")
Back to Journals » Drug Design, Development and Therapy » Volume 15
L-Cysteine Provides Neuroprotection of Hypoxia-Ischemia Injury in Neonatal Mice via a PI3K/Akt-Dependent Mechanism
Authors Li T, Li J, Li T, Zhao Y , Ke H, Wang S, Liu D, Wang Z
Received 19 November 2020
Accepted for publication 26 January 2021
Published 11 February 2021 Volume 2021:15 Pages 517—529
DOI https://doi.org/10.2147/DDDT.S293025
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Anastasios Lymperopoulos
Tingting Li,1,* Jiangbing Li,1,2,* Tong Li,3,* Yijing Zhao,1 Hongfei Ke,1 Shuanglian Wang,1 Dexiang Liu,4 Zhen Wang1
1Department of Physiology, School of Basic Medical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, Shandong, People’s Republic of China; 2Department of Cardiology, Shandong Provincial Hospital Affiliated to Shandong University, Jinan, Shandong, People’s Republic of China; 3Department of Neurosurgery Surgery, Qingdao Municipal Hospital, Shandong Province, People’s Republic of China; 4Department of Medical Psychology and Ethics, School of Basic Medicine Sciences, Cheeloo College of Medicine, Shandong University, Jinan, Shandong, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Dexiang Liu
Department of Medical Psychology and Ethics, School of Basic Medical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, Shandong, 250012, People’s Republic of China
Email [email protected]
Zhen Wang
Department of Physiology, School of Basic Medical Sciences, Cheeloo College of Medicine, Shandong University, 44 Wenhua Xi Road, Jinan, Shandong, 250012, People’s Republic of China
Email [email protected]
Background: Previous work within our laboratory has revealed that hydrogen sulfide (H2S) can serve as neuroprotectant against brain damage caused by hypoxia-ischemia (HI) exposure in neonatal mice. After HI insult, activation of the phosphatidylinositol-3-kinase (PI3K)/protein kinase B (Akt) signaling pathway has been shown to be implicated in neuro-restoration processes. The goal of the current study was to determine whether the neuroprotective effects of H2S were mediated by the PI3K/Akt signaling pathway.
Methods: The mouse HI model was built at postnatal day 7 (P7), and the effects of L-Cysteine treatment on acute brain damage (72 h post-HI) and long-term neurological responses (28 days post-HI) were evaluated. Nissl staining and Transmission electron microscopy were used to evaluate the neuronal loss and apoptosis. Immunofluorescence imaging and dihydroethidium staining were utilized to determine glial cell activation and ROS content, respectively.
Results: Quantitative results revealed that L-Cysteine treatment significantly prevented the acute effects of HI on apoptosis, glial cell activation and oxidative injury as well as the long-term effects upon memory impairment in neonatal mice. This protective effect of L-Cysteine was found to be associated with the phosphorylation of Akt and phosphatase and a tensin homolog deletion on chromosome 10 (PTEN). Following treatment with the PI3K inhibitor, LY294002, the neuroprotective effects of L-Cysteine were attenuated.
Conclusion: PTEN/PI3K/Akt signaling was involved in mediating the neuroprotective effects of exogenous H2S against HI exposure in neonatal mice.
Keywords: Akt, H2S, hypoxia-ischemia, neuroinflammation
Introduction
Neonatal hypoxia-ischemia (HI) brain injury is associated with mortality or severe neurological disabilities.1 Accordingly, understanding of the pathologic and survival mechanisms of HI injury on brain functions represents a critical area of investigation for the development of neuroprotective treatments.
Hydrogen sulfide (H2S) has been reported to be involved in modulating physiological and pathological processes within the central nervous systems.2,3 Normally, in mammalian cells, H2S originates from the metabolism of L-Cysteine through catalysis involving three enzymes, cystathionine-β-synthase (CBS), cystathionine-γ-lyase (CSE) and 3-mercaptopyruvate sulfurtransferase (3MST). These H2S-producing enzymes are highly expressed in various types of tissue and cells, including the brain.4 An exogenous administration of H2S has been shown to exert a neuroprotective effect through its capacity to affect anti-apoptotic, anti-inflammatory and anti-oxidative functions.5,6 Recently, we reported that L-Cysteine was able to suppress HI-induced microglial activation and neuroinflammation by releasing H2S.7,8 Although these reports indicate neuroprotective effects of H2S, the underlying mechanisms of this neuroprotection against brain damage resulting from HI have not been fully elucidated.
The phosphoinositol-3-kinases (PI3Ks)/protein kinase B (Akt) signaling pathway has been largely discussed with regard to its role in inflammation, metabolism, cell growth and survival.9 Interestingly, the PI3K/Akt pathway contributes to HI-induced brain damage through suppressing microglial activation, neuroinflammation and neuronal apoptosis.10 In addition, the protective effects of H2S treatment in myocardial11 and cerebral ischemia were positively associated with this PI3K/Akt signaling pathway.12 Based on these findings, we found that stimulation of Akt phosphorylation by L-Cysteine rescues brain function in neonatal mice exposed to HI insult.7 The aim of this study was to explore whether the neuroprotective effects of H2S on HI-induced brain injury were mediated by the PI3K/Akt pathway.
Materials and Methods
Experimental Animals
C57BL/6J female mice (postnatal day 3, P3) were purchased from the laboratory animal center of Shandong University. The mice were housed in a temperature- and light-controlled room and allowed free access to food and water. Animal experiments were conducted in accordance with the principles for animal research issued by the Council of International Organizations for Medical Sciences. The Animal Ethics and Welfare Committee of Shandong University approved this study.
HI Model and Treatments
The neonatal mouse HI model was a variation of the classic Rice–Vannucci model used to mimic human HIE as described previously.7 Briefly, postnatal day 7 (P7) mice were used as rodents at this age to display a brain maturity comparable to that of a 36–40 week old term human fetus.10,13 The mice were anesthetized with isoflurane, the right common carotid artery was exposed and permanently ligated using a 4–0 suture line. Mice were returned to their home cage for a 1 h recovery period followed by placement in a hypoxic environment (8% O2 + 92% N2) for 1.5 h to induce HI. The sham group was only anesthetized and the right common carotid artery was exposed.
