Back to Journals » OncoTargets and Therapy » Volume 9
Lapatinib induces autophagic cell death and differentiation in acute myeloblastic leukemia
Authors Chen Y ![]() , Fang L, Su W, Hsu W, Yang K, Huang H
, Fang L, Su W, Hsu W, Yang K, Huang H
Received 2 February 2016
Accepted for publication 3 May 2016
Published 20 July 2016 Volume 2016:9 Pages 4453—4464
DOI https://doi.org/10.2147/OTT.S105664
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Yu-Jen Chen,1–4 Li-Wen Fang,5 Wen-Chi Su,6,7 Wen-Yi Hsu,1 Kai-Chien Yang,1 Huey-Lan Huang8
1Department of Medical Research, 2Department of Radiation Oncology, Mackay Memorial Hospital, 3Institute of Traditional Medicine, School of Medicine, National Yang-Ming University, 4Institute of Pharmacology, Taipei Medical University, Taipei, 5Department of Nutrition, I-Shou University, Kaohsiung, 6Research Center for Emerging Viruses, China Medical University Hospital, 7Graduate Institute of Clinical Medical Science, China Medical University, Taichung, 8Department of Bioscience Technology, College of Health Science, Chang Jung Christian University, Tainan, Taiwan, Republic of China
Abstract: Lapatinib is an oral-form dual tyrosine kinase inhibitor of epidermal growth factor receptor (EGFR or ErbB/Her) superfamily members with anticancer activity. In this study, we examined the effects and mechanism of action of lapatinib on several human leukemia cells lines, including acute myeloid leukemia (AML), chronic myeloid leukemia (CML), and acute lymphoblastic leukemia (ALL) cells. We found that lapatinib inhibited the growth of human AML U937, HL-60, NB4, CML KU812, MEG-01, and ALL Jurkat T cells. Among these leukemia cell lines, lapatinib induced apoptosis in HL-60, NB4, and Jurkat cells, but induced nonapoptotic cell death in U937, K562, and MEG-01 cells. Moreover, lapatinib treatment caused autophagic cell death as shown by positive acridine orange staining, the massive formation of vacuoles as seen by electronic microscopy, and the upregulation of LC3-II, ATG5, and ATG7 in AML U937 cells. Furthermore, autophagy inhibitor 3-methyladenine and knockdown of ATG5, ATG7, and Beclin-1 using short hairpin RNA (shRNA) partially rescued lapatinib-induced cell death. In addition, the induction of phagocytosis and ROS production as well as the upregulation of surface markers CD14 and CD68 was detected in lapatinib-treated U937 cells, suggesting the induction of macrophagic differentiation in AML U937 cells by lapatinib. We also noted the synergistic effects of the use of lapatinib and cytotoxic drugs in U937 leukemia cells. These results indicate that lapatinib may have potential for development as a novel antileukemia agent.
Keywords: lapatinib, autophagic cell death, leukemia, differentiation, AML
Introduction
Acute myeloid leukemia (AML) is a group of heterogeneous diseases with abnormally active proliferation of hematopoietic precursors, which block patients’ normal hematopoiesis, causing neutropenia and anemia.1 Although more than 50% of adult AML patients show complete remission with conventional chemotherapeutic drugs, only 20%–30% of patients show long-term disease-free survival.2 Despite being associated with complicated genetic mutations, ~50% of AML cases correlate with mutations caused by the aberrant activation of receptor tyrosine kinases (eg, FLT3 and c-KIT) or downstream effectors (eg, Ras).1–3 Thus, in recent years, some small-molecule inhibitors, for example, sorafenib, gefitinib, erlotinib, and quizartinib (AC220) have been tested in preclinical studies as targeted therapies for AML.1,4,5
Lapatinib (GlaxoSmithKline plc, London, UK) is a small-molecule inhibitor against the receptor tyrosine kinase of epidermal growth factor receptor (EGFR) or ErbB/Her family members.6–8 EGFR/ErbB/Her superfamily members, including EGFR/ErbB1/Her1, ErbB2/Her2/Neu, ErbB3/Her3, and ErbB4/Her4, are common targets for anticancer drugs because their functions are commonly dysregulated in many cancers.9–11 Lapatinib, which is taken orally, was originally approved by the US Food and Drug Administration in combination with capecitabine for advanced breast cancers that have developed resistance to the anti-ErbB2 monoclonal antibody trastuzumab (Herceptin; Genentech, Inc., South San Francisco, CA, USA) or other drugs.6,10,12,13
Differentiation therapies play important roles in leukemia treatment. Differentiation therapies in oncology include changing malignant tumors to curable tumors or terminally differentiated cells that undergo no further proliferation.14 This includes the induction of cancer cell differentiation and tumor eradication by either stopping tumor cell proliferation or increasing drug sensitivity after differentiation. One successful method of differentiation therapy is treating acute promyelocytic leukemia (APL) with all-trans retinoic acid. All-trans retinoic acid changes malignant leukemia to treatable cell forms by inducing differentiation of these leukemia cells into drug-sensitive cells. Other drugs that induce differentiation include guanosine triphosphate, herbimycin A, and butyrates for erythroid differentiation of chronic myelogenous leukemia (CML),14–16 and 12-O-tetradecanoylphorbol-13-acetate (TPA), HIV-tat protein, thrombopoietin, and dimethyl sulfoxide (DMSO) for the megakaryocytic differentiation of CML or APL.17–20
In our previous studies, we found that lapatinib induced autophagic cell death in CML and hepatoma.21,22 In this study, we focused on lapatinib-induced cytotoxicity mechanisms in AML cells. We found that lapatinib inhibits the growth of several leukemia cell lines in a dose- and time-dependent manner via either autophagic or apoptotic cell death depending on the different cell types. The induction of autophagic cell death and macrophagic differentiation by lapatinib has been demonstrated in U937 cells. We also found a synergistic effect of lapatinib and other cytotoxic drugs in AML U937 cells. The results of our study suggest that lapatinib may be a potentially useful therapy for leukemia.
Materials and methods
Cell culture
The following human leukemia cell lines were used in our study: AML-derived HL-60 and U937 cells,23 CML-derived MEG-01 and KU812 cells, APL-derived NB4 cells,22 and Jurkat T lymphoma. These cell lines were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (Hyclone, Logan, UT, USA) and penicillin-streptomycin (Thermo Fisher Scientific, Waltham, MA, USA) at 37°C under 5% CO2. Genetic recombination experiments in our study were approved by the Biosafety Committee of Chang Jung Christian University.
Cell treatments
Lapatinib (GlaxoSmithKline) or erlotinib (Hoffman-La Roche Ltd., Basel, Switzerland) tablets were ground and dissolved in DMSO (Sigma-Aldrich Co., St Louis, MO, USA) to prepare a 1,000-fold stock solution. Autophagy inhibitor 3-methyladenine (3-MA) was dissolved in an RPMI 1640 medium. The pan caspase inhibitor Z-VAD-fmk (R&D Systems, Inc., Minneapolis, MN, USA), etoposide (Sigma-Aldrich Co.), paclitaxel (Sigma-Aldrich Co.), and autophagy inducer thapsigargin (Sigma-Aldrich Co.) at concentrations of 20 mM, 1 μM, 25 μM, and 150 μM, respectively, were dissolved in DMSO to prepare a 1,000-fold stock solution. Treatment with protein kinase C activator TPA (Sigma-Aldrich Co.) was used as the positive control for the differentiation of leukemia cells.24 The 10,000-fold TPA stock solution (1 mM) was prepared in DMSO.
