Back to Journals » Journal of Inflammation Research » Volume 14
Lack of Macrophage Migration Inhibitory Factor Reduces Susceptibility to Ventricular Arrhythmias During the Acute Phase of Myocardial Infarction
Authors Lyu J, Huang J, Wu J, Yu T, Wei X, Lei Q ![]()
Received 4 February 2021
Accepted for publication 16 March 2021
Published 7 April 2021 Volume 2021:14 Pages 1297—1311
DOI https://doi.org/10.2147/JIR.S304553
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Juanjuan Lyu,1 Jia Huang,2,3 Jin Wu,1 Tao Yu,3,4 Xinchuan Wei,2,3 Qian Lei2,3
1Department of Pediatrics, West China Second University Hospital, Sichuan University, Chengdu, 610041, People’s Republic of China; 2Department of Anesthesiology, Sichuan Provincial People’s Hospital, University of Electronic Science and Technology of China, Chengdu, 610072, People’s Republic of China; 3Chinese Academy of Sciences Sichuan Translational Medicine Research Hospital, Chengdu, 610072, People’s Republic of China; 4Department of Cardiac Surgery, Sichuan Provincial People’s Hospital, University of Electronic Science and Technology of China, Chengdu, 610072, People’s Republic of China
Correspondence: Qian Lei Email [email protected]
Background: Macrophages are involved in inflammatory responses and play a crucial role in aggravating ventricular arrhythmias (VAs) after myocardial infarction (MI). Macrophage migration inhibitory factor (MIF) participates in inflammatory responses during acute MI. In the present study, we hypothesized that knockout (KO) of MIF may prevent VAs during the acute phase of MI by inhibiting macrophage-derived pro-inflammatory mediators.
Methods and Results: We demonstrated that MIF-KO mice in a mouse model of MI exhibited a significant decrease in susceptibility to VAs both in vivo (84.6% vs 40.7%, P < 0.05) and ex vivo (86.7% vs 40.0%, P < 0.05) at day 3 after MI compared with that in wild-type (WT) mice. Both WT and MIF-KO mice presented similar left ventricular contractility, peri-infarct myocardial fibrosis and sympathetic reinnervation, and circulating and local norepinephrine levels during the acute phase of MI. Meanwhile, MIF-KO mice had inhibited macrophage aggregation, alleviated connexin 43 (Cx43) redistribution, and reduced level of pro-inflammatory mediators, including tumor necrosis factor-α and interleukin-1β (P < 0.05) at day 3 after MI. The differences in susceptibility to VAs, expression of pro-inflammatory mediators, and Cx43 redistribution after MI between WT and MIF-KO mice disappeared by macrophage depletion with clodronate liposomes in both groups. Furthermore, the pro-inflammatory activity of cultured peritoneal macrophages was inhibited by MIF deficiency and recovered with replenishment of exogenous MIF in vitro.
Conclusion: In conclusion, we found that lack of MIF reduced the susceptibility to VAs in mouse heart during the acute phase of MI by inhibiting pro-inflammatory activity of macrophages and improving gap-junction and electrical remodeling.
Keywords: myocardial infarction, ventricular arrhythmias, macrophage migration inhibitory factor, inflammatory mediators, sympathetic nerve
Introduction
Ventricular arrhythmias (VAs) are the main cause of sudden death following myocardial infarction (MI).1 The myocardial necrosis, fibrosis, and altered norepinephrine (NE) levels contribute to arrhythmogenesis.2–5 In addition, augmented sympathetic neural remodeling plays a key role in VAs.6,7 Moreover, infiltrating macrophages are involved in cardiac sympathetic hyperinnervation, which peaks at one week after MI, and have garnered much attention during the healing phase after acute MI.8,9 However, the mechanisms by which infiltrating macrophages contribute to VAs during the acute phase of MI have not been comprehensively understood.
Macrophages are the major immune cells in mediating inflammatory responses following MI.10,11 Therefore, macrophage mediates release of a variety of pro-inflammatory mediators. The mediators tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) contribute to the initiation of VAs after MI.12 Therefore, interfering with macrophages may represent a strategy to control VAs after MI. Macrophage migration inhibitory factor (MIF) regulates the activity of immune cells.13,14 Moreover, MIF has been reported to be involved in many inflammatory diseases, including cardiovascular diseases.13,15 Furthermore, myocardial ischemia may trigger the secretion of MIF expressed by cardiomyocytes.16 Many studies have reported a role of MIF in cardioprotection from myocardial ischemia-reperfusion injury and MI.13,17–21 In contrast, the role of MIF in myocardial-ischemia-induced or MI-induced VAs has rarely been investigated. TNF-α and IL-1β, primarily released from macrophages, have been shown to be closely associated with the incidence of VAs.12,22–24 Acute MI often induces prominent pro-inflammatory activation with concomitant VAs, followed by anti-inflammatory responses during the healing phase. Therefore, we hypothesized that increased MIF may promote VA by enhancing macrophage activation and inflammatory responses during the acute phase of MI.
Materials and Methods
Animal Care and MI Model
This study was approved by the medical ethics committees of Sichuan Provincial People’s Hospital (No. 2018–069 and 2019–252) and West China Second University Hospital (No. 2020–009), China. The animal experimental protocols adhered to NIH guidelines. MIF-knockout (KO) mice (Jackson, Bar Harbor, ME, USA) were produced with a background of C57BL/6 mice. We used C57BL/6 wild-type (WT) mice as controls. All mice were raised in an environment with 12/12-h light/dark cycles, and ample food and water were provided. Mice at 8–10 weeks old were used for MI or sham surgery. Mice were anesthetized with 2% isoflurane and then intratracheally intubated. The third intercostal space was incised to expose the heart. Ligation of the left anterior descending coronary artery was performed to induce MI. Sham-operated mice only underwent thoracotomies without coronary arterial ligation.
To further study the effect of infiltrating macrophages on VAs after MI, each mouse received a tail venous injection with clodronate (FormuMax Scientific, USA) or liposome-vehicle at 200 μL on day 1 before surgery, and days 1 and 3 after surgery.
Echocardiographic and Hemodynamic Assessments
Transthoracic echocardiography was conducted with a high-resolution imaging system (VisualSonics, Toronto, ON, Canada) before surgery and on day 3 after surgery. The standard parasternal long axis transection of the left ventricle (LV) was captured. Then, the data of LV end diastolic dimension (LVEDD), LV end systolic dimension (LVESD), ejection fraction (EF), fractional shortening (FS), and stroke volume (SV) were measured.
Hemodynamics was measured with a 1-Fr transducer catheter (Millar, Houston, TX, USA). Animals were anesthetized with 1.5% isoflurane. The transducer was introduced into the right common carotid artery, inserted into the LV via the aorta. Data was collected with Acqknowledge 3.8.2 software (Biopac, Goleta, CA, USA). Data of systolic arterial blood pressure (SBP), diastolic arterial blood pressure (DBP), LV peak systolic pressure (LVPSP), LV end diastolic pressure (LVEDP), LV +dP/dTmax, and LV –dP/dTmax were analyzed.
In vivo Cardiac Electrophysiology
We anesthetized the mice with 1.5% isoflurane at day 3 after surgery. Surface electrocardiograms in lead II were recorded with sub-dermal electrodes. As previously described,9 an octapolar catheter (Numed, Hopkinton, NY, USA) for electrophysiology was inserted into the right atrium and ventricle. Leads 1, 2 and 3, 4 of the catheter were used to deliver electrical stimulation and assess intracardiac electrograms, respectively. The stimulus was twice as intense as that of the capture threshold. A burst stimulation of 20 pulses at a frequency of 50 Hz was used to induce VAs. Electrical stimuli were delivered three times at 3-min stabilization intervals. VAs are defined as more than four continuous ventricular contractions or induction of ventricular fibrillation.25
The QT interval and RR interval were identified from lead II of surface electrocardiograms. Then, the formula QTc = QT/(RR0.5) was used to calculate the corrected QT interval (QTc). The effective refractory period was determined using a series of seven stimuli (S1) 20 ms less than the RR interval with one extra-stimulus (S2).
Ex vivo Cardiac Electrophysiology
Mice were anesthetized with 3% isoflurane and the hearts were excised. The isolated hearts were quickly cannulated to a horizontal Langendorff system and perfused with Krebs solution. The mechanical contraction of myocardium was inhibited with 2.3-butanedione monoxime (20 nmol/l). The field electrocardiograms were recorded with electrodes close to the right atrium, left atrium, and apex. A bipolar electrode was used to stimulate the right ventricular base. The stimuli were set as 2-ms pulse durations with twice the voltage magnitude as that of the capture threshold. Susceptibility to VAs was evaluated as same program as that described above for in vivo electrophysiology.
