Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Lactoferrin-modified PEGylated liposomes loaded with doxorubicin for targeting delivery to hepatocellular carcinoma
Authors Wei M, Guo X, Tu L, Zou Q, Li Q, Tang C, Chen B, Xu Y, Wu C
Received 20 April 2015
Accepted for publication 7 June 2015
Published 12 August 2015 Volume 2015:10(1) Pages 5123—5137
DOI https://doi.org/10.2147/IJN.S87011
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Lei Yang
Minyan Wei,1,2 Xiucai Guo,1,3 Liuxiao Tu,1 Qi Zou,1 Qi Li,1 Chenyi Tang,1 Bao Chen,1 Yuehong Xu,1 Chuanbin Wu1
1Department of Pharmaceutics, School of Pharmaceutical Sciences, Sun Yat-sen University, 2Department of Pharmaceutics, School of Pharmaceutical Sciences, Guangzhou Medical University, 3Department of Pharmacy, 12th People’s Hospital of Guangzhou City, Guangzhou, People’s Republic of China
Abstract: Lactoferrin (Lf) is a potential-targeting ligand for hepatocellular carcinoma (HCC) cells because of its specific binding with asialoglycoprotein receptor (ASGPR). In this present work, a doxorubicin (DOX)-loaded, Lf-modified, polyethylene glycol (PEG)ylated liposome (Lf-PLS) system was developed, and its targeting effect and antitumor efficacy to HCC was also explored. The DOX-loaded Lf-PLS system had spherical or oval vesicles, with mean particle size approximately 100 nm, and had an encapsulation efficiency of 97%. The confocal microscopy and flow cytometry indicated that the cellular uptake of Lf-PLS was significantly higher than that of PEGylated liposome (PLS) in ASGPR-positive cells (P<0.05) but not in ASGPR-negative cells (P>0.05). Cytotoxicity assay by MTT demonstrated that DOX-loaded Lf-PLS showed significantly stronger antiproliferative effects on ASGPR-positive HCC cells than did PLS without the Lf modification (P<0.05). The in vivo antitumor studies on male BALB/c nude mice bearing HepG2 xenografts demonstrated that DOX-loaded Lf-PLS had significantly stronger antitumor efficacy compared with PLS (P<0.05) and free DOX (P<0.05). All these results demonstrated that a DOX-loaded Lf-PLS might have great potential application for HCC-targeting therapy.
Keywords: asialoglycoprotein receptor, immunoliposome, PEGylated modification, post-insertion, hepatic cancer, active targeting
Introduction
Hepatocellular carcinoma (HCC) is the sixth most prevalent cancer and the third most frequent cause of cancer-related death worldwide.1 Treatments of HCC can be divided into curative and palliative.2 Curative treatments conventionally include surgical resection, liver transplantation, and percutaneous ablation.2,3 Patients with early HCC should be considered for curative treatments that may achieve long-term complete response and improved survival.3–5 Unfortunately, more than 80% of patients present with advanced or unresectable disease, and these patients are just suitable for palliative approaches.2–5 Systemic chemotherapy is commonly used as a palliative treatment for improved survival.2–4
Currently, doxorubicin (DOX), an anthracycline antibiotic, is one of the most important chemotherapeutic agents for HCC.6–8 DOX exerts its cytotoxicity by inhibiting the synthesis of nucleic acids within cancer cells.8 However, the systemic administration of DOX is severely obstructed by its limited therapeutic responses and undesirable systemic toxicities.6–8 Therefore, improving the selective accumulation of DOX in HCC tumor cells might be an effective method to enhance its antitumor efficacies and minimize its systemic toxicities.7,8
Polyethylene glycol (PEG)ylated liposomes (PLSs) are widely considered as potential anticancer chemotherapeutic agent carriers for cancer treatment.9–12 PLS exhibits preferential localization in the solid tumor tissue due to the enhanced permeability and retention effect (EPR effect), which relies on the PEGylated modification to extend the circulation time and avoid rapid clearance by the reticuloendothelial system.9,10 Furthermore, specific tumor-homing ligand modification of the PLS could significantly improve its therapeutic efficacy by enhancing the drug accumulation into cancer cells (because ligands have showed specific binding to the receptors overexpressed in tumor cells).9,10 So, a ligand-modified PLS system might be a promising approach to selectively deliver DOX to HCC cells for HCC treatment.
Asialoglycoprotein receptors (ASGPRs) are the promising targets for drug delivery in HCC treatment, due to their high expressions on the surface of HCC cell lines.11,13 Recently, lactoferrin (Lf), a mammalian cationic iron-binding glycoprotein belonging to the Tf family, has been demonstrated to bind ASGPR with high affinity in a galactose-independent manner.14–17 It might be suggested that Lf is a good ligand for ASGPR binding. With its specific binding, Lf has been applied to gene delivery successfully, and its ability to target hepatic tumor cells also has been confirmed.18–21 In our previous work, an Lf-modified PEGylated liposome (Lf-PLS) system was successfully constructed, and the results demonstrated that Lf-PLS might have great potential for HCC targeting, with low toxicity.21 However, the feasibility of whether this targeting delivery carrier loaded with chemotherapeutic agent could obtain an enhanced drug accumulation into HCC cells and achieve an increased antitumor effect still needs to be confirmed. Therefore, in this present work, the Lf-PLS system was applied as an active HCC-targeting drug carrier, for encapsulation of DOX.
The purposes of this study were to develop a DOX-loaded Lf-PLS system and to investigate its targeting effect and antitumor efficacy to HCC. PLSs were prepared by thin film method combined with PEG-lipid post-insertion.22 Lf was conjugated to the carboxyl terminal of 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG2000)-COOH previously anchored on liposomes. DOX was chosen as a model drug and encapsulated into the carrier by ammonium sulfate gradient method. Then the DOX-loaded Lf-PLS system was characterized in detail. In order to testify to the targeting effect of this system, cellular uptakes by different ASGPR-expressed cells were performed, by confocal microscopy observation and flow cytometry determination. In vitro cytotoxicity study was conducted to evaluate the antiproliferative effect of DOX-loaded Lf-PLS by MTT assay. In vivo antitumor study was further carried out on BALB/c nude mice bearing HepG2 xenografts, to investigate the antitumor efficacy of DOX-loaded Lf-PLS.
Material and methods
Reagents and cells
Soybean phosphatidylcholine (purity >92%) and DSPE-PEG2000 were purchased from Lipoid GmbH (Ludwigshafen, Germany). DSPE-PEG2000 (ammonium salt) (DSPE-PEG2000-COOH) was purchased from Avanti Polar lipids (Alabaster, AL, USA). Cholesterol, 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (EDC), N-hydroxysulfosuccinimide (NHS), bovine Lf, 4′,6-diamidino-2-phenylindole (DAPI), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), and 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) were purchased from Sigma-Aldrich Corp (St Louis, MO, USA). DOX hydrochloride was purchased from Beijing Huafeng United Technology Co., Ltd (Beijing, People’s Republic of China). The gel filtration mediums used in affinity chromatography, protein chromatography and gel filtration chromatography were Sephadex® G75 and Sepharose® CL-4B, which were purchased from GE Healthcare (Little Chalfont, UK). Dulbecco’s Modified Eagle’s Medium (DMEM), fetal bovine serum (FBS), and penicillin–streptomycin were acquired from GIBCO® (Life Technologies Corp, Carlsbad, CA, USA). Human HCC cell lines HepG2, BEL7402, SMMC7721, and mouse embryonic fibroblast cell line NIH 3T3 were obtained from the Shanghai Institute of Cell Biology (Chinese Academy of Sciences, Shanghai, People’s Republic of China). All other reagents were of analytical grade.
Animals
Male BALB/C nude mice (18–20 g) were obtained from the SLAC Laboratory Animal Co. Ltd. (Changsha, People’s Republic of China). All work performed with animals was in accordance with and approved by the Experiment Animal Administrative Committee of Sun Yat-sen University.
Preparation of drug-loaded liposomes
Preparation of DOX-loaded conventional liposomes
Liposomes of soybean phosphatidylcholine/cholesterol (3:2 molar ratio) were prepared by the thin film hydration method as described previously.23 Briefly, the above lipids were dissolved in dichloromethane/ethanol (1:2 V/V) and dried to form a thin lipid film in a rotary evaporator (EYELA, Tokyo, Japan). Then the film was hydrated with 155 mM ammonium sulfate, and the liposome suspension was extruded through a polycarbonate membrane (200 nm) for ten times using an extruder (Avestin, Ottawa, ON, Canada).
Afterward, DOX was remotely loaded into the liposomes via the ammonium gradient method.11,23 Liposomes were eluted through a Sephadex G 75 column (GE Healthcare) preequilibrated with a HEPES buffer (20 mM HEPES and 144 mM NaCl, pH 7.4). DOX was added to the liposomes and incubated at 60°C for 20 minutes. Free DOX was removed by Sepharose CL-4B column.
Preparation of DOX-loaded PLS
DOX-loaded PLSs were prepared using the PEG-lipid post-insertion technique.22 DSPE-PEG2000 (containing 5 mol% of the total lipid content) was added to the DOX-loaded liposome suspension, and the mixture was incubated at 60°C for 1 hour.