Viability analysis
After treatments, cell viability was analyzed using the trypan blue assay (Thermo Fisher Scientific) or the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) assay (Promega Corporation, Fitchburg, WI, USA) according to the manufacturer’s instructions. Cell numbers or optical density (OD) values of the control cells (DMSO vehicle-treated) were calculated as 100% viability. Data from triplicate (or more) experiments were presented as mean ± standard deviation (SD). P-values (P<0.05, P<0.01, P<0.001) were calculated by Student’s t-test between lapatinib-treated and DMSO control cells or as indicated.
Observation of morphology and LC3 aggregation by microscopy
After treatments with drugs for 3 days, the cells were centrifuged on slides using cytospin apparatus (Thermo Fisher Scientific), and were fixed and stained with Liu’s stain (Muto Pure Chemicals, Tokyo, Japan) according to the manufacturer’s instructions. Cell morphology was observed under a microscope. For the observation of LC3 aggregation, the stable expression of GFP-LC3 in U937 leukemia cells was induced by the transduction of a GFP-LC3-containing lentivirus and blasticidin selections as described previously.25
Flow cytometry
For the detection of apoptotic cells with DNA laddering (hypodiploid or sub-G1 phase) by flow cytometry (FACSCalibur; Becton Dickinson, Franklin Lakes, NJ, USA), treated cells were spun and resuspended in a hypotonic buffer (0.1% sodium citrate, 0.1% Triton X-100) with 5 μg/mL propidium iodide (Sigma-Aldrich Co.). Total dead cells (propidium iodide-positive cells) and apoptotic cells were simultaneously detected by flow cytometry.21,22 The percentage of acridine orange-positive cells was detected after cells were collected and stained with acridine orange (10 μg/mL, Sigma-Aldrich Co.) for 15 minutes. For determining the percentage of CD14- or CD68-positive cells, the cells were collected and stained with 0.5 μg/mL antibodies against CD14 (BD Pharmingen, San Diego, CA, USA) or CD68 (Dako, Hamburg, Germany) at 4°C for 20 minutes and stained with Alexa Fluor 488-conjugated anti-Mouse IgG secondary antibody (Thermo Fisher Scientific) at 4°C for another 20 minutes. After washing with phosphate-buffered saline (PBS), CD14- or CD68-positive cells were measured and analyzed by flow cytometry. For phagocytosis analysis, cells were treated, collected, washed with PBS, and mixed with green fluorescent latex beads (Sigma-Aldrich Co.), and then were split into two tubes. One tube was incubated on ice as a control, while the other tube was incubated at 37°C for 2 hours. After washing with PBS, fluorescent beads containing cells (with phagocytotic ability) were analyzed by flow cytometry. For the detection of intracellular reactive oxygen species (ROS) production, cells were treated with DMSO or different concentrations of lapatinib for 24 hours, and then incubated at 37°C for 30 minutes with a medium containing 2 μM 2′,7′-dichlorofluorescein diacetate (Molecular Probes, Eugene, OR, USA) for cytosolic hydrogen peroxide detection. ROS production was then measured by flow cytometry. All the flow cytometric data were analyzed using FlowJo software (Tree Star, Inc., Ashland, OR, USA).
Western blot analysis
After the drug treatment, U937 cells were spun and lysed in lysis buffer (Sigma-Aldrich Co.) with protease inhibitors (Hoffman-La Roche Ltd.). Cell lysates with 50 μg of protein were subjected to 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, followed by transferring to polyvinylidene fluoride membranes for immunoblotting. The following antibodies were used for immunoblotting: actin (Sigma-Aldrich Co.), ATG5 (Cell Signaling, Beverly, MA, USA), Beclin-1 (or ATG6, Cell Signaling), ATG7 (Santa Cruz Biotechnology Inc., Dallas, TX, USA), LC3 (or ATG8; Abgent, San Diego, CA, USA), and BNIP (Abcam, Cambridge, UK).
Transmission electron microscopy
After treatment, U937 cells were spun and then fixed with a 2.5% glutaraldehyde-containing cacodylate buffer (pH 7.4). Then the cells were stained with 1% osmium tetroxide, dehydrated, and embedded in Spurr’s resin according to the manufacturer’s instructions (Electron Microscopy Science, Fort Washington, PA, USA). The cell morphology was observed by transmission electron microscopy (JEM-1200EXII; JEOL, Tokyo, Japan).
Knockdown of autophagy-related genes by shRNA expression system
