by the electrostatic attraction of oppositely charged polyelectrolytes. Recent work has demonstrated the potential for LbL NPs to promote in vivo pharmacokinetics, the ability to control release of loading agents and the capability to enhance molecular-targeting capabilities.18,19 These commonly applied polyelectrolytes contain hyaluronic acid (HA),20 chitosan (CH),20 poly(L-lysine),21 poly(glutamic acid),21 poly(acrylic acid)22 and so on.23
In the present study, we designed a novel LbL NP platform equipped with active targeting ligand HA as a carrier for CIS and GEM delivery. As a prodrug of CIS, Pt (IV) complex with a carboxyl group was conjugated to the amino group of CH, resulting in a CH-Pt conjugation with positive charge. GEM with amino group was conjugated to the carboxyl group of HA, resulting in a HA-GEM conjugation with negative charge. The key strategies here were to use biocompatible and biodegradable polymeric prodrug (CH-Pt and HA-GEM) with the LbL technique. The LbL NPs consisted of the CH-Pt core and the HA-GEM layer, named as HA-GEM/CH-Pt NPs. We evaluated their physiochemical properties such as particle size and zeta potential, in vitro release kinetics. The antitumor efficacy was also evaluated both in vitro and in vivo on a CD44-overexpressed lung cancer cell line and mice tumor model, respectively.
Materials and methods
Materials and animals
CIS, GEM, dicyclohexylcarbodiimide (DCC), dimethylaminopyridine (DMAP), Dulbecco’s Modified Eagle’s Medium (DMEM) and 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma-Aldrich (St Louis, MO, USA). All chemicals and reagents used were of analytical grade or higher. NCl-H460 human non-small lung cancer cells (NCl-H460 cells) were obtained from the American Type Culture Collection (Rockville, MD, USA). BALB/c nude mice (4–6 weeks old, 18–22 g weight) were purchased from Shanghai Slack Laboratory Animal Co., Ltd. (Shanghai, People’s Republic of China). Experiments were performed according to the National Institutes of Health guide for the care and use of laboratory animals (NIH Publications No 8023, revised 1978); all the animal studies were approved by the Medical Ethics Committee of Shanghai Xuhui District Central Hospital (No FDU-XHH-1002017001011302).
Synthesis of CH-Pt
CH-Pt was synthesized by linking the carboxyl groups of Pt (IV) complex with the amino groups of CH (Figure 1A).24 Briefly, CIS (100 mg) was treated with hydrogen peroxide to obtain dihydroxyl Pt (IV) derivative. Dihydroxyl Pt (IV) derivative (200 mg), DCC (20 mg) and DMAP (15 mg) were dissolved in dichloromethane (10 mL). CH (500 mg) was then added into the aforementioned organic solution and stirred at 600 rpm at room temperature for 24 h in the dark. The product was purified by repeated filtration. CH-Pt was dried under the vacuum to remove the solvent.
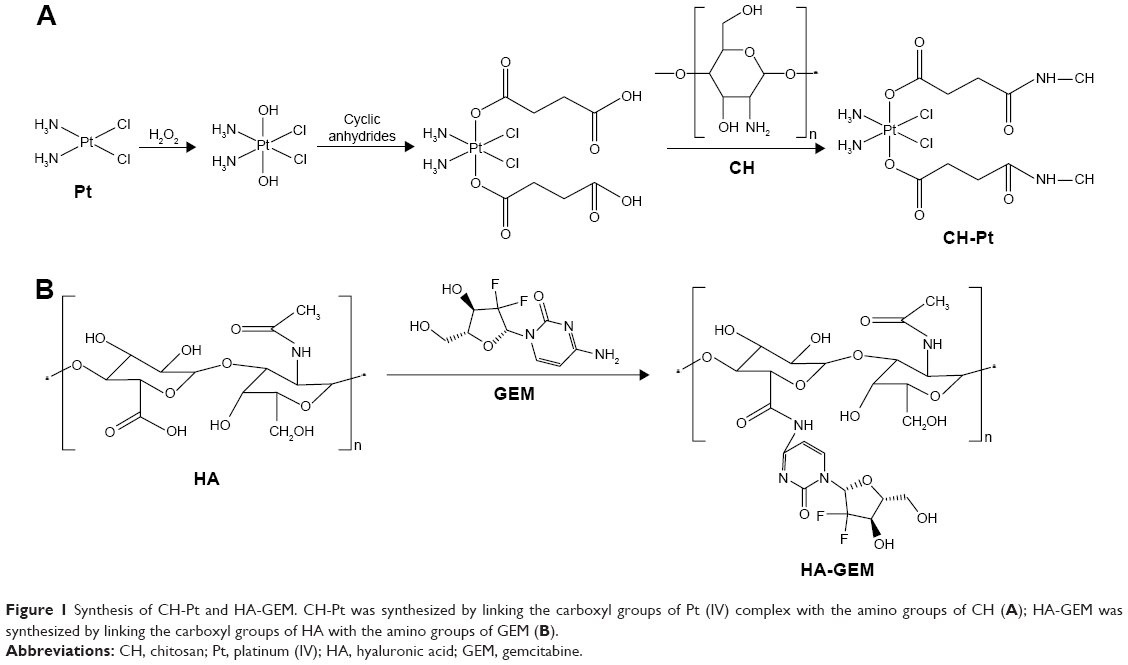 | Figure 1 Synthesis of CH-Pt and HA-GEM. CH-Pt was synthesized by linking the carboxyl groups of Pt (IV) complex with the amino groups of CH (A); HA-GEM was synthesized by linking the carboxyl groups of HA with the amino groups of GEM (B). |
Synthesis of HA-GEM
HA-GEM was synthesized by linking the carboxyl groups of HA with the amino groups of GEM (Figure 1B).25 Briefly, HA (100 mg) and GEM (25 mg) were dissolved in dimethyl sulfoxide (DMSO; 10 mL) under nitrogen gas. DCC (15 mg) and DMAP (10 mg) were then sequentially added into the DMSO solution. The reaction mixture was stirred at 600 rpm at room temperature for 24 h in the dark. The reaction was quenched using the excess cold methanol, and the product was purified by repeated filtration. HA-GEM was dried under vacuum to remove the solvent. CH-Pt and HA-GEM were characterized by 1H-NMR spectroscopy and FT-IR spectroscopy.
Preparation of HA-GEM/CH-Pt NPs
CH-Pt-loaded NPs without HA-GEM (CH-Pt NPs) were prepared by solvent diffusion technique.26 Briefly, CH-Pt (200 mg) was firstly dissolved in acetic acid solution (10 mL). The solution was added drop-by-drop into the Milli-Q water (40 mL) stirred at 600 rpm at room temperature. CH-Pt NPs were collected by centrifugation at 15,000 rpm for 10 min, resuspended in Milli-Q water and stored at 2°C–8°C.
