Back to Journals » Neuropsychiatric Disease and Treatment » Volume 17
Late-Onset Leukodystrophy Mimicking Hereditary Spastic Paraplegia without Diffuse Leukodystrophy on Neuroimaging
Authors Zhang T, Yan C, Liu Y, Cao L, Ji K, Li D, Chi L, Zhao Y
Received 19 December 2020
Accepted for publication 12 April 2021
Published 12 May 2021 Volume 2021:17 Pages 1451—1458
DOI https://doi.org/10.2147/NDT.S296424
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yu-Ping Ning
Tongxia Zhang,1,2 Chuanzhu Yan,1,3 Yiming Liu,1 Lili Cao,1 Kunqian Ji,1 Duoling Li,1 Lingyi Chi,2,4,5 Yuying Zhao1
1Research Institute of Neuromuscular and Neurodegenerative Diseases and Department of Neurology, Qilu Hospital, Shandong University, Jinan, People’s Republic of China; 2School of Basic Medical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, People’s Republic of China; 3Mitochondrial Medicine Laboratory, Qilu Hospital (Qingdao), Shandong University, Qingdao, People’s Republic of China; 4Brain Science Research Institute, Qilu Hospital, Shandong University, Jinan, People’s Republic of China; 5Department of Neurosurgery, Qilu Hospital, Shandong University, Jinan, People’s Republic of China
Correspondence: Lingyi Chi
Department of Neurosurgery, Qilu Hospital, Shandong University, No. 107 Wenhua West Road, Jinan City, Shandong Province, 250012, People’s Republic of China
Tel +86-18560085730
Email [email protected]
Yuying Zhao
Department of Neurology, Qilu Hospital, Shandong University, No. 107 Wenhua West Road, Jinan City, Shandong Province, 250012, People’s Republic of China
Tel +86-18560085530
Fax +86-531-82169217
Email [email protected]
Purpose: Leukodystrophies are frequently regarded as childhood disorders, but they can occur at any age, and the clinical and imaging patterns of the adult-onset form are usually different from the better-known childhood variants. Several reports have shown that various late-onset leukodystrophies, such as X-linked adrenoleukodystrophy and Krabbe disease, may present as spastic paraplegia with the absence of the characteristic white matter lesions on neuroimaging; this can be easily misdiagnosed as hereditary spastic paraplegia. The objective of this study was to investigate the frequency of late-onset leukodystrophies in patients with spastic paraplegia.
Patients and Methods: We performed genetic analysis using a custom-designed gene panel for leukodystrophies in 112 hereditary spastic paraplegia-like patients.
Results: We identified pathogenic mutations in 13 out of 112 patients, including five patients with adrenomyeloneuropathy, three with Krabbe disease, three with Alexander disease, and two with cerebrotendinous xanthomatosis. In terms of clinical manifestations, in addition to spastic paraplegia, three adrenomyeloneuropathy probands also had adrenocortical insufficiency, two Alexander disease probands developed urinary retention, one CTX proband developed cataracts and chronic diarrhea and the other presented with chronic diarrhea and mild tendon xanthomatosis. None of the patients had evidence of diffuse leukodystrophy on neuroimaging.
Conclusion: Patients with late-onset spastic paraplegia should be screened for underlying leukodystrophies, irrespective of the presence of additional complicating symptoms and neuroimaging abnormalities.
Keywords: targeted next-generation sequencing, pathogenic mutations, genetic causes, leukodystrophy frequency, magnetic resonance imaging
Introduction
Leukodystrophies are frequently regarded as disorders of childhood, but they can affect humans of any age, and the clinical and imaging patterns of the adult-onset form are usually different from the better-known childhood variants.1 In routine clinical practice, white matter lesions on magnetic resonance imaging (MRI) are frequently regarded as a hallmark of leukodystrophies; however, these lesions can be very mild or even absent in some late-onset patients. Several reports have shown that various late-onset leukodystrophies, such as X-linked adrenoleukodystrophy (ALD) and Krabbe disease (KD), may present as spastic paraplegia (SP) without leukodystrophy on neuroimaging and be easily misdiagnosed as hereditary spastic paraplegia (HSP) on clinical grounds.2–5
Currently, no effective treatment is available for HSP, but recent breakthroughs in the treatment of leukodystrophy, such as enzyme replacement therapy and hematopoietic stem cell transplantation,6–8 have provided more opportunities for improving the clinical outcome of patients with leukodystrophy. Therefore, the early differentiation of leukodystrophy from HSP is of great importance in improving patients’ prognosis and in providing early treatment opportunities for other pedigree members.
