Back to Journals » Neuropsychiatric Disease and Treatment » Volume 13
Korsakoff’s syndrome: a critical review
Authors Arts NJM, Walvoort SJW, Kessels RPC ![]()
Received 18 July 2017
Accepted for publication 4 October 2017
Published 27 November 2017 Volume 2017:13 Pages 2875—2890
DOI https://doi.org/10.2147/NDT.S130078
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Nicolaas JM Arts,1,2 Serge JW Walvoort,1 Roy PC Kessels1,3,4
1Centre of Excellence for Korsakoff and Alcohol-Related Cognitive Disorders, Vincent van Gogh Institute for Psychiatry, Venray, 2Neuropsychiatry Center Thalamus, Institution for Integrated Mental Health Care Pro Persona, Wolfheze, 3Department of Neuropsychology and Rehabilitation Psychology, Donders Institute for Brain, Cognition and Behaviour, Radboud University, 4Department of Medical Psychology, Radboud University Medical Center, Nijmegen, the Netherlands
Abstract: In this review, we present a survey on Korsakoff’s syndrome (KS), a residual syndrome in patients who suffered from a Wernicke encephalopathy (WE) that is predominantly characterized by global amnesia, and in more severe cases also by cognitive and behavioral dysfunction. We describe the history of KS and its definition, its epidemiology, and the lack of consensus criteria for its diagnosis. The cognitive and behavioral symptoms of KS, which include anterograde and retrograde amnesia, executive dysfunction, confabulation, apathy, as well as affective and social-cognitive impairments, are discussed. Moreover, recent insights into the underlying neurocognitive mechanisms of these symptoms are presented. In addition, the evidence so far on the etiology of KS is examined, highlighting the role of thiamine and alcohol and discussing the continuity hypothesis. Furthermore, the neuropathology of KS is reviewed, focusing on abnormalities in the diencephalon, including the mammillary bodies and thalamic nuclei. Pharmacological treatment options and nonpharmacological interventions, such as those based on cognitive rehabilitation, are discussed. Our review shows that thiamine deficiency (TD) is a crucial factor in the etiology of KS. Although alcohol abuse is by far the most important context in which TD occurs, there is no convincing evidence for an essential contribution of ethanol neurotoxicity (EN) to the development of WE or to the progression of WE to KS. Future research on the postmortem histopathological analysis of brain tissues of KS patients is crucial for the advancement of our knowledge of KS, especially for associating its symptoms with lesions in various thalamic nuclei. A necessary requirement for the advancement of studies on KS is the broad acceptance of a comprehensive definition and definite diagnostic criteria. Therefore, in this review, we propose such a definition of KS and draft outlines for prospective diagnostic criteria.
Keywords: Korsakoff’s syndrome, alcohol amnestic disorder, Wernicke encephalopathy, thiamine deficiency, ethanol neurotoxicity, thalamus, memory, executive function, history
Introduction
Korsakoff’s syndrome (KS) is a residual syndrome in patients who suffered from a Wernicke encephalopathy (WE), but did not receive immediate and adequate treatment with thiamine replacement therapy. The most conspicuous symptom of KS is global amnesia, which can be very profound. In combination with other cognitive and behavioral deficits, usually present in more severe forms of KS, this may have far-reaching effects on daily life.1–3 Studies that have examined the extent, pattern, and nature of the anterograde episodic memory deficits in KS have contributed greatly to the delineation of concepts regarding human memory formation and to the realization that memory is not a unitary function. Moreover, the study of KS has brought to the fore that diencephalic structures play a critical role in memory function, thus stimulating the search for separate and distinctive brain structures and neural circuits underlying the component mnemonic processes.4,5
The neuropsychological sequelae of KS have been studied since the nineteenth century, leading to an increasing refinement of our comprehension of both amnesia and executive dysfunction.1 However, affective and volitional disorders, although reported in KS from the onset,6–9 have rarely been studied. Moreover, papers on the histopathological lesions underlying KS have not been published since 2000, notwithstanding the fact that several of the most fundamental and intriguing problems involved have not been resolved adequately.10–12
There is no generally accepted definition of KS, and there are no generally accepted criteria for the diagnosis of KS.13 DSM-5 classifies KS as “alcohol-induced major neurocognitive disorder, amnestic confabulatory type.”14 This classification is misleading, as KS in itself is not alcohol-induced. Moreover, this classification does not allow for a diagnosis or classification of KS in non-alcoholic patients and obscures the close relationship of KS with WE. In ICD-10, both alcoholic and non-alcoholic KS are classified, but in different groups.15 Non-alcoholic KS is classified as an organic mental disorder (F04) and alcoholic KS as a mental disorder due to substance abuse (F10.26), complicating the use of these ICD-10 criteria. One of the aims of this paper, therefore, was to propose a comprehensive definition of KS and draft outlines for prospective diagnostic criteria.
History
KS is named after the Russian neuropsychiatrist Sergei Korsakoff, although the first detailed description of the syndrome was probably in a paper by Robert Lawson in 1878.16 However, Korsakoff was the first to provide a comprehensive account of the syndrome in a series of papers published between 1887 and 1891.6–8,17 He observed that the syndrome was usually associated with peripheral nerve inflammation (alcoholic polyneuritis; first described by Moeli in 188418) and presumed that it resulted from a toxin. Therefore, he called the syndrome “polyneuritic psychosis or cerebropathia psychica toxaemica.” In 1897, the German psychiatrist Friedrich Jolly proposed that the syndrome was better named “Korsakoff’s symptom complex” or “Korsakoff’s syndrome.”19
A few years before Korsakoff published his first papers on the syndrome, the German psychiatrist Carl Wernicke described “polioencephalitis superior haemorrhagica” in the second volume of his Lehrbuch der Gehirnkrankheiten.9 In this classical description of what was later named “Wernicke encephalopathy,” he characterized the disease by the fairly distinctive triad of eye movement disturbances, ataxia, and mental confusion. We have known since the 1960s that in most cases of histopathologically confirmed WE, only an uncharacteristic mental confusion occurs and that the clinical diagnosis of WE can be challenging and may be missed – in some studies even in the majority of cases.20–22 Although the new operational criteria for the clinical diagnosis of WE, established by Caine et al23 (henceforth referred to as the “Caine criteria”; see Table 1), may lead to a remarkable improvement of the clinical diagnosis,23–25 the diagnosis of WE is in essence a histopathological one.20,21,26
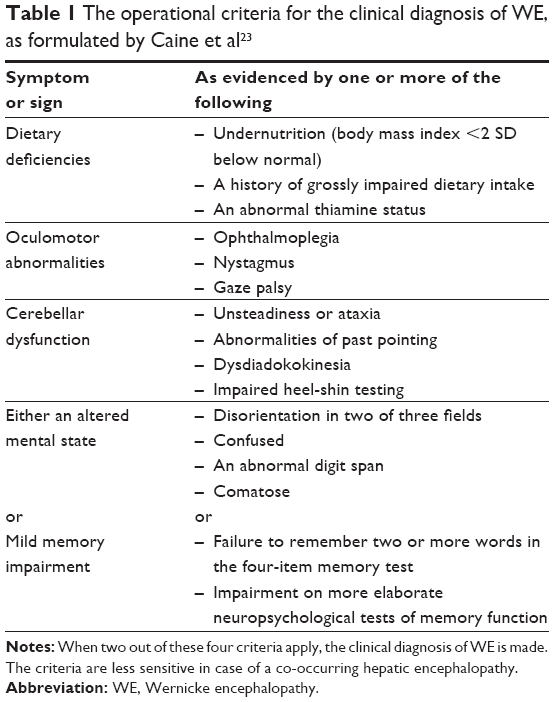 | Table 1 The operational criteria for the clinical diagnosis of WE, as formulated by Caine et al23 |
The German neuropsychiatrist Karl Bonhoeffer was probably the first to realize that WE and KS belong together. He observed that all cases who survived the WE phase developed KS as a residual syndrome.27,28 Jolly observed that a similar disorder of memory could also be observed in patients with “senile dementia” and other diseases.19,29 Consistent with these findings, Bonhoeffer proposed to distinguish henceforth between “Korsakoff’s psychosis” (after WE; what is now called KS) and “Korsakoff’s syndrome” (in dementia and other diseases). Soon both labels were used interchangeably. In the modern literature, they are used as synonyms, although the use of “Korsakoff’s psychosis” as a label is fading.
Korsakoff, Wernicke, and Bonhoeffer knew nothing about the real cause of WE and KS. Only after the concept of “vital amines” or “vitamins” had evolved (in 1912), and thiamine was isolated (in 1926) and synthesized (in 1936), the etiology of WE and KS could be fully clarified (in 1952).29,30 This knowledge eventually led to a highly effective therapy of WE: thiamine replacement. Although synthetic thiamine had been available for medical use in the 1940s,29 and proof for the therapeutic effectiveness of thiamine replacement therapy was delivered in 1952, it was not until 1966 that a clear recommendation for thiamine replacement therapy in WE was made.31 This therapy increased the survival of WE patients immensely and created a much larger population of chronic KS patients than was known previously.
