Back to Journals » Degenerative Neurological and Neuromuscular Disease » Volume 9
Lambert-Eaton Myasthenic syndrome: early diagnosis is key
Authors Ivanovski T ![]() , Miralles F
, Miralles F ![]()
Received 29 October 2018
Accepted for publication 18 March 2019
Published 13 May 2019 Volume 2019:9 Pages 27—37
DOI https://doi.org/10.2147/DNND.S192588
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Müller
Trajche Ivanovski,1 Francesc Miralles2
1Neurology Department, Hospital Universitari Son Llatzer, Palma de Mallorca, Balearic Islands, Spain; 2Neurology Department, Hospital Universitari Son Espases, Palma de Mallorca, Balearic Islands, Spain
Abstract: Lambert-Eaton myasthenic syndrome (LEMS) is an uncommon disorder of neuromuscular transmission with distinctive pathophysiological, clinical, electrophysiological and laboratory features. There are two forms of LEMS. The paraneoplastic (P-LEMS) form is associated with a malignant tumor that is most frequently a small cell lung carcinoma (SCLC), and the autoimmune (A-LEMS) form is often related to other dysimmune diseases. Approximately 90% of LEMS patients present antibodies against presynaptic membrane P/Q-type voltage-gated calcium channels (VGCC). These antibodies are directly implicated in the pathophysiology of the disorder, provoke reduced acetylcholine (ACh) at the nerve terminal and consequently lead to muscle weakness. LEMS is clinically characterized by proximal muscle weakness, autonomic dysfunction and areflexia. In clinically suspected cases, diagnoses are confirmed by serological and electrodiagnostic tests. The detection of P/Q-type VGCC antibodies is supportive when there is clinical suspicion but should be carefully interpreted in the absence of characteristic clinical or electrodiagnostic features. Typical electrodiagnostic findings (ie, reduced compound motor action potentials (CMAPs), significant decrements in the responses to low frequency stimulation and incremental responses after brief exercise or high-frequency stimulation) reflect the existence of a presynaptic transmission defect and are key confirmatory criteria. Diagnosis requires a high level of awareness and necessitates the initiation of a prompt screening and surveillance process to detect and treat malignant tumors. In clinically affected patients without cancer and after cancer treatment, symptomatic treatment with 3,4-diaminopyridine or immunosuppressive agents can significantly improve neurologic symptoms and the quality of life. We present a detailed review of LEMS with special emphasis on the pathophysiological mechanisms, clinical manifestation and diagnostic procedure.
Keywords: neuromuscular transmission, paraneoplastic disorder, muscle weakness, voltage-gated calcium channels, electrodiagnostic test
Introduction
Lambert-Eaton myasthenic syndrome (LEMS) is an uncommon neuromuscular junction (NMJ) disorder with distinctive pathophysiological, clinical, electrophysiological and laboratory features. More than a half of cases present a paraneoplastic form (P-LEMS) associated with a malignant tumor that is usually a small cell lung carcinoma (SCLC). The remaining cases are considered autoimmune (A-LEMS) and frequently overlap with other dysimmune diseases. LEMS is characterized by the presence of antibodies against presynaptic P/Q-type voltage-gated calcium channels (VGCC) that cause a reduction in the level of acetylcholine (ACh) released from the nerve terminal and consequent muscle weakness. Other common clinical findings are general fatigue, autonomic dysfunction and areflexia. The description of the electrophysiological criteria in the 1950s and the discovery of anti-VGCC antibodies in 1983 were substantial breakthroughs that improved our understanding of the pathophysiological mechanisms and facilitated early diagnosis. Given that LEMS is uncommon but frequently a paraneoplastic disorder associated with cancer in the initial stages, awareness and a high degree of suspicion are essential factors for an early diagnosis that can lead to the optimal management of these patients.
History
The name Lambert-Eaton syndrome is a tribute to Dr. Edward H. Lambert and Dr. Lee M. Eaton who were two distinguished American neurologists from the Mayo Clinic. In 1956, they described 6 patients with neuromuscular disorders resembling Myasthenia Gravis (MG), although some different clinical and electrophysiological features were present.1 In three of these patients, malignant tumors were detected, and x-ray imaging in two additional patients suggested the existence of intrathoracic malignant disease. Soon after, these neurologists published a novel article in which they precisely described the electrophysiological features of the newly recognized disorder of neuromuscular transmission that they named “myasthenic syndrome associated with malignant tumors”.2 From this point on, and particularly prior to the discovery of the anti-VGCC antibodies, these electrophysiological criteria have served as the foundation of the diagnosis of this syndrome and prevented possible confusion with MG. In the mid-1960s and the beginning of the 1970s, several case reports of LEMS with coexisting autoimmune diseases led to the hypothesis of an immuno-mediated disorder.3,4,5 This hypothesis became more credible in 1983 when the passive transfer of immunoglobulin G (IgG) from affected patients to mice reproduced the electrophysiological features of the disease.6 In the following years, Fukunaga et al suggested that calcium channels of the neuromuscular junction could be the targets of the pathogenic autoantibodies in LEMS and reproduced the presynaptic membrane lesions seen in patients with LEMS using the passive transfer model in mice.7,8 Further studies showed that antibodies with specificity for P/Q-type calcium channels are the determinant type in the pathophysiological process.9 Since then, additional advances have occurred in the understanding of this NMJ disorder, but our comprehension has mainly been enhanced as a consequence of numerous clinical studies that have improved screening programs and general patient management.