To investigate the frequency of late-onset leukodystrophies in patients with SP and to compare their phenotypic spectra with HSP in the Chinese population, in this study, 112 patients with late-onset spasticity lacking common HSP-causal genes and diffuse leukodystrophy on neuroimaging were screened for underlying genetic causes associated with leukodystrophies.
Patients and Methods
Patient Recruitment
Our cohort included 112 patients with progressive lower limb spasticity admitted to Qilu Hospital of Shandong University for diagnosis or management between January 2016 and June 2020. Written informed consent was obtained from all study participants for genetic analysis, and the study was carried out with institutional ethical approval.
The inclusion criteria were as follows: (1) age of onset of spastic gait ≥ 14 years; (2) evidence of a detailed examination, including blood tests, blood amino acid and fatty carnitine screening, urine organic acid screening, cerebrospinal fluid, and neuroimaging, to exclude other potential etiologies including metabolic derangements (including vitamin B12 and folic acid deficiency, hepatic myelopathy, methylmalonic acidemia, urea cycle disorders, and phenylketonuria), infection, inflammation, vascular abnormalities, and spinal cord compression; (3) brain MRI showed no evidence of diffuse white matter lesions; (4) no causative genes were found during the gene screening for SPG4 and SPG 11, which are the most common types of HSP. A flow chart describing the selection procedure of the cohort is shown in Supplementary Figure 1.
Targeted Next-Generation Sequencing
Firstly, a custom-designed probe library containing 130 known pathogenic genes reported to be associated with hereditary leukoencephalopathies was synthesized (Supplementary Table 1). Genomic DNA was extracted from peripheral blood lymphocytes and next-generation sequencing (NGS) was performed. The sets of variants were classified into “pathogenic”, “likely pathogenic”, “variants of uncertain significance” (VUS), “likely benign”, or “benign” according to guidelines from the American College of Medical Genetics and Genomics (ACMG). The “pathogenic” variants consistent with the clinical phenotypes were determined to be responsible for pathogenic mutations. Subsequently, three different web tools were used to predict the functional effects of the “likely pathogenic” and “VUS” variants: polyphen2 (http://genetics.bwh.harvard.edu/pph2/), Mutation Taster (http://www.Mutationtaster.org/), and SIFT (http://sift.jcvi.org/www/SIFT_enst_submit.html). The “likely benign” and “benign” variants were excluded because of their low pathogenic risk. Finally, candidate variants were confirmed by Sanger sequencing. Segregation tests were performed if the DNA of the proband’s immediate family member was available, and the final pathogenic mutations were determined in combination with the clinical information.
Biochemical Examination and Clinical Investigation
If the results were indicative of inborn metabolic disorders, we conducted additional biochemical examinations, including enzymatic activity and very long-chain fatty acids (VLCFA), to confirm the diagnosis. Clinical data, including neurological and psychiatric assessments, neuroimaging results, and neuro-electrophysiological features were collected to predict genotypic and phenotypic characteristics.
Results
We identified pathogenic mutations in 13 of the 112 patients, including five patients with adrenomyeloneuropathy (AMN), three with KD, three with Alexander disease (AxD), and two with cerebrotendinous xanthomatosis (CTX). Each proband’s variants and the predictions are listed in Table 1. Four probands with a positive family history were found through family investigation.