Definition, diagnosis, and epidemiology
There is no generally accepted definition of KS in the literature, and the implicitly accepted conceptions have changed over time. This has caused many problems that have never been debated or properly investigated. In the older literature, all definitions of the syndrome are purely cross-sectional in nature, without any requirement regarding its duration. The definition of Victor et al is a clear example: KS is
[…] an abnormal mental state in which memory and learning are affected out of all proportion to other cognitive functions in an otherwise alert and responsive patient.16,32
It is still cited in modern reviews,2,12,33 probably because it captures the distinguishing characteristics of KS very well. Recently, Kopelman et al slightly but importantly amended this definition by adding “resulting from nutritional depletion, ie, thiamine deficiency.”2 However, the absence of any criterion with regard to duration or reversibility in this definition had important consequences, not often recognized today. Victor et al diagnosed KS in any patient with memory problems, after the acute WE phase with confusion had subsided, regardless whether these were irreversible or transient. Patients would even receive this diagnosis when the memory disorder recovered completely within a few weeks (in one case even within 9 days).16,32 Nowadays, these quickly resolving memory disorders would probably be interpreted as part of the recovery phase of WE or as a comorbid and quickly remitting instance of alcoholic encephalopathy (AE). In order to receive the diagnosis of KS today, the memory disorder should be irreversible;34 in typical cases, only minor improvement may occur.35 Although the definition of Victor et al has survived, the concept of KS has changed, which should lead to a precautious use of the results of older studies. Another shortcoming of the older definitions is that the focus lies entirely on the severe memory disorder, while KS is a syndrome with many cognitive and behavioral symptoms.
The current inability to separate the consequences of thiamine deficiency (TD) from those of ethanol neurotoxicity (EN) is not due to some insurmountable obstacle. Nonetheless, it has led to hybrid diagnoses and umbrella terms like “alcohol-related dementia”36–38 or “alcohol-related brain injury.”39 These current “alcohol-related” concepts and diagnoses hamper the clinical diagnosis of KS and complicate its scientific study. We would like to argue that there are no valid reasons why a reliable definition and reliable diagnostic criteria for KS would be harder to devise than definitions or criteria for, say, Alzheimer’s disease (AD). Clinically, the diagnosis of AD is not easier to make than a KS diagnosis. In both, memory disorders are variously accompanied by other cognitive and behavioral problems. Neuropathologically though, WE is easier to diagnose than AD. Intricate problems, like differentiating AD from normal aging and discerning AD among other comorbidities contributing to dementia, such as dementia with Lewy bodies or vascular dementia, have never stopped both researchers and clinicians in the field from studying Alzheimer-specific biomarkers and defining disease-specific clinical criteria.40
A comprehensive definition of KS and reliable and valid diagnostic criteria for KS can be formulated based on the evidence collected in the past few decades. A useful definition of KS might have the following phrasing: “KS is a largely irreversible residual syndrome, caused by severe thiamine deficiency and occurring after incomplete recovery from a Wernicke encephalopathy, predominantly in the context of alcohol abuse and malnutrition, characterized by an abnormal mental state in which episodic memory is affected out of all proportion to other cognitive functions in an otherwise alert and responsive patient, whose psychological make-up may be further distinguished by executive dysfunction, flattened affect, apathy, lack of illness insight, and possibly by fantastic confabulations in the early stage.” In milder forms of KS, especially in non-alcoholic KS, executive dysfunction and other cognitive or behavioral disorders (apart from amnesia) may be absent.41,42 Therefore, it is premature to require the presence of these symptoms in all instances of KS as long as there is no consensus on the severity and duration of the memory disorder required for the diagnosis of KS.
Diagnostic criteria for KS should be modeled in analogy with the other research criteria (eg, the research criteria43 or the IWG-2 criteria44 for the diagnosis of AD). Therefore, future KS criteria should include 1) a description of the specific clinical phenotype of KS (for instance, a modified version of the here-proposed definition of KS), ideally including the requirement for a minimal severity of memory dysfunction, expressed in evidence-based cutoff scores for memory tests (such as the California Verbal Learning Test or the Rivermead Behavioral Memory Test); 2) in vivo evidence for WE pathology, either clinical (eg, the operational criteria by Caine et al;23 Table 1), neuroradiological (ie, using MRI; see also “Neuroimaging” section), or in lab reports (very low serum thiamine); and 3) a set of exclusion criteria. These diagnostic criteria would mean an immense improvement in the diagnosis of KS and a huge stimulus for scientific research.
Little is known about the current incidence and prevalence of KS. In 1987, Blansjaar conducted a survey on the prevalence of KS in The Hague, the Netherlands. He then found a prevalence of 4.8 per 10,000 inhabitants.45 Based on admission data in the Province of North-Holland in the Netherlands, Schnabel found a lower prevalence rate of about 3 patients per 10,000 inhabitants in 1992.45 Ramayya and Jauhar46 reported an annual incidence of KS of around 0.5 per 10,000 between 1990 and 1995 in Glasgow, Scotland. They cited six older studies, which found annual incidence rates between 0.01 and 0.65 per 10,000. Studies on the incidence of KS in other parts of the world are to our knowledge lacking altogether.
Etiology and pathogenesis
Though it has been doubted by some authors,16,47 KS is probably always preceded by WE.26 The uncertainty that has arisen over this issue can be explained by the fact that the clinical diagnosis of WE is very difficult and therefore often missed.20–22
Wernicke encephalopathy
In essence, the diagnosis of WE is a histopathological diagnosis, made after a postmortem examination of the brain.20–22 Although the incidence has always been highest in undernourished alcoholics, WE has also been described in patients with hyperemesis gravidarum, starvation, and all kinds of gastrointestinal diseases, more recently also in AIDS and after bariatric surgery.25,48–51
WE can be treated effectively by the intravenous or intramuscular infusion of large quantities of thiamine.52 When this is done promptly, that is, within hours after the development of the encephalopathy, a full recovery is likely to occur. Even after a few days have elapsed, a complete recovery is still possible after thiamine replacement. When treatment is further delayed, however, the patient may die or recover only partially, with KS and nutritional cerebellar degeneration (NCD) as the most conspicuous residual syndromes.33,53 Consistent with this incomplete recovery, KS patients show permanent damage to several structures around the third and fourth ventricles of the brain, especially in the diencephalon,51 while NCD patients show permanent damage in the cerebellum, especially in the vermis.53,54
KS and NCD are the most conspicuous residual syndromes following the acute WE phase, but there are almost certainly more residual conditions or syndromes. In the most recent series of histopathological studies on WE and KS, all arising from the New South Wales Tissue Resource Centre (NSWTRC; Table 2), all authors describe a subgroup of patients with a residual syndrome that is characterized by irreversible histopathological damage to several structures (the mammillary bodies, the thalamus, and other diencephalic structures) and with residual cognitive dysfunctions (especially executive and visuoconstructive dysfunction), but without damage to the anterior thalamic nuclei and without severe memory dysfunction, and thus without KS.26,54–61 Keeping in mind that an irreversible, severe amnesia is required for the diagnosis of KS,34 this subgroup with less severe histopathological damage and less severe cognitive dysfunctions deserves in-depth scientific study. Patients with these hitherto neglected residual syndromes following WE may have contaminated control groups of “uncomplicated” alcoholics in studies on cognitive dysfunction in KS and created the illusion of irreversible residual disturbances of cognition due to EN in long detoxified alcoholics.24 This misconception may also have led to the continuity hypothesis62–64 (see “Dismissing the continuity hypothesis” section). Pitel et al assessed whether long-abstinent alcoholics with neuropsychological deficits, but without KS, fulfilled one or more of the Caine criteria, retrospectively applied to the patient’s history after having reviewed the medical records.24 They found that patients meeting zero criteria did not differ from healthy controls, those meeting one criterion presented mild-to-moderate deficits on some of the functional domains, and those meeting two or more criteria had the most severe deficits on each of the domains examined. They concluded that the presence of signs of WE explains, at least partially, the heterogeneity of alcoholism-related cognitive and motor deficits. In other words, the presence of neuropsychological deficits in long abstinent alcoholics without KS is strongly associated with signs of WE.