Preparation of DOX loaded Lf-PLS
The DOX loaded Lf-PLS were prepared by covalent coupling with Lf to the surface of liposomes via the free amino groups of Lf and carboxyl functional groups of the PEG-lipid DSPE-PEG2000-COOH.24 Briefly, DSPE-PEG2000 and DSPE-PEG2000-COOH (lipid/DSPE-PEG2000/DSPE-PEG2000-COOH in a 95:4.5:0.5 molar ratio) were suspended into DOX-loaded liposomes and incubated at 60°C for 1 hour as mentioned earlier. Then both EDC and NHS (EDC/NHS/DSPE-PEG2000-COOH as 30:30:3, molar ratio) were added, and the mixture was stirred for 10 minutes at room temperature. Afterward, Lf (Lf/DSPE-PEG2000-COOH in a 1:40 molar ratio) was added into the liposome suspension, and then the suspension was incubated at 37°C for another 3 hours. Lf-modified PLS was separated from free Lf and DOX using Sepharose CL-4B.
Characterization of liposomes
Transmission electron microscopy (TEM)
The morphology and size of liposomes were observed by TEM (JEM-100CX II; JEOL, Tokyo, Japan). Briefly, the liposome dispersion was applied to a copper grid and allowed a 5-minute incubation at room temperature. Then the grid was stained with 1% uranyl acetate, and the image was taken after drying.
Particle size and zeta potential
The particle size and zeta potential of the DOX-loaded PLS and Lf-PLS were measured using a Malvern Zetasizer Nano ZS90 (Malvern Instruments, Malvern, UK). All the particle sizes were performed by dynamic light scattering (DLS) analysis at 25°C and at a scattering angle of 90°. The zeta potential values were calculated using the Smoluchowski equation.21
Encapsulation efficiency (EE)
To determine the EE of DOX in the liposome formulations, liposomes were separated from free DOX by Sepharose CL-4B. The DOX concentration was quantified by a TU-1901 UV-Vis spectrophotometer (Purkinje General, Beijing, People’s Republic of China) at 480 nm, after lysis of the liposomes with 1% (V/V) Triton™ X-100. The percent of DOX EE (EE %) was calculated based on the amount ratio of liposomal drug and total drug added.
In vitro release study
The in vitro release kinetics of DOX from PLS and Lf-PLS was measured by a dialysis method. Briefly, free DOX, and DOX-loaded PLS and Lf-PLS were placed in the dialysis bags (molecular weight [Mw] of 8,000–14,000 Da) and dialyzed against phosphate-buffered saline (PBS) with 10% FBS under continuous stirring at 37°C for 24 hours. At designated time points, aliquots were withdrawn from the incubation medium and replaced with equal volume of the fresh medium. The concentrations of DOX were then measured by using a model LS55 fluorescence spectrophotometer (Perkin-Elmer, Boston, MA, USA). The excitation and emission wavelengths were set at 480 nm and 590 nm, respectively.
Cell culture
Human HCC cell lines HepG2, BEL7402, and SMMC7721, and mouse embryonic fibroblast cell line NIH 3T3 were cultured in DMEM supplemented with 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin in a 37°C humidified incubator with 5% carbon dioxide.
Confocal microscopy studies
Cellular binding and internalization of the liposomes were analyzed by confocal microscopy. HepG2, BEL7402, SMMC7721, and NIH 3T3 cells were cultured on the cover slips in six-well plates for 24 hours. Then the cells were exposed to free DOX, and DOX-loaded PLS and Lf-PLS for 4 hours at 37°C, with a final DOX concentration of 40 μg/mL, respectively. After the incubation, the cover slips were washed with cold PBS (pH7.4) three times, and the cells were fixed with 4% p-formaldehyde for 20 minutes, followed by cell nuclei staining with DAPI for another 15 minutes. The fluorescence images were analyzed by laser scanning confocal microscope (LSM 710; Carl Zeiss Microscopy GmbH, Jena, Germany).
Flow cytometry analysis
The cellular uptake of DOX-loaded PLS and Lf-PLS was also assessed by flow cytometry analysis. HepG2, BEL7402, SMMC7721, and NIH 3T3 cells were seeded onto 24-well plates at a density of 5×105 cells per well and cultured for 24 hours. Then cells were incubated with DOX-loaded PLS and Lf-PLS at a DOX concentration of 40 μg/mL for 4 hours at 37°C, respectively. After the incubation, the cells were washed with cold PBS three times, trypsinized and collected by centrifugation, and then resuspended in PBS. The cell association of the fluorescence was analyzed by a Beckman Coulter EPICS XL flow cytometer (Beckman Coulter, Fullerton, CA, USA) equipped with an argon laser (488 nm) and 530/30 nm band pass filters for emission measurements. Approximately 10,000 events were counted for each sample. Each data point was collected in triplicate. The cells without any treatment were used as the blank controls.
In vitro cytotoxicity assay
MTT assay was applied to evaluate the in vitro cytotoxicity of the various DOX formulations on HepG2, BEL7402, and SMMC7721 cells. Briefly, cells were seeded into 96-well plates at a density of 5×103 cells per well and cultured for 24 hours. Then free DOX or DOX-loaded liposome formulations of various DOX concentrations were added, and the cells were incubated for 24, 48, and 72 hours, respectively. After the incubation, 20 μL MTT (5 mg/mL) were added to each well, and the cells were incubated for another 4 hours at 37°C. Finally, formazan crystals were solubilized by 150 μL of dimethyl sulfoxide (DMSO), and the absorbance was determined at 570 nm on an EXL 800 plate reader (BioTek, Winooski, VT, USA). The cell viability was calculated and compared with the untreated control. The 50% inhibitory concentration (IC50) was calculated from semilogarithmic dose–response plots. Experiments were repeated for three times, and data were presented as mean ± standard deviation (SD).
In vivo antitumor study
The in vivo antitumor efficacy was studied on the HepG2 cells-bearing male BALB/c nude mice in an armpit tumor model. Briefly, HepG2 cells (2×106 cells/mouse) were subcutaneously transplanted into the flanks of nude mice. Tumor volume was measured by a vernier caliper and calculated as
Volume = Length × (Width)2/2. | (1) |
The mice were randomly assigned to four groups (eight for each) after the tumor volume reached a volume of 50 mm3, and the treatment was also started at that time.
Saline, free DOX, and DOX-loaded PLS and Lf-PLS were injected intravenously at a dose of 5 mg DOX/kg body weight via the tail vein, respectively. Each group was treated every 7 days for 3 weeks (ie, three times). Tumor volumes were measured with calipers every 2 days and calculated by equation 1. Relative tumor volume (RTV) was calculated by the formula
RTV = Vn/V0, | (2) |
where Vn is the tumor volume measured at the corresponding day, and V0 is the tumor volume measured at the day before treatment.25 Changes in body weight of each mouse were monitored during the treatment to evaluate possible toxic effects of the therapy.
At the end point (day 21 after treatment), the mice were sacrificed, and the tumors were measured by calipers and the RTV calculated. The tumor volume inhibition (TVI) was calculated from the formula
TVI = (CRTV − TRTV)/CRTV, | (3) |
where TRTV represents the RTV of the treatment groups, and CRTV represents the RTV of the saline group. Then the postmortem tumor was harvested, washed, photographed, and weighted.25 The tumor weight inhibition (TWI) was calculated from the formula
TWI = (WC − WD)/WC, | (4) |
where WC and WD represents the tumor weight of animals from the saline and treatment groups, respectively.26
Statistical analysis
Data were presented as the mean ± SD. Statistical comparisons were performed by Student’s t-test or analysis of variance (ANOVA), followed by Fischer’s least significant difference (LSD) post hoc test using SPSS 13.0 software (SPSS Inc, Chicago, IL USA). A value of P<0.05 was considered statistically significant.