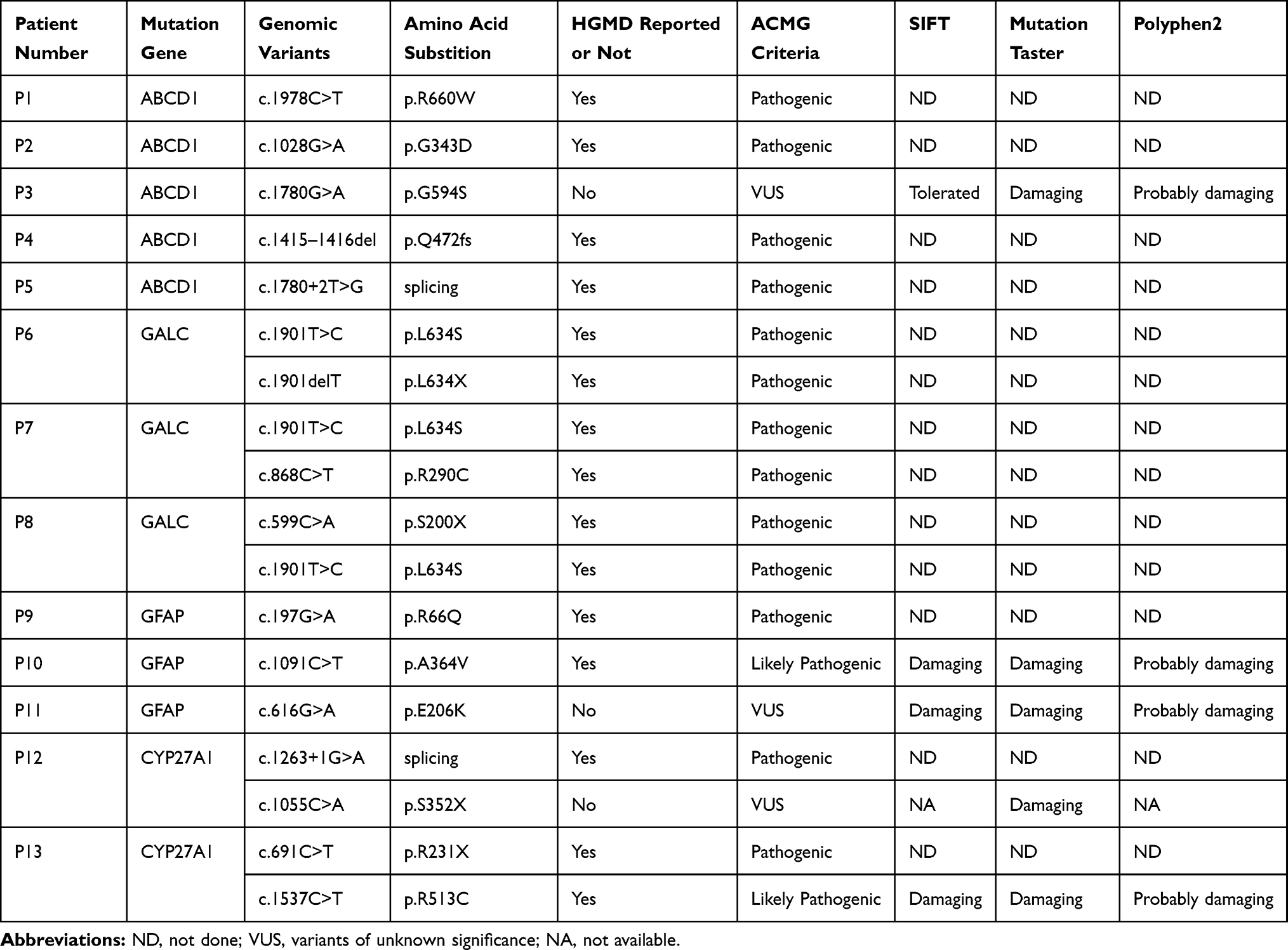 |
Table 1 Genomic Features of the 13 Patients |
Clinical Characteristics of the 13 Patients
Among the 13 patients, 11 were male and 2 were female. The age at onset of spasticity varied between 16 and 70 years old, with an average age of 39 years. Spastic paraplegia was the main finding in all patients, but in clinical and laboratory examinations, we found three AMN probands presenting with adrenocortical insufficiency characterized by elevated adrenocorticotropic hormone levels. Two AxD probands developed urinary retention. One CTX proband developed cataracts and chronic diarrhea, the other CTX proband presented with chronic diarrhea and mild tendon xanthomatosis.
Plasma VLCFA levels of AMN probands were high, while GALC enzymatic activity of KD probands was low, which were all consistent with the genetic diagnosis. The clinical and neuroimaging characteristics of the patients are shown in Table 2 and Figure 1, respectively.