Epidemiology
Underdiagnoses and frequent misdiagnoses interfere with accurate epidemiological estimations of LEMS. As observed in a study from the Netherlands, 58% of all cases received an incorrect diagnosis before LEMS was established.10 In the same study, the annual incidence was 0.4 per million with equal proportions of LEMS associated with SCLC and LEMS without SCLC. The estimated prevalence was 2.5 per million inhabitants, and there was a lower prevalence of P-LEMS that was probably due to poor survival in this group. The prevalence in the United States was indirectly estimated to be 1 in 100,000 based on the prevalence of small cell lung cancer.11 In a large and more recent North American epidemiologic study of LEMS, the annual incidence of confirmed cases was similarly estimated at 0.6 per million, and the prevalences were estimated at 2.8 and 3.8 per million for confirmed and probable cases, respectively. The P-LEMS form is more frequent among men (65%) and occurs at a median age of 60 years. In the Netherlands study, 58% of all LEMS cases were men, but men were far more frequent (reaching 76%) in the P-LEMS group. The presentation of A-LEMS is more balanced between males and females. The median age in this study was 55 years (range 11–73) and did not significantly differ between groups. The median duration of the disease prior to diagnosis among all LEMS cases was 11 months and was significantly shorter among the patients with P-LEMs than those with A-LEMS (3.5 vs 17 months, respectively).10 A similar age of onset and duration until diagnosis were obtained in a study conducted in the Unites States Veteran’s Affairs population.12 A diagnosis of LEMS anticipates tumor detection in most P-LEMS cases.13 Titulaer et al observed that a SCLC diagnosis preceded LEMS identification in only 6% of P-LEMS cases. SCLCs were identified in 92% of these patients within 3 months and in 96% within a year.14
Clinical manifestations
LEMS has an insidious onset and a slowly progressive clinical course. Muscle weakness, fatigue, autonomic dysfunction and areflexia are the most common clinical manifestations.15 Weakness usually begins to involve the proximal leg muscles, which are also the most severely affected muscle group, and arm weakness appears soon after.16 In the early stages, weakness is generally restricted to proximal muscle groups, later extends distally to involve the hand and foot muscles and finally spreads to the oculobulbar muscles. The extension speed is more rapid in P-LEMS cases than in A-LEMS cases.16 The excessive weakness and fatigue referred to by most LEMS patients are commonly disproportionate to the severity of weakness found on physical examination. Evaluations of muscle strength should be performed after rest to avoid the potentiation created by muscle testing.
The ocular or bulbar symptoms are less frequent and pronounced compared with those seen in MG but this difference should not be used as a distinctive clinical feature to differentiate MG from LEMS. Rarely, these symptoms can occur during the initial manifestation of the disease.17–21 The reported prevalence of oculobulbar manifestations varies according to different works and increases during the evolution of the disease, and such manifestations are present in approximately one-third of patients at 3 months and half of cases one year after onset.22 Ptosis and diplopia are the most common ocular symptoms, and dysphagia and dysarthria are the most frequent bulbar manifestations; however, in contrast to MG, these symptoms are often mild and transient.
Neurologists should be receptive to coexisting autonomic dysfunctions, which should be actively sought. Few patients spontaneously mention such dysfunctions, probably because they are not perceived as the most troublesome symptoms or do not have important repercussions in daily activities as in the case of leg weakness and fatigue. Dysautonomia has been reported in 80–96% of LEMS patients during the evolution of the disease.15,16,22 Dry mouth is the most commonly reported symptom followed by impotence in males and constipation. Dry eyes, orthostatic dizziness, blurred vision and altered perspiration are less frequent.
Deep tendon reflexes are characteristically decreased or absent but can be amplified after muscular contraction. As in the case of muscle weakness, tendon reflexes should be tested after a period of rest. Cerebellar ataxia, sensory neuropathy and limbic encephalitis are extremely uncommon manifestations that are almost exclusively associated with P-LEMS.16,23,24
Pathophysiology
Ca2+ ions play a central role in neurotransmission. They not only trigger the exocytosis of vesicles that are docked on the cytoplasmic side of the presynaptic membrane and are ready to be released (ie, the “readily releasable pool” or RRP) but also influence short-term synaptic plasticity and probably influence the mechanisms that restore the RRP after presynaptic activation.25
Ca2+ ions diffuse into the presynaptic terminal through VGCCs. These molecular structures are integral parts of the so-called “active zones” (AZs), which are the locations at which synaptic vesicles (SVs) are docked. AZs were first identified in freeze-fracture preparations that were examined with an electronic microscope. This technique, and more recently, electron microscope tomography, has revealed the ultrastructure of the rodent AZ, which consists of double rows of 80-nm-long intramembranous particles (which are believed to include the VGCCs) with two SVs between them.26 The presynaptic terminals of rodent NMJ have approximately 900 AZs.26
Figure 1 illustrates the most important presynaptic steps of neuromuscular transmission. Presynaptic depolarization opens the VGCCs, and Ca2+ ions pass through them to create a brief and circumscribed increase in calcium in the AZ that is termed a “calcium nanodomain”.25 This local increase in calcium allows the binding of several Ca2+ ions to the vesicle protein synaptotagmin. In vertebrate NMJs, synaptotagmin probably binds four Ca2+ ions, and this phenomenon is believed to trigger the fast synchronous release of SVs during presynaptic depolarization.27 The main determinants of the amplitude of the end-plate potential (epp) are the number of SVs released and, in cases of postsynaptic disorders of the NMJ such as MG, the number of ACh receptors present in the postsynaptic membrane.