The mice were randomly assigned to one of the four experimental groups: 1) Sham, 2) HI, 3) HI + L-Cysteine and 4) HI + L-Cysteine + LY294002. These compounds were administered intraperitoneally (ip) at 24, 48 or 72 h post-HI. For the HI + L-Cysteine + LY294002 group, LY294002 was administered at 30 min prior to the L-Cysteine injection. The L-Cysteine (could evoke H2S release) and LY294002 (an inhibitor of PI3K) were dissolved in phosphate-buffered saline (PBS). The 5 mg/kg dose of L-Cysteine was used based on our previous study.7 The 0.1 mg/kg, 1 mg/kg, 5 mg/kg LY294002 were used to test its effect on brain damage. Animals were euthanized at the post-HI times as indicated.
Brain Water Content
Wet and dry weights of the right hemisphere were recorded to assess brain water content. At 72 h after HI insult the entire brain was quickly removed and the right hemisphere was measured to determine the wet weight. Then the right hemisphere was placed in a drying oven at 105°C for 24 h, to determine the dry weight. Quantitative measurement of brain water content (%) = (wet weight of right hemisphere - dry weight of right hemisphere)/wet weight of right hemisphere × 100%.
Immunofluorescence Imaging
At 72 h post-HI, the brains were collected, sectioned into 4 µm slices in the region containing the infarct lesion. All brain slices were obtained from independent experiments and assays were performed from brain sections of similar anatomical locations. Following our previous procedures,7 These slices were stained with adaptor molecule-1 (Iba-1) (ab178847, Abcam, Cambridge, UK) or glial fibrillary acidic protein (GFAP) (60190-1-Ig, Proteintech Group, Rosemont, IL, USA). The slices were incubated with a fluorescently labeled secondary antibody for 30 min at 37°C in the dark followed by DAPI staining. Fluorescent images were observed using fluorescent microscopy and Image-Pro Plus 6.0 software was used to analyze the number of Iba-1+ and GFAP + cells in the right cortex.
Measurement of Infarct Size
At 72 h following HI insult, the brains were subjected to infarct staining with 2, 3, 5-triphenyl tetrazolium chloride (TTC). Brains were cut into 1 mm coronal slices and stained with 2% TTC solution (Sigma-Aldrich, Hamburg, Germany) at 37°C for 20 min. The infarct size was measured using Image-Pro Plus 6.0 software.

Nissl Staining
Nissl staining was conducted to evaluate brain tissue loss as described previously.14 Brain sections (4 μm thickness) were stained with 0.5% cresyl violet acetate for 20 min, rinsed with distilled water and then dehydrated with ethanol. The number of surviving neurons within different areas was counted at × 200 magnifications. Results are expressed as the number of surviving neuron within each group relative to that of the Sham group.8,15
Detection of Brain Atrophy
For determinations of brain atrophy, the brains from PND 35 mice (28 days post-HI) were removed. Left hemispheres and right hemispheres were weighed independently using a precision electronic balance. The percent of brain weight loss was calculated as follows: brain weight loss (%) = (weight of the left hemisphere - weight of the right hemisphere)/weight of the left hemisphere × 100%.
Transmission Electron Microscopy (TEM)
At 72 h after HI exposure, 1 mm thick sections were obtained from identical locations within right cortex. For TEM, 50 nm thickness sections were obtained using an Ultra microtome (EM UC 7, Leica, Germany) and then stained with uranyl acetate. Electron microscopic images were photographed using a Hitachi H-7500 TEM.
DHE Analysis
Dihydroethidium (DHE) staining was performed to detect ROS production in the right hemisphere as previously described.8 Coronal sections were incubated with 10 μM DHE (Sigma) in a dark for 30 min. DHE-positive images were quantified using Image-Pro Plus 6.0 software.
Y-Maze Test
The Y-maze test is widely used to evaluate short-term memory in animals.16 This test consisted of training and test trails. For the training trial, the novel arm was closed and the animal was pseudorandomly placed in a start arm and allowed to explore the start and other arms for 5 min. Then, mouse was removed and the Y-maze was cleaned. For the test trail, the mice were allowed to freely explore all three arms for 5 min. The SMART tracking system was used to record their activity. The percent of time spent and number of total arm entries in the novel arm were recorded and analyzed.
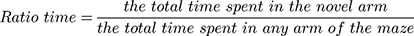
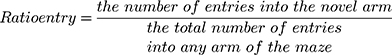
Western Blot
The right cortex was dissolved in RIPA buffer containing protease/phosphatase inhibitors and PMSF. A sample from the supernatant was removed for protein determination using the BCA protein assay kit. An equal amount of protein was fractionated by SDS-PAGE and transferred onto a PVDF membrane that was blocked at room temperature for1 h with 5% skimmed milk, followed by incubation at 4°C with primary antibodies for 12 h. Primary antibodies included Akt (1:1000, #9272S, Cell Signaling Technology, MA, USA), p-Akt (1:1000,#9271S, Cell Signaling Technology), Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) (1:1000, #9552, Cell Signaling Technology), p-PTEN (1:1000, #9551, Cell Signaling Technology) and β-actin (1:1000, #TA-09, ZSGB, Peking, China). The membranes were incubated for 1 h with the secondary antibody. The blots were developed with use of ECL kit reagents (Millipore Corporation).
Statistical Analysis
The SPSS 24.0 software program was used for performing the statistical analysis. Data were expressed as the means ± standard deviations. Unless otherwise noted, data among the different groups were compared using the one-way ANOVA followed by – the Bonferroni corrections test for post hoc analysis. The correlation between the total number of arm entries and Y-maze alterations were compared by Pearson test. p value <0.05 was considered as statistically significant.
Results
Inhibition of PI3K Attenuates the Effects of L-Cysteine on HI-Induced Damage
As reported previously,7,8 L-Cysteine administration significantly decreased the water content (p < 0.01) and infarct volumes (p < 0.01) in HI neonatal mice. PI3K/Akt inhibition by LY294002 at a dose of 5 mg/kg reversed H2S’s neuroprotective effects evidenced by the results obtained with water content (p < 0.01) and infarct volume (p < 0.01) determinations (Figure 1). Thus, we chose the 5 mg/kg dose of LY294002 for use in this study.