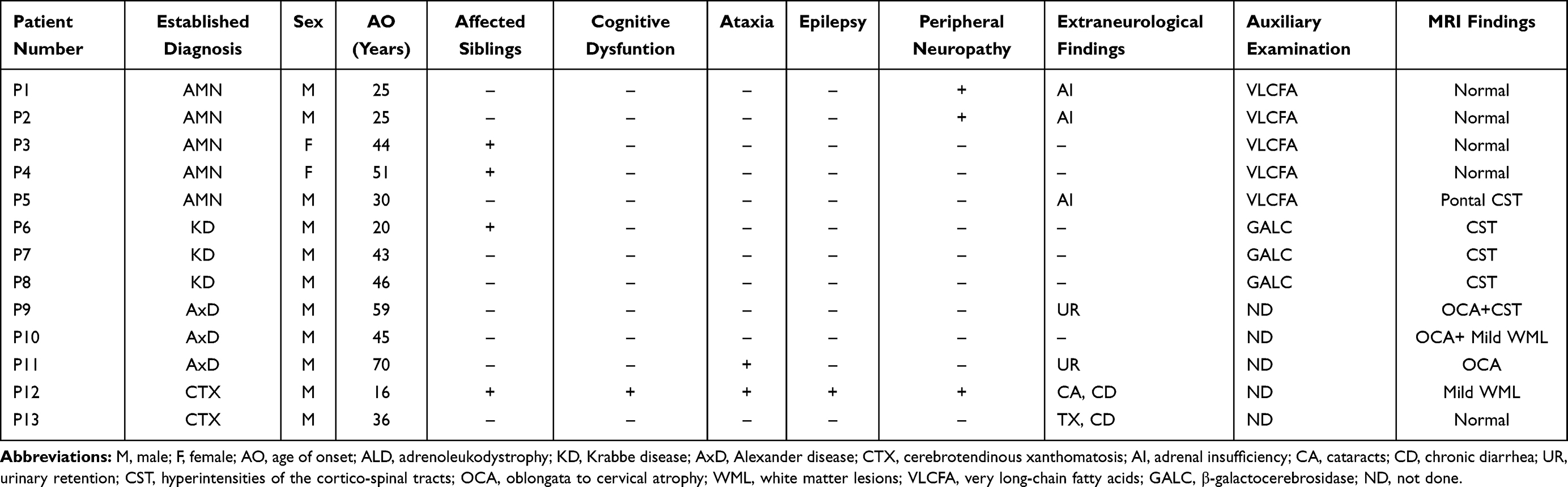 |
Table 2 Clinical Features of the 13 Patients |
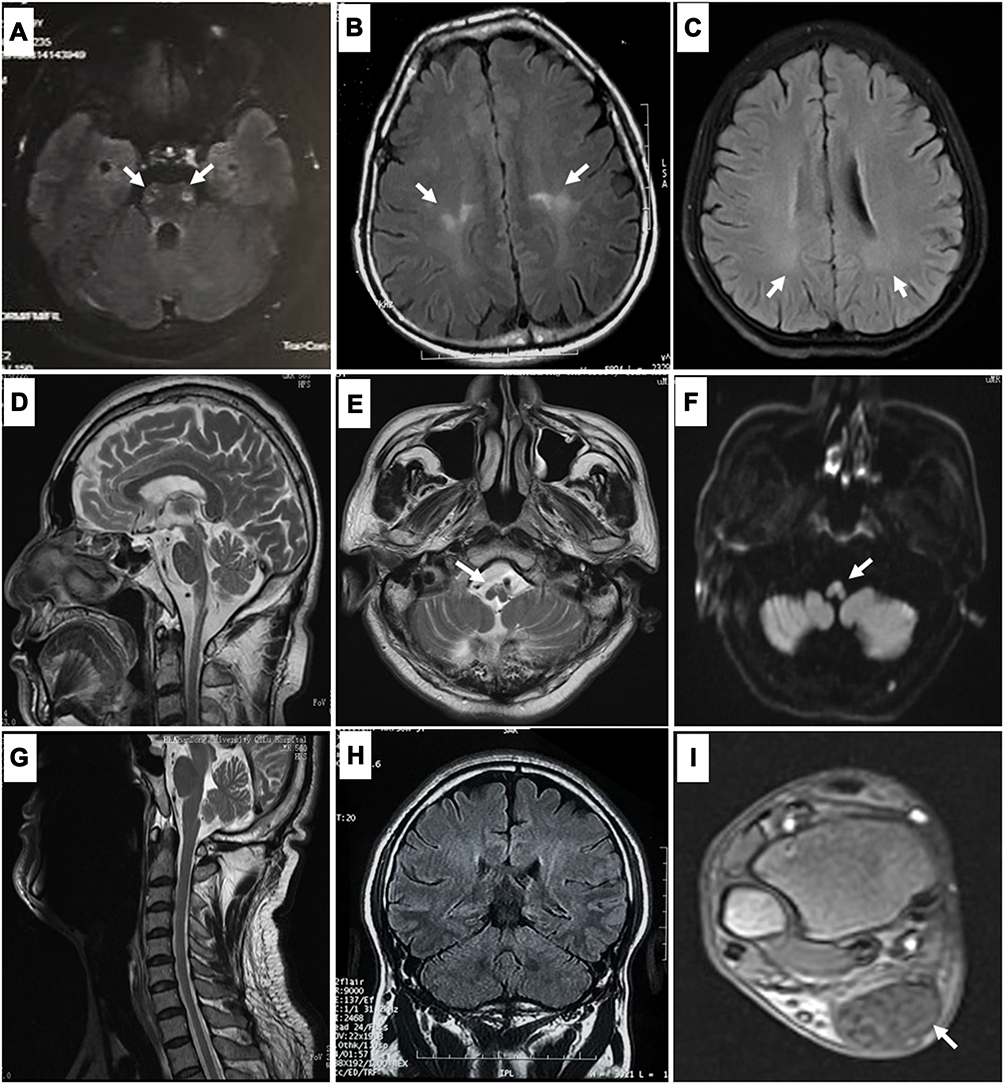 |
Figure 1 Magnetic resonance imaging (MRI) findings in patients with spastic paraplegia underlying leukodystrophy: Hyperintensities of the corticospinal tracts in patients with adrenomyeloneuropathy (AMN) ((A), patient P5 in Table 2) and Krabbe disease (KD) ((B), patient P8 in Table 2). Mild and nonspecific hyperintensities of the white matter are observed in a patient with cerebrotendinous xanthomatosis (CTX) ((C), patient P12 in Table 2). Atrophy from the medulla oblongata to the cervical spinal cord (D), with hyperintensities of the medulla oblongata on T2-weighted (E) and diffusion-weighted MRI (F), are observed in patients with Alexander disease (AxD) (patient P9 in Table 2). Cervical MRI (G) and coronal brain MRI (H) are normal, but enlargement of the Achilles tendons (I) is found in the patient with CTX (patient P13 in Table 2). |
Case Presentation
White matter changes on MRI are frequently regarded as a hallmark of leukodystrophies. However, the white matter lesions of the patients in our series were very mild or even absent. Therefore, “normal MRI appearance or mild white matter lesions” does not exclude leukodystrophy as a diagnosis. A more detailed description of the representative cases in this study is shown below, to increase awareness among neurologists about our study findings.
Case 1 (AMN, P3 in Table 2)
A 48-year-old woman presented with increasing stiffness in her legs, which initially affected the left leg prior to affecting both legs, and walking difficulty for four years. Her father had gait disturbance since he was 70 years old, while her brother and son were reported healthy.