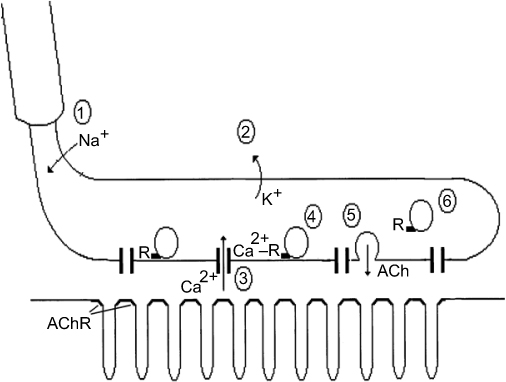 | Figure 1 Presynaptic events in neuromuscular transmission. (A) The active depolarization of a motor axon caused by the opening of heminodal (the transition between the myelinated and non-myelinated parts of the axon) voltage-gated Na+ channels is passively transmitted to the presynaptic terminal. (B) The presynaptic terminal is covered by glial cells (not represented), and its membrane contains voltage-gated K+ and Ca2+ channels. (C) The opening of the voltage-gated Ca2+ channels located in the active zones allows the entry of extracellular Ca2+ into the terminal, which creates a local increase in the Ca2+ concentration. (D)Four or five Ca2+ ions bind to synaptotagmin, which is the Ca2+ receptor that triggers the fast and synchronous release of acetylcholine (ACh). (E) Fusion of the docked vesicles at the active zones and the release of ACh into the intersynaptic space. (F) Replenishment of the empty active zones by vesicles from the recycling pool. This step is believed to be regulated by the intraterminal Ca2+ concentration. |
The number of SVs released by a train of presynaptic potentials depends both on the synaptic type and the length and frequency of the stimulating train. In NMJs in which the probability of the release of a SV after a stimulus is high (this probability is defined as p according to the quantal theory of neurotransmission), as is the case in human NMJs, successive epps are of lower amplitudes than the first epp. This phenomenon is called “synaptic depression”. The main factor responsible for this behavior is believed to be the depletion of the RRP of the SV caused by the previous stimuli because a reduction in p due to, for example, a reduction in extracellular calcium, supposes not only a reduction in the amplitude of the first epp but also a reduction in the magnitude of depression. When p is sufficiently low, it is possible that successive epps during a train may show progressive increases in amplitude, at least for some time. This phenomenon is known as “synaptic facilitation”, and it is believed to be related to the progressive rise in the presynaptic calcium concentration that is produced during a repetitive stimulation.28 This calcium would bind to receptors other than those that produce the phasic release of SVs.29 Between presynaptic depolarizations, the RRP would be restored by SVs that pass from the recycling pool to the AZ where they undergo the molecular changes that convert them into docked vesicles that are ready to be released. As previously mentioned, this transition rate would also probably be governed by the presynaptic calcium concentration.25
According to the simplest form of the quantal theory of synaptic transmission, the number of quanta released by a synaptic potential (one quantum = one SV) follows a binomial distribution that is defined by two parameters: the release probability of a quantum after a presynaptic action potential (p) and the number of SVs in the RRP (n). The mean number of SVs released by a stimulus, also known as the “quantal content” (denoted by m), is the product p ∙ n. This simple formulation of quantal theory does not explain the experimentally encountered variations in the estimates of p and n during trains of stimuli (ie, the effects of short-term synaptic plasticity). In contrast, both synaptic facilitation and synaptic depression may be accounted for by considering that synaptic transmission follows a compound binomial model in which p is nonstationary during a stimulating train and that p is, in fact, the product of two different probabilities, ie, the probability that an AZ is occupied by a quantum multiplied by the probability that this quantum is released by a presynaptic depolarization.27,30
The peculiar electromyographic characteristics of myasthenic syndrome allowed Lambert and Eaton to hypothesize that the number of quanta released by a stimulus is greatly decreased. This hypothesis was confirmed by Lambert and Elmqvist while studying the NMJs of patients with myasthenic syndrome in vitro.31 Their studies also showed that increasing the extracellular Ca2+ concentration greatly increased the very low basal amplitudes of epps until they were sufficiently high to elicit a muscle action potential. These observations led Lambert and Elmqvist to speculate “that some, as yet unknown substance, interferes with the utilization of calcium in the motor nerve terminals”.31 Several studies performed in the 80s and 90 s established that patients suffering from LEMS have autoantibodies against presynaptic VGCCs that reduce the inward Ca2+ current during presynaptic depolarization that triggers the synchronous release of SVs. This process supposes a reduction in the quantal content and the generation of an epp of abnormally low amplitude.
Recently, a new electromyographic characteristic has been added to those classically described by Lambert; the partial recovery of a compound muscle action potential (CMAP) that is typically observed in myasthenia gravis during low-frequency repetitive nerve stimulation (the so-called “U-pattern”) is not present in Lambert-Eaton myasthenic syndrome.32 This late recovery during low-frequency stimulating trains was also observed in subjects treated with atracurium (an antagonist of nicotinic receptors) at doses that produce a neuromuscular blockade.33 Moreover, the magnitude of the recovery of the CMAP amplitude, as well the maximum decrease in the CMAP amplitude attained during a train, increase as the frequency of stimulation is increased in both myasthenia gravis and atracurium-treated patients.33 Results from a computer model of human neuromuscular transmission suggest that this phenomenon is due to the fact that the mechanism of recovery from synaptic depression is Ca2+-dependent, and its efficacy depends on the intraterminal residual Ca2+ concentration. Because the amount of residual Ca2+ depends both on the stimulating frequency and the duration of the train, the extent of the recovery of the CMAP amplitude (which reflects a progressive increase in the epp amplitude) increases with the increase in the frequency of stimulation, and it is observed at the end of a low-frequency stimulating train. The absence of this late recovery in LEMS could only be simulated by the model if both the efficacy of the Ca2+-dependent mechanism of recovery from depression and the magnitude of synaptic facilitation, which is also thought to be linked to residual Ca2+ were reduced. These results from the computer model are consistent with the fact that Ca2+ entry during a presynaptic depolarization is greatly reduced in LEMS due to the presence of antibodies against presynaptic VGCCs.33
Electromyography
The electrophysiological criteria described by Lambert and Eaton have served as the basis of LEMS diagnosis since its identification as a distinctive NMJ disorder.
Repetitive nerve stimulation (RNS) is the electrophysiological test of choice, and the classical triad of electrophysiological findings consists of the following:
- Significantly and uniformly reduced CMAPs in motor nerve conduction studies at rest that usually reach less than 50% of the inferior limit of normal (Figure 2A). This is a common finding in all presynaptic NMJ disorders and is observed in up to 96% of LEMS cases.34

Figure 2 Classical electrodiagnostic findings in a patient with a paraneoplastic LEMS caused by an oat-cell lung carcinoma. (A) 1. Low basal CMAP amplitude (3.7 mV; lower limit of normality: 6 mV). 2. Increase (111%) in the CMAP amplitude after a brief (15 s) maximal-contraction post-tetanic potentiation. (B) Progressive reduction of the CMAP amplitude during a 2-Hz repetitive nerve stimulation without any recovery of this amplitude at the end of the train (16% decrease in amplitude).Abbreviations: APB: abductor pollicis brevis muscle. LEMS: Lambert-Eaton Myasthenic Syndrome. CMAP: compound muscle action potential.