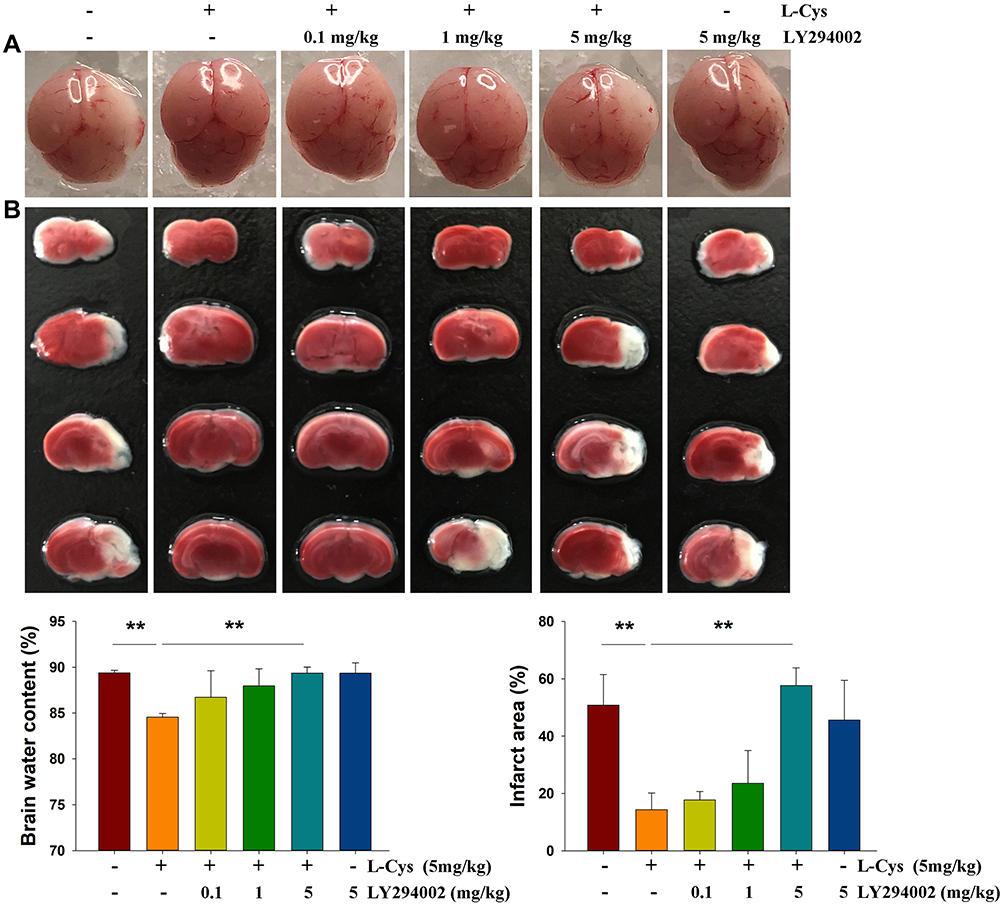 |
Figure 1 Akt inhibition attenuates the neuroprotective effects of L-Cysteine in HI. Representative brain images as obtained from each group at 72 h post-HI. (A) Brain water content in the right hemispheres and (B) Representative samples stained with TTC to determine quantified infarct volume (N =4/group). Values represent the mean ± SD, **p < 0.01 according to ANOVA. |
L-Cysteine Treatment Increases Akt and PTEN Phosphorylation in the Right Cortex Following HI Insult
PTEN is a prominent regulator of PI3K signaling.17 We found that the dephosphorylation of PTEN (p < 0.01) (Figure 2B) was accompanied with a dephosphorylation of Akt (p < 0.05) in the right cortex at 72 h post-HI injury (Figure 2A). However, the phosphorylation level of Akt was substantially increased following L-Cysteine treatment (p < 0.05), and reversed by LY294002 (p < 0.05) (Figure 2A). Moreover, phosphorylation of PTEN was substantially upregulated by L-Cysteine (p < 0.05), which was reversed by LY294002 (p < 0.05) (Figure 2B). Uncropped Western blots for all replicates were showed in Figure S2.
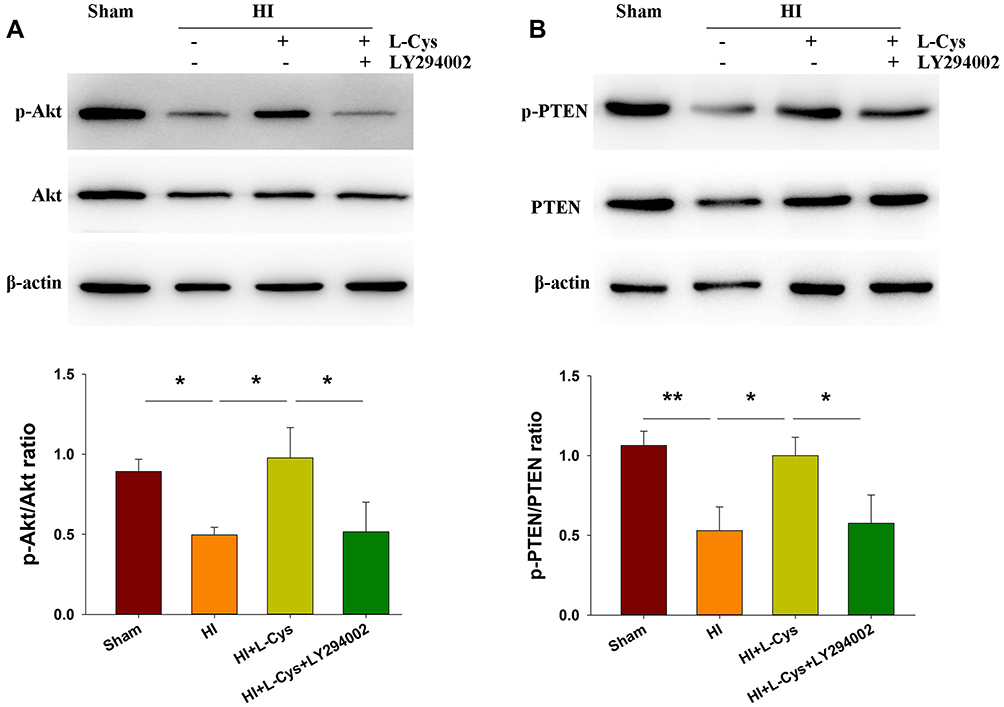 |
Figure 2 L-Cysteine treatment increases phosphorylated Akt and PTEN following HI. At 72 h post-HI the right cortex was collected for determinations of (A) Akt and phosphorylated Akt (p-Akt) and (B) PTEN and phosphorylated PTEN (p-PTEN) levels with Western blotting assays (N=3/group). Values represent the mean ± SD, *p < 0.05, **p < 0.01 according to ANOVA. |
Inhibition of PI3K Reverses the Neuroprotective Effects of L-Cysteine on HI-Induced Neuronal Loss
Neuronal loss at 72 h post-HI was investigated by Nissl staining. L-Cysteine treatment significantly reduced HI-induced neuronal loss in the prefrontal cortex (p<0.01), CA1 (p<0.05), CA3 (p<0.01) and DG (p<0.01) regions. This protective effect of L-Cysteine against neuronal loss was partially blocked with LY294002 within the prefrontal cortex (p<0.001), CA1 (p<0.05), CA3 (p<0.01) and DG (p<0.01) regions (Figure 3).