On physical examination, she had a spastic gait, normal cranial nerve function, and hypertonia as well as hyperreflexia in the lower limbs, associated with extensor plantar responses and hyperreflexia in the upper limbs. No cerebellar signs were observed. Brain and spinal MRI and cerebrospinal fluid examination were normal (not shown).
NGS indicated a heterozygous c.1780G>A (p.G594S) mutation in the ABCD1 gene, which has never been reported in ALD/AMN patients. This variant has been predicted as “tolerated” by SIFT, “damaging” by Mutation Taster, “probably damaging” by PolyPhen2. The same mutation was found in her father and son, but not in her brother. To further confirm the diagnosis, plasma VLCFA level testing was performed in the proband, which exhibited a mildly elevated concentration of C26:0 and C26:0/C22:0 ratio. No adrenal insufficiency was found. Her father’s plasma VLCFA levels were also tested, which exhibited markedly raised C26:0 and C24:0 levels. The C24:0/C22:0 and C26:0/C22:0 ratios were also elevated. The patient’s son consented to a neurological examination and was found to have hyperreflexia in all four limbs but normal muscle strength. Therefore, we assumed that he was a subclinical AMN patient. The plasma VLCFA values, genomic analysis results, and the pedigree chart are shown in Figure 2.
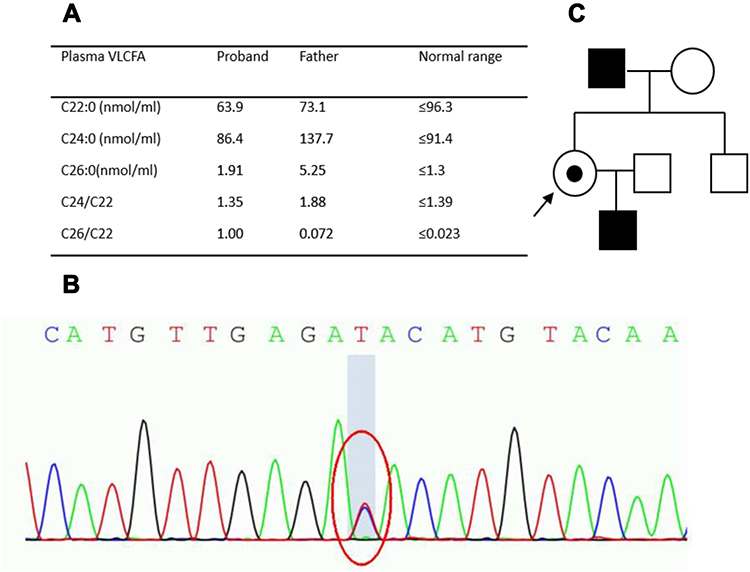 |
Figure 2 Plasma very long-chain fatty acid (VLCFA) values (A), genomic analysis results (B), and pedigree chart (C) of patient P3 in Table 2 (need color printing). |
Case 2 (KD, P8 in Table 2)
A 49-year-old man who experienced slowly progressive gait disturbance for three years was admitted to our hospital. His past medical history and family history were unremarkable. His parents were non-consanguineous. Neurological examination revealed spastic paraplegia, exaggerated patellar tendon reflexes, and positive Babinski and Chaddock signs. He exhibited no cognitive dysfunction, sensory loss, ataxia, or autonomic dysfunction.