Intracellular action potentials were recorded with a patch clamp amplifier (Inbio, Wuhan, China). The glass microelectrodes were filled with potassium chloride (3 mmol/l) and the tips were tapped into the epicardial myocytes. As previously described,9 the left middle (L-M), left apex (L-A), right middle (R-M), and right apex (RV-A) regions around the infarct border were selected for recording. The action potentials recorded from the four regions were used for analysis. The 90% action potential duration (APD90) was averaged from 90% repolarization time of five stable action potentials. The spatial dispersion of APD90, eg, coefficient of variation (CV), was calculated as standard deviation divided by mean.
Fibrosis Analysis in the Peri-Infarct Zone
After ex vivo electrophysiological experiments, the interstitial fibrosis percentage was measured by Masson’s trichrome staining. Each heart was perfused with 5 mL of 10% potassium chloride via the aortic root. A short-axis ring of the middle LV containing infarct area was fixed with 4% paraformaldehyde overnight. Then, the paraffin-embedded cross sections at 5 μm were sequentially stained in Weigert's iron hematoxylin, ponceau acid fuchsin and aniline blue solutions. Bright field images were captured and the views in the peri-infarct zone were analyzed using Image-Pro Plus 6.0 (NIH, Bethesda, MD, USA) with a fixed color threshold. We randomly choose six non-overlapping fields per section for quantitative analysis. Results are here reported as the average percentage of positive staining against tissue area.
Harvesting of Blood and Heart Tissues
Mice were anesthetized with isoflurane and then euthanized to harvest blood and heart tissue after in vivo electrophysiological experiments. Later, 2 mL of blood was collected in a tube with 2% ethylene diamine tetra acetic acid for NE measurement. Afterward, the hearts were excised and rinsed with phosphate-buffered saline. The tissues in peri-infarct zones of MI mice and the corresponding zones of sham-operated mice were harvested and then quickly stored in liquid nitrogen.
Measurement of Plasma and Peri-Infarct NE Levels
The blood was centrifuged at 3000 ×g for 15 min. Then, the plasma was stored at –80°C for NE analysis. The heart tissues were homogenized in a 5× volume of perchloric acid (0.1 mol/l) and reduced glutathione (5 mmol/l). The homogenates were centrifuged at 3000 ×g for 15 min. Then, the supernatant was stored at –80°C for NE analysis. NE levels were determined using enzyme-linked immunosorbent assays (ELISAs). The protein content of heart tissues was measured using the bicinchoninic acid method.
Immunofluorescence
The excised hearts were fixed with 4% paraformaldehyde. The paraffin-embedded heart sections were then incubated with rabbit anti-mouse anti-F4/80 (1:200, Abcam), anti-connexin 43 (Cx43; 1:200, Abcam), anti-tyrosine hydroxylase (anti-TH; 1:200, Abcam), and anti-troponin T (TnT; 1:400, Abcam) for 1 h. Thereafter, the tissue sections with anti-F4/80, anti-Cx43, and anti-TH were incubated with the fluorescent secondary antibodies for 1 h. The Alexa Fluor 488-conjugated donkey anti-rabbit IgG (1:200, Servicebio) and Cy3-conjugated goat anti-rabbit IgG (1:200, Servicebio) were used. Nuclei were counterstained with DAPI (1:1000, Invitrogen). The density of F4/80-positive stained cells was used to evaluate the degree of macrophage infiltration. The densities of Cx43- and TH-positive staining were used to evaluate the degrees of gap-junction redistribution and sympathetic reinnervation, respectively. Pictures were captured from six sections with 300-μm intervals in each heart. Six random non-overlapping fields per section were chosen for quantitative analysis. The densities of F4/80-positive cells, Cx43-positive proteins, and TH-positive fibers were calculated with ImageJ 1.52s software (NIH, Bethesda, MD, USA). The data are here presented as the relative number of positive cells or the ratio of positive area over the whole area (cells/mm2 or μm2/mm2). Furthermore, six random longitudinal TnT and Cx43 co-stained fields were chosen for Cx43 lateralization analysis. The total area of Cx43 (AreaT) and the area of Cx43 perpendicular to myocardial fiber bundle (AreaP) were measured. The degree of Cx43 lateralization calculated as (AreaT–AreaP)/AreaT.
Cell Harvesting and Culturing
Each mouse peritoneal cavity was injected with 1 mL of 3% Brewer thioglycolate medium. The abdomen was incised after 3 days. Then, 10 mL of Dulbecco’s Phosphate Buffered Saline was injected into the mouse peritoneal cavity and then aspirated. The peritoneal fluid with exudate cells was centrifuged at 400 ×g for 10 min. Then, the cell concentration was adjusted to 1 × 106 cells/well. The isolated peritoneal macrophages were cultured in DMEM/F12 medium with 10% fetal bovine serum for 3 days. To verify the effects of MIF on the expression and production of pro-inflammatory mediators, the culture medium of peritoneal macrophages from MIF-KO mice was added with 20 ng/mL recombinant MIF (rMIF; R&D). The levels of TNF-α and IL-1β were measured by quantitative real-time polymerase chain reaction (qPCR) and ELISAs.
qPCR
Total RNA extracted from either myocardial tissues or cultured peritoneal macrophages was reversely transcribed with a PrimeScript reagent kit (TaKaRa, Beijing, China). The mRNA levels of TNF-α and IL-1β were normalized to β-actin within each same sample. The following primers were used for PCR amplification (Table 1).
 |
Table 1 Primers Used for qPCR |
ELISAs
NE content in plasma and tissue supernatant, as well as TNF-α and IL-1β levels in the cellular supernatant, were determined using the corresponding ELISA kits (Cusabio, Houston, TX, USA) according to the product descriptions.
Statistical Analysis
Categorical data were indicated as numbers (percentages). Continuous data were reported as mean ± standard error of mean (SEM). Categorical data were analyzed via Chi-square or Fisher’s exact tests. For continuous data, Student’s t-tests or one-way analyses of variance (ANOVAs) followed by Tukey’s tests were used for comparisons between two groups or among more than two groups. All statistical analyses were conducted with SPSS 23.0 (IBM, USA). Statistical significance was defined as P < 0.05.
Results
Baseline Parameters of Echocardiography and Hemodynamics
The echocardiographic and hemodynamic parameters were measured before surgery. There was no statistically significant difference in any values of echocardiography or hemodynamics between WT and MIF-KO mice (Table 2).
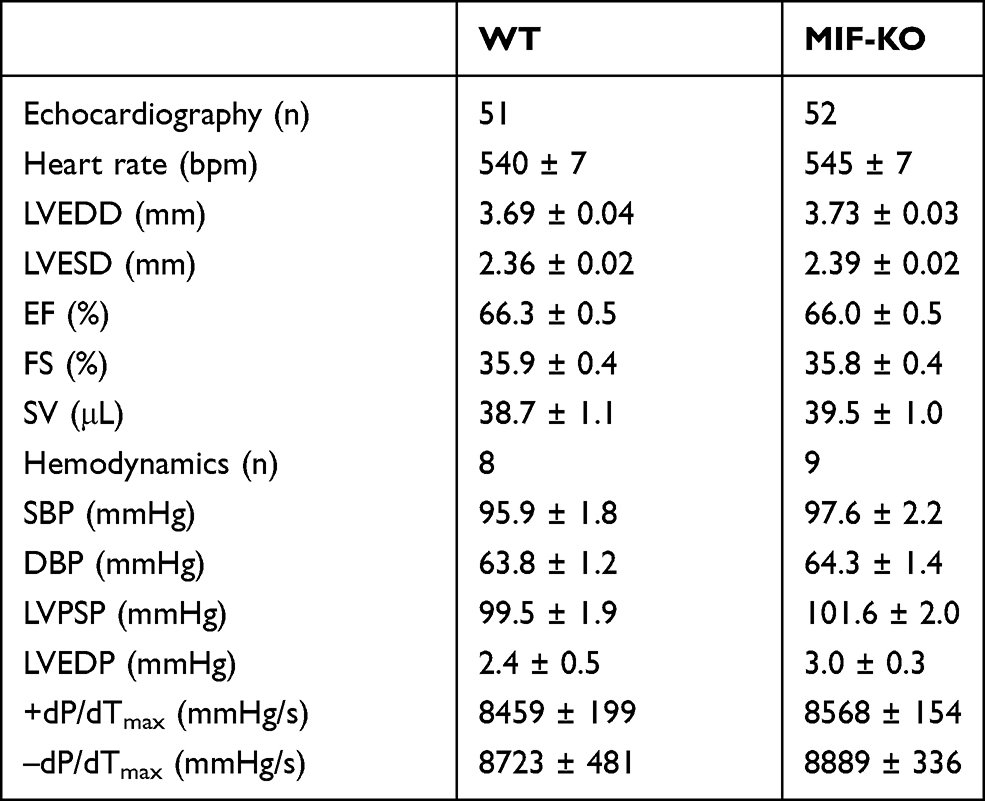 |
Table 2 Baseline Echocardiographic and Hemodynamic Parameters |
Susceptibility to VAs in vivo and ex vivo
To determine the effects of MI and MIF on susceptibility to VAs, in vivo (Figure 1A and B) and ex vivo (Figure 1G and H) examinations were performed. Results indicated that the arrhythmias were more inducible both in vivo (22/26, 84.6%) (Figure 1C) and ex vivo (13/15, 86.7%) (Figure 1I) in WT mice undergoing MI. However, only 11 in 27 (40.7%) mice (Figure 1C) and 6 in 15 (40.0%) isolated hearts (Figure 1I) exhibited induced VAs in MIF-KO MI mice. MIF deficiency lowered the incidence of VAs induced by burst stimuli after MI by approximately 50% both in vivo and ex vivo. Additionally, the average VA duration was shortened both in vivo (Figure 1D) and ex vivo (Figure 1J) in MIF-KO MI mice.