Results
Preparation of DOX-loaded liposomes
The procedure of DOX-loaded Lf-PLS preparation was illustrated in Figure 1. Conventional liposomes were prepared by thin film hydration, and DOX was loaded onto the liposomes by the ammonium sulfate gradient method. Then the outer surface of the liposomes was modified by PEG-lipid post-insertion. Finally, DOX-loaded Lf-PLS was synthesized by a coupling reaction between the amino groups of Lf and the carboxyl groups of PEG-lipid on the liposomes (Figure 1). The amount of Lf conjugated to the liposomes was measured by a bicinchoninic acid (BCA) protein assay, and the Lf binding efficiency was approximately 13.37 μg Lf/μmol PL, which was earlier reported.21
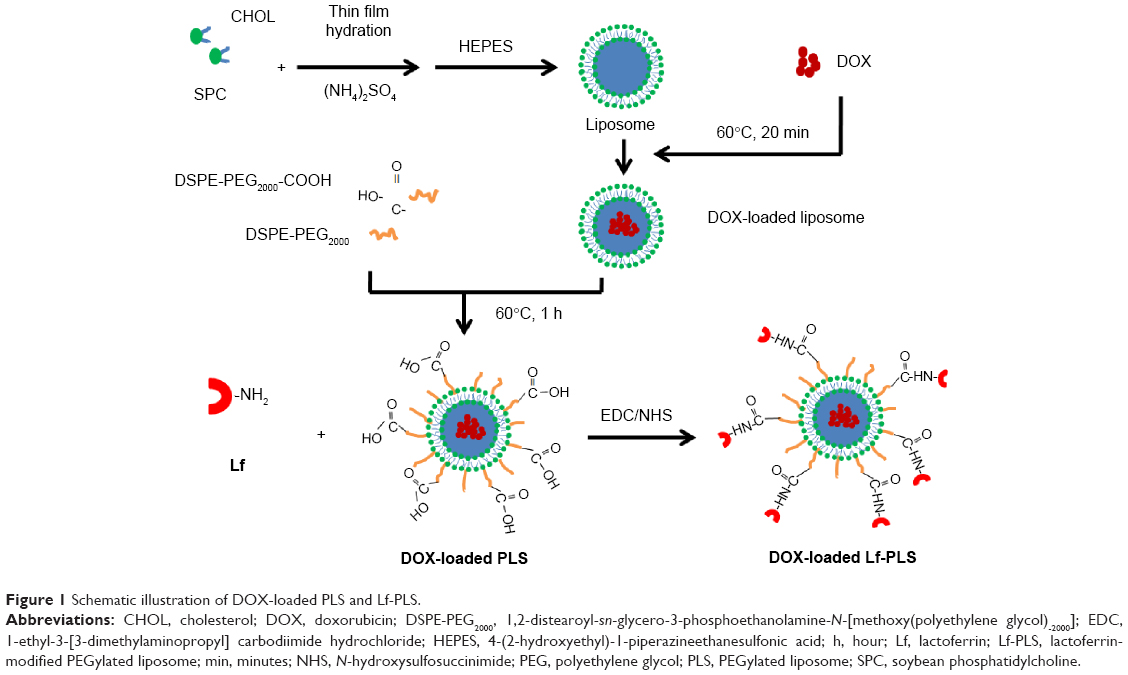 | Figure 1 Schematic illustration of DOX-loaded PLS and Lf-PLS. |
Characterization of DOX-loaded liposomes
DOX-loaded liposomes were found to be spherical or oval vesicles under TEM observation (Figure 2). The mean particle sizes of the DOX-loaded liposomes analyzed by DLS were approximately 100 nm with a polydispersity index less than 0.2. These results appeared to be consistent with those observed by TEM, which indicated that DOX-loaded liposomes had small particle sizes with a narrow size distribution (Table 1 and Figure 2). Both DOX-loaded PLS and Lf-PLS exhibited negative zeta potential values approximately −10 mV (Table 1), which might be because the negatively charged PEG-lipid insertion decreased the zeta potential of the liposomes.27,28 And the slightly increased zeta potential value of Lf-PLS might have resulted from the positively charged Lf attachment.29
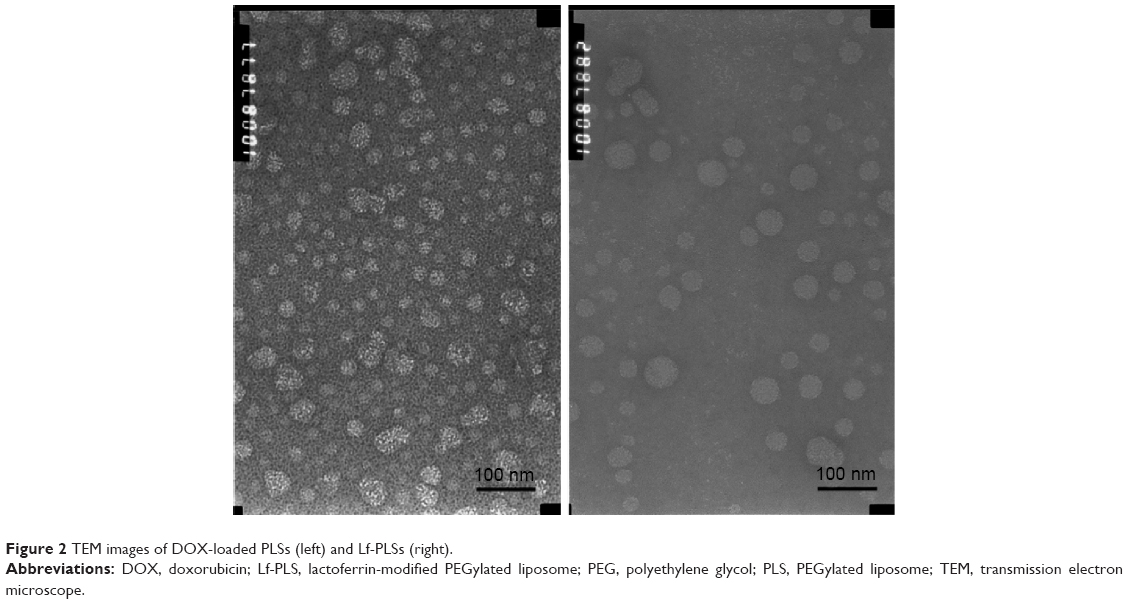 | Figure 2 TEM images of DOX-loaded PLSs (left) and Lf-PLSs (right). |
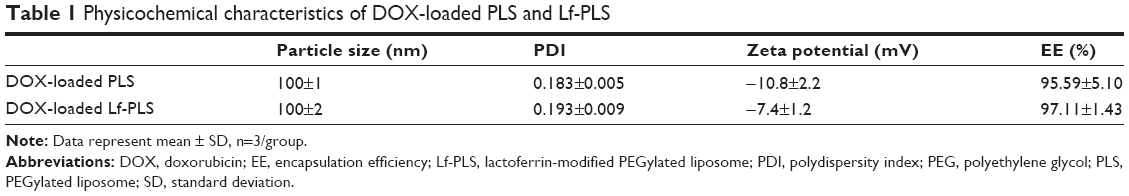 | Table 1 Physicochemical characteristics of DOX-loaded PLS and Lf-PLS |
The DOX EE of all the liposomes was above 90% (Table 1). This result indicated that there was no significant difference in the EE of PLS and Lf-PLS (P>0.05), implying that the modification of Lf did not affect the drug-entrapment ability of the liposomes.
In vitro release study of DOX-loaded liposomes
In order to investigate the stability and extended circulation character of the liposomes, pH 7.4 PBS with 10% FBS was chosen as the release medium to simulate the blood environment in the in vitro release study. The in vitro release profiles of DOX from different formulations were showed in Figure 3. More than 80% of the free DOX was found in release medium after approximately 4 hours, which suggested that free drug could freely diffuse through the dialysis membrane. In contrast with the free DOX, there was a sustained release of DOX from Lf-PLS. Only approximately 12% of DOX was released from Lf-PLS after 24 hours. And the release behavior of DOX from PLS was similar to that of Lf-PLS (P>0.05). The in vitro release study demonstrated that both liposomal formulations showed good stability and an extended circulation time characteristic to avoid drug clearance before arrival at the target cells.
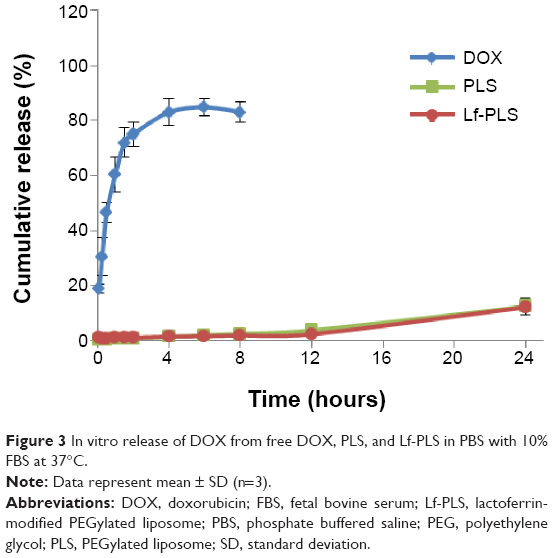 | Figure 3 In vitro release of DOX from free DOX, PLS, and Lf-PLS in PBS with 10% FBS at 37°C. |
Cellular uptake of Lf-modified liposomes
In order to investigate the targeting effect of DOX-loaded Lf-PLS, cellular uptake by different ASGPR-expressed cells, including ASGPR-positive HepG2, BEL7402, and SMMC7721 cells, and ASGPR-negative NIH 3T3 cells was carried out.11,30,31 Confocal microscopy observation was employed to evaluate the cellular uptake and distribution of the internalized DOX.
As shown in Figure 4, no matter the cell lines, overexpressed ASGPR or not, free DOX exhibited the highest fluorescence intensities in all of the DOX formulations, and the DOX fluorescence was found to be almost overlapped with the DAPI-stained cell nuclei. The phenomenon that cells treated with free DOX showed the highest cellular uptake was not unexpected because the cellular uptake behavior of free DOX was different from that of liposomes. Free DOX could readily diffuse across the cell membranes and then easily located into the cell nuclei, while liposomes entered the cells by receptor-meditated and nonspecific endocytosis, and then DOX released from the carriers and exited the endocytic compartment before locating into the cell nuclei.32–34 The different cellular uptake behaviors made the cellular uptake of free DOX easier.