Brain MRI revealed hyperintensity lesions along the upper parts of the bilateral pyramidal tracts in fluid-attenuated inversion recovery (FLAIR) images (Figure 1B). The NCS showed normal motor and sensory conduction velocities. NGS revealed two heterozygous mutations of the GALC gene, c.599C>A (p.S200X) and c.1901T>C (p.L634S), both have been reported as disease-causing.9,10 Lymphocyte GALC enzymatic activity decreased to 1.0 nmol/17 h/mg Pr (normal range, 19.0–68.2), so the patient received a diagnosis of KD.
Case 3 (AxD, P9 in Table 2)
This case describes a 64-year-old man referred to our hospital with a five-year history of progressive walking difficulties and dysuria. The patient’s family and personal clinical histories were unremarkable. Neurological examination revealed spastic gait with dysarthria, slight weakness of the left leg, and bilateral hyperactive deep tendon reflexes. The volume of residual urine in the patient’s bladder was 140 mL. Anal sphincter electromyography was normal. MRI revealed severe atrophy from the medulla oblongata to the cervical cord (Figure 1D). Interestingly, hyperintensity lesions with diffusion restriction were observed on DWI in the ventral part of the medulla oblongata, which remained prominent over five years in the follow-up MRI (Figure 1E and F). NGS revealed a heterozygous pathogenic mutation of c.197G>A (p.R66Q) in the GFAP gene.11
Case 4 (CTX, P13 in Table 2)
A 42-year-old man presented with a six-year history of leg weakness and gait disturbance. He had enlargement of tendons. His medical history included mild chronic diarrhea and surgery to excise an elbow lipoma. No relevant family history was reported. Neurological examination revealed hypertonia in his lower limbs, sustained ankle clonus, diffuse hyperreflexia, and extensor plantar responses associated with a pes cavus deformity. His upper limbs appeared normal except for symmetrically brisk tendon reflexes. Blood cholesterol level was normal, but the triglycerides were elevated (3.25 mmol/L, normal range 0.3–1.7 mmol/L). Spinal cord and brain MRI were all normal (Figure 1G and H). No abnormality was found in nerve conduction studies. Xanthomas were found in his bilateral Achilles tendons on MRI (Figure 1I). NGS revealed heterozygous mutations in c.691C>T (p.R231*) and c.1537C>T (p.R513C) in the CYP27A1 gene, which were previously reported.12,13
Discussion
Progressive spasticity of the lower limbs is a diagnostic challenge in adult neurology, especially when commonly acquired causes have been excluded. Leukodystrophies are frequently regarded as disorders of childhood with varied patterns of leukodystrophies in neuroimaging; as a result, they are rarely considered in the differential diagnosis of spasticity. However, leukodystrophies can appear at any age, and in late-onset patients, the white matter lesions can be very mild or even absent, in contrast to the better-known early-onset leukodystrophy variants. In our cohort study, leukodystrophies were found to account for approximately 11.6% (13/112) of cases of adult-onset lower limb spasticity with an unknown cause, which indicates that it is essential to include leukodystrophies in the differential diagnosis of adult-onset spasticity in order to increase the likelihood of making an accurate diagnosis.
ALD/AMN was the most frequent pathology encountered in our study (5/13). It should be noted that two female patients with AMN presented with isolated spastic paraplegia, without any additional complications in the clinical background or any abnormalities on MRI, which could have been easily misdiagnosed as pure HSP. Although ALD is an X-linked disease, it may manifest as spasticity in female carriers due to X–inactivation with a milder phenotype and later onset than affected males. ALD/AMN is usually diagnosed by elevated VLCFA concentrations and increased C24:0/C22:0 and C26:0/C22:0 ratios, but in 15–20% of affected female carriers, false-negative results occur.14 A relevant family history could indicate the diagnosis, but it is usually absent. Furthermore, adrenal involvement is usually absent and no specific findings are found on the brain and spinal MRI scans.15 Therefore, mutation screening of the ABCD1 gene is considered the most reliable method for the carriers’ diagnosis.16
Our cases show that KD may present with pure spasticity with age of onset after the fourth decade of life. Notably, the involvement of the pyramidal tract is characteristic in adult-onset KD, and its incidence as high as 82–94%, which is different from childhood-onset forms where signal changes of the Hilus of dentate and thalamus are usually observed.17–19 Besides, rare cases resembling HSP with normal neuroimaging have been reported.3 Therefore, lysosomal enzyme testing, especially galactocerebrosidase activity, as part of the standard workup of sporadic and autosomal recessive spastic paraplegia is a promising method to improve diagnostic accuracy with lower costs.