HA-GEM/CH-Pt NPs were prepared by self-assembly technique. Briefly, HA-GEM (200 mg) was dissolved in Milli-Q water (10 mL), added drop-by-drop into the CH-Pt NP suspension and stirred at 600 rpm at room temperature. HA-GEM/CH-Pt NPs were collected by centrifugation at 15,000 rpm for 10 min, resuspended in Milli-Q water and stored at 2°C–8°C.
HA-coated CH-Pt NPs without GEM were prepared by the same method using HA instead of HA-GEM, named HA/CH-Pt NPs.
HA-GEM-coated CH NPs without Pt were prepared by the same method using CH instead of CH-Pt, named HA-GEM/CH NPs.
Blank NPs without Pt and GEM were prepared by the same method using HA instead of HA-GEM, and CH instead of CH-Pt, named blank HA/CH NPs.
Particle morphology, particle size and zeta potential
The morphology of HA-GEM/CH-Pt NPs was analyzed using transmission electron microscopy (TEM).27 An aliquot of the NP suspensions was placed onto a clean copper grid and stained using 2% phosphotungstic acid for 2 min. The stained samples were then imaged using a Hitachi H-7000 transmission electron microscope (Hitachi, Tokyo, Japan). Particle size, polydispersity (PDI) and zeta potential of different samples were determined with Zeta Sizer Nano ZS apparatus (Malvern, Southborough, MA, USA).
Drug encapsulation and loading efficiency
The drug encapsulation efficiency (EE) and drug loading efficiency (DL) were measured after the drugs were removed from the NPs by ultrafiltration (Millipore, Boston, MA, USA).28 The amount of Pt in NPs was measured by using the HITACHI P-4010 inductively coupled plasma mass spectrometry (Hitachi Ltd, Kyoto, Japan).29 The amount of GEM in NPs was determined by HPLC (LC-20A; Shimadzu, Kyoto, Japan).25 Chromatographic separations were carried out using the Inertsil® ODS-3V (250×4.6 mm). The mobile phase was methanol and water (90:10, v/v); the detection wavelength was 248 nm. The EE and DL were calculated as follows:
|

|

Serum stability of NPs
The stability of NPs in serum was evaluated by incubating the NPs with 10% (v/v) fetal bovine serum (FBS) at 37°C for 72 h.30 Samples (100 μL) were diluted in Milli-Q water at predetermined time points. The variation in size, PDI and EE was measured to determine the stability of the particles.
In vitro drug release
In vitro GEM and Pt release from NPs was measured by dialysis technique in phosphate-buffered saline (PBS) (pH 7.4) at 37°C.31 HA-GEM/CH-Pt NPs and other NPs were added into the dialysis tube with a molecular weight cutoff of 3 kDa and dialyzed against 15 mL of PBS in a thermo-controlled shaker with a stirring speed of 100 rpm at 37°C for 72 h. The amount of drugs released was determined by the method mentioned in section “Drug encapsulation and loading efficiency”.
In vitro cytotoxicity and synergistic effects analysis
In vitro cytotoxicity of HA-GEM/CH-Pt NPs was determined using MTT assay.32 Briefly, NCl-H460 cells were seeded in 96-well plates at a density of 5,000 cells per well at 37°C in a 5% CO2 incubator 24 h prior to drug treatment. Subsequently, cells were treated with HA-GEM/CH-Pt NPs, HA-GEM/CH NPs, HA/CH-Pt NPs, CH-Pt NPs, free GEM, free Pt, free GEM and PT combination (free GEM/Pt), blank HA/CH NPs, 0.9% saline control and incubated for 72 h. After treatment, 20 μL MTT (5 mg/mL in PBS) reagent was added to each well and incubated for an additional 4 h at 37°C. The medium was discarded, the formed formazan crystals were dissolved in 200 μL of DMSO and absorbance was read at 570 nm. Cell viability and half-maximal inhibitory concentration (IC50) were then calculated for each sample.
Synergistic effects of the double drug-contained systems were evaluated by combination index (CI) analysis based on the Chou and Talalay’s method.33 CI values for GEM and Pt combinations were calculated according to the following equation: CI = (D)GEM/(Dx)GEM + (D)Pt/(Dx)Pt, where (D)GEM and (D)Pt are the concentrations of GEM and Pt in the combination system at the ICx value; (Dx)GEM and (Dx)P are ICx value of GEM alone and Pt alone. CIx <1 represents synergism and CIx >1 represents antagonism. In this study, CI50 values were applied and the IC50 values were used for calculation.
In vivo tissue distribution analysis
In vivo tissue distribution analysis were conducted on NCl-H460 cells of mouse models.34 NCl-H460 cells (1×107 cells in 200 μL of DMEM) were subcutaneously injected in the right flank regions of mice. When the tumor volumes reached around 100 mm3, the mice were randomly distributed into 9 groups with 8 mice in each group, and intravenously injected with HA-GEM/CH-Pt NPs, HA-GEM/CH NPs, HA/CH-Pt NPs, CH-Pt NPs, free GEM/Pt, free GEM, free Pt, blank HA/CH NPs and 0.9% normal saline (the dosage of GEM and/or Pt is 5 μg per g mice). At 12 and 72 h after intravenous injection, mice were sacrificed and the tumor and other main tissue samples (heart, liver, spleen, lung and kidney) were collected, weighed and homogenized with physiological saline to determine the amount of drugs in each tissue. The amount of drugs was determined by the method mentioned in section “Drug encapsulation and loading efficiency”.
In vivo antitumor effect and systemic toxicity
In vivo antitumor effect and systemic toxicity were conducted on NCl-H460 cells of mouse models as mentioned in section “In vivo tissue distribution analysis”.35 When the tumor volumes reached around 100 mm3, HA-GEM/CH-Pt NPs, HA-GEM/CH NPs, HA/CH-Pt NPs, CH-Pt NPs, free GEM/Pt, free GEM, free Pt, blank HA/CH NPs and 0.9% normal saline were administered intravenously every 3 days until 3 weeks (the dosage of GEM and/or Pt is 5 μg per g mice). The tumor sizes, tumor weights and body weights were closely monitored every 3 days. The tumor volume for each time point was calculated using the following formulation: length × width2/2.
Statistical analysis
Student’s t-test or analysis of variance was utilized to determine statistical significance among different groups. A P-value less than 0.05 was considered to be statistically significant.