- A decrement in the CMAP response upon low-frequency (2–5 Hz) RNS that produces a successive decline in CMAP amplitude from its normal baseline (Figures 2B and 3A). A CMAP decrease greater than 10% is required to be considered abnormal, and such a decrease is found in 94–98% of patients with LEMS.34,35
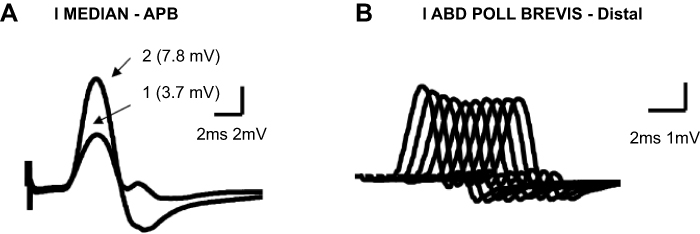
This electrophysiologic phenomenon can appear in both presynaptic and postsynaptic NMJ disorders, although it appears with a distinctive decremental pattern according some authors.32,36
In MG, with a train of 8–10 stimuli, the decrement in the response is usually maximal at the fourth or fifth stimulation and followed by increments in the responses with subsequent stimuli, although these responses never reach the size of the initial response. The pattern of responses has a characteristic morphology with an asymmetric U-shape. Unlike MG patients, a low-frequency RNS in LEMS patients leads to a progressive decremental response (Figures 2 and 3).
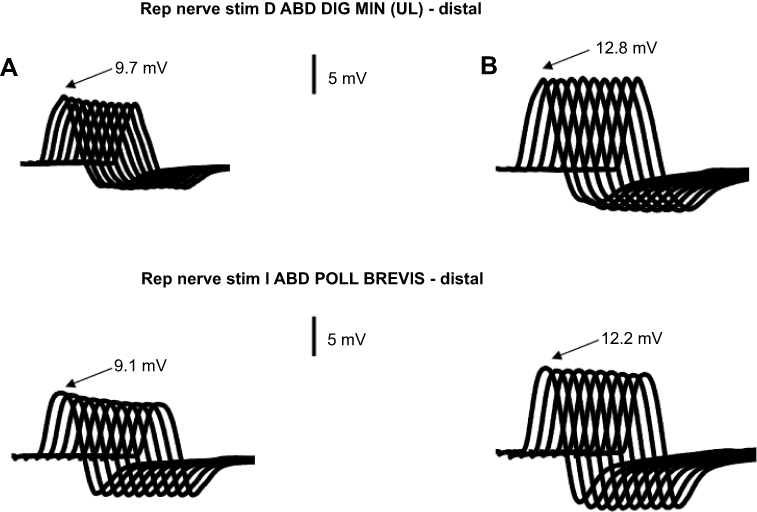 | Figure 3 Electrodiagnostic findings in a patient with a LEMS without evidence of cancer. (A) Progressive reductions of the CMAP amplitudes during 3-Hz repetitive nerve stimulations in two different hand muscles. Note the absence of any late increases in the CMAP amplitudes. The initial CMAP amplitudes were well above the lower limit of normality in both muscles (6 mV for the abductor pollicis brevis and 5 mV for the abductor digiti minimi). (B) After a brief (15 s) maximal contraction, there was an increase in the initial CMAP amplitude, and there was a complete or a near-complete absence of a reduction in the CMAP amplitude during a 3-Hz repetitive nerve stimulation.Abbreviations: ABD DIG MIN, abductor digiti minimi muscle; ABD POLL BREVIS, abductor pollicis brevis muscle; LEMS, Lambert-Eaton Myasthenic Syndrome; CMAP, compound muscle action potential. |
Hatanaka and Oh showed that longer exercises lead to a progressive decrease in the increment. These authors demonstrated significantly higher diagnostic sensitivity using a 10-second exercise compared with a 30-second exercise.37 The same authors later found that, compared with VGCC-negative LEMS, VGCC-positive LEMS is associated with a lower basal CMAP and a greater increment response.38 Their findings suggested that the RNS pattern was more severely altered in VGCC-positive LEMS and thus more indicative of LEMS. This observation further promoted the use of the 60% increment criterion, which is especially critical for the diagnosis of seronegative LEMS. These authors also concluded that the effect of post-exercise exhaustion on RNS does not have diagnostic value for LEMS.39
Single-fiber electromyography generally does not distinguish between presynaptic and postsynaptic disorders of the NMJ, although differences can be demonstrated if they are intentionally sought. Notably, in cases of LEMS, markedly abnormal jitter values can be registered diffusely and independently of the muscle tested or the grade of clinical affect. Blocking is more pronounced, and like jitter values, increases remarkably with low-frequency (5–10 Hz) stimulation.40,41,42
It should be noted that early in the course of LEMS, when the baseline CMAP amplitudes are not yet notably reduced, the abnormal decremental response to slow RNS may be easier to demonstrate than the more characteristic incremental response. The combination of a normal or slightly reduced CMAP and a lack of incremental response can easily result in a missed diagnosis of LEMS or, in cases with an evident decremental response, lead to a misdiagnosis of MG (Figure 3).
Autoantibody testing
Antibodies against the P/Q-type VGCC are directly implicated in the pathophysiology of LEMS and were traditionally considered to be highly specific for this presynaptic NMJ disorder.9,44 P/Q-type VGCC antibodies are found in more than 90% of LEMS cases, and this percentage is higher in patients with SCLC. These antibodies are infrequently detected in patients with lung cancer without LEMS and sporadically detected in healthy individuals and patients with other autoimmune diseases.9,44,45 However, recent studies suggest a much lower specificity for P/Q-type VGCCs (36%) than has previously been reported.43
The N-type VGCCs are also a common target in LEMS and antibodies against N-type VGCCs are the second-most commonly detected antibodies in LEMS patients.