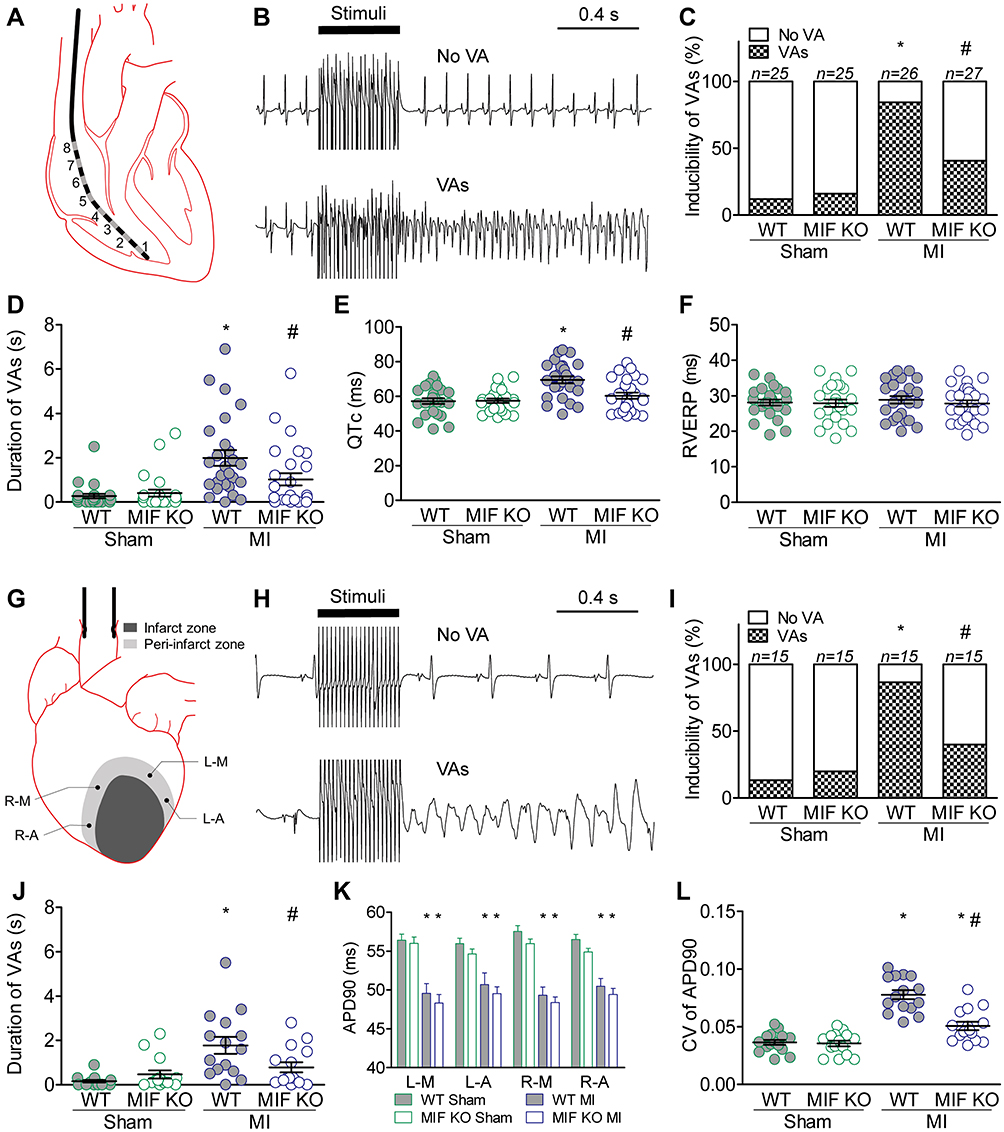 |
Figure 1 Electrophysiology in vivo and ex vivo. (A–F) in vivo. (A) Schematic showing intracardiac electrophysiology. (B) Representative intracardiac electrograms with and without VAs. (C) Incidence of VAs induced by stimuli in vivo. (D) The longest VA duration; one circle indicates data from one mouse. (E) QTc calculated as QT/(RR0.5). (F) Right ventricular effective refractory period (RVERP). WT sham group, n = 25; WT MI group, n = 26; MIF-KO sham group, n = 25; MIF-KO MI group, n = 27. (G–L) ex vivo. (G) Schematic showing intracellular recording at four peri-infarct regions within the isolated hearts: L-M, L-A, R-M, and R-A. (H) Representative field electrocardiograms with and without VAs. (I) Incidence of VAs induced by stimuli ex vivo. (J) The longest VA; one circle indicates data from one heart. (K) The APD90 at four peri-infarct regions. (L) Spatial dispersion of APD90 at four peri-infarct regions. Each group, n = 15. *P < 0.05 vs the sham-operated mice of the same type, #P < 0.05 vs the WT mice underwent the same operation. |
There was a significantly prolonged QTc in MI mice according to the surface electrocardiogram compared with that in sham-operated mice (Figure 1E); however, MI did not induce a statistically significant change in the effective refractory period of right ventricle (Figure 1F). MIF deficiency alleviated the MI-induced prolongation of QTc (Figure 1E) and decrease of the right ventricular effective refractory period (Figure 1F). The APD90 at four peri-infarct regions (L-M, L-A, R-M, and R-A) of the isolated hearts significantly decreased in the MI groups (Figure 1K). MIF deficiency reversed the decrease of APD90 and APD90 dispersion induced by MI at these four peri-infarct regions (Figure 1L).
Echocardiography and Hemodynamics After Surgery
Three days after surgery, no significant difference presented in the heart rates among four groups (Figure 2A). MI induced an increase in the LV chamber and a decrease in LV systolic function, but these changes were similar in WT and MIF-KO mice with MI (Figure 2B–E). However, the SV did not significantly decrease after MI in either WT or MIF-KO mice (Figure 2F). Nevertheless, the changes in arterial blood pressure, LV pressure and function following MI were similar in WT and MIF-KO groups (P < 0.05) (Figure 3).