A lentiviral short hairpin RNA (shRNA) expression system from the RNAi Consortium (TRC) was applied to knockdown the expression of the autophagy-related genes: ATG5, ATG7 (clone #1 or #2, TRCN0000007584 or TRCN0000007587, respectively), Beclin-1 (or ATG6, clone #1 or #2, TRCN0000033549 or TRCN0000033552, respectively), and RFP (as nontargeting shRNA control) as described previously.21 We obtained these pLKO.1-shRNA constructions from the National RNAi Core Facility in Taiwan. The shRNA construct preparation and lentiviral production and infection were performed according to the TRC and the Core Facility protocol as described previously.21,22 After puromycin selection for at least 1 week, cells were treated with lapatinib, and cell viability was validated by trypan blue and MTS assays as described earlier.
Results
Lapatinib-induced apoptotic and nonapoptotic cell death in several kinds of leukemia cells
In this study, we first performed a comparison of the effect of lapatinib on several leukemia cell lines. We used both MTS and trypan blue assays for viability validation. As shown in Figure 1, lapatinib reduced cell viability in a dose- and time-dependent manner in AML-derived U937, ALL Jurkat T, and CML-derived KU812 leukemia cells. Compared to other leukemia cell lines, KU812 is less sensitive to lapatinib-induced cytotoxicity. We also found lapatinib-induced mitochondrial collapse in U937 cells (Figure S1). These findings in relation to cell viability reduction and mitochondrial collapse are consistent with the cytotoxic effects of lapatinib in CML-derived K562 and MEG-01 and AML-derived HL-60 and NB4 cells observed in our previous study.22
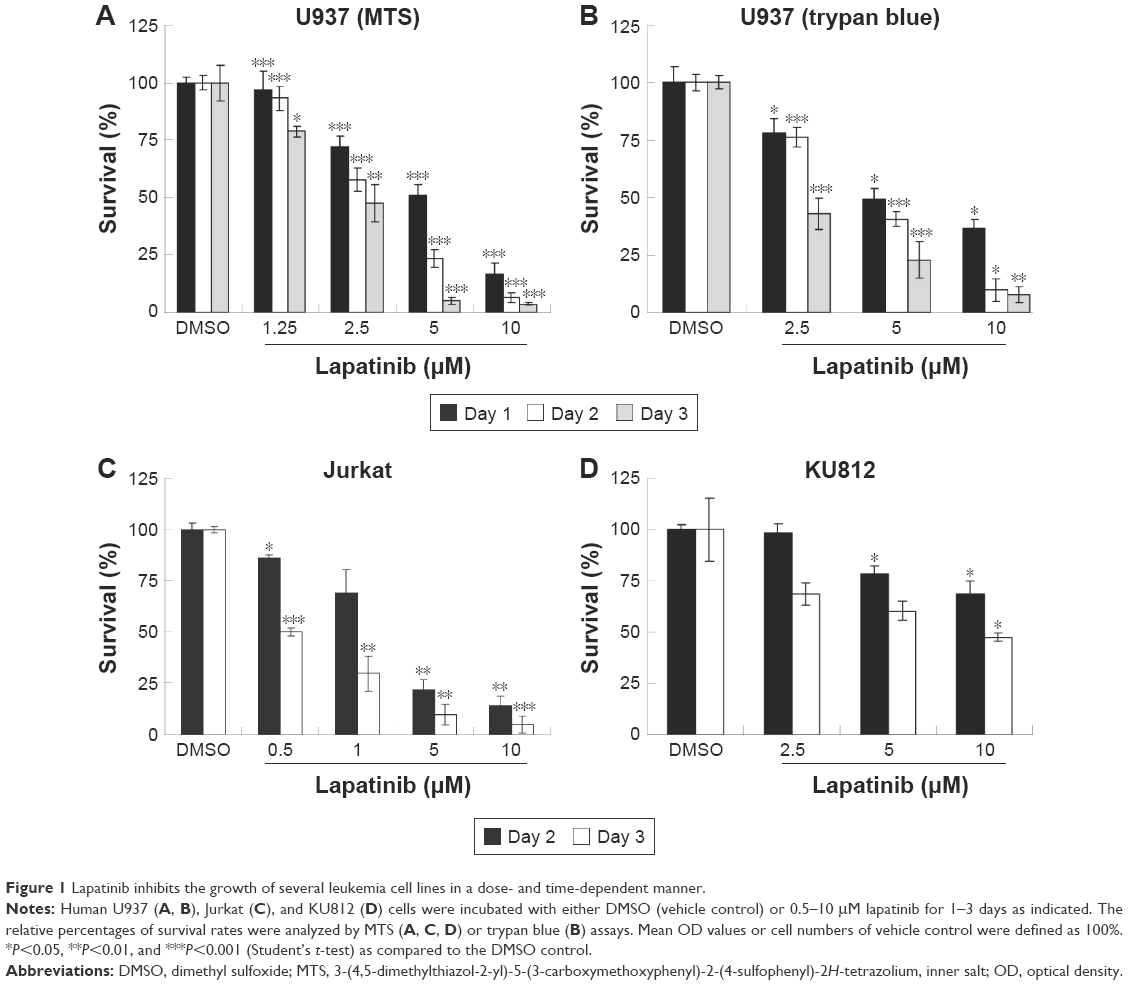 | Figure 1 Lapatinib inhibits the growth of several leukemia cell lines in a dose- and time-dependent manner. |
We then tested the mechanisms involved in lapatinib-induced cytotoxicity. We found a dramatic induction of apoptosis by lapatinib in HL-60 cells, and also induction of apoptosis and autophagic cell death in K562 cells. Approximately, 30% of the lapatinib-treated K562 cells showed DNA fragmentation.22 As shown in Figure 2A, in lapatinib-treated U937 cells, growth inhibition was mainly caused due to the presence of high percentages of dead cells, in contrast to the low percentages of sub-G1 cells. The low percentages of sub-G1 cells suggest nonapoptotic cell death in lapatinib-treated U937 cells. These data are similar to those obtained for CML-derived MEG-01 (Figure 2) and K562 cells.22 However, lapatinib induced production of massive amounts of sub-G1 cells in NB4 and Jurkat T cells (Figure 2E and F), like in AML-derived HL-60 cells.22 In addition, we confirmed the induction of apoptosis by lapatinib in U937 and Jurkat cells in the presence or absence of the broad-spectrum caspase inhibitor Z-VAD-fmk. As shown in Figure 2C and F, the percentages of sub-G1 cells were effectively reduced by the caspase inhibitor Z-VAD-fmk in Jurkat T cells but not in U937 cells. In agreement, we found that Z-VAD-fmk could not protect most of the U937 cells from lapatinib-induced cell death (Figure 2B), suggesting nonapoptotic cell death in lapatinib-treated U937 cells. Thus, according to our results (Figure 2, and our previous study),22 lapatinib induced apoptosis in AML HL-60, APL NB4, Jurkat T lymphoma, and small portions of CML K562 cells, but induced nonapoptotic cell death in the majority of K562, U937, and CML MEG-01 cells.
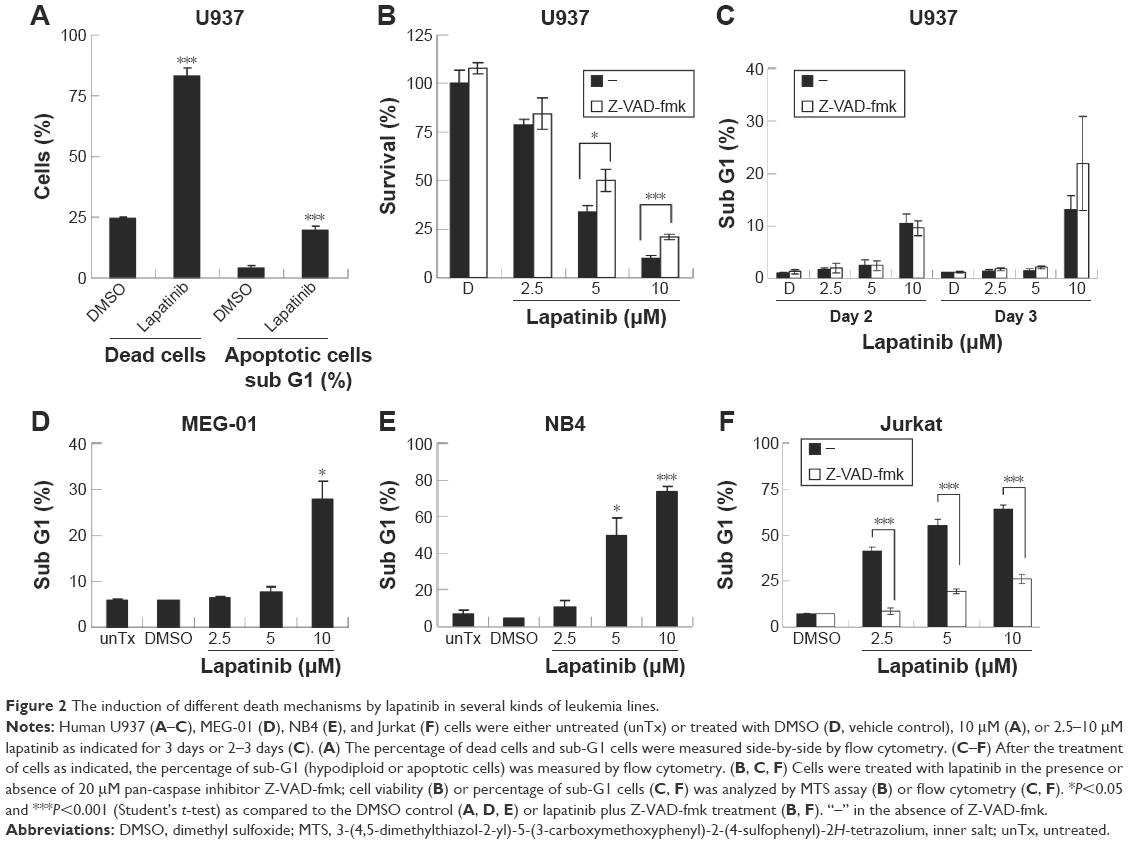 | Figure 2 The induction of different death mechanisms by lapatinib in several kinds of leukemia lines. |
Autophagic cell death in lapatinib-treated AML U937 leukemia cells
Since lapatinib effectively induces nonapoptotic cell death in AML U937 cells, we wondered whether lapatinib induced autophagic cell death, the second type of programmed cell death in U937 cells. As shown in Figure 3A, after treatment with lapatinib, massive cells with vacuoles (autophagic morphology) were observed. These morphological data were similar to those obtained for cells treated with the differentiation inducer TPA.24 To further confirm lapatinib-induced autophagic cell death in U937 cells, we used the GFP-LC3 overexpression system and fluorescence microscopy.26,27 In U937 cells, we detected cells with punctuated GFP-LC3 (LC3 aggregates) after lapatinib or autophagy inducer thapsigargin treatment (Figure 3B).