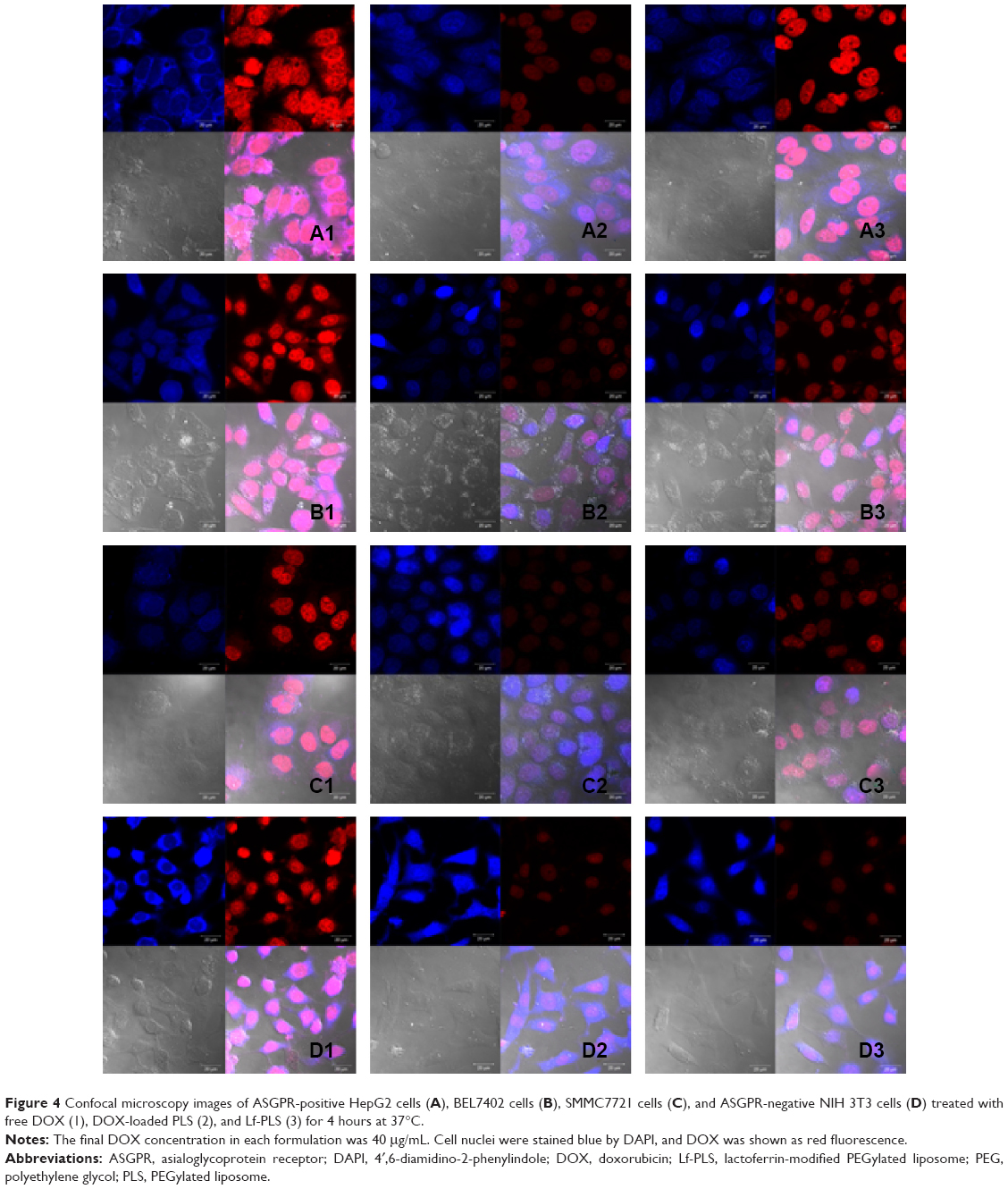 | Figure 4 Confocal microscopy images of ASGPR-positive HepG2 cells (A), BEL7402 cells (B), SMMC7721 cells (C), and ASGPR-negative NIH 3T3 cells (D) treated with free DOX (1), DOX-loaded PLS (2), and Lf-PLS (3) for 4 hours at 37°C. |
For the ASGPR-positive cells, including HepG2, BEL7402, and SMMC7721, the DOX fluorescence intensities of the Lf-PLS were much higher than those of PLS (Figure 4A–C). However, the results for ASGPR-negative cells NIH 3T3 were not the same (Figure 4D2 and D3). The fluorescence intensities of the both liposomes were quite low, showing no differences between them. These results suggested that Lf-PLS might enhance the cellular uptake by ASGPR-positive cells, which might be resulted from the specific binding of Lf to ASGPR on hepatic tumor cells.32–34 Liposomes also displayed strong fluorescence in cell nuclei, indicating that DOX was easily released from the both carriers and located on cell nuclei after internalization.35
Flow cytometry determination was also performed to quantify the cellular uptakes of the liposomes (Figure 5). As illustrated in Figure 5A–C, the DOX fluorescence intensities of Lf-PLS in ASGPR-positive cells, including HepG2, BEL7402, and SMMC7721, were twofold (26.93±4.40 vs 13.37±1.88) (P<0.05), 1.8-fold (35.73±5.01 vs 19.67±0.68) (P<0.05), and 1.3-fold (17.87±0.47 vs 13.93±0.45) (P<0.05) as much as those of PLS, respectively. However, Figure 5D showed that, in ASGPR-negative cells NIH 3T3, the fluorescence intensities showed no significant difference between PLS and Lf-PLS (14.07±0.85 vs 13.23±0.74) (P>0.05). These flow cytometry results confirmed the finding from the confocal microscopy observations that DOX-loaded liposomes could effectively target the ASGPR-positive cells via Lf modification.
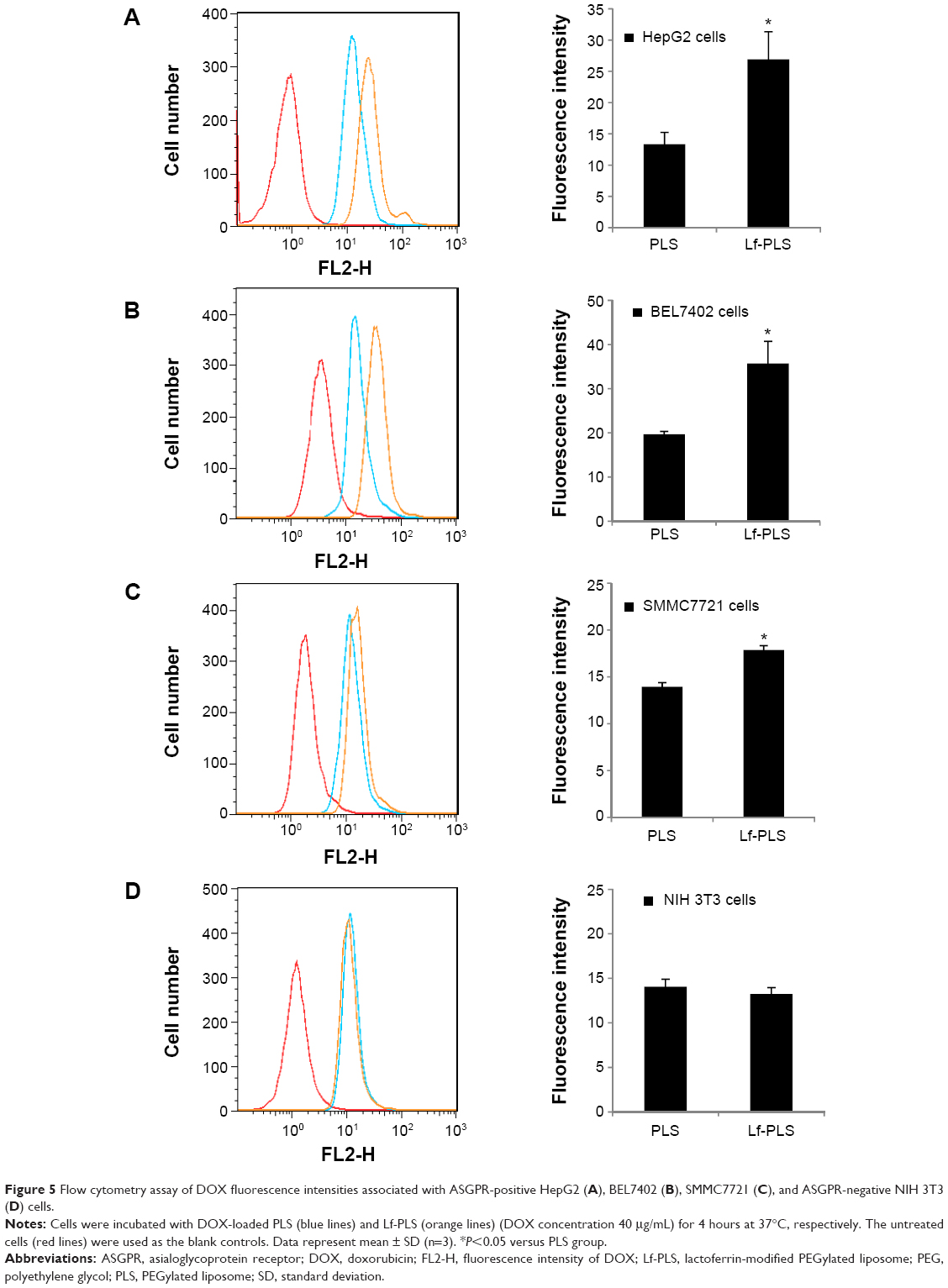 | Figure 5 Flow cytometry assay of DOX fluorescence intensities associated with ASGPR-positive HepG2 (A), BEL7402 (B), SMMC7721 (C), and ASGPR-negative NIH 3T3 (D) cells. |
It was demonstrated ASGPRs were overexpressed in the HCC cell lines including HepG2, BEL7402, and SMMC7721 cells, but negatively expressed in NIH 3T3 cells.36,37 The different cellular uptake of Lf-PLS in both HCC cell lines and NIH 3T3 cells might be mainly dependent on the different ASGPR expression on cell membranes.13,36,37 Similar results were also obtained in the previous study of coumarin-6-loaded Lf-PLS binding to HepG2.21
The cellular uptake results also suggested that more Lf-PLSs were endocytosed into HepG2 and BEL7402 cells than into SMMC7721 cells. The positive ASGPR expression of the HCC cell lines was demonstrated to be different.37 Of the three different cell lines, the ASGPR expression in BEL7402 cells was found to be the highest, followed by SMMC7721, and the ASGPR expression in HepG2 cells were showed to be the lowest.37 The cellular uptake results showing that more Lf-PLSs were endocytosed into HepG2 and BEL7402 cells than into SMMC7721 cells were not consistent with the ASGPR expression in the HCC cells.