These antibodies have been reported to be found in 33–49% of LEMS patients and are notably more frequent in cases of P-LMES; according some studies, they are present in up to 73% of patients in this group.9,44 These findings suggest that N-type VGCC-positive LEMS patients are more likely to have an underlying malignancy. Motonura et al reported that all N-type VGCC antibodies-positive LEMS patients were also positive for P/Q-type VGCC antibodies, which provides the impression that these antibodies could be a result of cross-reactivity between the P/Q- and N-type channels or the expression of multiple types of VGCCs by underlying SCLCs in P-LEMS.44,46
Antibodies against SOX-1 were also proposed as a serological tumor marker with diagnostic value for discriminating between P-LEMS and A-LEMS. According to some authors, these antibodies have been found in 64% of patients with P-LEMS and only 0–5% of A-LEMS patients and are therefore able to identify LEMS patients with a greater risk of malignancy.47,48 These antibodies have also been described in significant percentages of patients with SCLC without LEMS and in patients with other paraneoplastic neurological disorders, which indicates that a potential pathogenic role is very unlikely.
Approximately 10–15% of LEMS patients are seronegative but share practically identical electrophysiological features with seropositive LEMS patients, and the passive transfer of either seronegative or seropositive LEMS sera to mice appears to reproduce similar electrophysiological changes. These observations suggest that subthreshold concentrations of known antibodies or the existence of antibodies against unrevealed targets could be the cause in seronegative LEMS. Some of the recently studied targets are synaptotagmin-1, which is a VGCC-associated protein that is implicated in fast ACh release, and the M-type presynaptic muscarinic ACh receptor, which is implicated in the modulation of cholinergic neuromuscular transmission via linkage to P/Q-type VGCCs. Antibodies against these potential targets have been found in both seropositive and seronegative LEMS, although further studies are necessary to confirm their potential pathogenic roles.49,50
Diagnosis
Clinical suspicion is the first and most important step in the diagnostic process. LEMS should be considered in any patient presenting with proximal muscle weakness, especially if it is associated with hypo- or areflexia, dry mouth, constipation or oculobulbar symptoms.
In cases with a compatible clinical constellation, the diagnosis is confirmed with electrophysiological and serological tests. In suspected patients, typical electrodiagnostic findings remain the key confirmatory criteria, and the finding of a significant incremental response is practically diagnostic. The detection of diffusely reduced CMAPs in the absence of an alternative explanation should also raise the alarm. In such cases, an experienced neurologist or neurophysiologist should perform RNS or PET to confirm the existence of a presynaptic disorder. Anti-P/Q-type VGCC antibodies are helpful and provide additional diagnostic certainty but are complementary and cannot replace electrodiagnosis.
We propose a diagnostic algorithm (Figure 4) wherein we consider 3 possible electrodiagnostic scenarios for clinically suspected cases: 1. The patient exhibits a typical pattern (a significantly reduced CMAPs, a decremental CMAP response on low-frequency RNS and an incremental response after PET or HFS) that is confirmatory for LEMS. In such cases, the detection of specific antibodies differentiates between seropositive and seronegative LEMS (Figure 2). 2. The patient has suggestive electrophysiological findings (a normal or mildly reduced CMAPs and an abnormal decremental response without a significant incremental response) (Figure 3). This pattern is a possible scenario during the initial stages of LEMS; therefore, to avoid underdiagnosis, we recommend a repetition of the electrophysiological study. In such cases, the detection of anti-P/Q-type VGCC antibodies is helpful and increases the probability of the diagnosis. 3. In the third and last electrodiagnostic scenario, there are no electrophysiological findings suggestive of LEMS, or the findings do not reach the required values. If there is high clinical suspicion, the detection of specific antibodies could warrant the repetition of the test; otherwise, the diagnosis of LEMS is highly improbable.
 | Figure 4 Diagnostic algorithm for clinically suspected LEMS. Notes: *Electrophysiological studies: nerve conduction study, low-and high-frequency repetitive nerve stimulations and a post-exercise test.Abbreviations: LEMS, Lambert-Eaton Myasthenic Syndrome; VGCC, voltage-gated calcium channels. |
Evaluation for malignant disease and treatment
The active search for a malignant tumor is of great importance and should be started immediately after diagnosis. A diagnosis of LEMS anticipates tumor detection in most cases with P-LEMS.13 Using a screening program, SCLCs were identified in 91% of P-LEMS cases within 3 months and in 96% within one year. Some studies have reported improved survival in patients with P-LEMS associated with a SCLC compared with patients with a SCLC without LEMS.51,52 In a more recent prospective study, Maddison et al demonstrated that the presence of P-LEMS with a SCLC conferred a significant survival advantage independently of other prognostic variables.53
For newly diagnosed LEMS without cancer, the initial evaluation should be made with a thoracic CT scan and 18F-fluorodeoxyglucose positron emission tomography (FDG-PET) if the CT scan is non-diagnostic. A simple clinical scoring system based on age, weight loss, smoking, bulbar involvement, erectile dysfunction and Karnofsky performance status called the Dutch-English LEMS tumor association prediction (DELTA-P) score was developed and validated in 2011. This scoring system indicates the presence of a SCLC with very high accuracy and therefore aids physicians in identifying high-risk patients and optimizing the screening process and follow-up.54 SOX-1 antibodies have also been demonstrated to be an independent predictor of SCLC but were omitted from the DELTA-P for practical reasons. If the initial cancer evaluation is negative, screening with thoracic CT or FDG-PET should be repeated after 3–6 months depending on the DELTA-P score.22
The treatment of LEMS involves the removal or treatment of the underlying cancer in patients with P-LEMS and symptomatic treatment in clinically affected patients independent of the LEMS form. In patients with symptoms that interfere with daily activities, 3,4-diaminopyridine (3,4-DAP) is usually recommended as an initial therapy.31 3,4-DAP is a potassium channel blocker that prolongs presynaptic nerve terminal depolarization and consequently increases calcium entry through VGCCs, which ultimately leads to increased ACh release.
The beneficial effects of 3,4-DAP, which include improvements in muscle strength score or autonomic symptoms and increases in CMAP amplitude, have been demonstrated in several randomized controlled trials.55,56,57,58,59 In a recent study, Sanders and colleagues demonstrated that 3,4-DAP is an effective maintenance treatment for LEMS.60 A Cochrane review from 2011 based on four randomized and controlled trials in 54 LEMS patients supported the notion that oral 3,4-DAP showed a significant benefit and was generally well tolerated.61 3,4-DAP is approved in the European Union and was also recently approved in the United States where, until November 2018, it was only available for compassionate use.