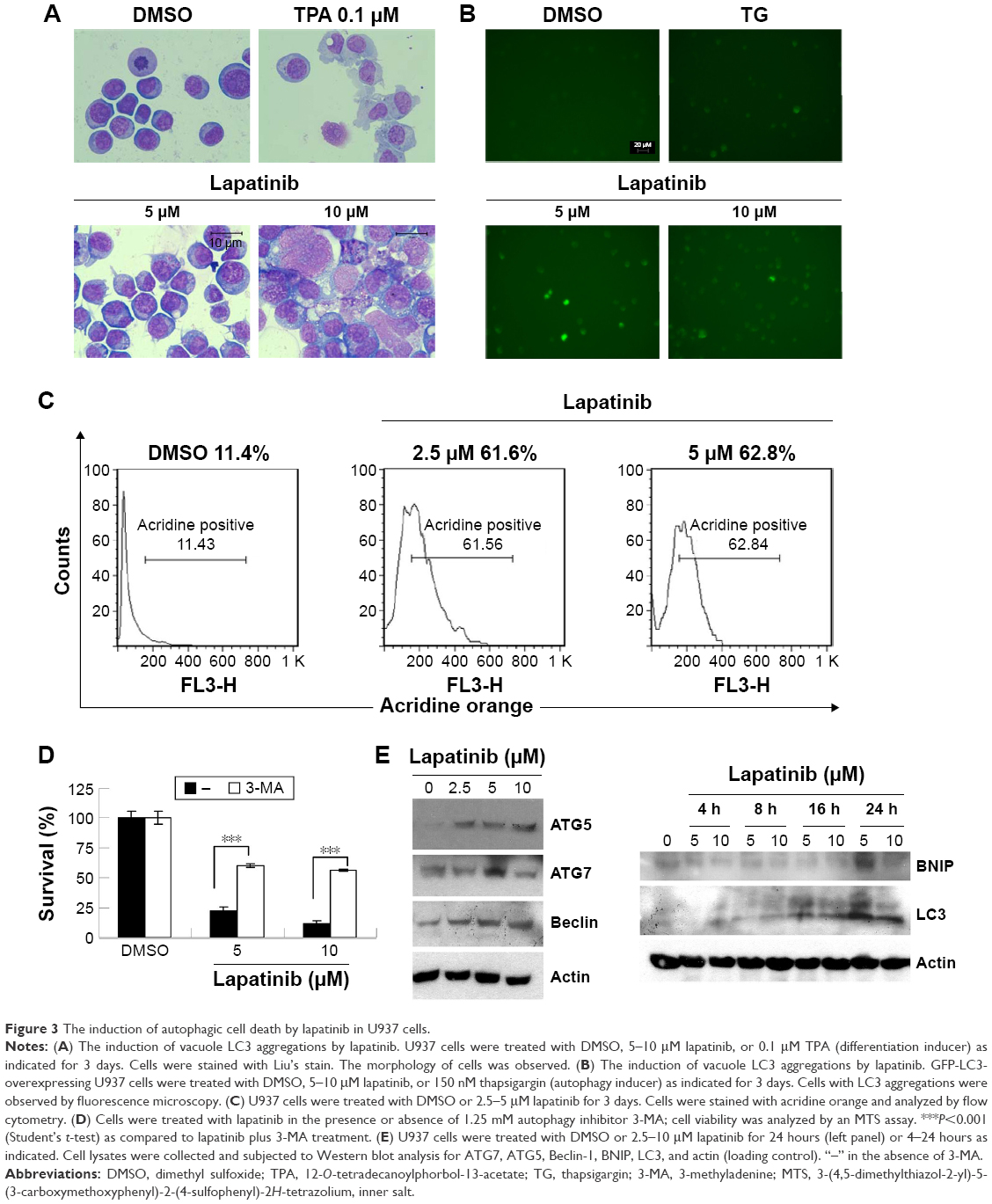 | Figure 3 The induction of autophagic cell death by lapatinib in U937 cells. |
To further confirm the autophagic cell death of U937 cells, we used acridine orange staining. As shown in Figure 3C, high numbers of acridine orange-positive cells were detected in lapatinib-treated cells. We also determined whether lapatinib-induced cytotoxicity was blocked in the presence of the autophagy inhibitor 3-MA. As shown in Figure 3D, 3-MA effectively blocked lapatinib-induced cytotoxicity in U937 cells. In addition, we checked the expression levels of these autophagy-related proteins. As shown in Figure 3E, we found a clear increase in Beclin-1 (ATG6), ATG5, ATG7, LC3, and BNIP in lapatinib-treated leukemia by immunoblotting.
Upon further examination of lapatinib-induced autophagic cell death by transmission electronic microscope (TEM), we found massive cells with vacuoles enclosing a lot of unhealthy mitochondria (Figure 4). Furthermore, we performed autophagy inhibition by using U937 cells with shRNA expression corresponding to autophagy regulatory proteins ATG7, ATG5, and Beclin-1 (Figure S2). U937 leukemia cells were transducted with shRNAs containing plasmids from TRC as described in the “Materials and methods” section and previous reports.21,22 As shown in Figure 5, the expression of shRNA against ATG7, ATG5, and Beclin-1 (but not nonspecific shRNA against RFP) inside the cells dramatically decreased lapatinib sensitivity according to examination by the MTS (Figure 5A) and trypan blue assays (Figure 5B). These data, showing the expression of shRNA, further confirmed lapatinib-induced autophagic cell death in AML U937 cells.