Results
Synthesis and characterization of CH-Pt and HA-GEM
The formation of CH-Pt and HA-GEM was confirmed by 1H-NMR and FT-IR spectroscopy. The 1H-NMR spectra (DMSO-d6, 300 MHz) of CH-Pt showed the peaks at δ (ppm) =2.05 (–NH2 of Pt), 2.45 (–CH2–CO–N–), 3.21 (–CH–O–), 4.26–4.78 (peak of H on the sugar rings of CH), 8.03 (–CO–NH–) (Figure 2). The 1H-NMR spectra of HA-GEM showed the peaks at δ (ppm) =2.01 (–OH), 3.32 (–CH2–CO–N–), 3.21 (–CH–O–C), 3.61 (–CH–OH), 3.86 (–CH–CO–N–), 4.53 (–CH–OH), 5.22 (–CH–O–C), 5.73 (–N–CH–O–), 6.67 (–N–CH=C–), 7.96 (–CO–NH–) (Figure 3). The relevant peaks were marked in the spectra as well as in the structural formula. The FT-IR spectrum of CH-Pt and HA-GEM showed a peak stretching at 1,657 cm−1 and 1,644 cm−1 representing carbonyl bond of amide (–CO–NH–), respectively.
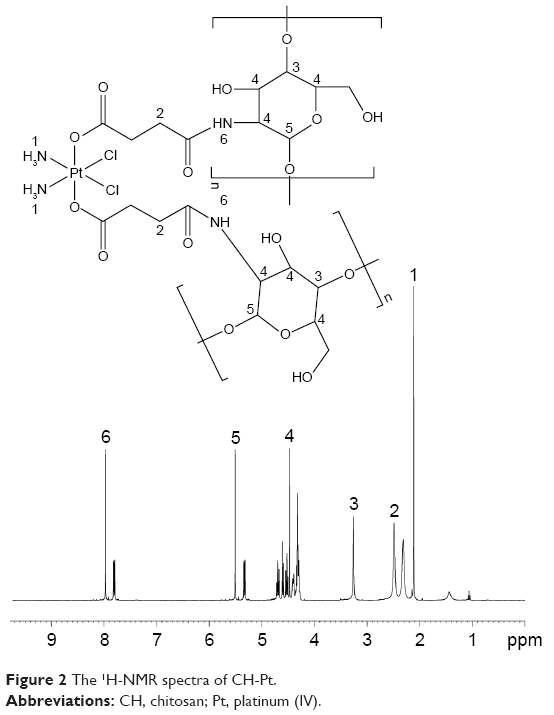 | Figure 2 The 1H-NMR spectra of CH-Pt. |
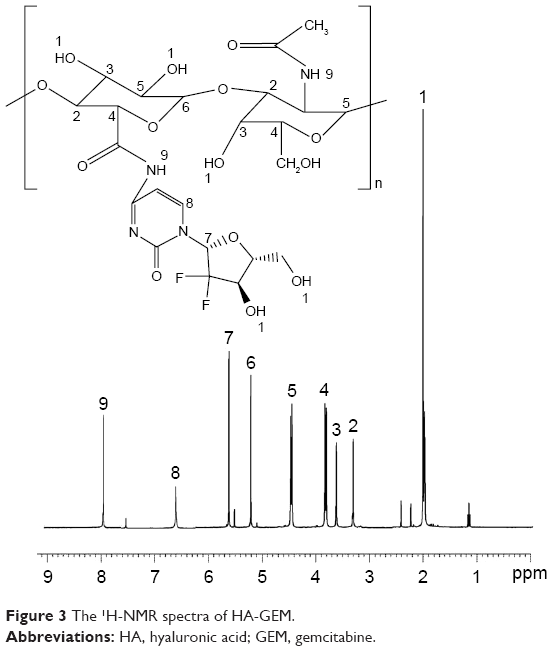 | Figure 3 The 1H-NMR spectra of HA-GEM. |
Physicochemical properties of NPs
Physicochemical properties of NPs including particle morphology, particle size, zeta potential, EE and DL were characterized. TEM image of HA-GEM/CH-Pt NPs displayed a core-shell structural spherical morphology and a size of around 200 nm (Figure 4). The size of HA-GEM/CH-Pt NPs, HA-GEM/CH NPs, HA/CH-Pt NPs, blank HA/CH NPs and CH-Pt NPs was 187, 186, 186, 175 and 113 nm, respectively (Figure 5). The size was slightly increased when loading with drugs. However, coating of HA shell enlarged the particles to a large extent. The zeta potential of CH-Pt NPs was 28 mV, which shifted to −21 mV after coating of HA layer to form HA-GEM/CH-Pt NPs. The EE of both GEM and Pt was about 90% for all the formulations tested. The DL of GEM and Pt was between 2%–4%.
 | Figure 4 TEM image of HA-GEM/CH-Pt NPs. |
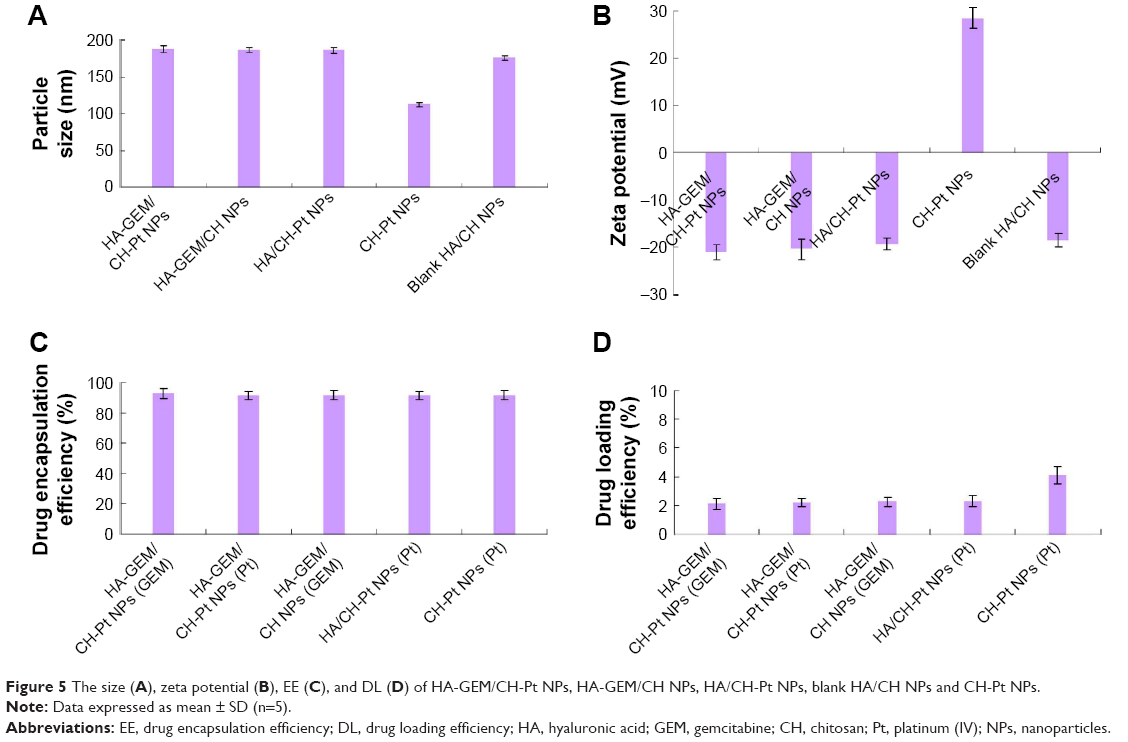 | Figure 5 The size (A), zeta potential (B), EE (C), and DL (D) of HA-GEM/CH-Pt NPs, HA-GEM/CH NPs, HA/CH-Pt NPs, blank HA/CH NPs and CH-Pt NPs. |
Serum stability
Serum stability of NPs was evaluated by measuring the size, PDI and EE of NPs in the presence of FBS. HA-GEM/CH-Pt NPs, blank HA/CH NPs and CH-Pt NPs exhibited no significant changes in size, PDI or EE (Figure 6). So, the NPs prepared were considered stable during the period of 72 h.