 |
Figure 3 Akt inhibition reverses the neuroprotective effects of L-Cysteine in HI. (A) Representative images of Nissl staining from each group at 72 h post-HI (Scale bar = 1000 µm). (B) Magnified views of boxed regions in A showing neuronal cell loss (Scale bar = 50 µm). Data are expressed as the number of surviving neuronal cells within each group relative to that of the Sham group within the different areas (N=5/group). Values represent the mean ± SD, *p < 0.05, **p < 0.01, ***p < 0.001 according to ANOVA. |
Inhibition of PI3K Attenuates the Neuroprotective Effects of L-Cysteine on HI-Induced Apoptosis
Ultrastructural assessments of neurons at 72 h post-insult were performed in the right cortex using electron microscopy. Neuronal membranes of the Sham group were intact and organelles, including rough endoplasmic reticulum and ribosomes, were clearly identified. In contrast, the integrity of neurons within the HI group was fragmented, organelles were absent and vacuoles were present. L-Cysteine treatment attenuated this HI-induced morphological damage (Figure 4), while LY294002 reversed these positive effects of L-Cysteine.
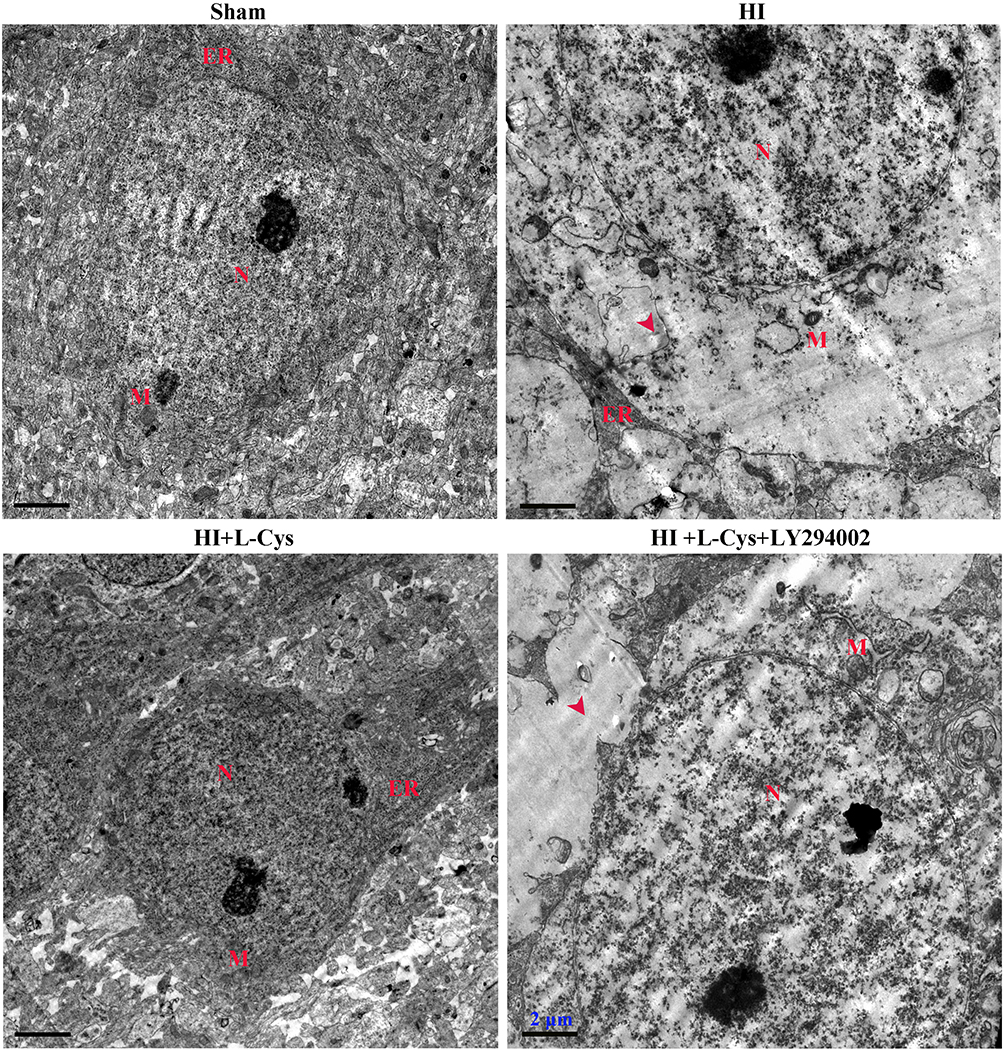 |
Figure 4 L-Cysteine decreases neuronal apoptosis via the Akt pathway in HI. Ultra-microstructure of neurons in the right cortex as obtained using TEM (Scale bar = 2 µm; N=4/group). Representative images demonstrated nucleus (N), mitochondria (M) and rough endoplasmic reticulum (ER). Red arrowhead indicates site of severe cytoplasmic edema. |
Inhibition of PI3K Attenuates the Neuroprotective Effects of L-Cysteine on HI-Induced ROS Content
HI exposure markedly increased ROS content in the right cortex (p<0.001), whereas L-Cysteine treatment suppressed this HI-induced ROS production (p<0.01). The combination of L-Cysteine and LY294002 partially blocked this protective effect of L-Cysteine on ROS content (p<0.01) (Figure 5).