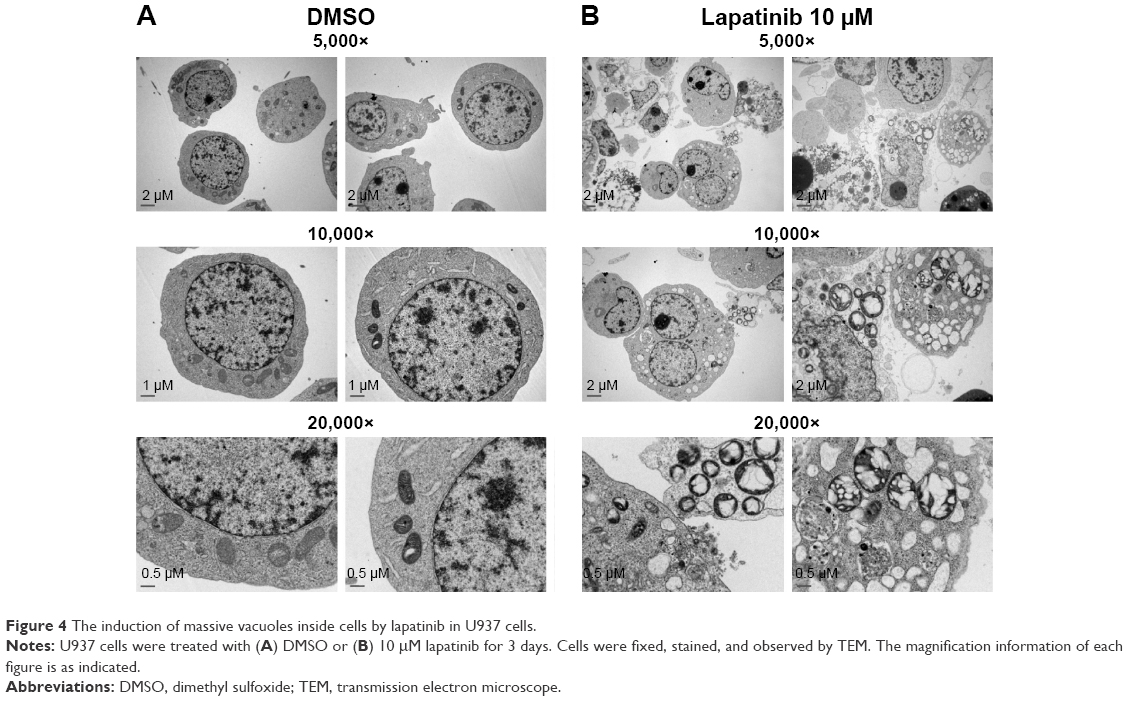 | Figure 4 The induction of massive vacuoles inside cells by lapatinib in U937 cells. |
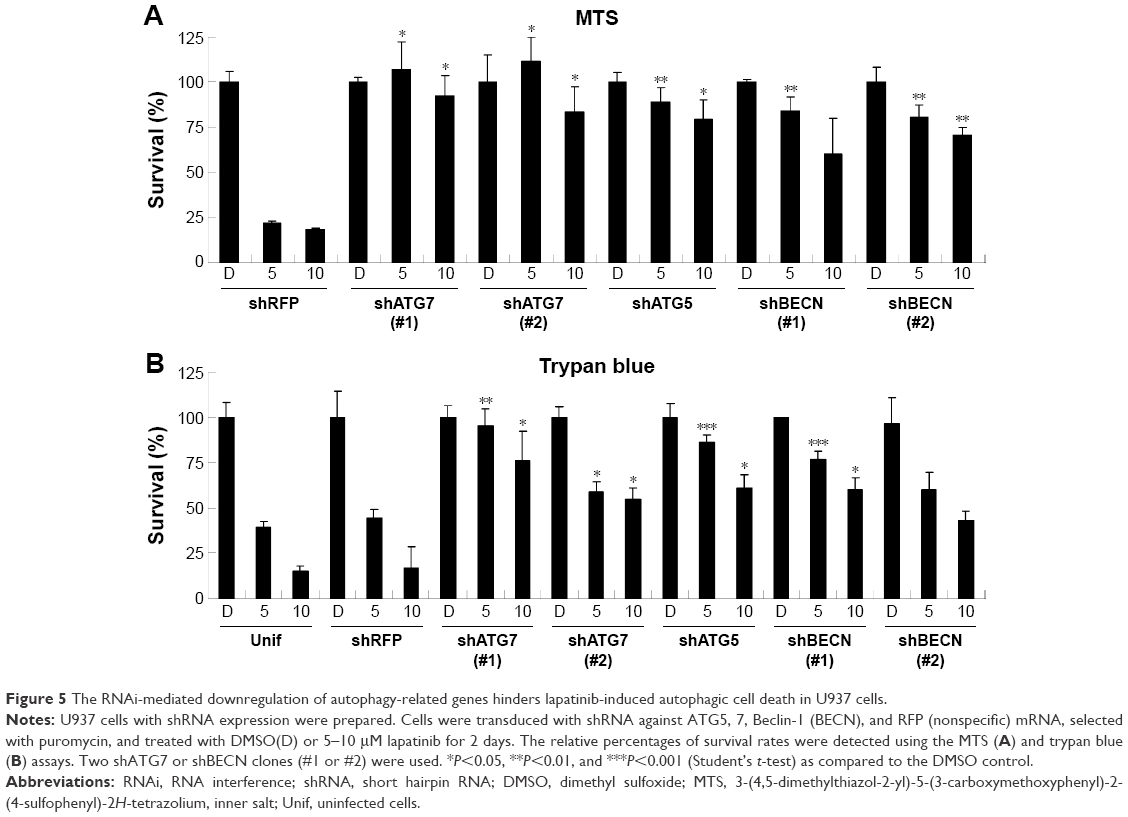 | Figure 5 The RNAi-mediated downregulation of autophagy-related genes hinders lapatinib-induced autophagic cell death in U937 cells. |
Differentiation induction by lapatinib in AML leukemia cells
Since we have previously demonstrated the induction of megakaryocytic differentiation marker CD61 by lapatinib in K562 cells,22 we determined whether lapatinib could also cause differentiation in U937 cells. As shown in Figure 6A and B, we found lapatinib induced the upregulation of CD14 and CD68 surface markers in U937 cells. This is similar to the effects caused by treatment with the differentiation inducer TPA.28–30 TPA induces the differentiation of AML cells into monocyte/macrophage-like cells or polymorphonuclear neutrophil cells.31 For further confirmation of differentiation in lapatinib-treated cells, phagocytic activity and ROS production assays were performed.24 As shown in Figure 6C and D, phagocytosis ability was dramatically increased with lapatinib treatment, similar to the positive-control TPA treatment. The cellular ROS level was also increased in lapatinib-treated U937 cells (Figure 6E). The upregulation of CD14 and CD68 surface markers and cellular ROS level suggests the induction of monocyte/macrophage differentiation by lapatinib treatment in AML U937 cells, similar to the effects of TPA treatment.
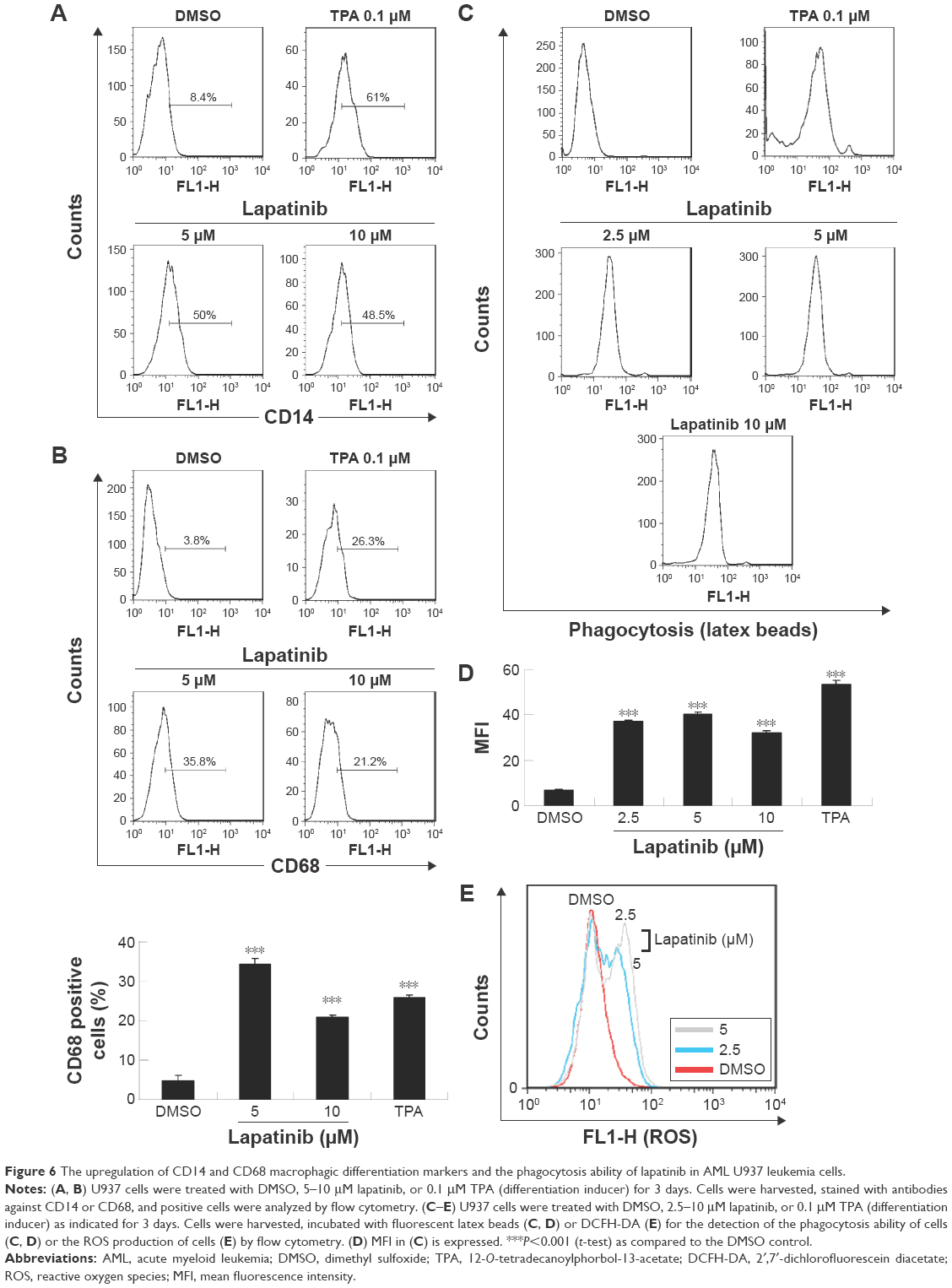 | Figure 6 The upregulation of CD14 and CD68 macrophagic differentiation markers and the phagocytosis ability of lapatinib in AML U937 leukemia cells. |
Synergistic effects of lapatinib and other anticancer drugs in U937 leukemia cells
To reduce the side effects and increase the therapeutic effects, cotreatment is a useful strategy for clinical therapies. We, therefore, wondered whether a combination of lapatinib and other anticancer drugs would show enhanced cytotoxic effects in leukemia. We first choose erlotinib, a targeted therapy drug and EGFR inhibitor that shows off-target effects in AML leukemia cells and patients.4,5,32–38 As shown in Figure 7 and consistent with previous reports, erlotinib alone failed to effectively inhibit U937 cell growth. However, in