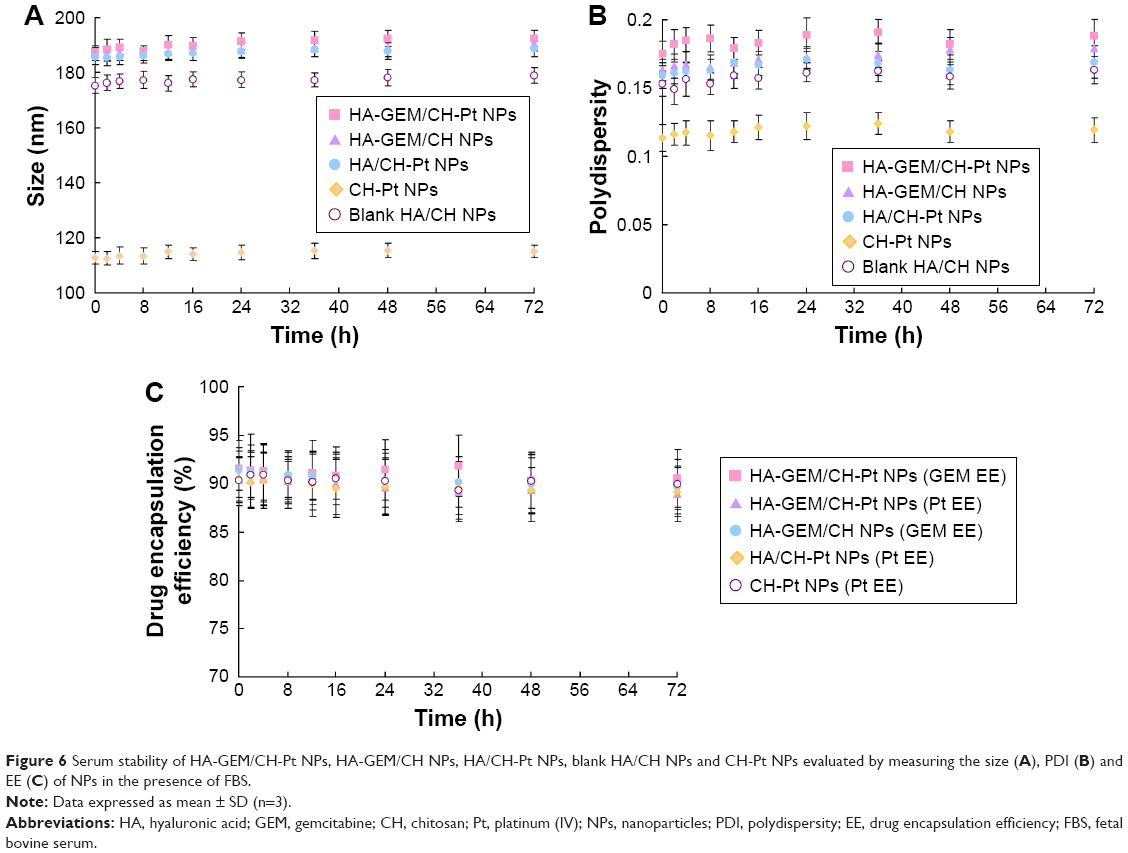 | Figure 6 Serum stability of HA-GEM/CH-Pt NPs, HA-GEM/CH NPs, HA/CH-Pt NPs, blank HA/CH NPs and CH-Pt NPs evaluated by measuring the size (A), PDI (B) and EE (C) of NPs in the presence of FBS. |
In vitro drug release
In vitro GEM and/or Pt release from HA-GEM/CH-Pt NPs and other NPs was studied and the profiles were described (Figure 7). The data indicated that both drugs were released from the NPs in a sustained manner and no burst release was observed. The GEM release from HA-GEM/CH-Pt NPs and HA-GEM/CH-NPs was faster than Pt release from the NPs (P<0.05). The release of Pt from HA-GEM/CH-Pt NPs and HA/CH-Pt NPs was slower than that from CH-Pt NPs (P<0.05).
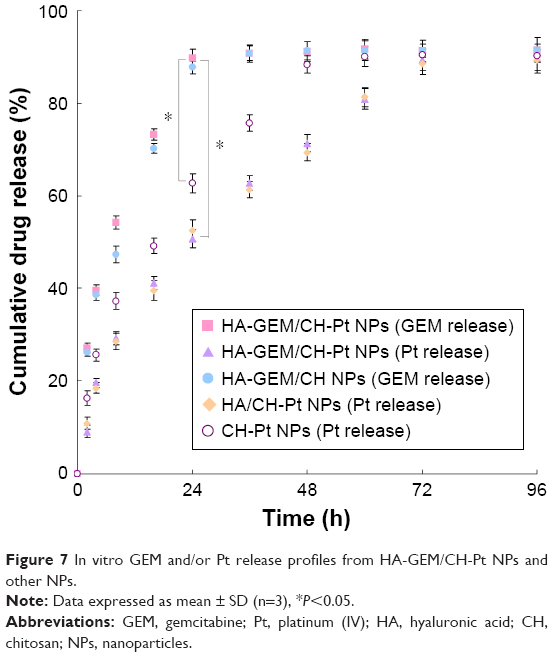 | Figure 7 In vitro GEM and/or Pt release profiles from HA-GEM/CH-Pt NPs and other NPs. |
In vitro cytotoxicity and synergistic effects
In vitro cytotoxicity of HA-GEM/CH-Pt NPs and other NPs was evaluated on NCl-H460 cells by MTT assay. At all the studied drug concentrations, the cytotoxicity of drug-loaded NPs was better than free drug solutions (P<0.05) (Figure 8A). To select the suitable GEM to Pt ratio to get the best synergism effect, CI50 values were calculated in HA-GEM/CH-Pt NPs. HA-GEM/CH-Pt NPs showed the best synergism when GEM to Pt ratio was 1/1 (Figure 8B). So, this ratio was used for the preparation and evaluation of HA-GEM/CH-Pt NPs.