Adult-onset AxD is a rare leukodystrophy that typically presents with progressive spasticity, weakness, bulbar dysfunction, and other brainstem features. Palatal myoclonus is characteristic, as is its autonomic involvement, which mimics more common neurodegenerative disorders.11,20 Neither case in our series had palatal myoclonus or prominent ataxia; the common findings in these cases were progressive spasticity and bladder disturbance with residual urine in the bladder. Each patient was eventually diagnosed based on the characteristic MRI findings of severe atrophy from the medulla oblongata to the upper cervical cord; of note, the extensive cerebral white matter abnormalities often observed in infantile and juvenile cases were not common in our patients.21
In CTX, the typical MRI findings of symmetrical cerebral and cerebellar white matter lesions are expected. Spinal xanthomatosis is a rare CTX variant in which patients present with symptoms of corticospinal and dorsal column tract dysfunction with specific T2-weighted post-gadolinium MRI changes in the lateral and dorsal tracts of the spinal cord.22 However, as our cases demonstrate, nearly normal MRI appearances should not detract from a possible diagnosis of spinal xanthomatosis. The presence of non-neurological complications, such as early-onset cataracts (present in 96% of patients), tendon xanthomas (50%), and chronic diarrhea (35%) are indicative but not present in all patients.23 The diagnosis should be confirmed by measuring serum cholestenol and urinary bile alcohol levels as well as molecular genetic analysis for mutations in CYP27A1.
To the best of our knowledge, this is the first study to use targeted NGS for leukodystrophies in the late-onset “HSP-mimic” population in China. The ABCD1, GALC, GFAP, and CYP27A1 genes should be regularly included in the HSP panel to increase diagnostic accuracy. The introduction of NGS to screen for genetic disorders associated with progressive SP will allow an earlier diagnosis and treatment of these patients and will be particularly useful in atypical cases such as those described in our article.
Conclusion
Spastic paraplegia is frequently encountered in adult neurology with heterogeneous causes. After the elimination of common acquired causes, genetic causes must be sought, and “normal MRI appearances or mild white matter lesions” should not detract from a possible diagnosis of leukodystrophy, especially AMN, KD, AxD, and CTX, as described in our series. Plasma VLCFA, serum cholestenol levels, and lysosomal enzyme activity should be included in the routine test of HSP-like patients and the ABCD1, GALC, GFAP, and CYP27A1 genes should be regularly included in the HSP genetic panel. This could greatly increase diagnostic accuracy and expand our knowledge of the genetic and clinical spectra of spasticity and leukoencephalopathy.
Ethics Approval and Informed Consent
This study was conducted in accordance with the Declaration of Helsinki and approved by the ethics committee of Qilu Hospital, Shandong University. Written informed consent was obtained from all subjects in this study for publication of the case details and any accompanying images.