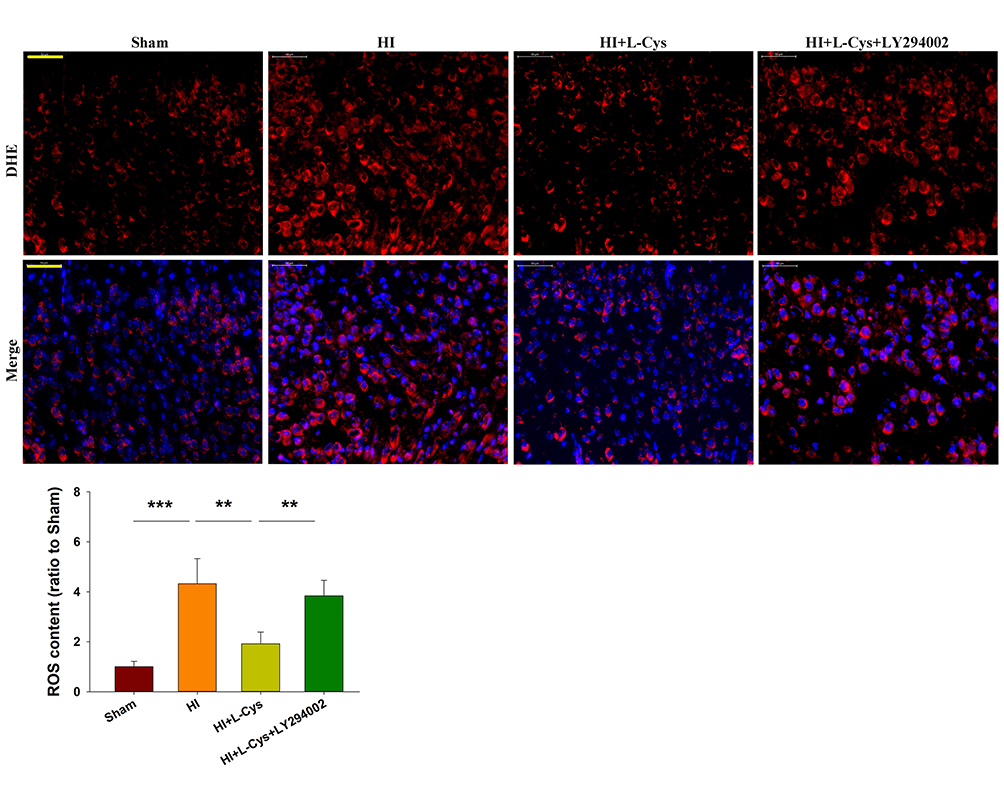 |
Figure 5 L-Cysteine suppresses HI-induced increases in ROS content through the Akt pathway. Representative photographs of DHE staining in the right cortex at 72 h post-HI. Quantification of ROS content in each group (Scale bar = 50 μm; N=4/group). Six randomly selected images (× 20) were captured from each section. Values represent the mean ± SD, **p < 0.01, ***p < 0.001according to ANOVA. |
Suppression of PI3K Attenuates the Neuroprotective Effects of L-Cysteine on HI-Induced Glial Activation
Results from immunostaining revealed that HI exposure significantly increased the number of Iba-1+ microglia/macrophages (p<0.01) (Figure 6A) and GFAP+ astrocytes (p<0.001) (Figure 6B), which were similar to that of a previous report.8 In response to L-Cysteine treatment the number of Iba-1+ microglia/macrophages (p<0.01) and GFAP+ astrocytes (p<0.001) were significantly decreased. The suppression of L-Cysteine on activation of microglia and astrocytes was attenuated by LY294002 (Figure 6). The qRT-PCR results showed that L-Cysteine administration significantly decreased the expression of TNF-α (p<0.001), CD32 (p<0.001), IL-1β (p<0.01) and increased the expression of IL-10 (p<0.05) in HI neonatal mice. While, LY294002 reversed these positive effects of L-Cysteine following HI insult (Figure S1).
 |
Figure 6 L-Cysteine treatment suppresses HI-induced glia activation through the Akt pathway. Representative photographs indicating quantified numbers of cells within the right cortex at 72 h post-HI for (A) Iba-1 and (B) GFAP staining (Scale bar = 50 μm; N=4/group). Values represent the mean ± SD, **p < 0.01, ***p < 0.001 according to ANOVA. |
Inhibition of PI3K Attenuates the Neuroprotective Effects of L-Cysteine on HI-Induced Cerebral Atrophy
When examining the long-term brain damage resulting from HI as assessed in PND 35 mice (28 days after HI insult), we found significant atrophy and liquefaction (Figure 7A), along with significant decreases in brain weight (p<0.001) (Figure 7B). This atrophy and decreased brain weights in the HI group were attenuated by L-Cysteine administration (p< 0.01), while LY294002 attenuated these beneficial effects of L-Cysteine on this cerebral atrophy following HI insult (p< 0.05).