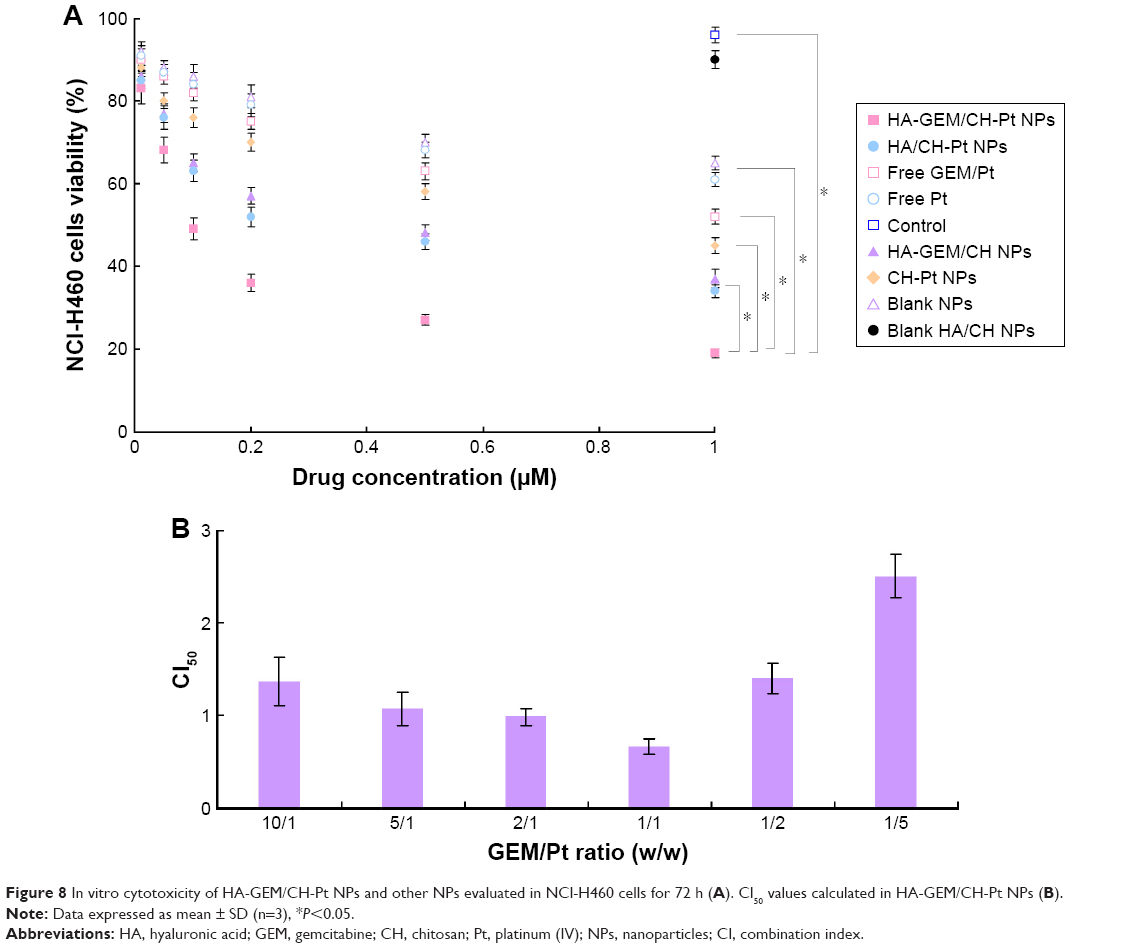 | Figure 8 In vitro cytotoxicity of HA-GEM/CH-Pt NPs and other NPs evaluated in NCl-H460 cells for 72 h (A). CI50 values calculated in HA-GEM/CH-Pt NPs (B). |
In vivo tissue distribution
In vivo GEM and Pt distribution of drug-loaded NPs were higher than drugs solutions in tumor tissues (P<0.05) (Figure 9). Drugs solutions were distributed in a higher quantity in the heart and kidneys; on the contrary, the drug-loaded NPs were mainly distributed in the lungs and liver. Distribution of drug solution was relatively high when tested at 12 h. However, it decreased dramatically at 72 h post-injection.
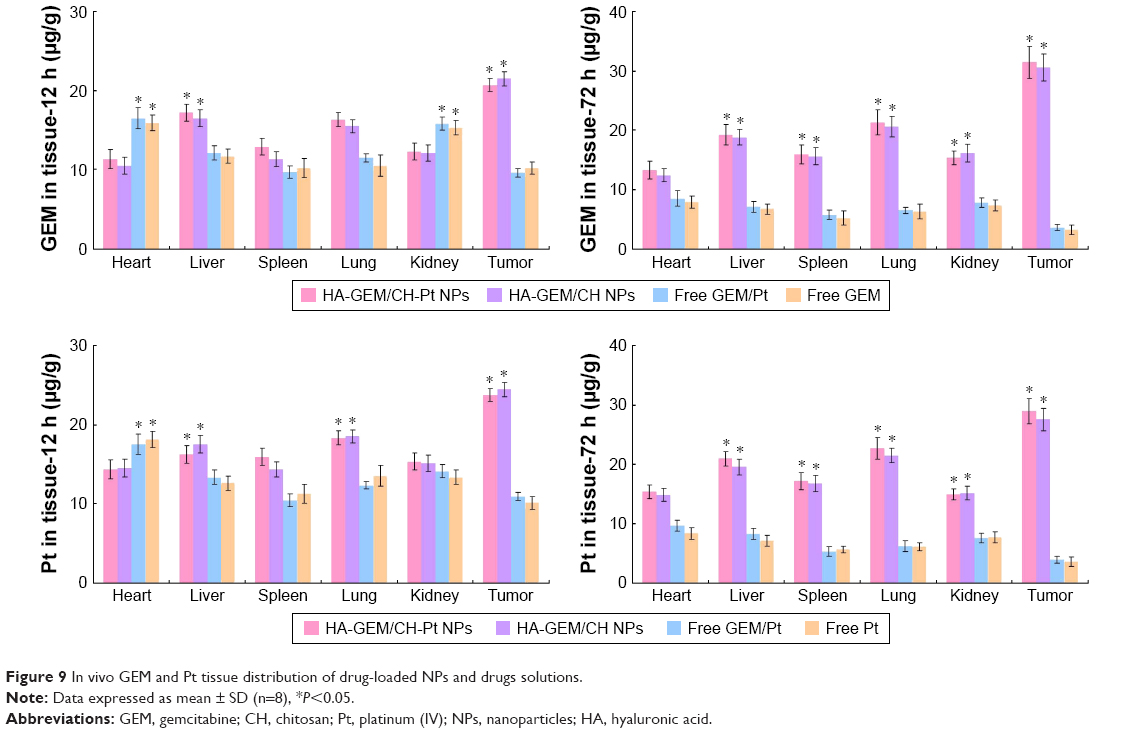 | Figure 9 In vivo GEM and Pt tissue distribution of drug-loaded NPs and drugs solutions. |
In vivo antitumor effect and systemic toxicity
In vivo antitumor effect and systemic toxicity were conducted on NCl-H460 cells of mouse models (Figure 10). The tumors treated with HA-GEM/CH-Pt NPs were significantly smaller than those treated with single-drug-loaded NPs and drug solutions (P<0.05). By contrast, the tumors treated with blank NPs were similar to those treated with saline (Figure 10A). Body weight changes were observed to determine the systemic toxicity of the systems (Figure 10B). Obviously, increases in mean body weights were found in 0.9% saline and blank NPs groups, and weight decrease was found in drug solution groups. No obvious changes of body weights were found in drug-loaded NPs groups.