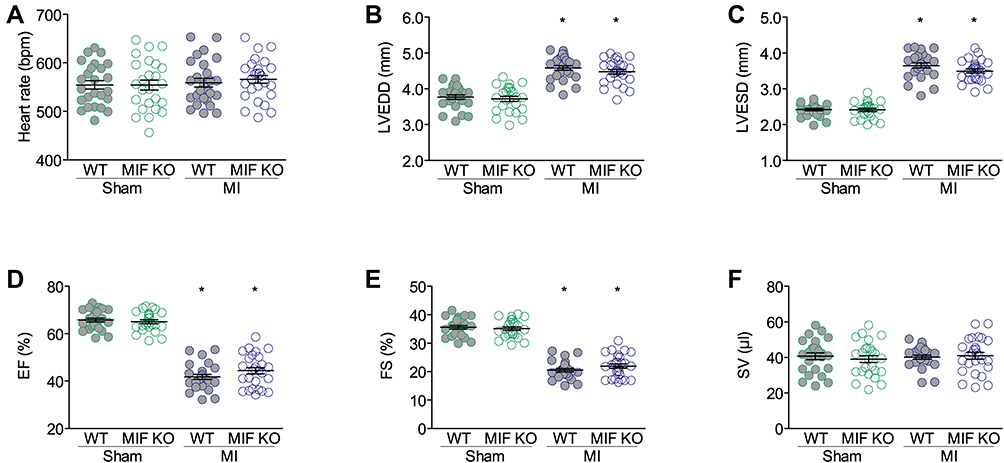 |
Figure 2 Echocardiographic parameters at three days after surgery. (A) Heart rates. (B) Left ventricle end diastolic dimensions (LVEDDs). (C) Left ventricle end systolic dimensions (LVESDs). (D) Ejection fractions (EFs). (E) Fractional shortening (FS). (F) Stroke volumes (SVs). WT sham group, n = 25; WT MI group, n = 26; MIF-KO sham group, n = 25; MIF-KO MI group, n = 27. *P < 0.05 vs the sham-operated mice of the same type. |
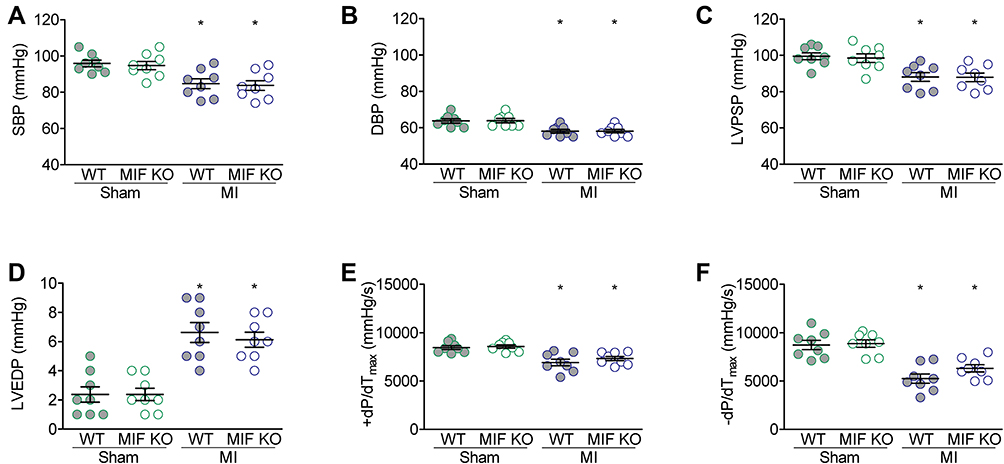 |
Figure 3 Hemodynamic measurements of blood pressure and left ventricle pressure after MI surgery. (A) Systolic arterial blood pressure (SBP). (B) Diastolic arterial blood pressure (DBP). (C) Left ventricle peak systolic pressure (LVPSP). (D) Left ventricle end diastolic pressure (LVEDP). (E) Left ventricle +dP/dTmax (+dP/dTmax). (F) Left ventricle –dP/dTmax (–dP/dTmax). For each group, n = 8. *P < 0.05 vs the sham-operated mice of the same type. |
Interstitial Fibrosis and NE Levels Following MI
The average interstitial fibrosis ratios in the peri-infarct zone were 4.5% in WT mice, but it was 4.1% in MIF-KO mice. The difference between the two was not statistically significant (P > 0.05), but both values were statistically significantly higher than the corresponding values in the sham-operated mice (P < 0.05) (Figure 4A and B). NE levels were measured via ELISAs to explore whether circulating and local NE content affected susceptibility to VAs during the acute phase of MI. The NE levels increased in the plasma while decreased in the peri-infarct zone compared with the responding zone in sham group for both WT mice (3.87 ± 0.28 vs 1.91 ± 0.19 ng/mL; 0.97 ± 0.10 vs 1.70 ± 0.13 pmol/mg, P < 0.05) and MIF-KO mice (3.58 ± 0.24 vs 2.16 ± 0.25 ng/mL; 0.86 ± 0.12 vs 1.67 ± 0.20 μg/g, P < 0.05). However, MI-induced changes in NE levels were similar in WT and MIF-KO groups (Figure 4C and D).
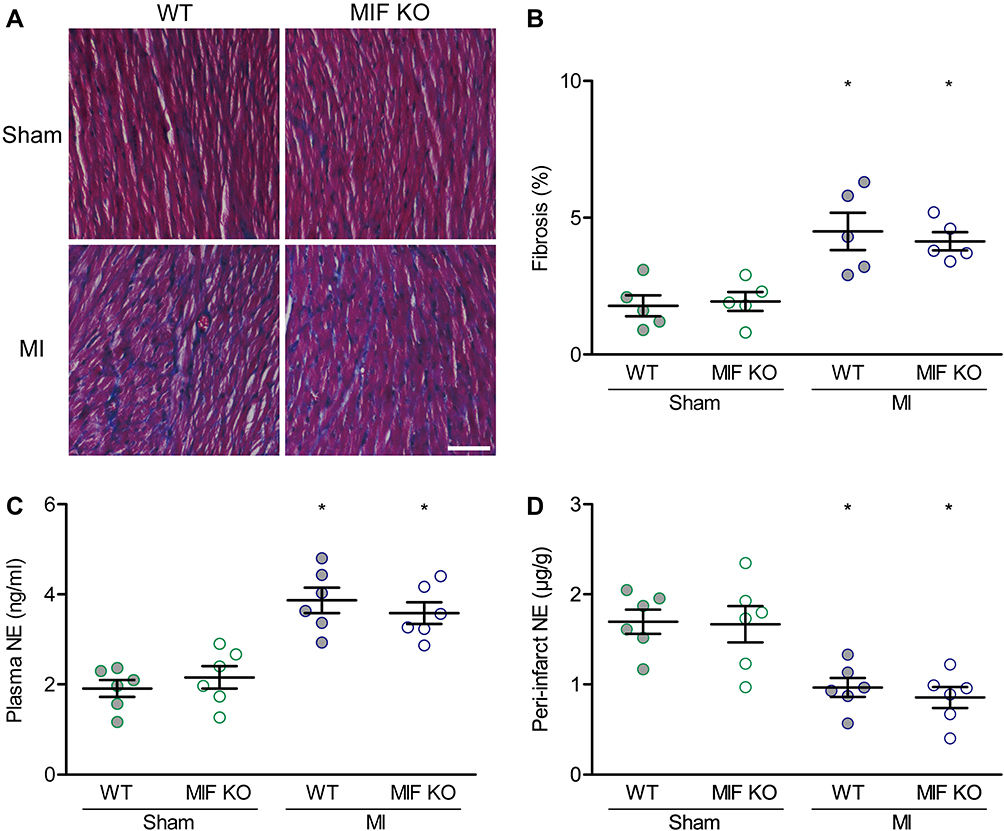 |
Figure 4 Effects of MIF-KO on interstitial fibrosis and norepinephrine (NE) levels. (A) Representative heart sections with Masson’s trichrome staining on day 3 after surgery. (B) Comparison of interstitial fibrosis between WT and MIF-KO groups. (C) NE level in plasma. (D) NE content in the peri-infarct myocardium. Each group for interstitial fibrosis identification, n = 5. Each group for NE content measurement, n = 6. *P < 0.05 vs the sham-operated mice of the same type. Scale bar, 100 μm. |
Effects of MIF Deficiency on Macrophage Infiltration, Cx43 Redistribution, Sympathetic Remodeling, and Inflammatory Mediators
Next, we investigated the influences of MIF deficiency on changes in inflammatory cells, gap-junction proteins, sympathetic reinnervation, and inflammatory mediators in the peri-infarct myocardium at 3 days after MI. First, we assessed the densities of macrophages, gap-junction proteins, and sympathetic nerves via immunofluorescent staining for the peri-infarct zones. Fluorescent staining for F4/80, Cx43, TH, and TnT indicated infiltrating macrophages, gap-junction proteins, sympathetic nerve fibers, and cardiomyocytes, respectively. MI led to obvious infiltration of macrophages, decrease and lateralization of Cx43, and reduction of TH. However, the macrophage count was 42.4% lower (P < 0.05, Figure 5A and B), Cx43 proportion 45.8% higher (P < 0.05, Figure 5C and D), and Cx43 lateralization 31.1% lower (P < 0.05, Figure 5C and E) in MIF-KO mice compared with these parameters in WT mice three days after MI, without changes in sympathetic reinnervation (Figure 5F and G). In sham-operated mice, there was no significant difference in macrophage density, Cx43, or TH morphometry between WT and MIF-KO mice (Figure 5, P > 0.05). Furthermore, qPCR results demonstrated that the mRNA levels of TNF-α and IL-1β in the peri-infarction zone were up-regulated in response to MI, whereas they were down-regulated in MIF-KO group compared with WT group (Figure 6A and B).