combination with low doses of lapatinib, both drugs showed dramatic growth inhibition of U937 cells. Again, a combination of low doses of lapatinib and etoposide or paclitaxel effectively increased the cytotoxic effects as compared to a single drug treatment. These results suggest the synergistic effects of lapatinib and other anticancer drugs in U937 leukemia cells. These synergistic effects of lapatinib and other anticancer drugs suggest the possible benefits of the development of combination therapies in leukemia treatment.
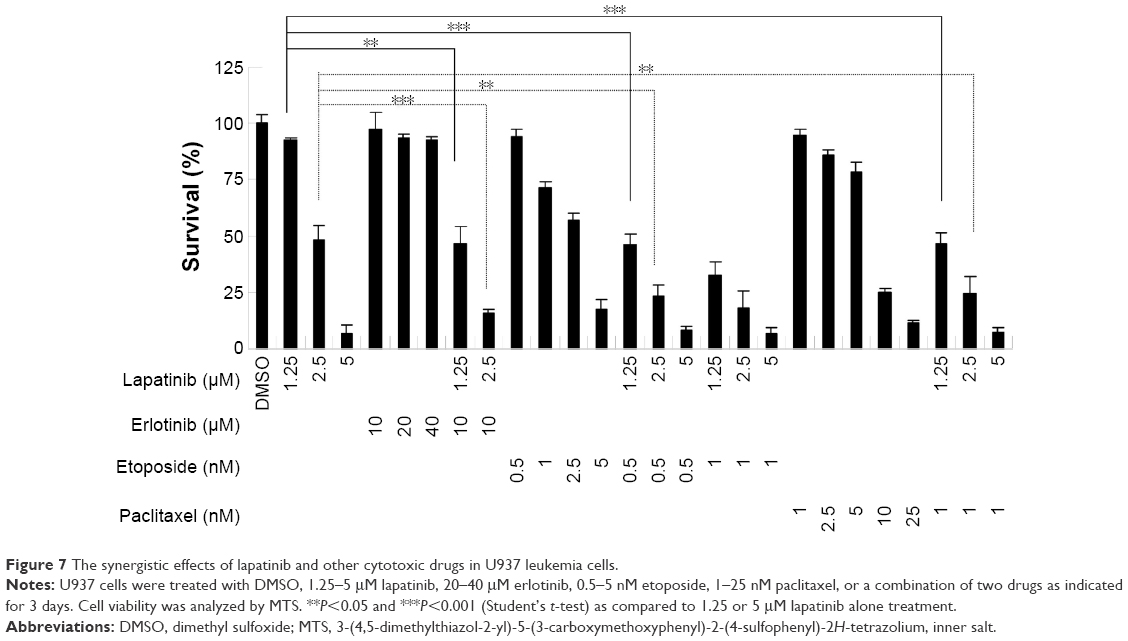 | Figure 7 The synergistic effects of lapatinib and other cytotoxic drugs in U937 leukemia cells. |
Discussion
In this study, we found lapatinib inhibits the growth of several leukemia cell lines in a dose- and time-dependent manner (Figure 1) either via autophagic or apoptotic cell death depending on the type of cell lines (Figure 2).22 This includes apoptosis induction in AML HL-60, APL NB4, Jurkat T lymphoma, and a minority of CML K562 cells, and the induction of nonapoptotic cell death in AML U937, CML MEG-01, and the majority of K562 cells by lapatinib. In U937 cells, autophagic cell death induced by lapatinib was demonstrated by massive vacuole formation observed by TEM (Figure 4) and microscopy (Figure 3A); LC3 aggregations (Figure 3B); acridine orange-positive cells (Figure 3C); cell death inhibition by 3-MA (Figure 3D) (but not Z-VAD-fmk [Figure 2B and C]); upregulation of the autophagy-related proteins ATG5, ATG7, Beclin, BNIP, and LC3 (Figure 3E); and cell death inhibition by knockdown of shRNA against ATG5, ATG7, and Beclin (Figure 5).26 Moreover, we also found induction of differentiation by lapatinib in U937 cells in correlation with the upregulation of monocyte/macrophage markers CD14 and CD68 (Figure 6A and B).23,36 Lapatinib also increased the phagocytosis ability of U937 cells, as shown by massive incorporation of fluorescent beads and higher ROS production (Figure 6C–E).23 We further found that the use of lapatinib and other cytotoxic drugs, including elortinib, etoposide, and paclitaxel, in U937 cells has a synergistic effect that increased tumor cytotoxicity (Figure 7). These findings suggest that a combination therapy using lapatinib and other anticancer drugs would further benefit anti-AML applications.