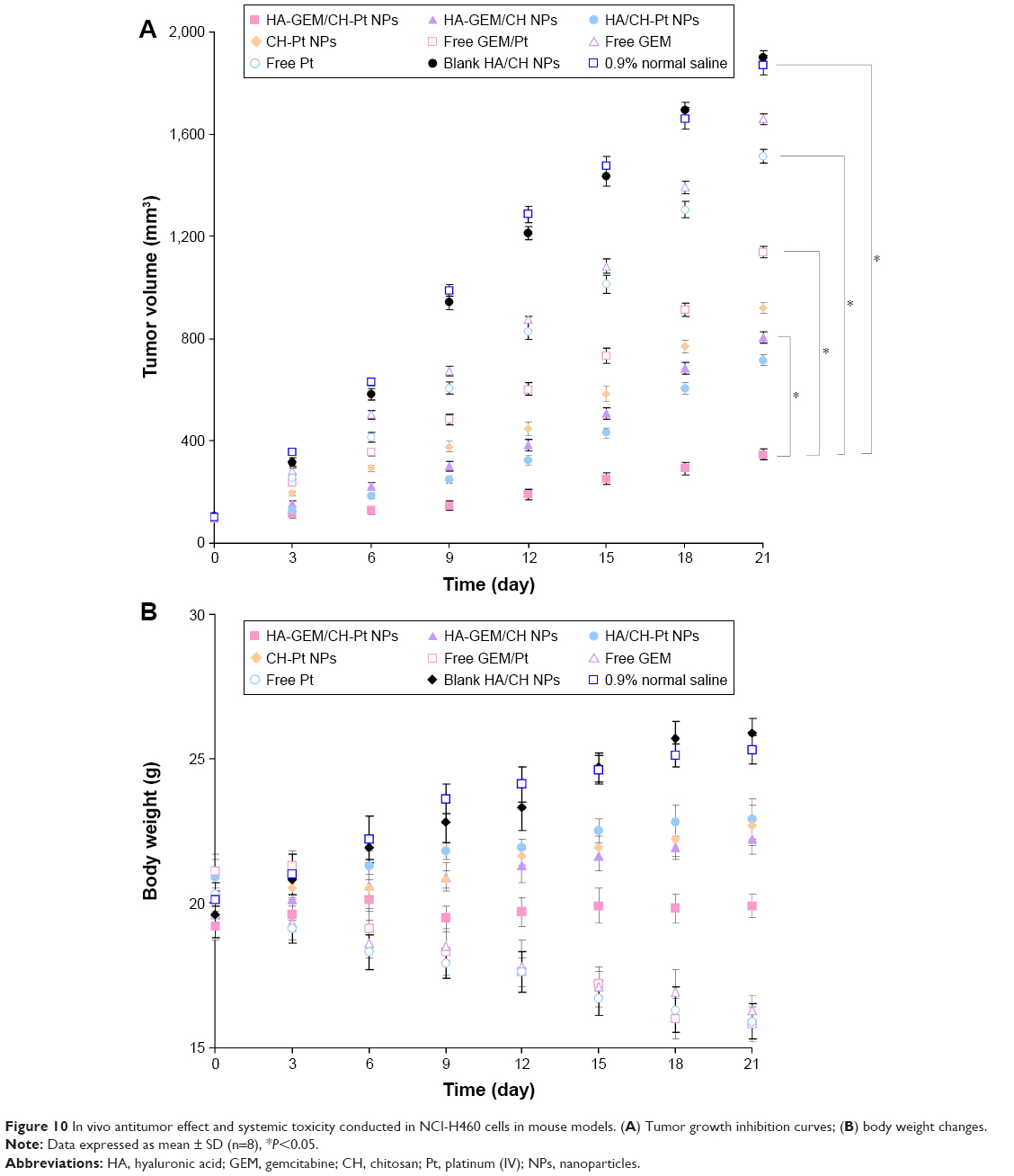 | Figure 10 In vivo antitumor effect and systemic toxicity conducted in NCl-H460 cells in mouse models. (A) Tumor growth inhibition curves; (B) body weight changes. |
Discussion
In this study, CH-Pt and HA-GEM prodrugs were synthesized. LbL NPs were prepared by using CH-Pt as positive charged core and HA-GEM as negatively charged shell. The physicochemical properties of the NPs and biological efficiency on cancer cells and cancer-bearing animal models were studied.
CH-Pt was synthesized by linking the carboxyl groups of Pt (IV) complex with the amino groups of CH. Pt (IV) prodrugs are typically designed based on cytotoxic Pt (II) constructs and commonly include CIS, carboplatin, oxaliplatin and [Pt(dach)Cl2] structures.24 Pt (II) could be oxidized by treatment with hydrogen peroxide or ozone. The trans-dihydroxo Pt (IV) derivative can be synthesized by treating the trans-dihydroxo complex with acid anhydrides or acid chlorides (Figure 1A). HA-GEM was synthesized by conjugating HA with GEM using amido linkage (Figure 1B). HA-GEM exhibited strongly negative charges due to the hydroxyl and carboxyl groups in HA.17
Enhanced permeability and retention (EPR) effect in tumor tissues could be utilized for nanosized drug delivery.36 Drug could accumulate at the tumor site and enhance tumor targeting based on the EPR effect, thereby reducing the drug dose needed to obtain a greater antitumor effect and minimizing toxicity. Particle sizes smaller than 200 nm are conducive to drug accumulation at the tumor site.37 The sizes of the HA-GEM/CH-Pt NPs, blank HA/CH NPs and CH-Pt NPs were less than 200 nm in this study. Zeta potential is an important parameter among the physicochemical characteristics of NPs. It affects the cellular and tissue uptake efficiency and toxic effect on cells. For positively charged NPs, cytotoxicity remains a problem especially in vivo.38 For instance, cationic NPs may cause cell shrinking, reduced number of cell mitoses and vacuolization of the cytoplasm. To overcome these limitations, anionic NPs were developed to reduce the cytotoxicity.39 The zeta potential of CH-Pt NPs was 28 mV, which shifted to −21 mV after coating of HA layer to form HA-GEM/CH-Pt NPs. EE was over 90% for the two drugs loaded in different NPs. High and comparable EE of each component is a prerequisite for controlled loading of several modalities in the same NP. Serum stability of NPs in 10% FBS was tested to simulate the in vivo hemocompatibility of systems.40 The size, PDI and EE of all kinds of particles in serum showed no obvious change during 72 h of incubation. This could be the evidence that the NPs are stable in the presence of serum, and the particles may not aggregate following in vivo administration.