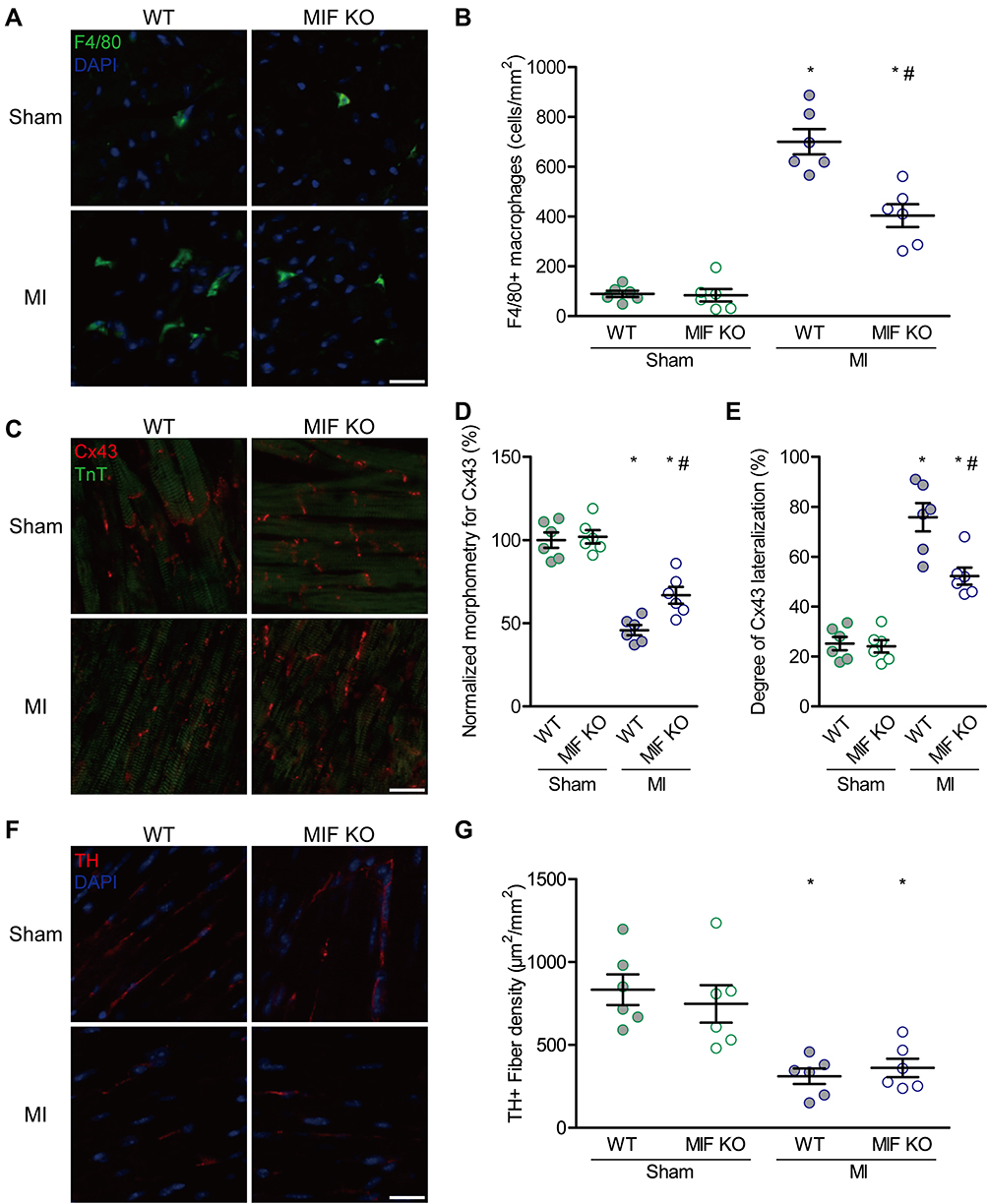 |
Figure 5 Effects of MIF on inflammatory cells, gap-junctions, and sympathetic reinnervation in the peri-infarction myocardium. (A) F4/80- and DAPI-stained ventricular sections and the statistical results. The green indicates stained macrophages. (B) Comparison of F4/80-stained macrophages among the four groups. (C) Cx43- and TnT-stained ventricular sections and the statistical results. The red indicates stained gap-junctions. (D) Comparison of morphometry for Cx43 among the four groups. (E) Comparison of Cx43 lateralization among the four groups. (F) TH- and DAPI-stained ventricular sections and the statistical results. The red indicates stained sympathetic nerves. (G) Comparison of TH-stained sympathetic nerve fibers among the four groups. Each group, n = 6. *P < 0.05 vs the sham-operated mice of the same type, #P < 0.05 vs the WT mice underwent the same operation. Scale bar, 20 μm. |
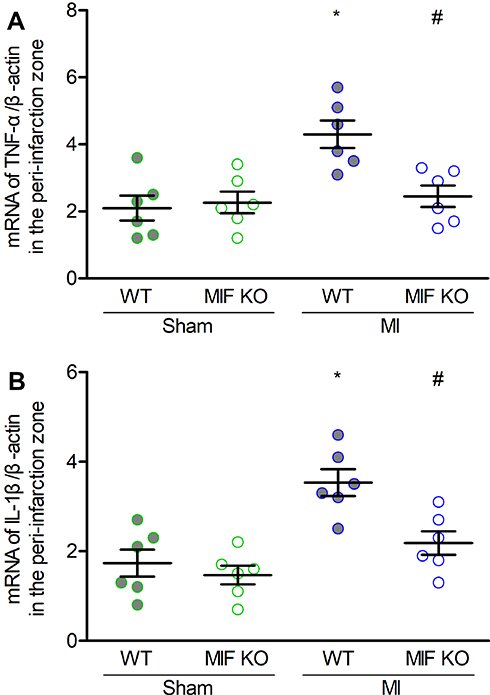 |
Figure 6 Effects of MIF on inflammatory mediators in the peri-infarction zone. (A) TNF-α, and (B) IL-1β mRNA levels in the four groups. Each group, n = 6. *P < 0.05 vs the sham-operated mice of the same type, #P < 0.05 vs the WT mice underwent the same operation. |
Macrophage Depletion Eliminates MIF-Induced Increases in VA Susceptibility and is Associated with Decreases in Both Cx43 Redistribution and Inflammatory Mediator Levels
To further study the role of macrophages in MIF-induced enhancement of VA susceptibility following MI, clodronate was used to deplete MI-induced macrophage infiltration. The incidence and duration of VAs after MI were reduced after macrophage depletion in both the in vivo and ex vivo WT groups (Figure 7A–D). The density of infiltrating macrophages was lower in both the WT and MIF-KO groups, but there was no significant difference between the two groups on day 3 after MI (Figure 7E and F). Furthermore, MI-induced differences in Cx43 expression (Figure 7E and G), Cx43 lateralization (Figure 7E and H) and mRNA levels of inflammatory mediators (Figure 7I and J) between WT and MIF-KO groups were no longer present following clodronate administration.
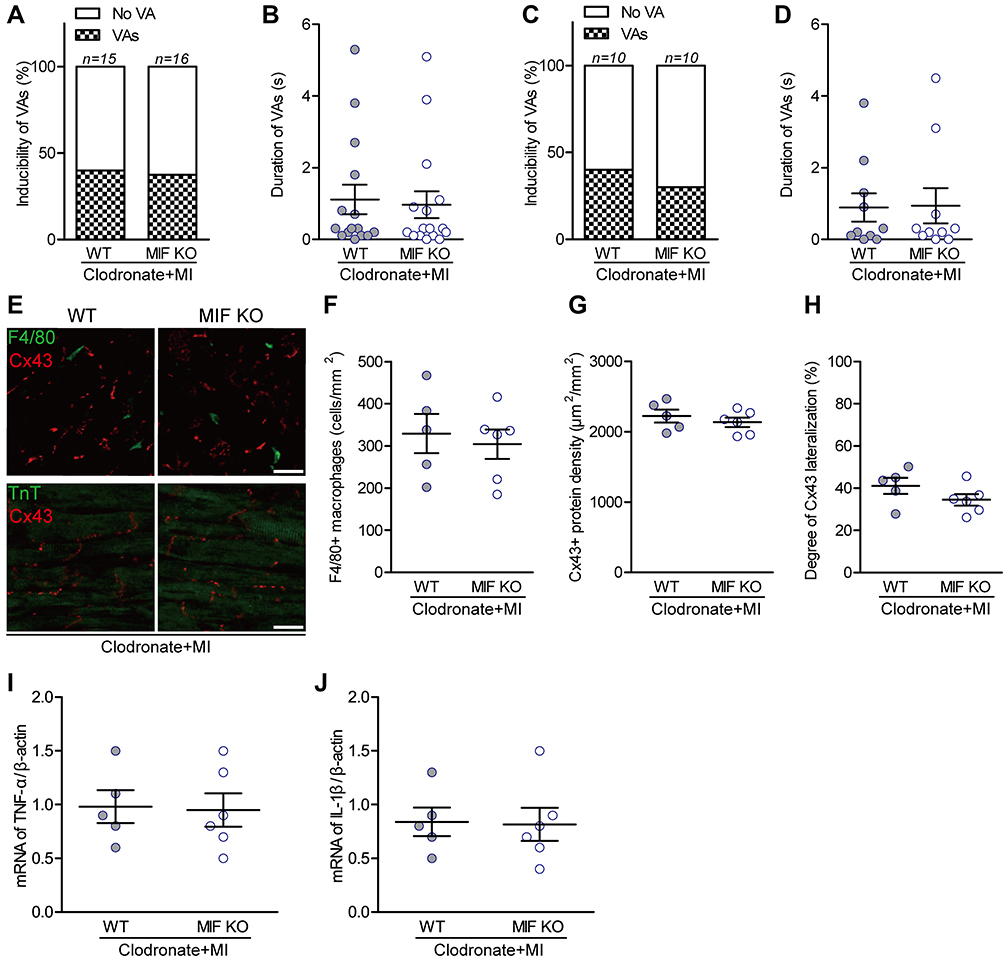 |
Figure 7 Effects of macrophage depletion on MI-induced ventricular arrhythmias (VAs) and their relationships with Cx43 and inflammatory mediators. (A) Incidence of VAs after macrophage depletion in vivo. (B) The longest VA durations in vivo; one circle indicates data from one mouse. (C) Incidence of VAs after macrophage depletion ex vivo. (D) The longest VA durations ex vivo; one circle indicates data from one heart. (E) Representative F4/80 and Cx43 co-stained ventricular sections (upper), and TnT and Cx43 co-stained ventricular sections (lower). (F) Statistical results of F4/80-stained macrophages. (G) Statistical results of Cx43-stained gap-junctions. (H) Statistical results of Cx43 lateralization. (I) TNF-α, and (J) IL-1β mRNA levels measured with qPCR between the two groups. For each group, n is reported in the corresponding column or is indicated by the number of circles. No significant difference was found (P > 0.05). |
Peritoneal Macrophage Results
Peritoneal macrophages were cultured for investigating the effects of MIF on pro-inflammatory activities of macrophages. MIF deficiency inhibited transcription and secretion of TNF-α (Figure 8A and C) and IL-1β (Figure 8B and D). However, the inhibited pro-inflammatory activity was restored after replenishment of exogenous MIF in vitro (Figure 8).