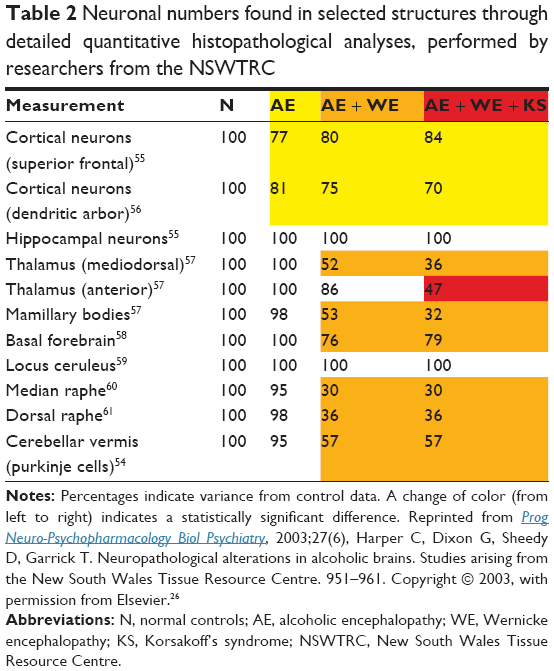 | Table 2 Neuronal numbers found in selected structures through detailed quantitative histopathological analyses, performed by researchers from the NSWTRC |
Neuropathology
Between 1892 and 2000, around 45 papers have been published on the neuropathology of KS, resulting in a succession of discoveries, in which each further addition did not challenge the involvement of the structures described before. The increasing sophistication of histopathological research and the dramatically increased survival of WE and KS patients since the general availability of thiamine replacement therapy in the 1960s are probably the most important factors leading to the addition of new structures to the list of lesions involved in the occurrence of KS. The last series of papers on clinic-anatomical correlation based on histopathology, by researchers of the NSWTRC26 (see also the “Wernicke encephalopathy” section and Table 2) are by far the most sophisticated histopathological studies on KS to date.51
Still, the current state of knowledge of KS is portrayed as controversial or conflicting,12,65 especially with regard to the question whether the critical lesion for the severe memory disorder is to be found in the dorsomedial or in the anterior thalamic nuclei. However, Harding et al, in one of the NSWTRC studies,57 presented clear evidence for the critical role of the anterior thalamic nuclei. They convincingly argued that earlier studies16,32 failed to detect the histopathological damage in the anterior thalamic nuclei, because these studies did not utilize unbiased stereological techniques. In their study, patients with severe damage to the mediodorsal thalamic nuclei but without damage to the anterior thalamic nuclei after WE did not suffer from severe memory impairment during life. That the anterior nuclei are the critical lesion site for the memory disorder in KS is widely supported by converging evidence from anatomical studies (where these nuclei are seen as part of the limbic-diencephalic memory circuit, like the hippocampus), animal studies, and studies on vascular lesions in the thalamus.66–71 In contrast, the mediodorsal nuclei are densely and reciprocally connected to the prefrontal cortex and the amygdala, and probably belong to the extended-amygdala system.67
Neuroimaging
In the acute phase of WE, characteristic alterations (enhanced signal intensity in one or more of the structures depicted in Figures 1 and 2 and enumerated in Table 2; see also Figure 3) appear in approximately half of the cases.25,72,73 Therefore, finding these alterations on MRI strongly supports a clinical diagnosis of WE,25 and when KS is suspected, their presence supports the clinical diagnosis (see also “Definition, diagnosis, and epidemiology” section).
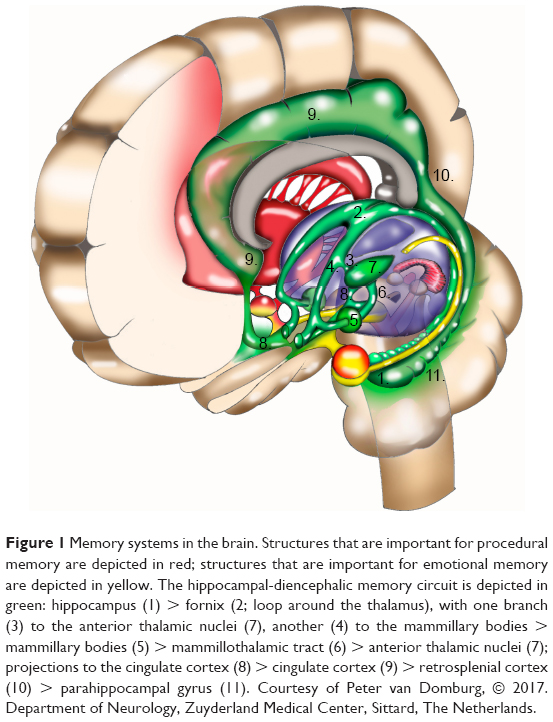 | Figure 1 Memory systems in the brain. Structures that are important for procedural memory are depicted in red; structures that are important for emotional memory are depicted in yellow. The hippocampal-diencephalic memory circuit is depicted in green: hippocampus (1) > fornix (2; loop around the thalamus), with one branch (3) to the anterior thalamic nuclei (7), another (4) to the mammillary bodies > mammillary bodies (5) > mammillothalamic tract (6) > anterior thalamic nuclei (7); projections to the cingulate cortex (8) > cingulate cortex (9) > retrosplenial cortex (10) > parahippocampal gyrus (11). Courtesy of Peter van Domburg, © 2017. Department of Neurology, Zuyderland Medical Center, Sittard, The Netherlands. |
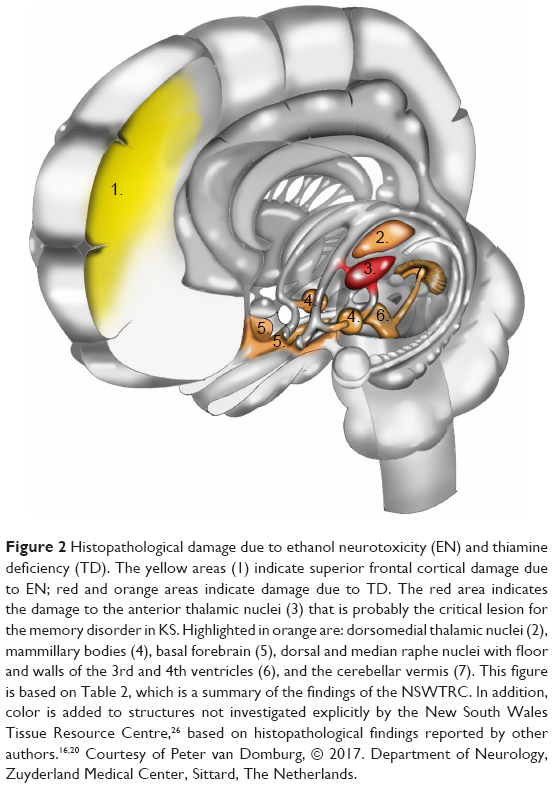 | Figure 2 Histopathological damage due to ethanol neurotoxicity (EN) and thiamine deficiency (TD). The yellow areas (1) indicate superior frontal cortical damage due to EN; red and orange areas indicate damage due to TD. The red area indicates the damage to the anterior thalamic nuclei (3) that is probably the critical lesion for the memory disorder in KS. Highlighted in orange are: dorsomedial thalamic nuclei (2), mammillary bodies (4), basal forebrain (5), dorsal and median raphe nuclei with floor and walls of the 3rd and 4th ventricles (6), and the cerebellar vermis (7). This figure is based on Table 2, which is a summary of the findings of the NSWTRC. In addition, color is added to structures not investigated explicitly by the New South Wales Tissue Resource Centre,26 based on histopathological findings reported by other authors.16,20 Courtesy of Peter van Domburg, © 2017. Department of Neurology, Zuyderland Medical Center, Sittard, The Netherlands. |
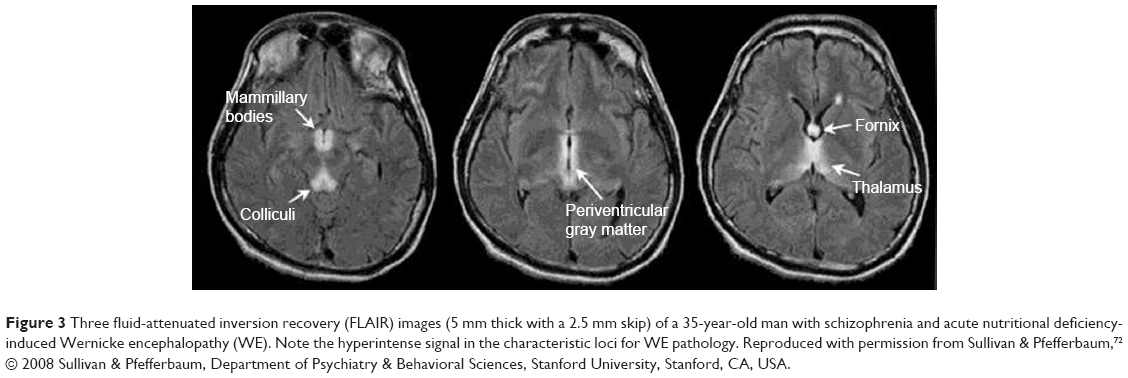 | Figure 3 Three fluid-attenuated inversion recovery (FLAIR) images (5 mm thick with a 2.5 mm skip) of a 35-year-old man with schizophrenia and acute nutritional deficiency-induced Wernicke encephalopathy (WE). Note the hyperintense signal in the characteristic loci for WE pathology. Reproduced with permission from Sullivan & Pfefferbaum,72 © 2008 Sullivan & Pfefferbaum, Department of Psychiatry & Behavioral Sciences, Stanford University, Stanford, CA, USA. |
MRI studies of alcoholic KS patients reveal that many of them have gray and white matter volume deficits, but also that these do not differ from the volume deficits observed in alcoholic patients without KS.74,75 The medial thalami and the mammillary bodies are probably more severely damaged in KS patients, though a consistent correlation between mammillary body volume and memory impairment has not been found.76 A more pronounced atrophy of these structures in KS is not an unexpected finding, for these structures are notably damaged in WE and KS.26 More surprising was the finding that KS patients show more volume loss in the genu (anterior part) of the corpus callosum compared to alcoholic patients without KS.74 Possibly, these differences reflect a higher incidence of undiagnosed Marchiafava–Bignami syndrome in KS, a rare disorder of the corpus callosum, in which a major contribution of nutritional deficiencies is suspected.51,77
In non-alcoholic KS patients, MRI scans do not show consistent gray or white matter volume reductions. In some of these patients, atrophy of the mammillary bodies, the thalamus, or the cerebellar vermis has been found. However, in the eleven best documented cases of non-alcoholic KS to date, in whom both MRI and extensive neuropsychological assessment were performed, no marked atrophy in any region or structure was found.41,42 Therefore, we argue that in individual cases, MRI does not enable us to reliably diagnose KS, although an MRI with characteristic lesions in the acute phase of WE (Figure 3) obviously supports a subsequent diagnosis of KS.