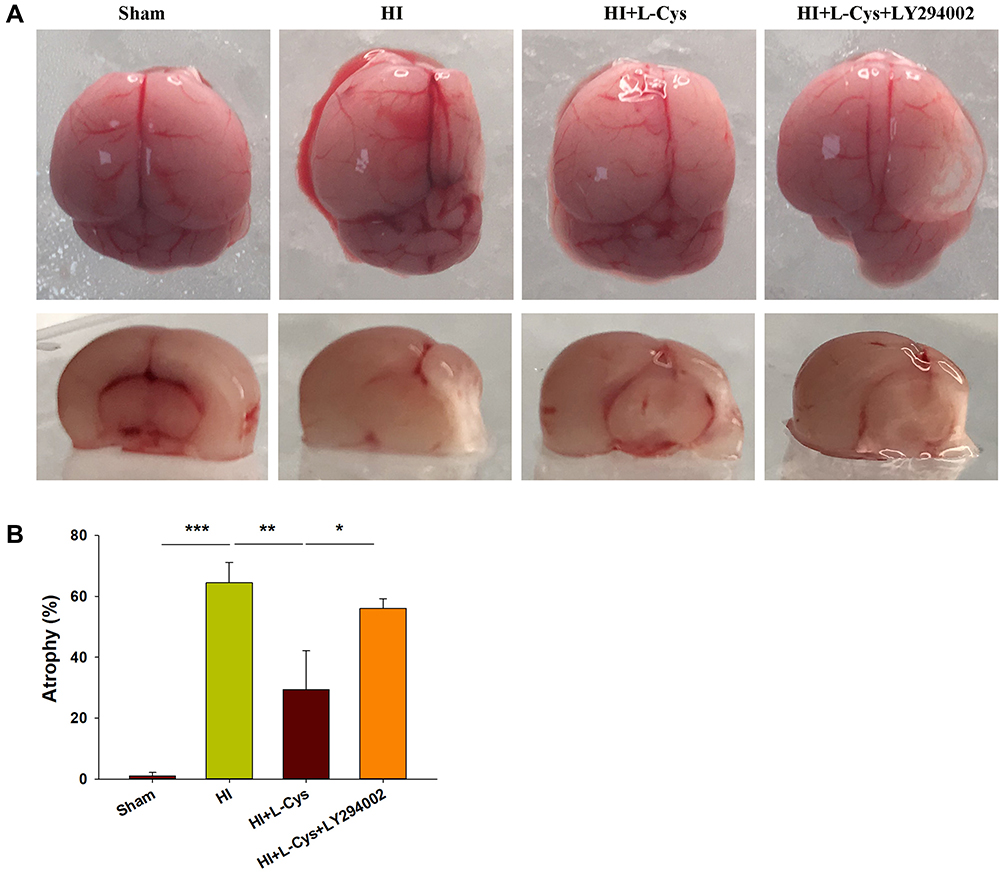 |
Figure 7 Akt inhibition attenuates neuroprotective effects of L-Cysteine on cerebral atrophy in HI. (A) Representative photographs of brain at 4 weeks after injury. (B) Left and right hemispheres were weighed independently, and the ratios of right to left weights were calculated (N=4/group). Values represent the mean ± SD, *p < 0.05, **p < 0.01, ***p < 0.001 according to ANOVA. |
Inhibition of PI3K Reverses the Neuroprotective Effects of L-Cysteine on HI-Induced Neurological Dysfunction
The effects of HI were also accompanied by obvious changes in behavioral responses in Y-maze performance as determined in PND 35 mice (28 days after HI insult). Following HI there were significant reductions in the amount of time and number of entries (p < 0.01 for both) in the novel arm of the maze as compared with that obtained in the Sham group (Figure 8A and B). However, L-Cysteine administration significantly increased both the time and entries (p < 0.05) in the novel arm as compared with that of the HI group (Figure 8A and B). The combination of LY294002 and L-Cysteine reversed these effects of L-Cysteine on improving Y-maze performance post-HI. In addition to the improvements in Y-maze performance following L-Cysteine administration, we also found that there was a statistically significant positive correlation between the time and entry preferences for the novel arm (correlation coefficient = 0.368, p < 0.05; N = 32) (Figure 8C).
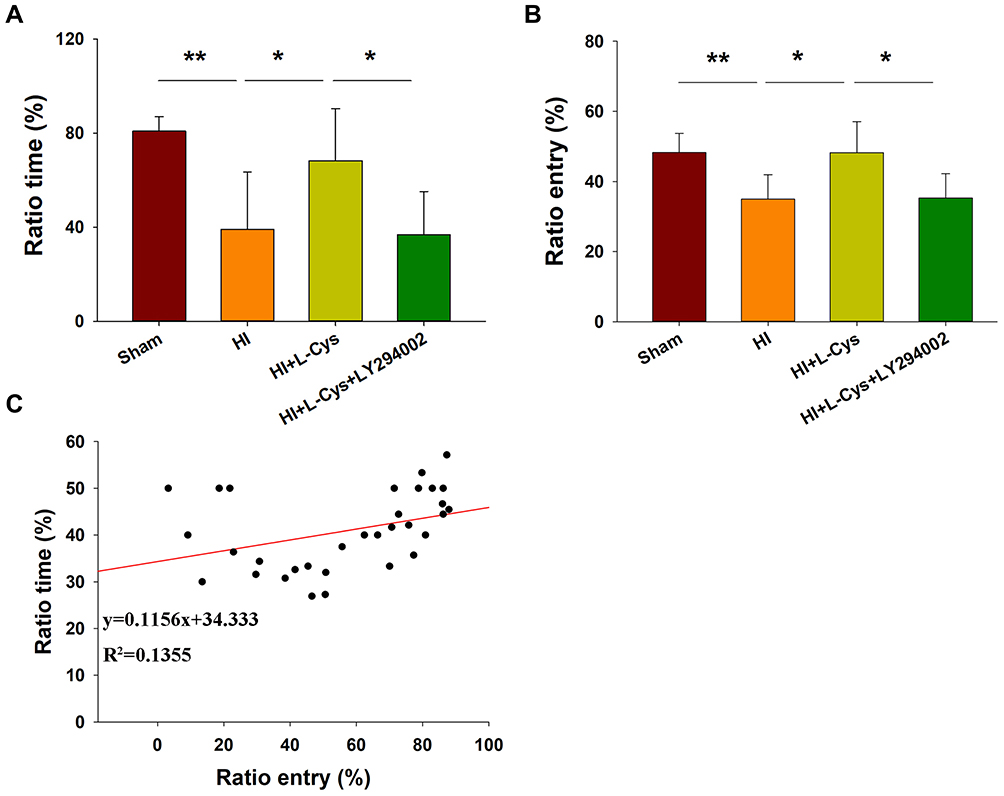 |
Figure 8 L-Cysteine improves HI-induced neurobehavioral responses via the Akt pathway. Percent of (A) time and (B) entries into the novel and other arms in the Y-maze test as performed by PND 35 mice (N = 8/group). (C) Scattered plot was generated for novel arm time and entry preferences. Values represent the mean ± SD, *p < 0.05, **p < 0.01 according to ANOVA. |
Discussion
In this study, the HI-induced neonatal mouse model was used to investigate the role of the PI3K/Akt pathway in brain damage resulting from HI. To examine this eventuality, L-Cysteine was used to activate the Akt signaling pathway. With L-Cysteine treatment, HI-induced brain damage was alleviated. Blocking of this Akt activity attenuated L-Cysteine’s protective role in HI mice, as indicated by increases in apoptosis, glial activation, ROS content and neurological dysfunction.