Acetylcholinesterase inhibitors, such as pyridostigmine, are generally not effective as monotherapies for LEMS, although benefits have been reported in some patients when combined with 3,4-DAP.13
Guanidine can also be combined with 3,4-DAP and was the first symptomatic agent used for LEMS; however, with very limited use due to its high toxicity.
In patients who do not achieve satisfactory control of their symptoms and in refractory patients who do not respond to the previously described therapies, immunomodulating treatment should be initiated.
Intravenous immunoglobulin (IVIG) in a typical regimen (2 g/kg total dose divided over two to five days) has been successfully employed for the treatment of several immune-mediated neurological diseases. IVIG can be used as an induction treatment in symptomatic patients or as a maintenance treatment in patients with a satisfactory initial response who experience a recurrence of the symptoms.62,63,64
The only randomized, double-blind, placebo-controlled crossover trial showed significant improvements in indices of limb, respiratory and bulbar muscle strength that were associated with reductions in serum VGCC antibodies.64 Prednisone alone or in combination with other corticosteroid-sparing immunosuppressive agents, such as azathioprine or mycophenolate, can be reasonable alternatives.
Rituximab or plasma exchange in combination with other immunosuppressive agents can be used in refractory LEMS patients.65,66,67
Conclusions
A diagnosis of a rare disorder supposes a complex process in which knowledge, awareness and a high degree of suspicion are critical factors for an early diagnosis.
In clinically suspected LEMS, diagnosis is straightforward and should be assured given that highly specific diagnostic tools are easily available. Clinical and electrodiagnostic criteria for LEMS are currently the mainstay in the diagnosis of LEMS. Elevated P/Q-type VGCC antibody titers are helpful when a strong clinical suspicion is present but need to be interpreted carefully when they are positive without clinical correlations.
Early recognition and the prompt initiation of the screening program are the priorities in the management of LEMS. The improved survival in patients with P-LEMS associated with SCLC and the improvement in the neurological symptoms following cancer treatment clearly suggest that the careful search for and treatment of a malignant tumor is essential in the prognosis of P-LEMS. In the case of A-LEMS, early recognition can improve disability and the quality of life.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Lambert EH, Eaton LM, Rooke ED. Defect of neuromuscular conduction associated with malignant neoplasms. Am J of Physiol. 1956;187(3):612–613.
2. Eaton LM, Lambert EH. Electromyography and electric stimulation of nerves in diseases of the motor unit: observations on a myasthenic syndrome associated with malignant tumors. JAMA. 1957;163(13):1117–1124. doi:10.1001/jama.1957.02970480021005
3. Norris FH. Neuromuscular transmission in thyroid disease. Ann Intern Med. 1966;64(1):81.
4. Brown JW, Nelson JR, Herrmann C. Sjogren’s syndrome with myopathic and myasthenic features. Bull Los Angel Neuro Soc. 1968;33(1):9–20.
5. Gutmann L, Crosby TW, Takamori M, Martin JD. The Eaton-Lambert syndrome and autoimmune disorders. Am J Med. 1972;53(3):354–356.
6.
7. Fukunaga H, Engel AG, Osame M, Lambert EH. Paucity and disorganization of presynaptic membrane active zones in the Lambert-Eaton myasthenic syndrome. Muscle Nerve. 1982;5(9):686–697. doi:10.1002/(ISSN)1097-4598
8. Fukunaga H, Engel AG, Lang B, Newsom-Davis J, Vincent A. Passive transfer of Lambert-Eaton myasthenic syndrome with IgG from man to mouse depletes the presynaptic membrane active zones. Proc Natl Acad Sci. 1983;80(24):7636–7640.
9. Lennon VA, Kryzer TJ, Griesmann GE, et al. Calcium-channel antibodies in the Lambert-Eaton syndrome and other paraneoplastic syndromes. N Engl J Med. 1995;332(22):1467–1475. doi:10.1056/NEJM199506013322203
10. Wirtz PW, van Dijk JG, van Doorn PA, et al. The epidemiology of the Lambert-Eaton myasthenic syndrome in the Netherlands. Neurology. 2004;63(2):397–398.
11. Sanders DB. Lambert-Eaton myasthenic syndrome: diagnosis and treatment. Ann N Y Acad Sci. 2003;998:500–508.
12. Abenroth DC, Smith AG, Greenlee JE, Austin SD, Clardy SL. Lambert-Eaton myasthenic syndrome: epidemiology and therapeutic response in the national veteran’s affairs population. Muscle Nerve. 2017;56(3):421–426. doi:10.1002/mus.25520
13. Tim RW, Massey JM, Sanders DB. Lambert-Eaton myasthenic syndrome: electrodiagnostic findings and response to treatment. Neurology. 2000;54(11):2176–2178.
14. Titulaer MJ, Wirtz PW, Willems LN, Kralingen KWV, Smitt PAS, Verschuuren JJ. Screening for small cell lung cancer: A follow-up study of patients with Lambert-Eaton myasthenic syndrome. J Clin Oncol. 2008;26(26):4276–4281. doi:10.1200/JCO.2008.17.5133
15. Oneill JH, Murray NMF, Newsom-Davis J. The Lambert-Eaton myasthenic syndrome. Brain. 1988;111(3):577–596.
16. Titulaer M, Wirtz P, Kuks J, et al. The Lambert-Eaton myasthenic syndrome 1988–2008: A clinical picture in 97 patients. J Neuroimmunol. 2008;201–202:153–158. doi:10.1016/j.jneuroim.2008.05.025
17. Burns TM, Russell JA, Lachance DH, Jones HR. Oculobulbar involvement is typical with Lambert-Eaton myasthenic syndrome. Ann Neurol. 2003;53(2):270–273. doi:10.1002/ana.10477
18. Wirtz PW, Sotodeh M, Nijnuis M, et al. Difference in distribution of muscle weakness between myasthenia gravis and the Lambert-Eaton myasthenic syndrome. J Neurol Neurosurg Psychiatry. 2002;73(6):766–768.