The contribution of alcohol
There is little doubt that chronic alcohol abuse has always been the most important factor in creating the context in which WE and KS could occur. Two of the three patients originally described by Wernicke9 and two thirds (30/46) of the patients described by Korsakoff6–8 were alcoholics. In modern cohorts, the proportion of alcoholics among the patients developing WE or KS has increased to at least 90%.78,79 However, it is still debated whether EN is in any sense essential for the occurrence of WE or essential for the development of KS following WE. Severe alcohol abuse certainly contributes to the development of WE: the high calorie content of alcohol suppresses the feeling of hunger and favors malnutrition, the combustion of alcohol requires extra thiamine pyrophosphate (a co-enzyme in energy-bound processes), alcoholic gastroenteritis impairs the absorption of thiamine, alcoholic liver diseases reduces thiamine storage in this organ, and alcohol may impair the utilization of thiamine.48,52,80
Still, none of these factors are essential for the development of WE or KS. WE develops when thiamine is below a critical level in gastrointestinal conditions, in cancer, in hyperemesis gravidarum, after chemotherapeutic treatments and in systemic diseases such as AIDS, renal diseases, thyrotoxicosis, and magnesium depletion – regardless of alcohol intake. The incidence of WE and KS in these conditions may be as high as the incidence in alcohol use disorders.50
Furthermore, the evidence that EN is essential for the progression of WE to KS is contestable. The progression rate of WE to KS in alcoholic WE patients is often considered to be around 84%, a figure that is mentioned in most of the review papers.2,67,81–83 However, this figure was derived from the monograph of Victor et al16,32 and was not intended as an estimation of the general progression rate of WE to KS in alcoholics. It was simply a description of their cohort, which was probably not very representative, chiefly because they used clinical definitions that have become obsolete now. For the diagnosis of WE, they required the classical triad, resulting in a selection bias favoring severe cases. The diagnosis of KS, on the other hand, was made when there was a memory disorder in the (sub)acute phase, even when it disappeared quickly (in one case it lasted only 9 days). In a subsequent – though rarely cited – study, Wood et al found a progression rate of alcoholic WE to KS of 56%.84 Moreover, in large studies based on autopsy, the prevalence of WE brain pathology is much higher than the prevalence or incidence of KS.21,22,48 Therefore, it is likely that the proposed progression rate of 85% in alcoholics is an unreliably high estimation. Conversely, although the progression rate of WE to KS in non-alcoholic WE patients has been suggested to be low (≤29%),85,86 later reviews have reported that in up to 56% of these patients, WE is followed by “clinically obvious memory impairment” (Scalzo et al,50 p 1365). This progression rate is identical to the one found by Wood et al in alcoholic WE patients.84
These conclusions find support from animal studies, where the interval between the onset of WE and start of thiamine replacement is a crucial factor in determining the severity of the ensuing residual damage and the remaining disturbances.67 Moreover, animal studies and well-designed clinical studies in humans found that the effects of EN on the brain are largely or completely reversible, unlike the effects of TD. A recent review on TD in laboratory animals concludes that
[…] the interaction between ethanol and thiamine deficiency does not produce more behavioral or neural pathology, with the exception of reduction of white matter, than long-term thiamine deficiency alone.67
Reduction in white matter is also the main effect of EN in humans. This atrophy of white matter in alcoholics is probably the result of demyelination, a largely or completely reversible process.87 On CT and MRI, white matter atrophy gradually diminishes in alcoholics who stopped drinking and remain abstinent, probably as a result of remyelination.77,88 Moreover, well-nourished and otherwise healthy alcoholics usually show minimal cognitive or behavioral effects of their chronic heavy alcohol use,89 often improve quickly after abstinence,90,91 and typically recover completely after an abstinence period of 1 or 2 years.92
There is some evidence, mostly from animal studies, that EN may also result in irreversible gray matter (neuronal) damage, but “the degree of neuronal loss in alcoholics without additional nutritional or metabolic pathologies is surprisingly subtle” (Sutherland et al,93 p 606). Moreover, the (favorable) outcomes of clinical studies suggest that neuronal damage is not severe in humans, or can be adequately compensated for.92
Dismissing the continuity hypothesis
The evidence discussed in the previous two sections amount to a dismissal of the continuity hypothesis, both in its original62,63,94 and in its revised64 forms. Basically, the continuity hypothesis states that cognitive disturbances in non-KS alcoholics and alcoholic KS patients lie along a continuum of mild to moderate deficits; due to EN, there is a correlation between cognitive performance and drinking history. According to the revised continuity theory by Pitel et al,64
[…] there is an “episodic memory continuity” between AL and KS and a “working memory continuity” among AL, KS, and alcohol dementia patients (“AL” refers to uncomplicated alcoholics).
However, this hypothesis holds only in specific situations. Uncomplicated alcoholics may suffer from cognitive disturbances, but these will diminish and even disappear after prolonged abstinence. Recently detoxified alcoholics may still suffer from cognitive disturbances, but probably only as long as the temporary effects of demyelination last and as long as re-myelination is not completed. All evidence indicates that memory deficits in alcoholics who did not suffer from TD and did not develop WE disappear within a few weeks. The recovery of executive and other cognitive disorders may require a few to several months. Moreover, there is no convincing evidence that EN alone may lead to “alcohol dementia.” So far, all patients who received this diagnosis eventually turned out to suffer from residual syndromes after WE, from AD, from other forms of dementia, or from other forms of alcohol-related co-occurring brain damage (pellagra, traumatic brain injury, pontine or extrapontine myelinolysis, Marchiafava–Bignami disease, and hepatic encephalopathy).16,51
It is true that a minority of purportedly “uncomplicated” alcoholics does not recover completely after 1 or 2 years of abstinence. However, the best explanation to account for irreversible cognitive disorders in a minority of “uncomplicated” alcoholics is TD. In other words, these “uncomplicated” alcoholics may not be uncomplicated after all.24 WE usually (in 50%–70%) has an unobtrusive symptomatology and course. Therefore, the diagnosis is missed more often than not.21,22 Moreover, while there is histopathological evidence for a continuum of irreversible TD effects in the brain, of which KS is only the most conspicuous and most severe variant,24,26,57 there is no convincing evidence for a continuum of irreversible EN effects.92,95 In all probability, AE is a temporary and (almost) completely reversible disorder.92,95 In the minority of chronic alcoholics with residual cognitive disturbances after 1 or 2 years of abstinence, it is important to exclude mild forms of WE (up to 19%, mostly undiagnosed79), Marchiafava–Bignami disease, pontine or extrapontine myelinolysis, pellagra, traumatic brain damage, and hepatic encephalopathy, before these cognitive effects can be ascribed to EN. Exclusion of these comorbid pathologies is often very difficult to achieve without histopathological postmortem examinations, which cannot be accomplished in clinical studies. Therefore, to date, there is little direct evidence for irreversible cognitive effects caused by EN. However, we fully acknowledge the reality of AE, that is, of the temporary, reversible (after prolonged abstinence) cognitive effects of EN.