A few studies have focused on lapatinib-induced autophagic cell death. This study is consistent with our previous findings regarding the induction of autophagic cell death in hepatoma cells, induction of autophagic cell death, apoptosis, and differentiation by lapatinib in CML K562 cells.21,22 In addition to our findings, some studies have also discussed the induction of autophagy by lapatinib, such as in HCT116 colon cancer cells39 and in lapatinib and obatoclax cotreated breast cancer cells.40,41
Consistent with the findings of our study, some EGFR inhibitors, for example, sorafenib, gefitinib, erlotinib, and quizartinib (AC220), have been reported as targeted therapies for AML treatment in preclinical studies.1,4,5 Irwin et al also found the antiproliferation effect of lapatinib in human ALL Z119 and Z181 cell lines with the Philadelphia chromosome and ErbB2 expression.42 Also, similar to the findings of our study, cytotoxic activity and differentiation induction were found for gefitinib and erlotinib and other targeted therapy drugs against EGFR, in leukemia cells and clinical responses.4,5,32–38,43 According to the previous findings and our preliminary results (data not shown), there are no clear expression patterns for EGFR or ErbB2 in AML or in some of the tested cell lines, such as HL-60,33,36,44,45 despite the demonstration of a EGF-EGFR autocrine loop in U937 cells.46 Thus, the real targets of lapatinib, as well as gefitinib and erlotinib EGFR inhibitors need to be further investigated.43,47
Conclusion
We found that lapatinib induced cytotoxicity in several leukemia cell lines via either autophagic or apoptotic cell death. Lapatinib-induced autophagic cell death was observed in AML U937 cells. We also found differential induction by lapatinib and that treatment with lapatinib and other cytotoxic drugs (in AML U937 cells) had a synergistic effect. Our study suggests that a combination of lapatinib and other anticancer drugs may potentially be used as an AML therapy for clinical application.
Acknowledgments
The authors thank Miranda Loney and Ronald Lovel for English editing, Dr King-Song Jeng for experimental assistance, and the National RNAi Core Facility, Academia Sinica, Taiwan, for the pLKO.1-shRNA constructions. This study was supported by grants MMH-E-102-13 and MMH-E-103-13 from Mackay Memorial Hospital, grants NSC 100-2314-B-309-004- and NSC 100-2314-B-195-007-MY3 from the National Science Council of Taiwan, and grants MOST 103-2313-B-309-001- and MOST 104-2320-B-309-001- from the Ministry of Science and Technology, Republic of China.
Disclosure
The authors report no conflicts of interest in this work.
References
Hatzimichael E, Georgiou G, Benetatos L, Briasoulis E. Gene mutations and molecularly targeted therapies in acute myeloid leukemia. Am J Blood Res. 2013;3(1):29–51. | ||
Tallman MS, Gilliland DG, Rowe JM. Drug therapy for acute myeloid leukemia. Blood. 2005;106(4):1154–1163. | ||
Kelly LM, Gilliland DG. Genetics of myeloid leukemias. Annu Rev Genomics Hum Genet. 2002;3:179–198. | ||
Boehrer S, Ades L, Galluzzi L, et al. Erlotinib and gefitinib for the treatment of myelodysplastic syndrome and acute myeloid leukemia: a preclinical comparison. Biochem Pharmacol. 2008;76(11):1417–1425. | ||
Boehrer S, Ades L, Braun T, et al. Erlotinib exhibits antineoplastic off-target effects in AML and MDS: a preclinical study. Blood. 2008;111(4):2170–2180. | ||
MacFarlane RJ, Gelmon KA. Lapatinib for breast cancer: a review of the current literature. Expert Opin Drug Saf. 2011;10(1):109–121. | ||
Rusnak DW, Alligood KJ, Mullin RJ, et al. Assessment of epidermal growth factor receptor (EGFR, ErbB1) and HER2 (ErbB2) protein expression levels and response to lapatinib (Tykerb, GW572016) in an expanded panel of human normal and tumour cell lines. Cell Prolif. 2007;40(4):580–594. | ||
Rusnak DW, Lackey K, Affleck K, et al. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol Cancer Ther. 2001;1(2):85–94. | ||
Holbro T, Hynes NE. ErbB receptors: directing key signaling networks throughout life. Annu Rev Pharmacol Toxicol. 2004;44:195–217. | ||
Kopper L. Lapatinib: a sword with two edges. Pathol Oncol Res. 2008;14(1):1–8. | ||
Ito Y, Tokudome N, Sugihara T, Takahashi S, Hatake K. Does lapatinib, a small-molecule tyrosine kinase inhibitor, constitute a breakthrough in the treatment of breast cancer? Breast Cancer. 2007;14(2):156–162. | ||
Tevaarwerk AJ, Kolesar JM. Lapatinib: a small-molecule inhibitor of epidermal growth factor receptor and human epidermal growth factor receptor-2 tyrosine kinases used in the treatment of breast cancer. Clin Ther. 2009;31(Pt 2):2332–2348. | ||
Ryan Q, Ibrahim A, Cohen MH, et al. FDA drug approval summary: lapatinib in combination with capecitabine for previously treated metastatic breast cancer that overexpresses HER-2. Oncologist. 2008;13(10):1114–1119. | ||
Spira AI, Carducci MA. Differentiation therapy. Curr Opin Pharmacol. 2003;3(4):338–343. | ||
Honma Y, Matsuo Y, Hayashi Y, Omura S. Treatment of Philadelphia-chromosome-positive human leukemia in SCID mouse model with herbimycin A, bcr-abl tyrosine kinase activity inhibitor. Int J Cancer. 1995;60(5):685–688. | ||
Morceau F, Dupont C, Palissot V, et al. GTP-mediated differentiation of the human K562 cell line: transient overexpression of GATA-1 and stabilization of the gamma-globin mRNA. Leukemia. 2000;14(9):1589–1597. | ||
Meshkini A, Yazdanparast R. Involvement of ERK/MAPK pathway in megakaryocytic differentiation of K562 cells induced by 3-hydrogenkwadaphnin. Toxicol In Vitro. 2008;22(6):1503–1510. | ||