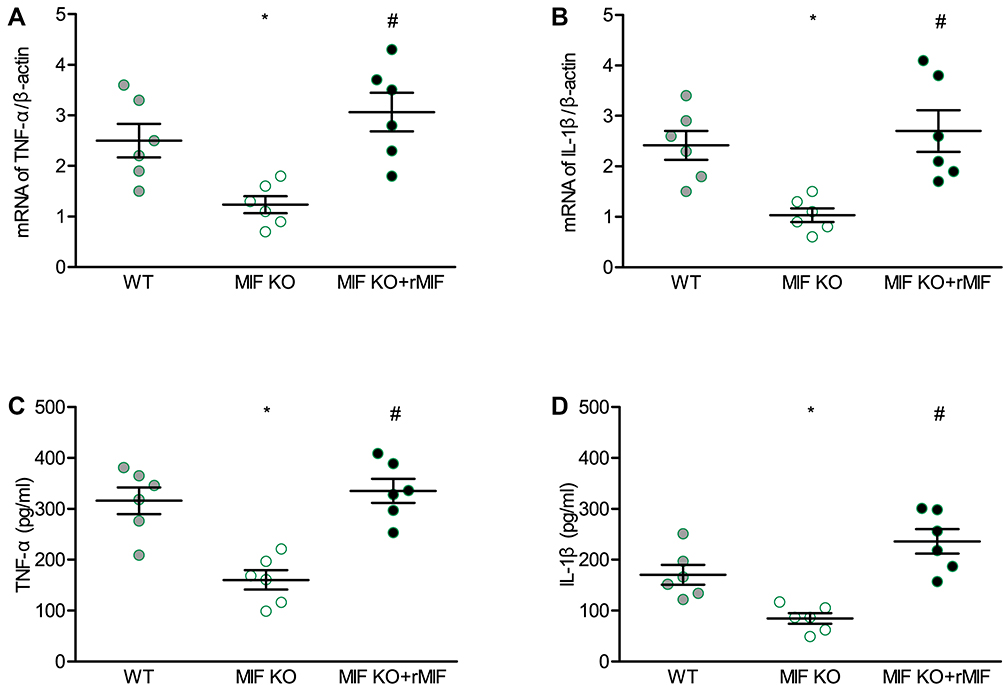 |
Figure 8 Effects of MIF deficiency and exogenous recombinant MIF replenishment on inflammatory mediators in cultured peritoneal macrophages from mice. (A) TNF-α, and (B) IL-1β mRNA levels measured with qPCR. (C) TNF-α, and (D) IL-1β secretion levels measured with ELISA. For each group, n = 6. *P < 0.05 vs the WT group, #P < 0.05 vs the MIF KO group. |
Discussion
Main Findings
The present study investigated the preventive effects and underlying mechanisms of MIF deficiency on VAs during the acute phase of MI. Our findings elucidated the following: (1) lack of MIF in mice decreased the susceptibility to VAs during the acute phase of MI, whereas it did not reduce the myocardial interstitial fibrosis or contractile injury; (2) MIF deficiency inhibited MI-induced inflammatory responses, as demonstrated by lower expression and production of pro-inflammatory mediators and decreased infiltrating macrophages; (3) MIF-KO mice had significantly improved Cx43 redistribution but similar sympathetic denervation in the peri-infarct zone, as well as similar NE levels, comparing to parameters in WT mice; and (4) pro-inflammatory mediators were suppressed in peritoneal macrophages from MIF-KO mice relative to WT mice. Taken together, these findings suggest that MIF elicits a strong pro-arrhythmia effect during the acute phase of MI via inducing a pro-inflammatory phenotype, but does not alter sympathetic reinnervation, NE levels, and myocardial interstitial fibrosis.
Causes of VAs During the Acute Phase of MI
VAs after MI have several causes, including myocardial ischemia, necrosis, fibrosis, alteration in catecholamine levels, autonomic neural remodeling and inflammatory mediator products. In the present study, similar LV function, hemodynamics, and interstitial fibrosis were observed in both WT and MIF-KO mice after MI. Arrhythmogenesis induced by catecholamines has been widely recognized.5,26 However, there are contradictory findings regarding alterations of NE levels during ischemia in the infarct myocardium.27–29 This discrepancy may attribute to reported values of catecholaminergic levels being associated with different detection methods, sampling areas, statuses of sympathetic neuronal function and degrees or phases of myocardial ischemia or infarction. The results of this study showed that although the level of circulating NE was increased in MI mice compared to that in sham-operated mice, the NE content in the peri-infarct myocardium was decreased. This phenomenon may be explained by sympathetic denervation after MI, as shown in Figure 5F and G. Nevertheless, the altered NE levels following MI were similar between WT and MIF-KO mice suggesting that NE levels do not confer different susceptibilities to VAs. Previous studies have shown that sympathetic hyperinnervation after MI increases automaticity, triggers activity, and aggravates electrophysiological heterogeneity, thus inducing VAs following MI.6,7,29–31 Usually, sympathetic reinnervation gradually presents after MI and peaks at one week after MI in animals. However, it has been reported that sympathetic hyperinnervation appears as early as day 3 after MI.32 In the present study, we found transient sympathetic denervation following MI in both WT and MIF-KO mice, which is in line with the results of previous studies.28,33 Thus, it is likely that sympathetic denervation does not yield different susceptibilities to VAs during the acute phase of MI. A previous study also verified that local transient denervation did not change the susceptibility to VAs after MI. Therefore, inhibition of macrophage infiltration and a concomitant reduction in inflammatory mediators in the local myocardium associated with MIF deficiency may have contributed to the lowered susceptibility to VAs in MIF-KO MI mice. Moreover, we used the mouse model of ventricular arrhythmia induced by electrical stimulation, rather than the spontaneous ventricular arrhythmia model, to study the effect and mechanism of MIF, which may be different from those of MIF in other animal models or patients.
Electrical Remodeling and Inflammatory Response After MI
MI usually triggers a significant inflammatory response with resultant VAs. The pro-arrhythmic effects of the macrophage-derived inflammatory mediators, TNF-α and IL-1β, have been verified to be associated with abnormalities in APD and Ca2+ buffering, as well as alterations in Cx43 expression and lateralization.23,24 Cx43 alterations were also observed after MI in the present study. Furthermore, the prolonged QTc intervals in vivo and the increased spatial dispersion of APD90 ex vivo indicate that MIF deficiency improved electrical remodeling in the peri-infarct zone. This improved electrical remodeling may produce an electrophysiological substrate for decreasing the incidence of VAs.