19. Rudnicki SA. Lambert-Eaton myasthenic syndrome with pure ocular weakness. Neurology. 2007;68(21):1863–1864. doi:10.1212/01.wnl.0000264798.56174.df
20. Titulaer MJ, Wirtz PW, Wintzen AR, Verschuuren JJ. Re: lambert-Eaton myasthenic syndrome with pure ocular weakness. Neurology. 2008;70(1):86–87. doi:10.1212/01.wnl.0000295708.62369.59
21. Katyal N, Govindarajan R. Pure ocular weakness as the initial manifestation of Lambert-Eaton myasthenic syndrome. Cureus. 2017;12(9):e2007.
22. Titulaer MJ, Lang B, Verschuuren JJ. Lambert-Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies. Lancet Neurol. 2011;10(12):1098–1107. doi:10.1016/S1474-4422(11)70245-9
23. Mason WP, Graus F, Lang B, et al. Small cell lung cancer, paraneoplastic cerebellar degeneration and the Lambert-Eaton myasthenic syndrome. Brain. 1997;120(8):1279–1300.
24. Cho JJ, Wymer JP. Paraneoplastic Lambert-Eaton myasthenic syndrome with limbic encephalitis: clinical correlation with the coexistence of anti-VGCC and anti-GABAB receptor antibodies. J Clin Neuromuscul Dis. 2017;19(2):84–88. doi:10.1097/CND.0000000000000192
25. Neher E, Sakaba T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron. 2008;59(6):861–872. doi:10.1016/j.neuron.2008.08.019
26. Homan AE, Meriney SD. Active zone structure-function relationships at the neuromuscular junction. Synapse. 2018;72(11):e22057. doi:10.1002/syn.2018.72.issue-11
27. Slater CR. The functional organization of motor nerve terminals. Prog Neurobiol. 2015;134:55–103. doi:10.1016/j.pneurobio.2015.09.004
28. Jackman SL, Regehr WG. The mechanisms and function of synaptic facilitation. Neuron. 2017;94(3):447–464. doi:10.1016/j.neuron.2017.02.047
29. Jackman SL, Turecek J, Belinsky JE, Regehr WG. The calcium sensor synaptotagmin 7 is required for synaptic facilitation. Nature. 2016;529(7584):88–91. doi:10.1038/nature16507
30. Sakaba T. Kinetics of transmitter release at the calyx of Held synapse. Proc Jpn Acad Ser B Phys Biol Sci. 2018;94(3):139–152. doi:10.2183/pjab.94.010
31. Lambert EH, Elmqvist D. Quantal components of end-plate potentials in the myasthenic syndrome. Ann N Y Acad Sci. 1971;183(1):183–199.
32. Baslo MB, Deymeer F, Serdaroglu P, Parman Y, Ozdemir C, Cuttini M. Decrement pattern in Lambert-Eaton myasthenic syndrome is different from myasthenia gravis. Neuromuscul Disord. 2006;16(7):454–458. doi:10.1016/j.nmd.2006.05.009
33. Miralles F. Modeling the response to low-frequency repetitive nerve stimulation of myasthenia gravis and Lambert-Eaton myasthenic syndrome. Med Biol Eng Comput. 2016;54(11):1761–1778. doi:10.1007/s11517-016-1462-4
34. Tim RW, Massey JM, Sanders DB. Lambert-Eaton myasthenic syndrome (LEMS): clinical and electrodiagnostic features and response to therapy in 59 patients. Ann N Y Acad Sci. 1998;841(1MYASTHENIA GR):823–826.
35. Oh SJ, Kurokawa K, Claussen GC, Ryan HF. Electrophysiological diagnostic criteria of Lambert-Eaton myasthenic syndrome. Muscle Nerve. 2005;32(4):515–520. doi:10.1002/mus.20389
36. Luigetti M, Modoni A, Monaco ML. Low-rate repetitive nerve stimulation in Lambert-Eaton myasthenic syndrome: peculiar characteristics of decremental pattern from a single-center experience. Clin Neurophysiol. 2013;124(4):825–826. doi:10.1016/j.clinph.2012.08.026
37. Hatanaka Y, Oh SJ. Ten-second exercise is superior to 30-second exercise for post-exercise facilitation in diagnosing Lambert-Eaton myasthenic syndrome. Muscle Nerve. 2008;37(5):572–575. doi:10.1002/mus.20979
38. Oh SJ, Hatanaka Y, Claussen GC, Sher E. Electrophysiological differences in seropositive and seronegative Lambert-Eaton myasthenic syndrome. Muscle Nerve. 2007;35(2):178–183. doi:10.1002/mus.20672
39. Chiou-Tan FY, Gilchrist JM. Repetitive nerve stimulation and single-fiber electromyography in the evaluation of patients with suspected myasthenia gravis or Lambert-Eaton myasthenic syndrome: review of recent literature. Muscle Nerve. 2015;52(3):455–462. doi:10.1002/mus.24745
40. Trontelj JV, Stålberg E. Single motor end-plates in myasthenia gravis and LEMS at different firing rates. Muscle Nerve. 1991;14(3):226–232. doi:10.1002/mus.880140305
41. Chaudhry V, Watson DF, Bird SJ, Cornblath DR. Stimulated single-fiber electromyography in Lambert-Eaton myasthenic syndrome. Muscle Nerve. 1991;14(12):1227–1230. doi:10.1002/mus.880141215
42. Todisco V, Cirillo G, Capuano R, Dambrosio A, Tedeschi G, Gallo A. Stimulated single-fiber electromyography (sSFEMG) in Lambert-Eaton syndrome. Clin Neurophysiol Prac. 2018;3:148–150. doi:10.1016/j.cnp.2018.07.001
43. Motomura M, Johnston I, Lang B, Vincent A, Newsom-Davis J. An improved diagnostic assay for Lambert-Eaton myasthenic syndrome. J Neurol Neurosurg Psychiatry. 1995;58(1):85–87.