An explanation for the interesting cellular uptake results might be that besides the different ASGPR expression in the HCC cells, Lf-PLS might exploit different uptake mechanisms to enter the different HCC cell lines.38 To clarify these phenomena, the specific endocytotic pathways for Lf-PLS in different HCC cell lines should be explored.39
In vitro cytotoxicity
The cytotoxicity of DOX-loaded Lf-PLS against ASGPR-positive cells was evaluated using MTT assay. The effects on cell viability of free DOX and its liposomal formulations in HepG2, BEL7402, and SMMC7721 cells were graphically represented in Figure 6. The IC50 values were also showed in Table 2.
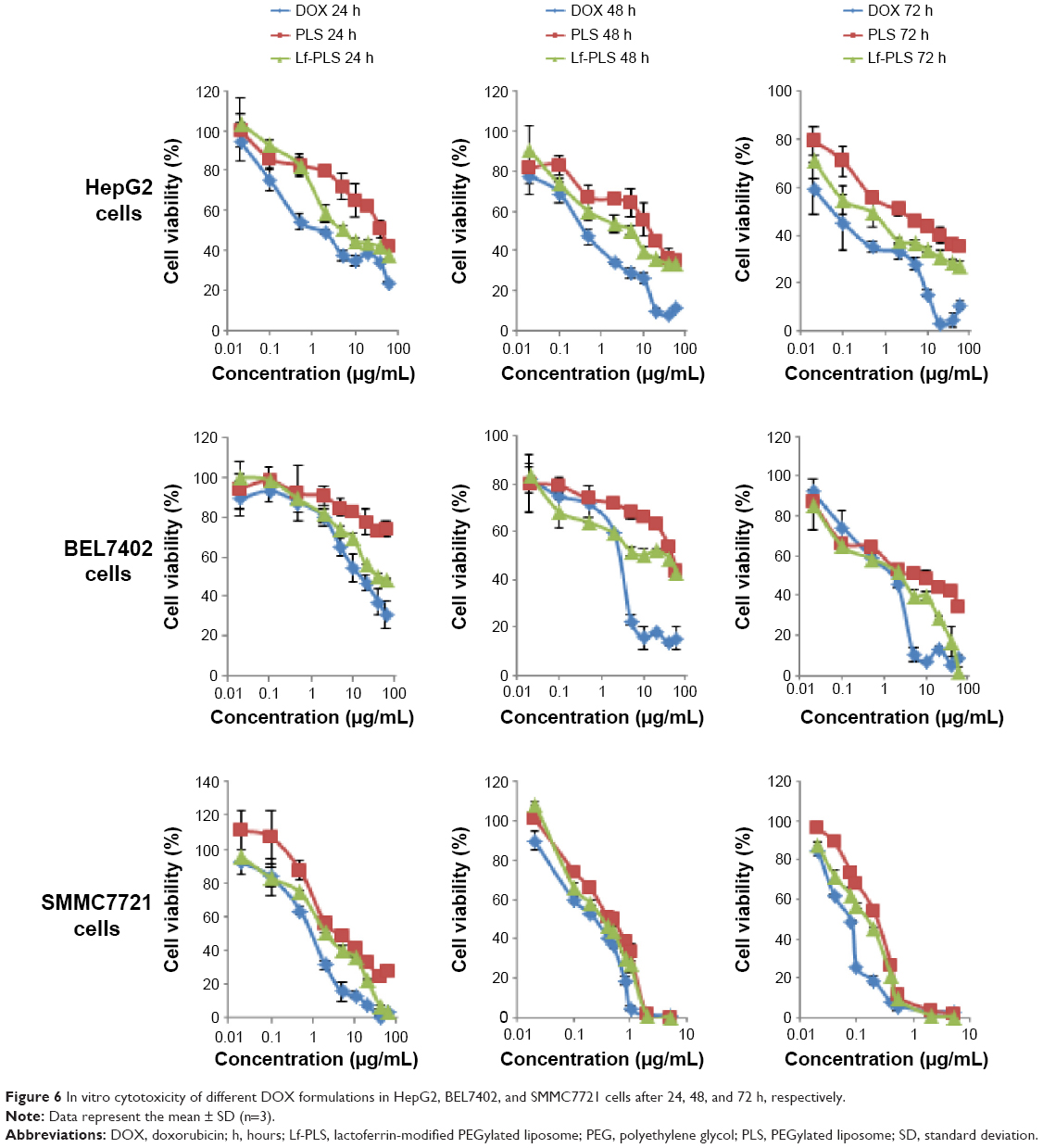 | Figure 6 In vitro cytotoxicity of different DOX formulations in HepG2, BEL7402, and SMMC7721 cells after 24, 48, and 72 h, respectively. |
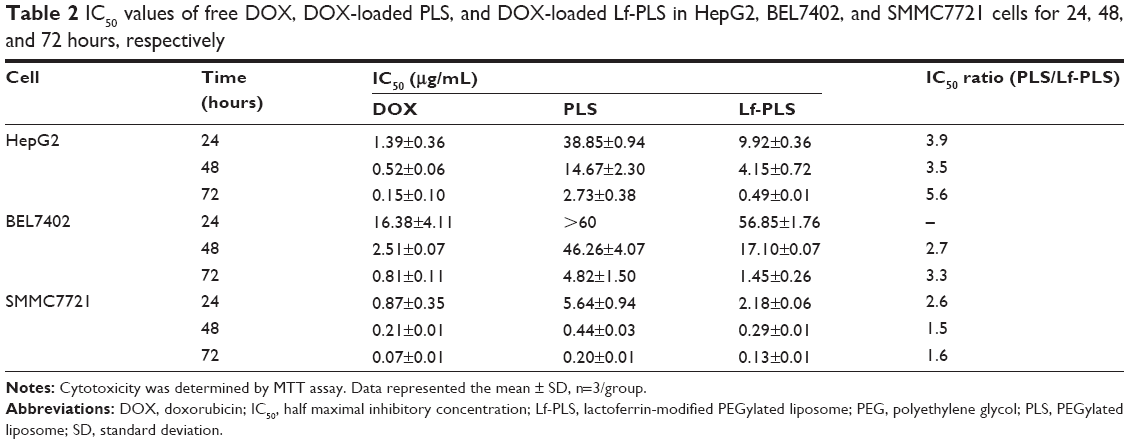 | Table 2 IC50 values of free DOX, DOX-loaded PLS, and DOX-loaded Lf-PLS in HepG2, BEL7402, and SMMC7721 cells for 24, 48, and 72 hours, respectively |
As showed in Figure 6, the cytotoxic effects of free DOX and DOX-loaded liposomes in ASGPR-positive cells were remarkably increased, in a dose- and time-dependent manner.
The IC50 values of free DOX, and DOX-loaded PLS and Lf-PLS against HepG2 cells for 72 hours were 0.15±0.10 μg/mL, 2.73±0.38 μg/mL, and 0.49±0.01 μg/mL, respectively (Table 2). These revealed that the cytotoxicity of free DOX was significantly higher than when loaded on liposomes. Similar results were found in BEL7402 and SMMC7721 cells. The good inhibitory effects of free DOX might be attributed to its good binding ability to nuclear DNA when exposed to tumor cells. The cytotoxicity of DOX-loaded Lf-PLS against ASGPR-positive cells was found to be approximately two- to sixfold higher than that of PLS, suggesting that DOX-loaded Lf-PLS exhibited superior antiproliferative effects compared with PLS (Table 2). The improved cytotoxicity effects of DOX-loaded Lf-PLS against ASGPR-positive HCC cells might have resulted from the enhanced cellular uptake of Lf-PLS mediated by Lf, which is in good agreement of the flow cytometry determinations and the confocal microscopy observations.