Sustained release of drugs from both HA-GEM/CH-Pt NPs and CH-Pt NPs was observed in the in vitro drug release study. The core-shell structured NPs affected the release behavior of the systems. The GEM release from HA-GEM/CH-Pt NPs was the fastest, since GEM was released from the outer shell of the NPs. The release of Pt from HA-GEM/CH-Pt NPs was slower than that from CH-Pt NPs, which indicate that the HA-GEM layer at the interface of the CH-Pt core acts as a molecular fence that helps to retain the drugs inside the NPs.31 The sustained release manner of HA-GEM/CH-Pt NPs could protect the drugs for a relatively longer time from being degraded in the circulation system, prevent premature drug release prior to reaching the tumor sites and thus may perform persistent therapeutic effect.41
The in vitro cytotoxicity of both drug-loaded NPs and free drugs reduced cell viability in a concentration-dependent manner. Higher cytotoxicity of drug-loaded NPs was better than free drug, indicating that NPs delivery systems can enhance the cytotoxicity in vitro. The single-drug-loaded NPs show similar effect to free dual drugs GEM/Pt in vitro; this may be due to the synergistic effects of the two drugs used together. However, the double/single-drug-loaded NPs show significant effect to the free double/single-drug groups (P<0.05). In addition, blank NPs showed negligible toxicity (data not shown). To validate the synergistic effect of drugs co-loaded with NPs, the CI was further determined using the isobologram equation of Lv et al.42 HA-GEM/CH-Pt NPs displayed an overall CI value <1 when GEM to Pt ratio was 1/1; in this ratio, the combined antitumor effect of the dual-drug-loaded NPs was better than the single-drug-loaded ones and could develop the ability of the drugs to a large extent.
Solid tumors have leakage microvasculatures. Nanosized particles may be delivered to the tumor tissue owing to the EPR effects.29 EPR effects also made the entry of the NPs into the tumor more easily, thus prevented the entry of the system in the normal tissue. In vivo tissue distribution results showed that the administration of drug-loaded NPs led to a dramatic increase of drug accumulation in the tumor tissue than the free drug solutions. NPs of this size are typically found in the liver and spleen, and have low signal in other organs. The high signal from kidney is in the 72 h of the drugs loaded NPs may prove the long circulation time of the NPs and the disintegrate happened at the end of the 72 h after injection. This behavior may help with the performance of systems for the in vivo antitumor therapy.
In vivo tumor growth was more prominently inhibited by HA-GEM/CH-Pt NPs than the single-drug-loaded NPs and drug solutions. This could be the evidence that dual-drug-loaded NPs could be synergetic in the treatment of lung cancer in vivo, which is in accordance with the results of the in vitro cytotoxicity and synergistic effects studies. To be noticed, improved anticancer activity of HA-GEM/CH-Pt NPs was obtained along with a lower toxicity than the drugs injected alone or mixed together, which could be observed by the loss of body weight of the animals.36 According to the tissue distribution results, drug solutions were distributed in a higher quantity in the heart and kidneys; on the contrary, the drug-loaded NPs were mainly distributed in the tumor, lungs and liver. Drug distribution in the heart and kidney may cause systemic toxicity; distribution in a lower quantity in the heart and kidneys could decrease the side effects and lead to better antitumor therapeutic efficiency. These studies demonstrated that dual drugs co-loaded lay-by-layer with HA-GEM/CH-Pt NPs, with the most significant synergistic therapeutic efficacy, exhibited no significant toxicity to major organs and tissues. Therefore, the combination therapy system is a promising strategy in enhancing the lung cancer chemotherapy.
Conclusion
In the present study, LbL NP co-loading GEM and Pt (IV) prodrugs were designed for synergistic combination therapy of lung cancer. The LbL NPs consisted of the CH-Pt core and the HA-GEM layer. LbL HA-GEM/CH-Pt NPs exhibited the most significant synergistic therapeutic efficacy but showed no significant toxicity to major organs and tissues. Therefore, the combination therapy system is a promising strategy in enhancing the lung cancer chemotherapy.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. | ||
Shi C, Yu H, Sun D, et al. Cisplatin-loaded polymeric nanoparticles: characterization and potential exploitation for the treatment of non-small cell lung carcinoma. Acta Biomater. 2015;18:68–76. | ||
Reck M, Heigener DF, Mok T, Soria JC, Rabe KF. Management of non-small-cell lung cancer: recent developments. Lancet. 2013;382(9893):709–719. | ||
Heger Z, Polanska H, Krizkova S, et al. Co-delivery of VP-16 and Bcl-2-targeted antisense on PEG-grafted oMWCNTs for synergistic in vitro anti-cancer effects in non-small and small cell lung cancer. Colloids Surf B Biointerfaces. 2017;150:131–140. | ||
Greco F, Vicent MJ. Combination therapy: opportunities and challenges for polymer-drug conjugates as anticancer nanomedicines. Adv Drug Deliv Rev. 2009;61(13):1203–1213. | ||
Mörth C, Valachis A. Single-agent versus combination chemotherapy as first-line treatment for patients with advanced non-small cell lung cancer and performance status 2: a literature-based meta-analysis of randomized studies. Lung Cancer. 2014;84(3):209–214. | ||
Masters GA, Temin S, Azzoli CG, et al; American Society of Clinical Oncology Clinical Practice. Systemic therapy for stage IV non-small-cell lung cancer: American society of clinical oncology clinical practice guideline update. J Clin Oncol. 2015;33(30):3488–3515. | ||
Scagliotti GV, Parikh P, von Pawel J, et al. Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non-small-cell lung cancer. J Clin Oncol. 2008;26(21):3543–3551. | ||
Cartei G, Binato S, Sacco C, et al. Simplified gemcitabine and platin regimen in patients with advanced or metastatic non-small cell lung cancer (NSCLC) to be proposed as neoadjuvant therapy. Ann Oncol. 2006;17 (Suppl 5):v47–v51. | ||