Therefore, the continuity hypothesis amounts to little more than the redundant statement that the temporary effects of EN and the largely irreversible effects of TD often co-occur and may produce different combinations of cognitive disturbances. These disturbances usually diminish after thiamine replacement and abstinence from alcohol, due to a small improvement of TD effects and a (nearly) complete recovery from EN effects, typically in the course of a few months. In short, the continuity hypothesis has become obsolete now.
Cognitive and behavioral symptoms
KS is generally known as a disorder of memory, but from the very first reports, it was obvious that it is a syndrome with many cognitive and behavioral symptoms. In addition to amnesia, Korsakoff,7,8 Gudden,96 and Bonhoeffer27,28 already listed apathy, flattened effect, and confabulations as characteristic symptoms. The concept of “executive functions” has fully crystallized only recently,97 but Grünthal98 and Van der Horst99 already isolated, described, and characterized the symptoms in patients with KS that we would now characterize as disorders of executive function. In this section, we present an overview of the main symptoms of KS, based on the most recent research.
Episodic memory dysfunction
Memory dysfunction is the only symptom of KS that has been studied comprehensively in the past 125 years. In the earlier studies, investigators were puzzled by apparent contradictions in the memory performance of KS patients,3,100,101 but these contradictions could be resolved in the 1970s and 1980s, after the discovery of multiple memory systems that can be disrupted independently by pathological processes.102–104
The severe memory impairment as a core characteristic for KS primarily relates to declarative memory. Within this declarative memory domain, both episodic memory, related to explicitly remembered personally experienced events specific to time and place, and semantic memory, related to facts, are affected. In each of these subdomains, the anterograde memory processes are typically more severely affected than retrograde memory processes.1,5,105–107
A more recent development in KS studies is the theory that anterograde episodic memory impairment in KS is due primarily to a deficit in contextual memory.108–112 KS amnesics often encode temporal1,12,113 and spatial113–115 contextual information less efficiently than normal controls and consequently have great difficulties in source memory.109,116,117 Source memory is conceptualized as a core component of episodic recall. It allows for the reconstruction of contextual details characterizing the acquisition of episodic events.
In KS, anterograde amnesia is surely one of the most prominent symptoms, but remote memory is affected as well. The resulting retrograde amnesia comprises both non-personal information – like news facts – and events from autobiographical memory.110,113,118,119 Remote memory loss in KS patients can extend back many years, even several decades,110,120 but early (eg, childhood) memories are relatively spared. This disproportionate loss of more recent memories in KS and other pathologies is called a “temporal gradient.” This phenomenon was first described by Théodule Ribot (in 1881) and is since then known as Ribot’s law. In KS, the majority of studies have found evidence for a steep temporal gradient, but some have observed more uniform remote memory impairments across all past time periods.121 Methodological difficulties may be a partial explanation for this inconsistency, but, more importantly, there were also differences in the kind of memory studied: non-personal versus autobiographical memory. Because autobiographic memories are difficult to assess, they are studied less often than non-personal memories.122 Autobiographical episodic memory refers to personal events that a person is able to re-experience in a detailed spatial and temporal context, for example, remembering how you met your first love in the bar of the swimming pool on a warm Friday evening. In a recent study, Rensen et al122 showed that the temporal gradient extended to episodic autobiographical memory, replicating previous findings.2,110,119,123
Semantic memory dysfunction
Semantic memory refers to the memory of language, concepts, and well-rehearsed facts (eg, “Paris is the capital of France”), independent of the recall of any particular episode or incident specific in time and place.102 Semantic memory is relatively preserved in KS. Unlike dementia patients, KS patients show near-normal performances on semantic memory tests.124,125 The ability to retrieve information from preexisting semantic memory seems to be preserved.1,3,126–128 However, Pitel et al129 demonstrated that, compared to uncomplicated alcoholics, KS patients are impaired in new category and feature learning, which is independent of episodic memory. This specific impairment may therefore reveal a genuine deficit of semantic memory in KS. Autobiographical semantic memory has also been studied in KS. The semantic aspect refers to knowledge that is not linked to single events, for example, knowing that you used to swim on Fridays or what your home address was during childhood. A temporal gradient has been repeatedly demonstrated in semantic autobiographical memory in KS.2,110,119,122
Procedural memory
Procedural memory is the nondeclarative memory system underlying verbal, motor, and other cognitive skills,130 resulting in learning without awareness (ie, implicit learning). Several studies of procedural learning in KS patients showed preserved learning,131–136 while others reported impaired learning.137–141 These differences may be largely explained by the type of material to be acquired, complicated by the fact that many procedural tasks also rely on explicit memory and executive functions that are impaired in KS. In recent reviews, it is therefore concluded that patients with KS show maximum procedural learning potential when the task is minimally dependent on other cognitive domains than procedural learning, when feedback is given during the task, and when the task itself is restricted in response options.142,143
Executive dysfunction
The concept of executive dysfunction did not exist before the 1970s.97 “Executive function” is an umbrella term for planning, response inhibition, response generation, concept shifting, and working memory updating.144,145 Grünthal correctly observed (in 1924) that KS patients have great difficulty in recalling the context of memories.98 He also realized that confabulations could not always be explained as automatic or unconscious attempts to fill memory gaps. They are often better understood as failures in context reconstruction. He therefore characterized this problem as an “alignment disorder” (Einstellungsstörung). Van der Horst was the first (in 1932) to observe that KS patients have great difficulty in organizing their memories in the appropriate temporal sequence.99 They lose track of the order of events, a symptom that is known since 1929 as “achronogenesis.”146 Still, it took considerable time to realize that KS patients might also have severe problems in planning and task organization, possibly because these problems are difficult to comprehend without some apprehension of executive functions.147
The executive dysfunction in KS is unremitting and affects all executive processes, including response inhibition, response generation, working memory updating, planning, and concept shifting.147,148 Lack of illness insight is related to executive dysfunction. Alcoholic KS patients have been found to report no cognitive complaints on self-report measures of subjective cognitive problems, despite their poor performance on several cognitive tests.149 There is evidence that in non-alcoholic KS, awareness of cognitive dysfunction may be preserved.41,42
Confabulations
Confabulations can be defined as “false or erroneous memories arising involuntarily (ie, not deliberately) in the context of a neurological amnesia”.12 The involuntary and unconscious nature of confabulations is well captured in the colloquial description “honest lying.”150 Korsakoff devoted a whole paper to this symptom of KS.17 Bonhoeffer was the first to use the word “confabulation” [Konfabulation] in this context.27
Bonhoeffer already distinguished between momentary or embarrassment confabulation [Augenblickskonfabulation, Verlegenheitskonfabulation] and fantastic or productive confabulation [phantastische Konfabulation, produktive Konfabulation].27,28 Following Bonhoeffer, a distinction is made between provoked (or reactive, or momentary) and spontaneous (or fantastic) confabulations.151–153 In provoked confabulations, the patient confabulates in an inconsistent fashion. Usually these confabulations are triggered and the erroneous memory communicated is generally plausible. In the spontaneous or fantastic type, the confabulations are presented and elaborated without provocation. They are less variable than the provoked confabulations and generally have a grandiose content.152,154
Kopelman,153 after reviewing many opinions on the phenomenology, mechanisms, and neuropathology of confabulation, concluded that provoked confabulations are probably fleeting disorders or intrusion errors, due to failing memory, also seen in healthy subjects when their memory is weak.153,155,156 Therefore, they do not necessarily imply an underlying pathology or a specific anatomical basis.157
The underlying mechanisms of confabulations are under debate, but have been linked to intrusions on memory tests,158 temporal context confusion,159,160 both temporal context confusion and executive dysfunction,161,162 and poor strategic and memory-monitoring capacities.163,164 The spontaneous confabulations in KS usually disappear within weeks after the development of KS, but provoked confabulations can often be found in its chronic stage.41,158
Apathy
Apathy can be defined as “an absence of responsiveness to stimuli as demonstrated by a lack of self-initiated action.”165 This is just one of the many possible definitions. Modern definitions are broader than those of a century ago – when apathy was exclusively seen as a disorder of volition – and comprise cognitive and affective symptoms.166,167 This probably led to an unwarranted overlap with executive dysfunction and depression.168
That apathy is a characteristic and fundamental symptom of alcoholic KS has been established from the earliest descriptions of this syndrome.7,8,18,27,96,169 However, in modern studies on KS, this symptom has rarely attracted any attention so far. The only paper we were able to find on apathy in KS is that written by Talland in 1960.169 Apathy might be less conspicuous or even absent in non-alcoholic KS.41,42
Although modern studies on the neuropathology underlying apathy in KS are lacking, all recent studies on the neuropathological basis of apathy, abulia, avolition, athymhormia, or akinetic mutism point to the prefrontal anterior cingulate cortico-subortical circuit,170–173 which connects the anterior cingulate with the basal ganglia and the mediodorsal thalamic nuclei. Lesions in other cortico-subcortical circuits rarely – if ever – cause apathy or any other diminishment of volition.