Williams CA, Mondal D, Agrawal KC. The HIV-1 Tat protein enhances megakaryocytic commitment of K562 cells by facilitating CREB transcription factor coactivation by CBP. Exp Biol Med (Maywood). 2005;230(11):872–884. | ||
Rouyez MC, Boucheron C, Gisselbrecht S, Dusanter-Fourt I, Porteu F. Control of thrombopoietin-induced megakaryocytic differentiation by the mitogen-activated protein kinase pathway. Mol Cell Biol. 1997;17(9):4991–5000. | ||
Tetteroo PA, Massaro F, Mulder A, Schreuder-van Gelder R, von dem Borne AE. Megakaryoblastic differentiation of proerythroblastic K562 cell-line cells. Leuk Res. 1984;8(2):197–206. | ||
Chen YJ, Chi CW, Su WC, Huang HL. Lapatinib induces autophagic cell death and inhibits growth of human hepatocellular carcinoma. Oncotarget. 2014;5(13):4845–4854. | ||
Huang HL, Chen YC, Huang YC, et al. Lapatinib induces autophagy, apoptosis and megakaryocytic differentiation in chronic myelogenous leukemia k562 cells. PLoS One. 2011;6(12):e29014. | ||
Chen YJ, Huang WP, Yang YC, et al. Platonin induces autophagy-associated cell death in human leukemia cells. Autophagy. 2009;5(2):173–183. | ||
Datta R, Yoshinaga K, Kaneki M, Pandey P, Kufe D. Phorbol ester-induced generation of reactive oxygen species is protein kinase cbeta-dependent and required for SAPK activation. J Biol Chem. 2000;275(52):41000–41003. | ||
Su WC, Chao TC, Huang YL, Weng SC, Jeng KS, Lai MM. Rab5 and class III phosphoinositide 3-kinase Vps34 are involved in hepatitis C virus NS4B-induced autophagy. J Virol. 2011;85(20):10561–10571. | ||
Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8(4):445–544. | ||
Kroemer G, Galluzzi L, Vandenabeele P, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16(1):3–11. | ||
Schmittgen TD, Lee EJ, Jiang J, et al. Real-time PCR quantification of precursor and mature microRNA. Methods. 2008;44(1):31–38. | ||
Mollinedo F, Lopez-Perez R, Gajate C. Differential gene expression patterns coupled to commitment and acquisition of phenotypic hallmarks during neutrophil differentiation of human leukaemia HL-60 cells. Gene. 2008;419(1–2):16–26. | ||
Collins SJ, Ruscetti FW, Gallagher RE, Gallo RC. Normal functional characteristics of cultured human promyelocytic leukemia cells (HL-60) after induction of differentiation by dimethylsulfoxide. J Exp Med. 1979;149(4):969–974. | ||
Clerget M, Polla BS. Erythrophagocytosis induces heat shock protein synthesis by human monocytes-macrophages. Proc Natl Acad Sci U S A. 1990;87(3):1081–1085. | ||
Pitini V, Arrigo C, Altavilla G. Erlotinib in a patient with acute myelogenous leukemia and concomitant non-small-cell lung cancer. J Clin Oncol. 2008;26(21):3645–3646. | ||
Lindhagen E, Eriksson A, Wickstrom M, et al. Significant cytotoxic activity in vitro of the EGFR tyrosine kinase inhibitor gefitinib in acute myeloblastic leukaemia. Eur J Haematol. 2008;81(5):344–353. | ||
Miranda MB, Duan R, Thomas SM, et al. Gefitinib potentiates myeloid cell differentiation by ATRA. Leukemia. 2008;22(8):1624–1627. | ||
Ravindranathan M, Klementich FJ, Jones DV Jr. Potential interaction of chemotherapy and gefitinib in the induction of hematologic neoplasia. Leukemia. 2007;21(12):2546–2547. | ||
Stegmaier K, Corsello SM, Ross KN, Wong JS, Deangelo DJ, Golub TR. Gefitinib induces myeloid differentiation of acute myeloid leukemia. Blood. 2005;106(8):2841–2848. | ||
Takigawa N, Takeuchi M, Shibayama T, et al. Successful treatment of a patient with synchronous advanced non-small cell lung cancer and acute myeloid leukemia by a combination of gefitinib, low-dose cytarabine and aclarubicin. Anticancer Res. 2005;25(3c):2579–2582. | ||
Ennishi D, Sezaki N, Senoo T, Terui Y, Hatake K, Hino N. A case of acute promyelocytic leukemia during gefitinib treatment. Int J Hematol. 2006;84(3):284–285. | ||
Galluzzi L, Aaronson SA, Abrams J, et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 2009;16(8):1093–1107. | ||
Cruickshanks N, Tang Y, Booth L, Hamed H, Grant S, Dent P. Lapatinib and obatoclax kill breast cancer cells through reactive oxygen species-dependent endoplasmic reticulum stress. Mol Pharmacol. 2012;82(6):1217–1229. | ||
Tang Y, Hamed HA, Cruickshanks N, Fisher PB, Grant S, Dent P. Obatoclax and lapatinib interact to induce toxic autophagy through NOXA. Mol Pharmacol. 2012;81(4):527–540. | ||
Irwin ME, Nelson LD, Santiago-O’Farrill JM, et al. Small molecule ErbB inhibitors decrease proliferative signaling and promote apoptosis in philadelphia chromosome-positive acute lymphoblastic leukemia. PLoS One. 2013;8(8):e70608. | ||
Boehrer S, Galluzzi L, Lainey E, et al. Erlotinib antagonizes constitutive activation of SRC family kinases and mTOR in acute myeloid leukemia. Cell Cycle. 2011;10(18):3168–3175. | ||
Hahn CK, Berchuck JE, Ross KN, et al. Proteomic and genetic approaches identify Syk as an AML target. Cancer Cell. 2009;16(4):281–294. | ||
Allen H, Hsuan J, Clark S, et al. Expression of epidermal-growth-factor receptor in the K562 cell line by transfection. Altered receptor biochemistry. Biochem J. 1990;271(3):785–790. | ||
Eales-Reynolds LJ, Laver H, Modjtahedi H. Evidence for the expression of the EGF receptor on human monocytic cells. Cytokine. 2001;16(5):169–172. | ||
Lainey E, Sebert M, Thepot S, et al. Erlotinib antagonizes ABC transporters in acute myeloid leukemia. Cell Cycle. 2012;11(21):4079–4092. |
Supplementary materials
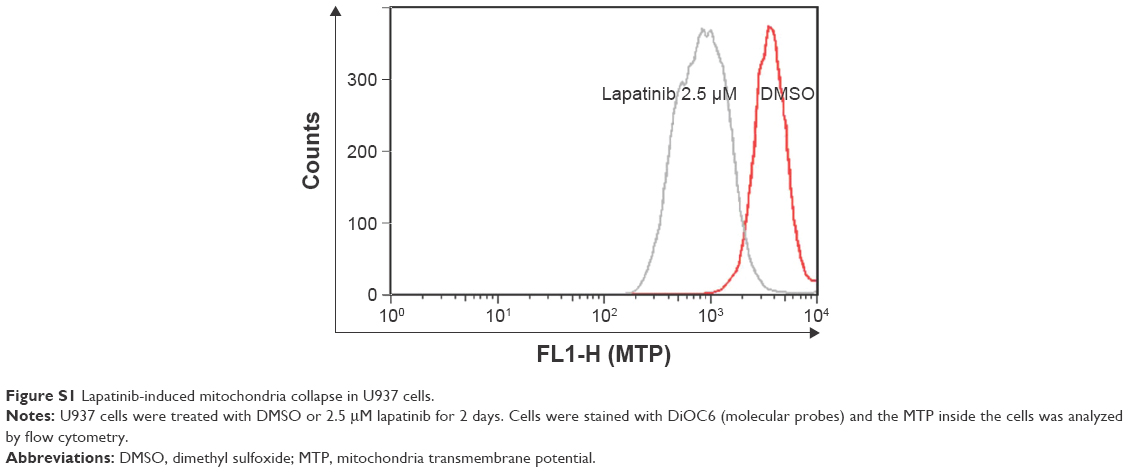 | Figure S1 Lapatinib-induced mitochondria collapse in U937 cells. |
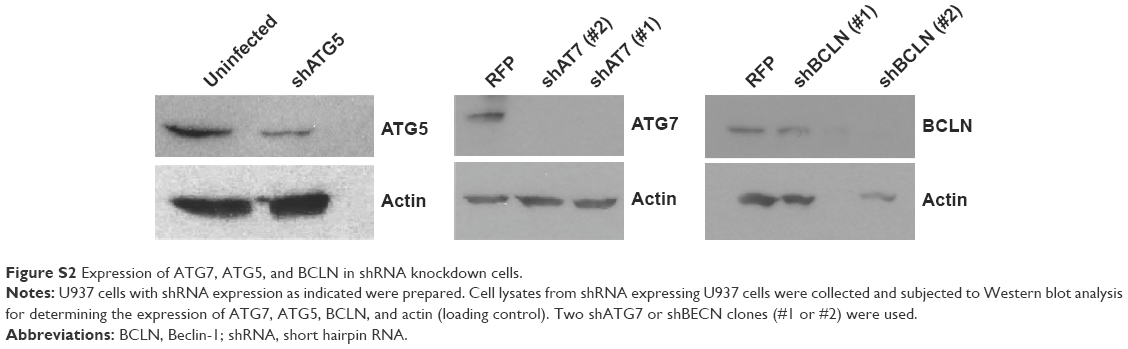 | Figure S2 Expression of ATG7, ATG5, and BCLN in shRNA knockdown cells. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.