However, the MTT study (Figure 6 and Table 2) showed that more cytotoxic effects of Lf-PS were found in SMMC7721 cells than in HepG2 and BEL7402 cells, which seems to conflict with the results of cellular uptake. Similarly, the MTT study (Figure 6 and Table 2) also showed that at the same incubation times, the cytotoxicity of DOX in SMMC7721 was found to be the strongest, followed by HepG2, and the cytotoxicity of DOX in BEL7402 was found to be the lowest in the three cell lines. Similar results were found in the DOX-loaded PLS incubation. These results suggested that the three different hepatoma cell lines showed different chemosensitivities to the model drug DOX.40–42 Of the three cell lines, BEL7402 cells were found to be the most resistant to DOX, followed by HepG2 cells, and SMMC7721 cells were the most sensitive to DOX. It was also suggested that although the transport mechanism for free DOX was different from that of DOX-loaded liposomes, the DOX chemosensitivities in hepatoma cell lines still did not change after encapsulation.
The conflicting evidence that cellular uptake showed that more Lf-PLS were endocytosed into HepG2 and BEL7402 than into SMMC7721 cells, while in vitro cytotoxicity results showed to be just the opposite might also have been caused by the different DOX sensitivities of the three hepatoma cells. Actually, the cytotoxicity results also showed that the IC50 ratio of PLS/Lf-PLS in HepG2 and BEL7402 cells were found to be significantly higher than in SMMC7721 cells, which was consistent with the results of cellular uptake. And similar results were also obtained by other researchers.39,43
In vivo antitumor study
The antitumor activity of the DOX formulations was evaluated in male BALB/C nude mice bearing a HepG2 tumor model. As showed in Figure 7A, the tumor growth was significantly inhibited by all of the DOX formulations compared with the saline group (P<0.05). Treatment with DOX-loaded Lf-PLS resulted in significantly greater growth retardation of HepG2 tumors compared with DOX-loaded PLS and free DOX (P<0.05).
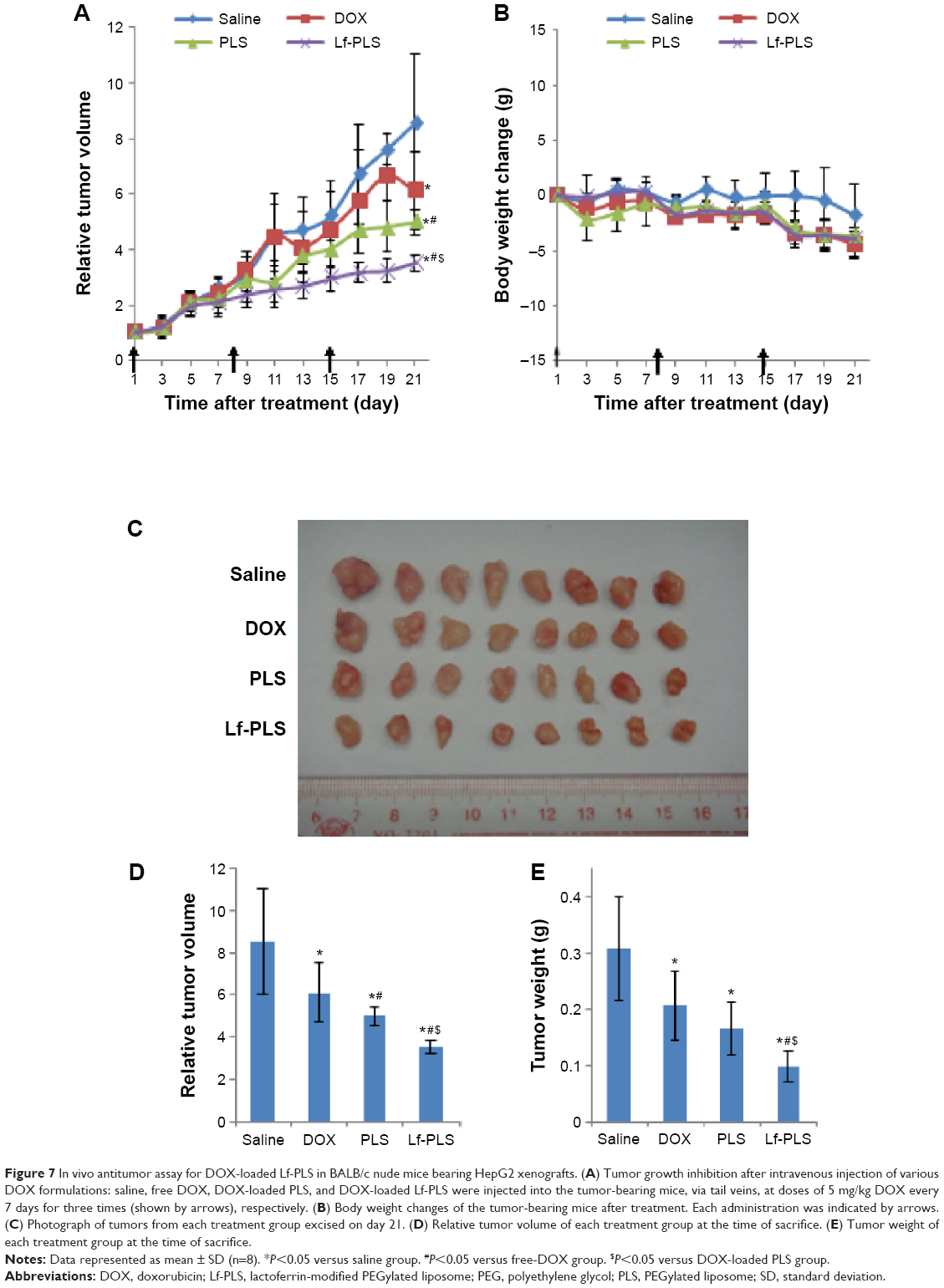 | Figure 7 In vivo antitumor assay for DOX-loaded Lf-PLS in BALB/c nude mice bearing HepG2 xenografts. (A) Tumor growth inhibition after intravenous injection of various DOX formulations: saline, free DOX, DOX-loaded PLS, and DOX-loaded Lf-PLS were injected into the tumor-bearing mice, via tail veins, at doses of 5 mg/kg DOX every 7 days for three times (shown by arrows), respectively. (B) Body weight changes of the tumor-bearing mice after treatment. Each administration was indicated by arrows. (C) Photograph of tumors from each treatment group excised on day 21. (D) Relative tumor volume of each treatment group at the time of sacrifice. (E) Tumor weight of each treatment group at the time of sacrifice. |
At the end of the experiment (day 21), DOX-loaded Lf-PLS treatment groups exhibited the strongest inhibitory effect on tumor growth (Figure 7D and Table 3). The TVI rate of DOX-loaded Lf-PLS was 60.93% compared with the saline group with its RTV of 3.52±0.29 – which was as much as 1.9-fold (P<0.05) and 1.4-fold (P<0.05) of those treated with free DOX and DOX-loaded PLS, respectively. Moreover, the tumor weight on day 21 showed the same trend, where DOX-loaded Lf-PLS showed the lightest tumor weight among all of the treatment groups (Figure 7C and E). Similarly, the TWI rate of the DOX-loaded Lf-PLS group was 68.22% compared with the saline group, with a tumor weight of 0.10±0.03 g, which was as much as 2.1-fold (P<0.05) and 1.5-fold (P<0.05) of those treated with free DOX and DOX-loaded PLS, respectively (Figure 7E and Table 3).
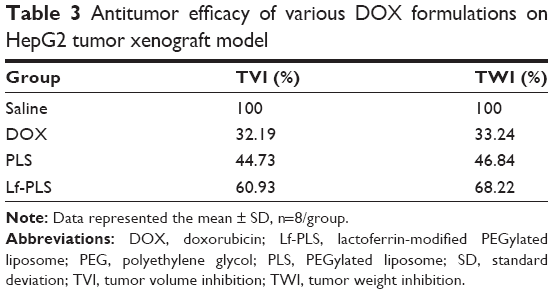 | Table 3 Antitumor efficacy of various DOX formulations on HepG2 tumor xenograft model |
The body weight changes of the tumor-bearing mice were also recorded as an indication of safety. Figure 7B shows that there were no significant body weight changes in all of the treatment groups, indicating the low toxicity of DOX-loaded Lf-PLS in vivo.
Discussion
Chemotherapy is one of the most important treatments for HCC. However, chemotherapy for HCC is obstructed by the poor specificity and unbearable toxicity of the cytotoxic drugs.1–3 Targeting drug delivery could improve the drug efficacy and minimize the side effects, by specific drug delivery to the HCC region.9–11 In the present work, a DOX-loaded Lf-PLS system was designed and constructed, where PLS was used as a drug carrier and Lf was chosen as a targeting ligand in order to deliver the chemotherapeutic agent DOX specifically to HCC, which overexpresses ASGRP.