Disorders of affect, emotion perception, and social cognition
From the earliest reports, alcoholic KS patients have been described as emotionally flat and affectively detached, but that is not the whole picture. In the early stages, KS patients are frequently irritable or euphoric, and they usually remain affectively unstable throughout the rest of their lives; suspicion and intense emotions are easily provoked.3,7,8,27 So far, affective impairments have rarely been studied in KS.174–176 Elaborating on the fairly recent recognition of executive dysfunction in KS patients,147 there is a tendency to explain affective impairments in KS as a consequence of a lack of reflective abilities rather than a primary lack of affective abilities.174,177,178 For instance, deficits in perspective taking, an important aspect of Theory of Mind, have been found to be related to executive dysfunction in KS, and not to any inability to infer the thoughts and feelings of others.179 Recently, this explanation has been challenged.176 Based on previous studies, these authors argue that a subtler exploration of affective abilities in KS suggests that they might be impaired. For instance, the evidence that KS patients have a serious impairment of affective prosody perception178 and facial expression recognition, with stronger difficulties for categories like fear, anger, and surprise,180 suggest that affective abilities are not unaffected in KS. These hypotheses are strengthened by the evidence that KS patients tend to overestimate the affective content of stimuli and show emotional overreaction or enhanced sensitivity in other situations.175,181
Pharmacological treatment and cognitive rehabilitation
As a residual syndrome after WE, KS can be seen as a form of acquired brain damage. After treating WE with thiamine replacement and after a convalescence phase, the effect of pharmacological interventions is necessarily confined to enhancing the skills that remain, or to suppress symptoms that interfere with normal functioning. In patients with traumatic brain injury, there is some evidence that these interventions may be effective.182 In KS, however, there is no evidence of any beneficial effect of pharmacological therapy. Several case studies and eight double-blind trials have been published, where KS patients were treated with clonidine, fluvoxamine, reboxetine, or rivastigmine. Together, these studies did not produce any consistent evidence for the efficacy of any of these interventions.183 Therefore, we may safely conclude that, until this date, no effective pharmacological treatment for KS is available.
The perspectives for rehabilitation programs are more promising than those of pharmacological interventions. The traditional view that KS is a static condition that does not permit further recovery is obsolete and therefore gradually abandoned, thanks to accumulating evidence that memory rehabilitation is possible in KS.143,184,185 Memory compensation techniques such as using agendas, memory cards, smartphones, and smartwatches are promising. Six studies on the use of traditional and digital assistive technologies (reviewed in Oudman et al143) produced evidence that these memory compensation techniques may be helpful in KS, provided that 1) the formulated goals are restricted, 2) much time is available to guide the patient, and 3) the use of these technologies is holistically embedded or combined with elaborated learning techniques.143
Interventions based on errorless learning are probably – at least in theory – best suited to the cognitive abilities and disabilities of KS. Its most important element is that the patient is not allowed to make errors during learning. All guessing is eliminated, in order that the patients’ procedural memory is not allowed to become familiar with an incorrect or unproductive strategy, which the defective episodic memory cannot correct or compensate for.186,187 So far, only a few studies have been published in which errorless learning has been compared to trial-and-error learning.134,188–190 The results of these studies are somewhat mixed, but errorless learning might be beneficial compared to trial-and-error learning in some learning situations. Moreover, the beneficial effects of errorless learning are not restricted to procedural learning but may also support semantic learning.143
Conclusion and further perspectives
In this narrative review, we summarized the current knowledge on KS. It differs in important respects from other modern reviews on KS and WE.2,48,81–83 That is, we argue that the contribution of alcohol abuse to the development of WE and KS is limited to creating the context in which TD is most likely to develop and in which lengthy patients’ and doctors’ delay are prone to occur. Alcohol abuse is the most important cause of malnutrition and TD, and the living conditions and socioeconomic status of alcoholics are responsible for extended treatment delays, thus favoring the progression of WE to KS. However, we have not been able to uncover convincing evidence for an essential contribution of EN to the development of WE or to the progression of WE to KS. The contribution of alcohol abuse to the development of WE and KS is massive, but seems to be circumstantial. Animal experiments and recent studies on the development of WE and KS in non-alcoholic patients have demonstrated that the occurrence of WE and KS can be fully explained by TD. Moreover, the best explanation for the progression of WE to KS is treatment delay. Furthermore, we concluded that there is little evidence to support the continuity hypothesis, neither in its original62,63,94 nor in its revised91 version.
The study of the complete spectrum of residual syndromes after WE is only possible when KS is adequately and comprehensively defined and when it can be diagnosed reliably. Therefore, there is a clear need for a generally accepted definition (for which we formulated a proposal in the “Definition, diagnosis, and epidemiology” section) and reliable diagnostic criteria. Moreover, the scattered information about the etiology, symptoms, treatment, and course of the many forms of collateral brain damage in alcoholics should be integrated in a comprehensive “taxonomy of alcohol-related brain damage.” A renewed interest in postmortem histopathological analysis of brain tissue of KS patients is an important prerequisite for the further development of our knowledge of KS, especially for the functional localization or association of its symptoms with damage in various thalamic nuclei. With neuroimaging techniques, the underlying neuropathology of KS cannot be rendered visible in a reliable way. This is a major drawback, not just for our knowledge of KS. KS is the most constant and consistent form of thalamic pathology. Improved knowledge of KS may help to answer vexing questions with regard to thalamic function: 1) What are the precise functions of the structures that are joined in the hippocampal-diencephalic memory circuit, such as the thalamus, mammillary bodies, fornix, retrosplenial cortex, hippocampus proper, and parahippocampal structures?191–193 2) What is the exact functional specialization of the separate thalamic nuclei?194–197 3) In which ways do the anterior and mediodorsal thalamic nuclei contribute to episodic memory?12,195,198–200
Disclosure
The authors report no conflicts of interest in this work.