Inflammatory response mediates a complex contribution to the pathogenesis of MI-associated arrhythmias.34,35 As one of the most important inflammatory cells, macrophages are a natural component of the heart and comprise approximately 8% of noncardiomyocytes.36 It has been widely recognized that macrophages are essential for recovery of the infarct during the healing phase.37–40 Additionally, macrophages also contribute to ventricular arrhythmogenesis after MI via promoting sympathetic hyperinnervation and producing inflammatory mediators.8,23,24 During dynamic observation of macrophage infiltration after MI, pro-inflammatory M1 macrophages have been shown to massively infiltrate in the peri-infarct myocardium and peak at day 3 post-MI, after which the amount of anti-inflammatory M2 macrophages gradually increases, peaking at day 7 post-MI and then gradually diminish thereafter.8,32,41 According to our present findings, we hypothesize that M1 macrophages participate in pro-inflammatory responses and VAs during the acute phase of MI, whereas sympathetic reinnervation is not obvious during this phase.
Macrophage-Mediated Pro-Arrhythmia Effect of MIF After MI
MIF is a multifunctional mediator involved in many cardiovascular diseases. Recent studies have revealed the important role of MIF in myocardial injury not only in animal models but also in clinical patients with acute myocardial ischemia or MI.17,42–45 Previous studies have shown that MIF can reduce ischemic/reperfusion injury.16,46 However, it has also been indicated that MIF deficiency presents cardioprotective effects in prolonged ischemic/reperfusion injury and MI.13,20 Interestingly, MIF exhibits an opposite effect following myocardial ischemic or MI. In addition, other investigators have suggested that MIF exhibits a bidirectional effect during acute MI.21 Nevertheless, the pro-inflammatory activity of MIF during the acute phase of MI has recently been clarified.47 Therefore, these controversial results indicate that inflammatory responses are necessary for myocardial healing but that they may become deleterious if excessively prolonged. Furthermore, MIF contributes to infiltration of macrophages and in initiating inflammatory responses after MI.15 However, few studies have investigated the arrhythmogenic effects of MIF during the acute phase of MI.
Here, we demonstrated a prominent decrease in susceptibility to VAs in MIF KO mouse hearts during the acute phase of MI. These preventive effects on VAs were mainly due to the inhibition of inflammatory activities, as evidenced by downregulation of TNF-α and IL-1β in MIF-KO mice compared with these levels in WT mice following MI. This effect was further verified by macrophage depletion in WT hearts. Moreover, MIF KO inhibited the release of inflammatory mediators from peritoneal macrophages.
Limitations
One of the limitations of this study is that only one time point during the early stage of MI was studied. The interaction between MIF and macrophages was not dynamically evaluated. Thus, it remains unclear whether the macrophage dynamics and phenotype affect the function of MIF and susceptibility to ventricular arrhythmias. In addition, the in vivo spatiotemporal methods for measurement of the extracellular NE level and the sympathetic activity in heart are limited. As the main element of catecholamine transmitters of cardiac sympathetic nerve, NE in plasma and myocardium measured with ELISA represented the extracellular catecholamines and sympathetic activity. In addition, the in vivo spatiotemporal method for catecholamine measurement in the heart is limited. We detected plasma and myocardial NE using ELISA as the extracellular level of catecholamines. Perchloric acid was used as protein precipitant and stabilizer of catecholamines in the process of myocardial tissue homogenization. Although perchloric acid may affect the absorbance of ELISA detection and lead to deviation of measurement results, we think it will not change the difference between groups.
Conclusions
In conclusion, we found that MIF KO reduced macrophage infiltration and the expression/production of pro-inflammatory mediators, and was associated a decrease in VA susceptibility. Genetic deletion of MIF reduced the inducibility of VAs in mice by predominantly suppressing pro-inflammatory responses. Furthermore, our present findings also highlight the importance of inhibiting excessive inflammatory responses and that MIF may present a therapeutic target for reducing susceptibility to VAs during the acute phase of MI. Taken together, our findings may be helpful for preventing and treating VAs during the early stage of MI.
Highlights
- MIF-KO in mice is associated with less induced VAs during the acute phase of MI.
- MIF-KO inhibited the pro-inflammatory response, Cx43 decrease and lateralization after MI.
- The effect of MIF-KO on VA susceptibility was inhibited by macrophage depletion.
- Exogenous MIF reversed the pro-inflammatory activity of macrophages from MIF-KO mice.
Abbreviations
VA, Ventricular arrhythmia; MI, Myocardial infarction; NE, Norepinephrine; TNF-α, Tumour necrosis factor-α; IL-1β, Interleukin-1β; MIF, Macrophage migration inhibitory factor; KO, Knockout; WT, Wild-type; LV, Left ventricle; LVEDD, LV end diastolic dimension; LVESD, LV end systolic dimension; EF, Ejection fraction; FS, Fractional shortening; SV, Stroke volume; SBP, Systolic arterial blood pressure; DBP, Diastolic arterial blood pressure; LVPSP, LV peak systolic pressure; LVEDP, LV end diastolic pressure; QTc, Corrected QT interval; L-M, Left middle; L-A, Left apex; R-M, Right middle; R-A, Right apex; APD, Action potential duration; CV, coefficient of variation; ELISA, Enzyme-linked immunosorbent assays; Cx43, Connexin 43; TH, Tyrosine hydroxylase; TnT, Troponin T; rMIF, Recombinant MIF; qPCR, Quantitative real-time polymerase chain reaction; SEM, Standard error of mean; ANOVA, One-way analyses of variance; RVERP, Right ventricular effective refractory period; DAPI, 4ʹ,6-Diamidino-2-phenylindole.
Ethical Approval
This study was approved by the West China Second University Hospital and Sichuan Provincial People’s Hospital, China.
Funding
This work was supported by the National Natural Science Foundation of China (81500281, 81601664), Sichuan Science and Technology Program (2019YJ0577, 2021YFS0097, 2021YFS0196), Science and Technology Project of the Health Planning Committee of Sichuan (20ZD011, 19PJ126), Science and Technology Project of Sichuan Overseas Students (2019-58) and Youth Research Project of Sichuan Medical Association (Q19017).
Disclosure
The authors report no conflicts of interest.
References
1. Solomon SD, Zelenkofske S, McMurray JJ, et al. Sudden death in patients with myocardial infarction and left ventricular dysfunction, heart failure, or both. N Engl J Med. 2005;352(25):2581–2588. doi:10.1056/NEJMoa043938
2. Liang C, Wang K, Li Q, Bai J, Zhang H. Influence of the distribution of fibrosis within an area of myocardial infarction on wave propagation in ventricular tissue. Sci Rep. 2019;9(1):14151. doi:10.1038/s41598-019-50478-5
3. Jalife J. Ventricular fibrillation: mechanisms of initiation and maintenance. Annu Rev Physiol. 2000;62:25–50. doi:10.1146/annurev.physiol.62.1.25
4. Nademanee K, Taylor R, Bailey WE, Rieders DE, Kosar EM. Treating electrical storm: sympathetic blockade versus advanced cardiac life support-guided therapy. Circulation. 2000;102(7):742–747. doi:10.1161/01.CIR.102.7.742
5. Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49. doi:10.1146/annurev.physiol.70.113006.100455
6. Cao JM, Fishbein MC, Han JB, et al. Relationship between regional cardiac hyperinnervation and ventricular arrhythmia. Circulation. 2000;101(16):1960–1969. doi:10.1161/01.CIR.101.16.1960
7. Chen PS, Chen LS, Cao JM, Sharifi B, Karagueuzian HS, Fishbein MC. Sympathetic nerve sprouting, electrical remodeling and the mechanisms of sudden cardiac death. Cardiovasc Res. 2001;50(2):409–416. doi:10.1016/S0008-6363(00)00308-4
8. Wernli G, Hasan W, Bhattacherjee A, van Rooijen N, Smith PG. Macrophage depletion suppresses sympathetic hyperinnervation following myocardial infarction. Basic Res Cardiol. 2009;104(6):681–693. doi:10.1007/s00395-009-0033-3