PLSs, whose liposomal surface is incorporated with PEG chains, are demonstrated to prolong the circulation time by avoiding reticuloendothelial system clearance and facilitate the passive accumulation in tumor tissue via the EPR effect.9,10 In bioconjugated modification, PEG chains also have been repeatedly used as linkers, providing functional groups for conjugating specific ligands, to achieve active targeting.9,10 Therefore, PLSs were chosen as the drug carriers in this study. However, simply increasing the circulation time by PEG-lipid insertion may be insufficient to enhance the therapeutic efficiency of the cytotoxic drugs.
The strategy of modifying specific ligands on PLSs via PEG chains may improve specific drug delivery by receptor-mediated binding. Repeated attempts have been made to deliver drugs specifically to HCC via ASGPR because of their high affinity and rapid internalization in hepatic tumor cells.11,13,14 Recent researches demonstrated that Lf might be a potential HCC ligand because of its specific binding with ASGPR.14,19–21 And Lf exhibits its active targeting ability for HCC cells by the ASGPR endocytosis process.14,19–21 Our previous work also confirmed the HCC-targeting effect of PLS modified with Lf.21 Thus, Lf was applied to be the ASGPR-targeting ligand in this work. The designed DOX-loaded Lf-PLS system was expected to improve the DOX therapeutic efficacy in HCC cells, via the combined effect of both passive and active targeting mechanisms.
The DOX-loaded Lf-PLS system displayed beneficial physicochemical characteristics, including uniform particle size, narrow particle-size distribution, negative charge, high EE, and sustained drug release. Compared with positively charged liposomes, neutral or negatively charged liposomes with particle size less than 200 nm exhibited a longer circulation time due to their weaker interaction with serum proteins, and they also displayed an increased accumulation in tumors by the EPR effect.10,28,44 The DOX-loaded Lf-PLS system showed as spherical or oval vesicles with a mean particle size approximately 100 nm and zeta potential approximately −10 mV. These small particle size and negative charge characteristics of DOX-loaded Lf-PLS provided a favorable condition for HCC transport. Meanwhile, the stability and sustained drug release of DOX-loaded Lf-PLS in 10% FBS was also confirmed by its long circulation characteristic.
In order to confirm the ability of Lf-PLS to target HCC cells, cellular uptake by the ASGPR-positive cells was compared with that of the negative control. Considering the ASGPR overexpression on HepG2, BEL7402, and SMMC7721 cells, these three HCC cell lines were chosen as the target cells. Meanwhile, NIH 3T3 cells were selected as the negative controls since these cells expressed ASGPR negatively and have been previously used as the ASGPR-negative controls in an ASGPR-targeting study.36
The cellular uptake of nanoparticles might be influenced by the size, shape, material, and the cell types and their surface receptors, etc.40 The same nanoparticles might exploit different uptake mechanisms to enter different cell types.38 Previously, Lf-PLS was demonstrated to result in significantly higher cellular uptake by HepG2 cells.21 Our previous study has also proved that Lf-PLS could enter HCC cells with active transport through ASGPR-mediated endocytosis, which might have been induced by Lf. In this current work, confocal microscopy images and flow cytometry assays showed that the cellular uptake of Lf-PLS was significantly higher than that of PLS in ASGPR-positive cells (P<0.05) but not in ASGPR-negative cells (P>0.05). The different cellular uptake of Lf-PLS by both ASGPR-positive and -negative cells might mainly depend on the different ASGPR expression in their cell membranes.13,36,37 The higher cellular uptake of Lf-PLS in ASGPR-positive cells than in ASGPR-negative cells demonstrated the targeting effect of Lf-PLS for HCC cells. The fact that more Lf-PLSs were endocytosed into HepG2 and BEL7402 cells than into SMMC7721 cells was not consistent with the ASGPR expression in HCC cells, suggesting that besides the different ASGPR expression, different uptake mechanisms for Lf-PLS might have occurred in the three HCC cell lines. Therefore, the specific endocytotic pathways for Lf-PLS in the different HCC cell lines should be explored.39
Similar to cellular uptake, cytotoxicity effects of nanoparticles strongly depend on the material, solubility and release of toxic drugs, cell types, and incubation times, etc.40,41 Previously, empty PLS and Lf-PLS were demonstrated to have good tolerability and low toxicity in target cells (HepG2) and normal cells (ECV 304).21 In this work, to confirm the antiproliferative effect of DOX-loaded Lf-PLS, the HCC cell lines, including HepG2, BEL7402, and SMMC7721, which highly express ASGPR, were chosen as the target cells for the cytotoxicity study.
The in vitro cytotoxicity study showed that the increased antiproliferative effects of the DOX formulations displayed in a dose- and time-dependent manner. And the results also showed that DOX-loaded Lf-PLS had significantly stronger inhibitory effects in ASGPR-positive HCC cells than in PLS without Lf modification, which was consistent with the cellular uptake data. The antiproliferative effects might be induced by DOX but not the liposomes because of the low toxicity of the carrier. Moreover, DOX showed different chemosensitivities in the three HCC cell lines, where the order of the chemosensitivities to DOX was SMMC7721 > HepG2 > BEL7402. Similar results were found in DOX-loaded PLS and Lf-PLS, implying that the DOX chemosensitivities in HCC cell lines did not change after encapsulation.
Both the confocal microscopy images and MTT assay showed that free DOX displayed superior cellular uptake and more cytotoxicity effects than did DOX encapsulated in liposomes; the superior cellular uptake and cytotoxicity effects of DOX might have resulted from different uptake mechanisms of the DOX formulations. Free DOX could be easily located into cell nuclei via passive diffusion and then exert its antitumor efficiency by intercalating with the double-stranded helix DNA in the nuclei.32–34,45 DOX loaded on PLS and Lf-PLS might be transported into HCC cells by nonspecific and receptor-mediated endocytosis, respectively, and then exert its therapeutic effect after being released from the carrier. These different uptake mechanisms make the cellular uptake of free DOX easier, which might also contribute to stronger cytotoxicity in tumor cells. Nevertheless, the in vivo tumor accumulation and antitumor efficacy of the free DOX is rather limited because it can be easily cleared by the reticuloendothelial system.46 The in vivo antitumor results showed that free DOX exhibited the weakest antitumor effect, which could provide additional confirmation. This is why PLS was chosen to deliver DOX in our study.
Previously, the pharmacokinetic behavior in Kunming mice showed that Lf-PLS displays a prolonged circulation time characteristic. The in vivo imaging demonstrated that Lf-PLS had a good in vivo targeting distribution in the hepatic tumor. And the cellular uptake study demonstrated that Lf-PLS exhibited an increased cellular uptake by HepG2 cells compared with PLS, indicating that the active specific binding of Lf-PLS might be mediated by Lf. In order to verify the antitumor efficacy, in vivo antitumor activity of DOX-loaded Lf-PLS was examined in a HepG2 tumor xenograft model. The results showed that the antitumor efficacy of DOX-loaded Lf-PLS was significantly stronger than that of PLS and free DOX. It was further confirmed that the DOX-loaded Lf-PLS drug delivery system could enhance the antitumor effect in HCC by the combination of active and passive targeting effects. However, much work should be carried out in future. For example, pharmacokinetics and biodistribution studies of DOX-loaded Lf-PLS should be carried out. The mechanism by which Lf-PLS was superior to PLS in HCC treatment might need to be confirmed. Administration routes of Lf-PLS, such as transhepatic arterial injection, should be further explored. And the targeting ability and side effects to brain should be also monitored.
Conclusion
In conclusion, DOX-loaded Lf-PLS was successfully constructed as a novel drug delivery system for HCC targeting. This liposomal system prepared possessed small particle size (approximately 100 nm), good particle-size distribution (polydispersity index <0.2), high EE (>95%), and good stability (drug release <15% for 24 hours). Furthermore, DOX-loaded Lf-PLS exhibited significantly enhanced cellular uptake and improved inhibitory effects in ASGPR-positive HCC cell lines, and achieved remarkable antitumor efficiency on BALB/c nude mice bearing HepG2 xenografts. The results of this research demonstrated that DOX-loaded Lf-PLS might be a potential drug-targeting delivery system for HCC treatment.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (grant number 81402881), the Natural Science Foundation of Guangdong Province, China (grant number S2013040014348), and the Scientific Research Project of Guangzhou Medical University (grant number 2012C74).
Disclosure
The authors report no conflicts of interest in this work.