References
Kopelman MD. The Korsakoff syndrome. Br J Psychiatry. 1995;166(2):154–173. | ||
Kopelman MD, Thomson AD, Guerrini I, Marshall EJ. The korsakoff syndrome: clinical aspects, psychology and treatment. Alcohol Alcohol. 2009;44(2):148–154. | ||
Talland G. Deranged Memory: A Psychonomic Study of the Amnesic Syndrome. New York: Academic Press; 1965. | ||
Delay J, Brion S. Le Syndrome de Korsakoff. Paris: Masson; 1969. | ||
Fama R, Pitel AL, Sullivan EV. Anterograde episodic memory in Korsakoff syndrome. Neuropsychol Rev. 2012;22(2):93–104. | ||
Korsakoff SS. [Disturbance of psychic function in alcoholic paralysis and its relation to the disturbance of the psychic sphere in multiple neuritis of nonalcoholic origin] (Ob alkogol’nom paraliche). Vestn Psikhiatrii. 1887;4(2):1–102. | ||
Korsakoff SS. [Mental disturbances in combination with polyneuritis] (psychosis polyneuritica, s. cerebropathia psychica toxaemica). Medizinskoje Obozr. 1889;32(13):3–18. | ||
Korsakoff SS. [Some cases of individual cerebropathy in polyneuritis] (cerebropathia psychica toxaemica). Ezhenedelnaja Klin Gaz. 1889;9(5–7):85–92, 115–121, 136–143. | ||
Wernicke C. Lehrbuch Der Gehirnkrankheiten Für Aerzte Und Studirende, Band II. Kassel: Fischer; 1881. | ||
Harper C. The neuropathology of alcohol-related brain damage. Alcohol Alcohol. 2009;44(2):136–140. | ||
Kril JJ, Harper CG. Neuroanatomy and neuropathology associated with Korsakoff’s syndrome. Neuropsychol Rev. 2012;22(2):72–80. | ||
Kopelman MD. What does a comparison of the alcoholic Korsakoff syndrome and thalamic infarction tell us about thalamic amnesia? Neurosci Biobehav Rev. 2015;54:46–56. | ||
Gerridzen IJ, Moerman-van den Brink WG, Depla MF, et al. Prevalence and severity of behavioural symptoms in patients with Korsakoff syndrome and other alcohol-related cognitive disorders: a systematic review. Int J Geriatr Psychiatry. 2017;32(3):256–273. | ||
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-5. Washington, DC: American Psychiatric Association; 2013. | ||
WHO, editor. ICD-10. 4th ed. Geneva: WHO; 2010. | ||
Victor M, Adams RD, Collins G. The Wernicke-Korsakoff Syndrome and Related Neurologic Disorders Due to Alcoholism and Malnutrition. 2nd ed. Philadelphia: Davis; 1989. | ||
Korsakoff SS. Erinnerungstäuschungen (Pseudoreminiscenzen) bei polyneuritischer Psychose. Allgem Zeitschr Psychiat. 1891;47:390–410. | ||
Meggendorfer F. Intoxikationspsychosen. In: Bumke O, ed. Handbuch Der Geisteskrankheiten, Vol. 7. 1st ed. Berlin: Springer; 1928:151–400. | ||
Jolly F. Ueber die psychischen Störungen bei Polyneuritis. Charité-Annalen. 1897;22:579–612. | ||
Colmant H. Enzephalopathien Bei Chronischem Alkoholismus. Stuttgart: Enke; 1965. | ||
Harper C, Giles M, Finlay-Jones R. Clinical signs in the Wernicke-Korsakoff complex: a retrospective analysis of 131 cases diagnosed at necropsy. J Neurol Neurosurg Psychiatry. 1986;49(4):341–345. | ||
Torvik A. Wernicke’s encephalopathy – prevalence and clinical spectrum. Alcohol Alcohol Suppl. 1991;1:381–384. | ||
Caine D, Halliday GM, Kril JJ, Harper CG. Operational criteria for the classification of chronic alcoholics: identification of Wernicke’s encephalopathy. J Neurol Neurosurg Psychiatry. 1997;62(1):51–60. | ||
Pitel AL, Zahr NM, Jackson K, et al. Signs of preclinical Wernicke’s encephalopathy and thiamine levels as predictors of neuropsychological deficits in alcoholism without Korsakoff’s syndrome. Neuropsychopharmacology. 2011;36(3):580–588. | ||
Chamorro AJ, Rosón-Hernández B, Medina-García J-A, et al. Differences between alcoholic and nonalcoholic patients with Wernicke encephalopathy: a multicenter observational study. Mayo Clin Proc. 2017;92(6):899–907. | ||
Harper C, Dixon G, Sheedy D, Garrick T. Neuropathological alterations in alcoholic brains. Studies arising from the New South Wales Tissue Resource Centre. Prog Neuro-Psychopharmacology Biol Psychiatry. 2003;27(6):951–961. | ||
Bonhoeffer K. Die Akuten Geisteskrankheiten Der Gewohnheitstrinker. Jena: Fischer; 1901. | ||
Bonhoeffer K. Der Korsakowsche Symptomenkomplex in seinen Beziehungen zu den verschiedenen Krankheitsformen. Allg Z Psychiat. 1904;61:744–752. | ||
Wuest H. The history of thiamine. Ann N Y Acad Sci. 1962;98:385–400. | ||
Phillips BGB, Victor M, Adams RD, Davidson CS. A study of the nutritional defect in Wernicke’s syndrome; the effect of a purified diet, thiamine, and other vitamins on the clinical manifestations. J Clin Invest. 1952;31(10):859–871. | ||
Victor M. Treatment of the neurologic complications of alcoholism. Mod Treat. 1966;3(3):491–501. | ||
Victor M, Adams RD, Collins G. The Wernicke-Korsakoff Syndrome. A Clinical and Pathological Study of 245 Patients, 82 with Post-Mortem Examinations. Philadelphia: Davis; 1971. | ||
Thomson AD, Guerrini I, Marshall EJ. The evolution and treatment of Korsakoff’s syndrome out of sight, out of mind? Neuropsychol Rev. 2012;22(2):81–92. | ||
Kessels R. Korsakoff syndrome. In: Weiner I, Craighead W, editors. The Corsini Encyclopedia of Psychology. New York: Wiley; 2010:902–904. | ||
Fujiwara E, Brand M, Borsutzky S, Steingass H-P, Markowitsch HJ. Cognitive performance of detoxified alcoholic Korsakoff syndrome patients remains stable over two years. J Clin Exp Neuropsychol. 2008;30(5):576–587. | ||
Ridley NJ, Draper B, Withall A. Alcohol-related dementia: an update of the evidence. Alzheimers Res Ther. 2013;5(1):3. | ||
Sachdeva A, Chandra M, Choudhary M, Dayal P, Anand KS. Alcohol-related dementia and neurocognitive impairment: a review study. Int J High Risk Behav Addict. 2016;5(3):e27976. | ||
Cheng C, Huang C-L, Tsai C-J, Chou P-H, Lin C-C, Chang C-K. Alcohol-related dementia: a systemic review of epidemiological studies. Psychosomatics. 2017;58(4):331–342. | ||
Bowden S, Bardenhagen F, Ambrose M, Whelan G. Alcohol, thiamin deficiency, and neuropsychological disorders. Alcohol Alcohol Suppl. 1994;2:267–272. | ||
Arts N. Alzheimer’s disease. In: Koehler P, Bruyn G, Pearce J, editors. Neurological Eponyms. Oxford: Oxford University Press; 2000:261–268. | ||
Nikolakaros G, Ilonen T, Kurki T, Paju J, Papageorgiou SG, Vataja R. Non-alcoholic Korsakoff syndrome in psychiatric patients with a history of undiagnosed Wernicke’s encephalopathy. J Neurol Sci. 2016;370:296–302. | ||
Gasquoine PG. A case of bariatric surgery-related Wernicke–Korsakoff syndrome with persisting anterograde amnesia. Arch Clin Neuropsychol. 2017;32(5):610–617. | ||
Dubois B, Feldman HH, Jacova C, et al. Research criteria for the diagnosis of Alzheimer’s disease: revising the NINCDS-ADRDA criteria. Lancet Neurol. 2007;6(8):734–746. | ||
Dubois B, Feldman HH, Jacova C, et al. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol. 2014;13(6):614–629. | ||
Wierdsma AI. Follow-up after involuntary mental healthcare: who cares? Emergency compulsory admission and continuity of care in Rotterdam, the Netherlands. 2008. Available from http://hdl.handle.net/1765/13551. Acessed October 31, 2017. | ||
Ramayya A, Jauhar P. Increasing incidence of Korsakoff’s psychosis in the east end of Glasgow. Alcohol Alcohol. 1997;32(3):281–285. | ||
Cutting J. The relationship between Korsakoff’s syndrome and “alcoholic dementia.” Br J Psychiatry. 1978;132:240–251. | ||
Sechi G, Serra A. Wernicke’s encephalopathy: new clinical settings and recent advances in diagnosis and management. Lancet Neurol. 2007;6(5):442–455. | ||
Isenberg-Grzeda E, Kutner HE, Nicolson SE. Wernicke-Korsakoff-syndrome: under-recognized and under-treated. Psychosomatics. 2012;53(6):507–516. | ||
Scalzo SJ, Bowden SC, Ambrose ML, Whelan G, Cook MJ. Wernicke-Korsakoff syndrome not related to alcohol use: a systematic review. J Neurol Neurosurg Psychiatry. 2015;86(12):1362–1368. | ||
Zahr NM, Kaufman KL, Harper CG. Clinical and pathological features of alcohol-related brain damage. Nat Rev Neurol. 2011;7(5):284–294. | ||
Thomson AD, Cook CCH, Touquet R, Henry JA. The Royal College of Physicians report on alcohol: guidelines for managing Wernicke’s encephalopathy in the accident and emergency department. Alcohol Alcohol. 2002;37(6):513–521. | ||