44. Motomura M, Lang B, Johnston I, Palace J, Vincent A, Newsom-Davis J. Incidence of serum anti-P/Q-type and anti-N-type calcium channel autoantibodies in the Lambert-Eaton myasthenic syndrome. J Neurol Sci. 1997;147(1):35–42.
45. Zalewski NL, Lennon VA, Lachance DH, Klein CJ, Pittock SJ, Mckeon A. P/Q- and N-type calcium-channel antibodies: oncological, neurological and serological accompaniments. Muscle Nerve. 2016;54(2):220–227. doi:10.1002/mus.25027
46. Benatar M. Presynaptic neuronal antigens expressed by a small cell lung carcinoma cell line. J Neuroimmunol. 2001;113(1):153–162.
47. Sabater L, Titulaer M, Saiz A, Verschuuren J, Gure AO, Graus F. SOX1 antibodies are markers of paraneoplastic Lambert-Eaton myasthenic syndrome. Neurology. 2007;70(12):924–928. doi:10.1212/01.wnl.0000281663.81079.24
48. Titulaer MJ, Klooster R, Potman M, et al. SOX antibodies in small cell lung cancer and Lambert-Eaton myasthenic syndrome: frequency and relation with survival. J Clin Oncol. 2009;27(26):4260–4267. doi:10.1200/JCO.2008.20.6169
49. Takamori M, Motomura M, Fukudome T, Yoshikawa H. Autoantibodies against M1 muscarinic acetylcholine receptor in myasthenic disorders. Eur J Neurol. 2007;14(11):1230–1235. doi:10.1111/j.1468-1331.2007.01931.x
50. Takamori M. Lambert-Eaton myasthenic syndrome: search for alternative autoimmune targets and possible compensatory mechanisms based on presynaptic calcium homeostasis. J Neuroimmunol. 2008;201–202:145–152. doi:10.1016/j.jneuroim.2008.04.040
51. Wirtz PW, Lang B, Graus F, et al. P/Q-type calcium channel antibodies, Lambert-Eaton myasthenic syndrome and survival in small cell lung cancer. J Neuroimmunol. 2005;164(1–2):161–165. doi:10.1016/j.jneuroim.2005.04.001
52. Maddison P, Newsom-Davis J, Mills KR, Souhami RL. Favorable prognosis in Lambert-Eaton myasthenic syndrome and small cell lung carcinoma. Lancet. 1999;353(9147):117–118. doi:10.1016/S0140-6736(05)76153-5
53. Maddison P, Gozzard P, Grainge MJ, Lang B. Long-term survival in paraneoplastic Lambert-Eaton myasthenic syndrome. Neurology. 2017;88(14):1334–1339. doi:10.1212/WNL.0000000000003794
54. Titulaer MJ, Maddison P, Sont JK, et al. Clinical Dutch-English Lambert-Eaton myasthenic syndrome (LEMS) tumor association prediction score accurately predicts small cell lung cancer in the LEMS. J Clin Oncol. 2011;29(7):902–908. doi:10.1200/JCO.2010.32.0440
55. McEvoy KM, Windebank AJ, Daube JR, Low PA. 3,4-Diaminopyridine in the treatment of Lambert-Eaton myasthenic syndrome. N Engl J Med. 1989;321:1567. doi:10.1056/NEJM198912073212303
56. Wirtz P, Verschuuren J, Dijk JV, et al. Efficacy of 3,4-diaminopyridine and pyridostigmine in the treatment of Lambert-Eaton myasthenic syndrome: A randomized, double-blind, placebo-controlled, crossover study. Clin Pharmacol Ther. 2009;86(1):44–48. doi:10.1038/clpt.2009.35
57. Sanders DB, Massey JM, Sanders LL, Edwards LJ. A randomized trial of 3,4-diaminopyridine in Lambert-Eaton myasthenic syndrome. Neurology. 2000;54(3):603.
58. Oh SJ, Claussen GG, Hatanaka Y, Morgan MB. 3,4-Diaminopyridine is more effective than placebo in a randomized, double-blind, cross-over drug study in LEMS. Muscle Nerve. 2009;40(5):795–800. doi:10.1002/mus.21422
59. Oh SJ, Shcherbakova N, Kostera-Pruszczyk A, et al. Amifampridine phosphate (Firdapse®) is effective and safe in a phase 3 clinical trial in LEMS. Muscle Nerve. 2016;53(5):717–725. doi:10.1002/mus.25070
60. Sanders DB, Juel VC, Harati Y, et al. 3,4-diaminopyridine base effectively treats the weakness of Lambert-Eaton myasthenia. Muscle Nerve. 2018;57(4):561–568. doi:10.1002/mus.26052
61. Keogh M, Sedehizadeh S, Maddison P. Treatment for Lambert-Eaton myasthenic syndrome. Cochrane Database Syst Rev. 2011;CD003279.
62. Rich MM, Teener JW, Bird SJ. Treatment of Lambert-Eaton syndrome with intravenous immunoglobulin. Muscle Nerve. 1997;20(5):614–615.
63. Muchnik S, Losavio AS, Vidal A, Cura L, Mazia C. Long-term follow-up of Lambert-Eaton syndrome treated with intravenous immunoglobulin. Muscle Nerve. 1997;20(6):674–678.
64. Bain PG, Motomura M, Newsom-Davis J, et al. Effects of intravenous immunoglobulin on muscle weakness and calcium-channel autoantibodies in the Lambert-Eaton myasthenic syndrome. Neurology. 1996;47(3):678–683.
65. Maddison P, McConville J, Farrugia ME, et al. The use of rituximab in myasthenia gravis and Lambert-Eaton myasthenic syndrome. J Neurol Neurosurg Psychiatry. 2011;82(6):671–673. doi:10.1136/jnnp.2009.197632
66. Newsom-Davis J, Murray NM. Plasma exchange and immunosuppressive drug treatment in the Lambert-Eaton myasthenic syndrome. Neurology. 1984;34(4):480.
67. Dau PC, Denys EH. Plasmapheresis and immunosuppressive drug therapy in the Eaton-Lambert syndrome. Ann Neurol. 1982;11(6):570–575. doi:10.1002/ana.410110604
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.