References
Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379(9822):1245–1255. | ||
Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362(9399):1907–1917. | ||
Varela M, Sala M, Llovet JM, Bruix J. Treatment of hepatocellular carcinoma: is there an optimal strategy. Cancer Treat Rev. 2003;29(2):99–104. | ||
Thomas MB, O’Beirne JP, Furuse J, Chan AT, Abou-Alfa G, Johnson P. Systemic therapy for hepatocellular carcinoma: cytotoxic chemotherapy, targeted therapy and immunotherapy. Ann Surg Oncol. 2008;15(4):1008–1014. | ||
Abou-Alfa GK, Venook AP. The impact of new data in the treatment of advanced hepatocellular carcinoma. Curr Oncol Rep. 2008;10(3):199–205. | ||
Yeo W, Mok TS, Zee B, et al. A randomized phase III study of doxorubicin versus cisplatin/interferon alpha-2b/doxorubicin/fluorouracil (PIAF) combination chemotherapy for unresectable hepatocellular carcinoma. J Natl Cancer Inst. 2005;97(20):1532–1538. | ||
Wang Q, Zhong YJ, Yuan JP, et al. Targeting therapy of hepatocellular carcinoma with doxorubicin prodrug PDOX increases anti-metastatic effect and reduces toxicity: a preclinical study. J Transl Med. 2013;11:192. | ||
Chen X, Ding G, Gao Q, et al. A human anti-c-Met Fab fragment conjugated with doxorubicin as targeted chemotherapy for hepatocellular carcinoma. PLoS One. 2013;8(5):e63093. | ||
Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65(1):36–48. | ||
Bozzuto G, Molinari A. Liposomes as nanomedical devices. Int J Nanomedicine. 2015;10:975–999. | ||
Zhou X, Zhang M, Yung B, et al. Lactosylated liposomes for targeted delivery of doxorubicin to hepatocellular carcinoma. Int J Nanomedicine. 2012;7:5465–5474. | ||
Yu KF, Zhang WQ, Luo LM, et al. The antitumor activity of a doxorubicin loaded, iRGD-modified sterically-stabilized liposome on B16-F10 melanoma cells: in vitro and in vivo evaluation. Int J Nanomedicine. 2013;8:2473–2485. | ||
D’Souza AA, Devarajan PV. Asialoglycoprotein receptor mediated hepatocyte targeting – strategies and applications. J Control Release. 2015;203:126–139. | ||
Mishra N, Yadav NP, Rai VK, et al. Efficient hepatic delivery of drugs: novel strategies and their significance. Biomed Res Int. 2013;2013:382184. | ||
McAbee DD, Bennatt DJ, Ling YY. Identification and analysis of a CA(2+)-dependent lactoferrin receptor in rat liver. Lactoferrin binds to the asialoglycoprotein receptor in a galactose-independent manner. Adv Exp Med Biol. 1998;443:113–121. | ||
McAbee DD, Jiang X, Walsh KB. Lactoferrin binding to the rat asialoglycoprotein receptor requires the receptor’s lectin properties. Biochem J. 2000;348 Pt 1:113–117. | ||
Ward PP, Uribe-Luna S, Conneely OM. Lactoferrin and host defense. Biochem Cell Biol. 2002;80(1):95–102. | ||
Pathak A, Vyas SP, Gupta KC. Nano-vectors for efficient liver specific gene transfer. Int J Nanomedicine. 2008;3(1):31–49. | ||
Weeke-Klimp AH, Bartsch M, Morselt HW, et al. Targeting of stabilized plasmid lipid particles to hepatocytes in vivo by means of coupled lactoferrin. J Drug Target. 2007;15(9):585–594. | ||
Oh ST, Rih JK, Kwon HS, Hwang DS, Kim SY, Yim JB. Lactoferrin as a gene delivery vehicle to hepatocytes. Exp Mol Med. 1997;29(2):111–116. | ||
Wei M, Xu Y, Zou Q, et al. Hepatocellular carcinoma targeting effect of PEGylated liposomes modified with lactoferrin. Eur J Pharm Sci. 2012;46(3):131–141. | ||
Li SD, Huang L. Nanoparticles evading the reticuloendothelial system: role of the supported bilayer. Biochim Biophys Acta. 2009;1788(10):2259–2266. | ||
Evjen TJ, Hagtvet E, Nilssen EA, Brandl M, Fossheim SL. Sonosensitive dioleoylphosphatidylethanolamine-containing liposomes with prolonged blood circulation time of doxorubicin. Eur J Pharm Sci. 2011;43(4):318–324. | ||
Ying X, Wen H, Lu WL, et al. Dual-targeting daunorubicin liposomes improve the therapeutic efficacy of brain glioma in animals. J Control Release. 2010;141(2):183–192. | ||
Li X, Xu H, Dai X, Zhu Z, Liu B, Lu X. Enhanced in vitro and in vivo therapeutic efficacy of codrug-loaded nanoparticles against liver cancer. Int J Nanomedicine. 2012;7:5183–5190. | ||
Luo LM, Huang Y, Zhao BX, et al. Anti-tumor and anti-angiogenic effect of metronomic cyclic NGR-modified liposomes containing paclitaxel. Biomaterials. 2013;34(4):1102–1114. | ||
Cui J, Li C, Wang C, et al. Development of pegylated liposomal vincristine using novel sulfobutyl ether cyclodextrin gradient: is improved drug retention sufficient to surpass DSPE-PEG-induced drug leakage? J Pharm Sci. 2011;100(7):2835–2848. | ||
Zhao W, Zhuang S, Qi XR. Comparative study of the in vitro and in vivo characteristics of cationic and neutral liposomes. Int J Nanomedicine. 2011;6:3087–3098. | ||
Xie H, Zhu Y, Jiang W, et al. Lactoferrin-conjugated superparamagnetic iron oxide nanoparticles as a specific MRI contrast agent for detection of brain glioma in vivo. Biomaterials. 2011;32(2):495–502. | ||
Xu Z, Chen L, Gu W, et al. The performance of docetaxel-loaded solid lipid nanoparticles targeted to hepatocellular carcinoma. Biomaterials. 2009;30(2):226–232. | ||
Ma P, Liu S, Huang Y, Chen X, Zhang L, Jing X. Lactose mediated liver-targeting effect observed by ex vivo imaging technology. Biomaterials. 2010;31(9):2646–2654. | ||
Yang X, Hong H, Grailer JJ, et al. cRGD-functionalized, DOX-conjugated, and 64Cu-labeled superparamagnetic iron oxide nanoparticles for targeted anticancer drug delivery and PET/MR imaging. Biomaterials. 2011;32(17):4151–4160. | ||
Xiang Y, Liang L, Wang X, Wang J, Zhang X, Zhang Q. Chloride channel-mediated brain glioma targeting of chlorotoxin-modified doxorubicine-loaded liposomes. J Control Release. 2011;152(3):402–410. | ||
Jung SH, Jung SH, Seong H, Cho SH, Jeong KS, Shin BC. Polyethylene glycol-complexed cationic liposome for enhanced cellular uptake and anticancer activity. Int J Pharm. 2009;382(1–2):254–261. | ||
Chen H, Qin Y, Zhang Q, et al. Lactoferrin modified doxorubicin-loaded procationic liposomes for the treatment of gliomas. Eur J Pharm Sci. 2011;44(1–2):164–173. | ||
Bitzer M, Lauer U, Baumann C, Spiegel M, Gregor M, Neubert WJ. Sendai virus efficiently infects cells via the asialoglycoprotein receptor and requires the presence of cleaved F0 precursor proteins for this alternative route of cell entry. J Virol. 1997;71(7):5481–5486. | ||
Mu H, Lin KX, Zhao H, et al. Identification of biomarkers for hepatocellular carcinoma by semiquantitative immunocytochemistry. World J Gastroenterol. 2014;20(19):5826–5838. | ||
dos Santos T, Varela J, Lynch I, Salvati A, Dawson KA. Effects of transport inhibitors on the cellular uptake of carboxylated polystyrene nanoparticles in different cell lines. PLoS One. 2011;6(9):e24438. | ||
Quan G, Pan X, Wang Z, et al. Lactosaminated mesoporous silica nanoparticles for asialoglycoprotein receptor targeted anticancer drug delivery. J Nanobiotechnology. 2015;13:7. | ||
Fröhlich E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int J Nanomedicine. 2012;7:5577–5591. | ||
Kirchner C, Liedl T, Kudera S, et al. Cytotoxicity of colloidal CdSe and CdSe/ZnS nanoparticles. Nano Lett. 2005;5(2):331–338. | ||
Lee TK, Lau TC, Ng IO. Doxorubicin-induced apoptosis and chemosensitivity in hepatoma cell lines. Cancer Chemother Pharmacol. 2002;49(1):78–86. | ||
Li T, Takeoka S. Enhanced cellular uptake of maleimide-modified liposomes via thiol-mediated transport. Int J Nanomedicine. 2014;9:2849–2861. | ||
Cai L, Wang X, Wang W, et al. Peptide ligand and PEG-mediated long-circulating liposome targeted to FGFR overexpressing tumor in vivo. Int J Nanomedicine. 2012;7:4499–4510. | ||
Hwang T, Han HD, Song CK, et al. Anticancer drug-phospholipid conjugate for enhancement of intracellular drug delivery. Macromol Symp. 2007;249–250(1):109–115. | ||
Tagami T, Ernsting MJ, Li SD. Efficient tumor regression by a single and low dose treatment with a novel and enhanced formulation of thermosensitive liposomal doxorubicin. J Control Release. 2011;152(2):303–309. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.