It has been established that H2S exerts neuroprotective effects as demonstrated in models of stroke and neurodegenerative diseases.18,19 A number of mechanisms had been proposed for these protective effects of H2S including suppression of oxidative stress and neuroinflammation, mediation of mitochondrial function and attenuation of apoptosis.20,21 Our present results revealed that L-Cysteine significantly inhibited the area of ischemic infarcts and glial activation followed by improving long-term neurological function, which were consistent with our previous studies.7,8 Moreover, results from these previous studies also revealed that L-Cysteine administration increased CBS/H2S levels in the right cortex and protected brain against HI insult via release of H2S in neonatal mice.
Activation of the PI3K/Akt pathway has been shown to increase cell growth and survival by inducing a balance between autophagy and apoptosis in central nervous system diseases.22 Phosphorylated Akt promotes cell survival by phosphorylating its downstream effectors like Bax, Bak, and BAD, caspase-9, NF-κB families, FoxO subfamilies and the p53 family.9 HI-induced brain damage could be treated by certain agents that act on the PI3K/Akt signaling pathway, such as granulocyte stimulating factor,23 insulin growth factor 1,24 environmental enrichment.25 In the current study, HI induced a dephosphorylation of Akt, suggesting that one of the damaging effects of HI involved activation of the PI3K/Akt signaling pathway. As PI3K/Akt signaling is negatively regulated by PTEN, a dephosphorylation of PTEN limits functioning of the PI3K/Akt signaling pathway.26,27 In ischemic stroke, PTEN has been shown to attenuate neuronal damage through phosphorylating Akt,26 while down-regulation of p-Akt after HI was accompanied with down-regulation of p-PTEN.27 Here, we showed that L-Cysteine exerted its protective effects by activation of p-Akt/Akt signaling, associated with dysregulation of p-PTEN/PTEN. Interestingly, this protection was reversed by PI3K/Akt inhibitor, LY294002. Results from a number of reports indicate that H2S protection is related to PI3K/Akt signaling. For example, H2S prevents oxidative damage by activating PI3K/Akt, a process that is attenuated in the aged rat.28 H2S also protects mice against cerebral ischemia and reperfusion injury via activation of the PI3K/Akt/FoxO3a pathway29 and against ischemia and reperfusion-induced liver injury through the Akt pathway.30 When collating these findings it seems that HI impairs PI3K/Akt signaling, while L-Cysteine, via augmenting phosphorylated PTEN, initiates activation of the PI3K/Akt signaling pathway to produce the neuroprotective effects.
There are some limitations of this study. First, it is possible that other pathways (such as NF-κB, ERK) may also be exerting significant impacts on the neuroprotection of L-Cysteine. Which pathways may be vital for involved in the L-Cysteine-effect in HI injury have not yet been investigated. Examining PI3K/Akt pathway under L-Cysteine treatment, the most important one, is necessary to further elucidate the mechanism in the future. Second, the long-term changes in PTEN/Akt after L-Cysteine administration were not examined. Third, limited ip L-Cysteine administrations (24, 48 and 72 h after HI insult) were tested in this study. Whether multiple treatments of L-Cysteine using different time courses or modes of administration would produce better effects against HI will require future investigations.
In summary, the findings of this study provide novel evidence that PI3K/Akt signaling is involved in HI-induced brain damage. Taken together, our findings suggest that exogenous H2S protects against HI-induced brain damage by activating the PI3K/Akt pathway.
Abbreviations
Akt, protein kinase B; CNS, central nervous system; DAPI, 4′,6′-diamidino-2-phenylindole dihydrochloride hydrate; DHE, 2-hydroethidium; HI, hypoxia-ischemic; Iba-1, ionized calcium binding adapter molecule 1; PBS, phosphate-buffered saline; PI3K, phosphoinositide 3-kinase; PTEN, phosphatase and tensin homolog deleted on chromosome 10; P-PTEN, phosphorylated PTEN; ROS, reactive oxygen species; TTC, 2,3,5-triphenyltetrazolium chloride monohydrate.
Acknowledgments
Research funding support for this work was from the National Natural Science Foundation of China (No. 82072535, 81873768 and 81671213) to Dr. Zhen Wang, the National Key Research and Development Program of China (No. 2017YFC0820203) to Dr. Dexiang Liu and the National Natural Science Foundation of China (81873865) to Dr. Shuanglian Wang.
Author Contributions
All authors made a significant contribution to the work, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
There are no competing financial interests. The authors report no conflicts of interest in this work.
References
1. Ferriero DM. Neonatal brain injury. N Engl J Med. 2004;351(19):1985–1995. doi:10.1056/NEJMra041996
2. Szabo C, Papapetropoulos A. International union of basic and clinical pharmacology. CII: pharmacological modulation of H2S levels: H2S donors and H2S biosynthesis inhibitors. Pharmacol Rev. 2017;69(4):497–564.