Haxton KJ, Burt HM. Polymeric drug delivery of platinum-based anticancer agents. J Pharm Sci. 2009;98(7):2299–2316. | ||
Najjar A, Rajabi N, Karaman R. Recent approaches to platinum(IV) prodrugs: a variety of strategies for enhanced delivery and efficacy. Curr Pharm Des. 2017;23(16):2366–2376. | ||
Johnstone TC, Suntharalingam K, Lippard SJ. The next generation of platinum drugs: targeted Pt(II) agents, nanoparticle delivery, and Pt(IV) prodrugs. Chem Rev. 2016;116(5):3436–3486. | ||
Marcato PD, Fávaro WJ, Durán N. Cisplatin properties in a nanobiotechnological approach to cancer: a mini-review. Curr Cancer Drug Targets. 2014;14(5):458–476. | ||
Dubey RD, Saneja A, Gupta PK, Gupta PN. Recent advances in drug delivery strategies for improved therapeutic efficacy of gemcitabine. Eur J Pharm Sci. 2016;93:147–162. | ||
Luo C, Sun J, Sun B, He Z. Prodrug-based nanoparticulate drug delivery strategies for cancer therapy. Trends Pharmacol Sci. 2014;35(11):556–566. | ||
Fang JY, Al-Suwayeh SA. Nanoparticles as delivery carriers for anticancer prodrugs. Expert Opin Drug Deliv. 2012;9(6):657–669. | ||
Noh I, Kim HO, Choi J, et al. Co-delivery of paclitaxel and gemcitabine via CD44-targeting nanocarriers as a prodrug with synergistic antitumor activity against human biliary cancer. Biomaterials. 2015;53:763–774. | ||
Morton SW, Poon Z, Hammond PT. The architecture and biological performance of drug-loaded LbL nanoparticles. Biomaterials. 2013;34(21):5328–5335. | ||
Ramasamy T, Haidar ZS, Tran TH, et al. Layer-by-layer assembly of liposomal nanoparticles with PEGylated polyelectrolytes enhances systemic delivery of multiple anticancer drugs. Acta Biomater. 2014;10(12):5116–5127. | ||
Suh MS, Shen J, Kuhn LT, Burgess DJ. Layer-by-layer nanoparticle platform for cancer active targeting. Int J Pharm. 2017;517(1–2):58–66. | ||
Zhou D, Xiao H, Meng F, et al. Layer-by-layer assembled polypeptide capsules for platinum-based pro-drug delivery. Bioconjug Chem. 2012;23(12):2335–2343. | ||
Ficai D, Sonmez M, Albu MG, Mihaiescu DE, Ficai A, Bleotu C. Antitumoral materials with regenerative function obtained using a layer-by-layer technique. Drug Des Devel Ther. 2015;9:1269–1279. | ||
Morton SW, Shah NJ, Quadir MA, Deng ZJ, Poon Z, Hammond PT. Osteotropic therapy via targeted layer-by-layer nanoparticles. Adv Healthc Mater. 2014;3(6):867–875. | ||
Chin CF, Wong DY, Jothibasu R, Ang WH. Anticancer platinum (IV) prodrugs with novel modes of activity. Curr Top Med Chem. 2011;11(21):2602–2612. | ||
Lu Z, Su J, Li Z, Zhan Y, Ye D. Hyaluronic acid-coated, prodrug-based nanostructured lipid carriers for enhanced pancreatic cancer therapy. Drug Dev Ind Pharm. 2017;43(1):160–170. | ||
Fabiano A, Bizzarri R, Zambito Y. Thermosensitive hydrogel based on chitosan and its derivatives containing medicated nanoparticles for transcorneal administration of 5-fluorouracil. Int J Nanomedicine. 2017;12:633–643. | ||
Babu A, Wang Q, Muralidharan R, Shanker M, Munshi A, Ramesh R. Chitosan coated polylactic acid nanoparticle-mediated combinatorial delivery of cisplatin and siRNA/Plasmid DNA chemosensitizes cisplatin-resistant human ovarian cancer cells. Mol Pharm. 2014;11(8):2720–2733. | ||
Ni S, Qiu L, Zhang G, Zhou H, Han Y. Lymph cancer chemotherapy: delivery of doxorubicin-gemcitabine prodrug and vincristine by nanostructured lipid carriers. Int J Nanomedicine. 2017;12:1565–1576. | ||
Li C, Ge X, Wang L. Construction and comparison of different nanocarriers for co-delivery of cisplatin and curcumin: a synergistic combination nanotherapy for cervical cancer. Biomed Pharmacother. 2017;86:628–636. | ||
Chen W, Guo M, Wang S. Anti prostate cancer using PEGylated bombesin containing, cabazitaxel loading nano-sized drug delivery system. Drug Dev Ind Pharm. 2016;42(12):1968–1976. | ||
Miao L, Guo S, Zhang J, Kim WY, Huang L. Nanoparticles with precise ratiometric co-loading and co-delivery of gemcitabine monophosphate and cisplatin for treatment of bladder cancer. Adv Funct Mater. 2014;24(42):6601–6611. | ||
Zhang J, Miao L, Guo S, et al. Synergistic anti-tumor effects of combined gemcitabine and cisplatin nanoparticles in a stroma-rich bladder carcinoma model. J Control Release. 2014;182:90–96. | ||
Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. | ||
Xiong Y, Zhao Y, Miao L, Lin CM, Huang L. Co-delivery of polymeric metformin and cisplatin by self-assembled core-membrane nanoparticles to treat non-small cell lung cancer. J Control Release. 2016;244(Pt A):63–73. | ||
Tai W, Mo R, Lu Y, Jiang T, Gu Z. Folding graft copolymer with pendant drug segments for co-delivery of anticancer drugs. Biomaterials. 2014;35(25):7194–7203. | ||
Zhu B, Yu L, Yue Q. Co-delivery of vincristine and quercetin by nanocarriers for lymphoma combination chemotherapy. Biomed Pharmacother. 2017;91:287–294. | ||
Prados J, Melguizo C, Ortiz R, et al. Doxorubicin-loaded nanoparticles: new advances in breast cancer therapy. Anticancer Agents Med Chem. 2012;12(9):1058–1070. | ||
Ye J, Wang A, Liu C, Chen Z, Zhang N. Anionic solid lipid nanoparticles supported on protamine/DNA complexes. Nanotechnology. 2008;19(28):285708. | ||
Torii Y, Sugimura N, Mitomo H, Niikura K, Ijiro K. pH-responsive coassembly of oligo(ethylene glycol)-coated gold nanoparticles with external anionic polymers via hydrogen bonding. Langmuir. 2017;33(22):5537–5544. | ||
Ma P, Li T, Xing H, et al. Local anesthetic effects of bupivacaine loaded lipid-polymer hybrid nanoparticles: in vitro and in vivo evaluation. Biomed Pharmacother. 2017;89:689–695. | ||
Poon C, He C, Liu D, Lu K, Lin W. Self-assembled nanoscale coordination polymers carrying oxaliplatin and gemcitabine for synergistic combination therapy of pancreatic cancer. J Control Release. 2015;201:90–99. | ||
Lv S, Tang Z, Li M, et al. Co-delivery of doxorubicin and paclitaxel by PEG-polypeptide nanovehicle for the treatment of non-small cell lung cancer. Biomaterials. 2014;35(23):6118–6129. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.