Acknowledgments
The study was supported by the Taishan Scholars Program of Shandong Province. We would also like to thank the patients and their families for their participation in this study.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bonkowsky JL, Nelson C, Kingston JL, et al. The burden of inherited leukodystrophies in children. Neurology. 2010;75:718–725. doi:10.1212/WNL.0b013e3181eee46b
2. Muller Vom Hagen J, Karlea KN, Schule R, et al. Leukodystrophies underlying cryptic spastic paraparesis: frequency and phenotype in 76 patients. Eur J Neurol. 2014;21:983. doi:10.1111/ene.12423
3. Bajaj NPS, Waldman A, Orrell R, et al. Familial adult onset of Krabbe’s disease resembling hereditary spastic paraplegia with normal neuroimaging. J Neurol Neurosurg Psychiatry. 2002;72:635–638. doi:10.1136/jnnp.72.5.635
4. Bargiela D, Eglon G, Horvath R, Chinnery PF. An under-recognised cause of spastic paraparesis in middle aged women. Pract Neurol. 2014;14:182–184. doi:10.1136/practneurol-2013-000662
5. Chen Y, Wang M, Dong E, et al. Chinese patients with adrenoleukodystrophy and Zellweger spectrum disorder presenting with hereditary spastic paraplegia. Parkinsonism Relat Disord. 2019;65:256–260. doi:10.1016/j.parkreldis.2019.06.008
6. Costello DJ, Eichler AF, Eichler FS. Leukodystrophies classification, diagnosis and treatment. Neurologist. 2009;15:319–328. doi:10.1097/NRL.0b013e3181b287c8
7. Kohler W. Leukodystrophies with late disease onset: an update. Curr Opin Neurol. 2010;23:234–241. doi:10.1097/WCO.0b013e328338313a
8. Peters C, Steward CG, Inborn NIWP. Hematopoietic cell transplantation for inherited metabolic diseases: an overview of outcomes and practice guidelines. Bone Marrow Transplant. 2003;31:229–239. doi:10.1038/sj.bmt.1703839
9. Yang Y, Ren X, Xu Q, et al. Four novel GALC gene mutations in two Chinese patients with Krabbe disease. Gene. 2013;519:381–384. doi:10.1016/j.gene.2013.02.010
10. Furuya H, Kukita Y, Nagano S, et al. Adult onset globoid cell leukodystrophy (Krabbe disease): analysis of galactosylceramidase cDNA from four Japanese patients. Hum Genet. 1997;100:450–456. doi:10.1007/s004390050532
11. Prust M, Wang J, Morizono H, et al. GFAP mutations, age at onset, and clinical subtypes in Alexander disease. Neurology. 2011;77(13):1287–1294. doi:10.1212/WNL.0b013e3182309f72
12. Garuti R, Croce MA, Tiozzo R, et al. Four novel mutations of sterol 27-hydroxylase gene in Italian patients with cerebrotendinous xanthomatosis. J Lipid Res. 1997;38:2322–2334. doi:10.1016/S0022-2275(20)34946-4
13. Chen C, Yue Z, Hui W, et al. Clinical and molecular genetic features of cerebrotendinous xanthomatosis patients in Chinese families. Metab Brain Dis. 2017;32:1609–1618. doi:10.1007/s11011-017-0047-8
14. Moser HW, Mahmood A, Raymond GV. X-linked adrenoleukodystrophy. Nat Clin Pract Neurol. 2007;3:140–151. doi:10.1038/ncpneuro0421
15. el-Deiry SS, Naidu S, Blevins LS, Ladenson PW. Assessment of adrenal function in women heterozygous for adrenoleukodystrophy. J Clin Endocrinol Metab. 1997;82:856–860. doi:10.1210/jcem.82.3.3802
16. Deon M, Sitta A, Barschak AG, et al. Oxidative stress is induced in female carriers of X-linked adrenoleukodystrophy. J Neurol Sci. 2008;266:79–83. doi:10.1016/j.jns.2007.08.043
17. Debs R, Froissart R, Aubourg P, et al. Krabbe disease in adults: phenotypic and genotypic update from a series of 11 cases and a review. J Inherit Metab Dis. 2013;36:859–868. doi:10.1007/s10545-012-9560-4
18. Farina L, Bizzi A, Finocchiaro G, et al. MR imaging and proton MR spectroscopy in adult Krabbe disease. AJNR Am J Neuroradiol. 2000;21:1478–1482.
19. Abdelhalim AN, Alberico RA, Barczykowski AL, Duffner PK. Patterns of magnetic resonance imaging abnormalities in symptomatic patients with Krabbe disease correspond to phenotype. Pediatr Neurol. 2014;50:127–134. doi:10.1016/j.pediatrneurol.2013.10.001
20. Farina L, Pareyson D, Minati L, et al. Can MR imaging diagnose adult-onset Alexander disease? AJNR Am J Neuroradiol. 2008;29:1190–1196. doi:10.3174/ajnr.A1060
21. Davide P, Roberto F, Caterina M, et al. Adult-onset Alexander disease: a series of eleven unrelated cases with review of the literature. Brain. 2008;131:2321–2331. doi:10.1093/brain/awn178
22. Verrips A, Nijeholt GJ, Barkhof F, et al. Spinal xanthomatosis: a variant of cerebrotendinous xanthomatosis. Brain. 1999;122:1589–1595. doi:10.1093/brain/122.8.1589
23. Verrips A, Hoefsloot LH, Steenbergen GC, et al. Clinical and molecular genetic characteristics of patients with cerebrotendinous xanthomatosis. Brain. 2000;123:908–919. doi:10.1093/brain/123.5.908
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.