3. Paul BD, Snyder SH. Gasotransmitter hydrogen sulfide signaling in neuronal health and disease. Biochem Pharmacol. 2018;149:101–109. doi:10.1016/j.bcp.2017.11.019
4. Kimura H. Hydrogen sulfide and polysulfides as biological mediators. Molecules. 2014;19(10):16146–16157. doi:10.3390/molecules191016146
5. Chan SJ, Wong PT. Hydrogen sulfide in stroke: protective or deleterious? Neurochem Int. 2017;105:1–10. doi:10.1016/j.neuint.2016.11.015
6. Calvert JW, Coetzee WA, Lefer DJ. Novel insights into hydrogen sulfide–mediated cytoprotection. Antioxid Redox Signal. 2010;12(10):1203–1217. doi:10.1089/ars.2009.2882
7. Liu S, Xin D, Wang L, et al. Therapeutic effects of L-Cysteine in newborn mice subjected to hypoxia-ischemia brain injury via the CBS/H2S system: role of oxidative stress and endoplasmic reticulum stress. Redox Biol. 2017;13:528–540. doi:10.1016/j.redox.2017.06.007
8. Xin D, Chu X, Bai X, et al. l-Cysteine suppresses hypoxia-ischemia injury in neonatal mice by reducing glial activation, promoting autophagic flux and mediating synaptic modification via H2S formation. Brain Behav Immun. 2018;73:222–234. doi:10.1016/j.bbi.2018.05.007
9. Downward J. PI 3-kinase, Akt and cell survival. Semin Cell Dev Biol. 2004;15(2):177–182. doi:10.1016/j.semcdb.2004.01.002
10. Li B, Semple BD, Blomgren K, et al. Brain-immune interactions in perinatal hypoxic-ischemic brain injury. Prog Neurobiol. 2017;159:50–68.
11. Liu J, Li J, Tian P, et al. H2S attenuates sepsis-induced cardiac dysfunction via a PI3K/Akt-dependent mechanism. Exp Ther Med. 2019;17(5):4064–4072. doi:10.3892/etm.2019.7440
12. Wen X, Qi D, Sun Y, et al. H(2)S attenuates cognitive deficits through Akt1/JNK3 signaling pathway in ischemic stroke. Behav Brain Res. 2014;269:6–14. doi:10.1016/j.bbr.2014.04.027
13. Semple BD, Blomgren K, Gimlin K, et al. Brain development in rodents and humans: identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol. 2013;106–107:1–16.
14. Xin D, Li T, Chu X, et al. Mesenchymal stromal cell-derived extracellular vesicles modulate microglia/macrophage polarization and protect the brain against hypoxia-ischemic injury in neonatal mice by targeting delivery of miR-21a-5p. Acta Biomater. 2020;113:597–613. doi:10.1016/j.actbio.2020.06.037
15. Chu X, Cao L, Yu Z, et al. Hydrogen-rich saline promotes microglia M2 polarization and complement-mediated synapse loss to restore behavioral deficits following hypoxia-ischemic in neonatal mice via AMPK activation. J Neuroinflammation. 2019;16(1):104. doi:10.1186/s12974-019-1488-2
16. Yau JL, McNair KM, Noble J, et al. Enhanced hippocampal long-term potentiation and spatial learning in aged 11beta-hydroxysteroid dehydrogenase type 1 knock-out mice. J Neurosci. 2007;27(39):10487–10496. doi:10.1523/JNEUROSCI.2190-07.2007
17. Li DM, Sun H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res. 1997;57(11):2124–2129.
18. Cui Y, Duan X, Li H, et al. Hydrogen sulfide ameliorates early brain injury following subarachnoid hemorrhage in rats. Mol Neurobiol. 2016;53(6):3646–3657. doi:10.1007/s12035-015-9304-1
19. Wu D, Wang J, Li H, et al. Role of hydrogen sulfide in ischemia-reperfusion Injury. Oxid Med Cell Longev. 2015;2015:186908. doi:10.1155/2015/186908
20. Gheibi S, Aboutaleb N, Khaksari M, et al. Hydrogen sulfide protects the brain against ischemic reperfusion injury in a transient model of focal cerebral ischemia. J Mol Neurosci. 2014;54(2):264–270. doi:10.1007/s12031-014-0284-9
21. Wang JF, Li Y, Song J-N, et al. Role of hydrogen sulfide in secondary neuronal injury. Neurochem Int. 2014;64:37–47. doi:10.1016/j.neuint.2013.11.002
22. Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med. 2005;9(1):59–71. doi:10.1111/j.1582-4934.2005.tb00337.x
23. Li L, McBride DW, Doycheva D, et al. G-CSF attenuates neuroinflammation and stabilizes the blood-brain barrier via the PI3K/Akt/GSK-3beta signaling pathway following neonatal hypoxia-ischemia in rats. Exp Neurol. 2015;272:135–144. doi:10.1016/j.expneurol.2014.12.020
24. Zhao B, Zheng Z. Insulin growth factor 1 protects neural stem cells against apoptosis induced by hypoxia through Akt/Mitogen-activated protein kinase/extracellular signal-regulated kinase (Akt/MAPK/ERK) pathway in hypoxia-ishchemic encephalopathy. Med Sci Monit. 2017;23:1872–1879. doi:10.12659/MSM.901055
25. Duran-Carabali LE, Arcego DM, Odorcyk FK, et al. Prenatal and early postnatal environmental enrichment reduce acute cell death and prevent neurodevelopment and memory impairments in rats submitted to neonatal hypoxia ischemia. Mol Neurobiol. 2018;55(5):3627–3641.
26. Zhao J, Qu Y, Wu J, et al. PTEN inhibition prevents rat cortical neuron injury after hypoxia-ischemia. Neuroscience. 2013;238:242–251. doi:10.1016/j.neuroscience.2013.02.046
27. Li D, Qu Y, Mao M, et al. Involvement of the PTEN-AKT-FOXO3a pathway in neuronal apoptosis in developing rat brain after hypoxia-ischemia. J Cereb Blood Flow Metab. 2009;29(12):1903–1913. doi:10.1038/jcbfm.2009.102
28. Chen X, Zhao X, Cai H, et al. The role of sodium hydrosulfide in attenuating the aging process via PI3K/AKT and CaMKKβ/AMPK pathways. Redox Biol. 2017;12:987–1003. doi:10.1016/j.redox.2017.04.031
29. Ji K, Xue L, Cheng J, et al. Preconditioning of H2S inhalation protects against cerebral ischemia/reperfusion injury by induction of HSP70 through PI3K/Akt/Nrf2 pathway. Brain Res Bull. 2016;121:68–74. doi:10.1016/j.brainresbull.2015.12.007
30. Lu M, Jiang X, Tong L, et al. MicroRNA-21-regulated activation of the Akt pathway participates in the protective effects of H2S against liver Ischemia–reperfusion injury. Biol Pharm Bull. 2018;41(2):229–238. doi:10.1248/bpb.b17-00769
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.