9. Lyu J, Wang M, Kang X, et al. Macrophage-mediated regulation of catecholamines in sympathetic neural remodeling after myocardial infarction. Basic Res Cardiol. 2020;115(5):56. doi:10.1007/s00395-020-0813-3
10. Lambert JM, Lopez EF, Lindsey ML. Macrophage roles following myocardial infarction. Int J Cardiol. 2008;130(2):147–158. doi:10.1016/j.ijcard.2008.04.059
11. Hulsmans M, Clauss S, Xiao L, et al. Macrophages facilitate electrical conduction in the heart. Cell. 2017;169(3):510–522 e520. doi:10.1016/j.cell.2017.03.050
12. Chen Y, Zhang Q, Liao YH, et al. Effect of tumor necrosis factor-alpha on neutralization of ventricular fibrillation in rats with acute myocardial infarction. Mediators Inflamm. 2011;2011:565238. doi:10.1155/2011/565238
13. Gao XM, Liu Y, White D, et al. Deletion of macrophage migration inhibitory factor protects the heart from severe ischemia-reperfusion injury: a predominant role of anti-inflammation. J Mol Cell Cardiol. 2011;50(6):991–999. doi:10.1016/j.yjmcc.2010.12.022
14. Calandra T, Bernhagen J, Metz CN, et al. MIF as a glucocorticoid-induced modulator of cytokine production. Nature. 1995;377(6544):68–71. doi:10.1038/377068a0
15. Zernecke A, Bernhagen J, Weber C. Macrophage migration inhibitory factor in cardiovascular disease. Circulation. 2008;117(12):1594–1602. doi:10.1161/CIRCULATIONAHA.107.729125
16. Miller EJ, Li J, Leng L, et al. Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart. Nature. 2008;451(7178):578–582. doi:10.1038/nature06504
17. Rassaf T, Weber C, Bernhagen J. Macrophage migration inhibitory factor in myocardial ischaemia/reperfusion injury. Cardiovasc Res. 2014;102(2):321–328. doi:10.1093/cvr/cvu071
18. Rossello X, Burke N, Stoppe C, Bernhagen J, Davidson SM, Yellon DM. Exogenous administration of recombinant MIF at physiological concentrations failed to attenuate infarct size in a Langendorff perfused isolated mouse heart model. Cardiovasc Drugs Ther. 2016;30(5):445–453. doi:10.1007/s10557-016-6673-2
19. Chan W, White DA, Wang XY, et al. Macrophage migration inhibitory factor for the early prediction of infarct size. J Am Heart Assoc. 2013;2(5):e000226. doi:10.1161/JAHA.113.000226
20. White DA, Su Y, Kanellakis P, et al. Differential roles of cardiac and leukocyte derived macrophage migration inhibitory factor in inflammatory responses and cardiac remodelling post myocardial infarction. J Mol Cell Cardiol. 2014;69:32–42. doi:10.1016/j.yjmcc.2014.01.015
21. Liehn EA, Kanzler I, Konschalla S, et al. Compartmentalized protective and detrimental effects of endogenous macrophage migration-inhibitory factor mediated by CXCR2 in a mouse model of myocardial ischemia/reperfusion. Arterioscler Thromb Vasc Biol. 2013;33(9):2180–2186. doi:10.1161/ATVBAHA.113.301633
22. Monnerat G, Alarcon ML, Vasconcellos LR, et al. Macrophage-dependent IL-1beta production induces cardiac arrhythmias in diabetic mice. Nat Commun. 2016;7:13344. doi:10.1038/ncomms13344
23. Petkova-Kirova PS, London B, Salama G, Rasmusson RL, Bondarenko VE. Mathematical modeling mechanisms of arrhythmias in transgenic mouse heart overexpressing TNF-alpha. Am J Physiol Heart Circ Physiol. 2012;302(4):H934–H952. doi:10.1152/ajpheart.00493.2011
24. Kalbitz M, Amann EM, Bosch B, et al. Experimental blunt chest trauma-induced myocardial inflammation and alteration of gap-junction protein connexin 43. PLoS One. 2017;12(11):e0187270. doi:10.1371/journal.pone.0187270
25. Rajab M, Jin H, Welzig CM, et al. Increased inducibility of ventricular tachycardia and decreased heart rate variability in a mouse model for type 1 diabetes: effect of pravastatin. Am J Physiol Heart Circ Physiol. 2013;305(12):H1807–H1816. doi:10.1152/ajpheart.00979.2012
26. Thomas D, Kiehn J, Katus HA, Karle CA. Adrenergic regulation of the rapid component of the cardiac delayed rectifier potassium current, I(Kr), and the underlying hERG ion channel. Basic Res Cardiol. 2004;99(4):279–287. doi:10.1007/s00395-004-0474-7
27. Lameris TW, de Zeeuw S, Alberts G, et al. Time course and mechanism of myocardial catecholamine release during transient ischemia in vivo. Circulation. 2000;101(22):2645–2650. doi:10.1161/01.CIR.101.22.2645
28. Parrish DC, Alston EN, Rohrer H, et al. Infarction-induced cytokines cause local depletion of tyrosine hydroxylase in cardiac sympathetic nerves. Exp Physiol. 2010;95(2):304–314. doi:10.1113/expphysiol.2009.049965
29. Li W, Knowlton D, Van Winkle DM, Habecker BA. Infarction alters both the distribution and noradrenergic properties of cardiac sympathetic neurons. Am J Physiol Heart Circ Physiol. 2004;286(6):H2229–H2236. doi:10.1152/ajpheart.00768.2003
30. Cao JM, Chen LS, KenKnight BH, et al. Nerve sprouting and sudden cardiac death. Circ Res. 2000;86(7):816–821. doi:10.1161/01.RES.86.7.816
31. Wu P, Vaseghi M. The autonomic nervous system and ventricular arrhythmias in myocardial infarction and heart failure. Pacing Clin Electrophysiol. 2020;43(2):172–180. doi:10.1111/pace.13856
32. Yin J, Hu H, Li X, et al. Inhibition of Notch signaling pathway attenuates sympathetic hyperinnervation together with the augmentation of M2 macrophages in rats post-myocardial infarction. Am J Physiol Cell Physiol. 2016;310(1):C41–C53. doi:10.1152/ajpcell.00163.2015
33. Parrish DC, Francis Stuart SD, Olivas A, et al. Transient denervation of viable myocardium after myocardial infarction does not alter arrhythmia susceptibility. Am J Physiol Heart Circ Physiol. 2018;314(3):H415–H423. doi:10.1152/ajpheart.00300.2017
34. De Jesus NM, Wang L, Herren AW, et al. Atherosclerosis exacerbates arrhythmia following myocardial infarction: role of myocardial inflammation. Heart Rhythm. 2015;12(1):169–178. doi:10.1016/j.hrthm.2014.10.007
35. Yu L, Wang Y, Zhou X, et al. Leptin injection into the left stellate ganglion augments ischemia-related ventricular arrhythmias via sympathetic nerve activation. Heart Rhythm. 2018;15(4):597–606. doi:10.1016/j.hrthm.2017.12.003
36. Wang Z, Lu YL, Zhao WT, et al. Distinct origins and functions of cardiac orthotopic macrophages. Basic Res Cardiol. 2020;115(2):8. doi:10.1007/s00395-019-0769-3
37. Peet C, Ivetic A, Bromage DI, Shah AM. Cardiac monocytes and macrophages after myocardial infarction. Cardiovasc Res. 2020;116(6):1101–1112. doi:10.1093/cvr/cvz336
38. Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. 2013;339(6116):161–166. doi:10.1126/science.1230719
39. Hilgendorf I, Gerhardt LM, Tan TC, et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res. 2014;114(10):1611–1622. doi:10.1161/CIRCRESAHA.114.303204
40. van der Laan AM, Ter Horst EN, Delewi R, et al. Monocyte subset accumulation in the human heart following acute myocardial infarction and the role of the spleen as monocyte reservoir. Eur Heart J. 2014;35(6):376–385. doi:10.1093/eurheartj/eht331
41. Tokutome M, Matoba T, Nakano Y, et al. Peroxisome proliferator-activated receptor-gamma targeting nanomedicine promotes cardiac healing after acute myocardial infarction by skewing monocyte/macrophage polarization in preclinical animal models. Cardiovasc Res. 2019;115(2):419–431. doi:10.1093/cvr/cvy200
42. Voss S, Kruger S, Scherschel K, et al. Macrophage Migration Inhibitory Factor (MIF) expression increases during myocardial infarction and supports pro-inflammatory signaling in cardiac fibroblasts. Biomolecules. 2019;9(2):38. doi:10.3390/biom9020038
43. Fan F, Fang L, Moore XL, et al. Plasma macrophage migration inhibitor factor is elevated in response to myocardial ischemia. J Am Heart Assoc. 2016;5(7). doi:10.1161/JAHA.115.003128.
44. Ruze A, Chen BD, Liu F, et al. Macrophage migration inhibitory factor plays an essential role in ischemic preconditioning-mediated cardioprotection. Clin Sci. 2019;133(5):665–680. doi:10.1042/CS20181013
45. Zhao Q, Men L, Li XM, et al. Circulating MIF levels predict clinical outcomes in patients with ST-elevation myocardial infarction after percutaneous coronary intervention. Can J Cardiol. 2019;35(10):1366–1376. doi:10.1016/j.cjca.2019.04.028
46. Qi D, Hu X, Wu X, et al. Cardiac macrophage migration inhibitory factor inhibits JNK pathway activation and injury during ischemia/reperfusion. J Clin Invest. 2009;119(12):3807–3816. doi:10.1172/JCI39738
47. White DA, Fang L, Chan W, et al. Pro-inflammatory action of MIF in acute myocardial infarction via activation of peripheral blood mononuclear cells. PLoS One. 2013;8(10):e76206. doi:10.1371/journal.pone.0076206
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.