Laureno R. Nutritional cerebellar degeneration, with comments on its relationship to Wernicke disease and alcoholism. Handb Clin Neurol. 2011;103:175–187. | ||
Baker KG, Harding AJ, Halliday GM, Kril JJ, Harper CG. Neuronal loss in functional zones of the cerebellum of chronic alcoholics with and without Wernicke’s encephalopathy. Neuroscience. 1999;91(2):429–438. | ||
Kril JJ, Halliday GM, Svoboda MD, Cartwright H. The cerebral cortex is damaged in chronic alcoholics. Neuroscience. 1997;79(4):983–998. | ||
Harper C, Corbett D. Changes in the basal dendrites of cortical pyramidal cells from alcoholic patients – a quantitative Golgi study. J Neurol Neurosurg Psychiatry. 1990;53(10):856–861. | ||
Harding A, Halliday G, Caine D, Kril J. Degeneration of anterior thalamic nuclei differentiates alcoholics with amnesia. Brain. 2000;123(1):141–154. | ||
Cullen KM, Halliday GM, Caine D, Kril JJ. The nucleus basalis (Ch4) in the alcoholic Wernicke-Korsakoff syndrome: reduced cell number in both amnesic and non-amnesic patients. J Neurol Neurosurg Psychiatry. 1997;63(3):315–320. | ||
Baker KG, Halliday GM, Harper CG. Effect of chronic alcohol consumption on the human locus coeruleus. Alcohol Clin Exp Res. 1994;18(6):1491–1496. | ||
Baker K, Halliday G, Kril J, Harper C. Chronic alcoholism in the absence of Wernicke-Korsakoff syndrome and cirrhosis does not result in the loss of serotonergic neurons from the median raphe nucleus. Metab Brain Dis. 1996;11(3):217–227. | ||
Baker KG, Halliday GM, Kril JJ, Harper CG. Chronic alcoholics without Wernicke–Korsakoff syndrome or cirrhosis do not lose serotonergic neurons in the dorsal raphe nucleus. Alcohol Clin Exp Res. 1996;20(1):61–66. | ||
Ryback RS. The continuum and specificity of the effects of alcohol on memory. A review. Q J Stud Alcohol. 1971;32(4):995–1016. | ||
Butters N, Brandt J. The continuity hypothesis: the relationship of long-term alcoholism to the Wernicke-Korsakoff syndrome. Recent Dev Alcohol. 1985;3:207–226. | ||
Pitel AL, Beaunieux H, Witkowski T, et al. Episodic and working memory deficits in alcoholic korsakoff patients: the continuity theory revisited. Alcohol Clin Exp Res. 2008;32(7):1229–1241. | ||
Kril JJ, Harper CG. Neuroanatomy and neuropathology associated with Korsakoff’s syndrome. Neuropsychol Rev. 2012;22(2):72–80. | ||
Aggleton JP, Sahgal A. The contribution of the anterior thalamic nuclei to anterograde amnesia. Neuropsychologia. 1993;31(10):1001–1019. | ||
Vetreno RP, Ramos RL, Anzalone S, Savage LM. Brain and behavioral pathology in an animal model of Wernicke’s encephalopathy and Wernicke-Korsakoff syndrome. Brain Res. 2012;1436:178–192. | ||
Van Der Werf YD, Jolles J, Witter MP, Uylings BM. Contributions of thalamic nuclei to declarative memory functioning. Cortex. 2003;39(4–5):1047–1062. | ||
Yoneoka Y, Takeda N, Inoue A, et al. Acute Korsakoff syndrome following mammillothalamic tract infarction. Am J Neuroradiol. 2004;25(6):964–968. | ||
Carlesimo GA, Lombardi MG, Caltagirone C. Vascular thalamic amnesia: a reappraisal. Neuropsychologia. 2011;49(5):777–789. | ||
Wolff M, Alcaraz F, Marchand AR, Coutureau E. Functional heterogeneity of the limbic thalamus: from hippocampal to cortical functions. Neurosci Biobehav Rev. 2015;54:120–130. | ||
Sullivan EV, Pfefferbaum A. Neuroimaging of the Wernicke-Korsakoff syndrome. Alcohol Alcohol. 2009;44(2):155–165. | ||
Lough ME. Wernicke’s encephalopathy: expanding the diagnostic toolbox. Neuropsychol Rev. 2012;22(2):181–194. | ||
Pitel AL, Chételat G, Le Berre AP, Desgranges B, Eustache F, Beaunieux H. Macrostructural abnormalities in Korsakoff syndrome compared with uncomplicated alcoholism. Neurology. 2012;78(17):1330–1333. | ||
Le Berre AP, Pitel AL, Chanraud S, et al. Chronic alcohol consumption and its effect on nodes of frontocerebellar and limbic circuitry: comparison of effects in France and the United States. Hum Brain Mapp. 2014;35(9):4635–4653. | ||
Jung YC, Chanraud S, Sullivan EV. Neuroimaging of Wernicke’s encephalopathy and Korsakoff’s syndrome. Neuropsychol Rev. 2012;22(2):170–180. | ||
Zahr N. Clinical and pathological features of alcohol-related brain damage. Handb Clin Neurol. 2014;125:275–290. | ||
Harper C. The incidence of Wernicke’s encephalopathy in Australia – a neuropathological study of 131 cases. J Neurol Neurosurg Psychiatry. 1983;46(7):593–598. | ||
Harper CG, Sheedy DL, Lara AI, Garrick TM, Hilton JM, Raisanen J. Prevalence of Wernicke-Korsakoff syndrome in Australia: has thiamine fortification made a difference? Med J Aust. 1998;168(11):542–545. | ||
Cook CC, Hallwood PM, Thomson AD. B Vitamin deficiency and neuropsychiatric syndromes in alcohol misuse. Alcohol Alcohol. 1998;33(4):317–336. | ||
Day E, Bentham PW, Callaghan R, Kuruvilla T, George S. Thiamine for prevention and treatment of Wernicke-Korsakoff syndrome in people who abuse alcohol. In: Day E, editor. Cochrane Database of Systematic Reviews. Chichester, UK: John Wiley & Sons, Ltd; 2013:CD004033. | ||
Thomson AD, Marshall EJ. The natural history and pathophysiology of Wernicke’s encephalopathy and Korsakoff’s psychosis. Alcohol Alcohol. 2006;41(2):151–158. | ||
Thomson AD, Marshall EJ. The treatment of patients at risk of developing Wernicke’s encephalopathy in the community. Alcohol Alcohol. 2006;41(2):159–167. | ||
Wood B, Currie J, Breen K. Wernicke’s encephalopathy in a metropolitan hospital. A prospective study of incidence, characteristics and outcome. Med J Aust. 1986;144(1):12–16. | ||
Homewood J, Bond NW. Thiamin deficiency and Korsakoff’s syndrome: failure to find memory impairments following nonalcoholic Wernicke’s encephalopathy. Alcohol. 1999;19(1):75–84. | ||
Lough ME. Wernicke’s encephalopathy: expanding the diagnostic toolbox. Neuropsychol Rev. 2012;22(2):181–194. | ||
Garavan H, Brennan K, Hester R, Whelan R. The neurobiology of successful abstinence. Curr Opin Neurobiol. 2013;23(4):668–674. | ||
Monnig MA, Tonigan JS, Yeo RA, Thoma RJMB. White matter volume in alcohol use disorders: a meta-analysis. Addict Biol. 2013;18:581–592. | ||
Smith S, Fein G. Cognitive performance in treatment-naïve active alcoholics. Alcohol Clin Exp Res. 2010;34:2097–2105. | ||
Mann K, Günther A, Stetter F, Ackermann K. Rapid recovery from cognitive deficits in abstinent alcoholics: a controlled test-retest study. Alcohol Alcohol. 1999;34(4):567–574. | ||
Pitel AL, Rivier J, Beaunieux H, Vabret F, Desgranges B, Eustache F. Changes in the episodic memory and executive functions of abstinent and relapsed alcoholics over a 6-month period. Alcohol Clin Exp Res. 2009;33(3):490–498. | ||
Stavro K, Pelletier J, Potvin S. Widespread and sustained cognitive deficits in alcoholism: a meta-analysis. Addict Biol. 2013;18(2):203–213. | ||
Sutherland GT, Sheedy D, Kril JJ. Neuropathology of alcoholism. Handb Clin Neurol. 2014;125:603–615. | ||
Bowden S. Separating cognitive impairment in neurologically asymptomatic alcoholism from Wernicke-Korsakoff syndrome: is the neuropsychological distinction justified? Psychol Bull. 1990;107(3):355–366. | ||
Fein G, McGillivray S. Cognitive performance in long-term abstinent elderly alcoholics. Alcohol Clin Exp Res. 2007;31(11):1788–1799. | ||
Gudden H. Klinische und anatomische Beiträge zur Kenntniss der multiplen Alkoholneuritis nebst Bemerkungen über die Regenerationsvorgänge im peripheren Nervensystem. Arch Psychiatr Nervenkr. 1896;28(3):643–741. | ||
Baddeley A, Hitch G. Working memory. In: Bower GH, editor. The Psychology of Learning and Motivation: Advances in Research and Theory; Vol. 8. New York: Academic Press; 1974:47–89. | ||
Grünthal E. Über das Symptom der Einstellungsstörung bei exogenen Psychosen. Z Ges Neurol Psychiat. 1924;92:255–266. | ||
Van der Horst L. Über die Psychologie des Korsakowsyndroms. Mschr Psychiat Neurol. 1932;83:65–84. | ||
Brion S. Korsakoff’s syndrome: clinico-anatomical and physiological considerations. In: Talland GA, Waugh N, editors. The Pathology of Memory. New York: Academic Press; 1969:29–40. | ||
Angelergues R. Le Syndrome Mental de Korsakow. Paris: Masson; 1958. | ||
Tulving E. Episodic and semantic memory. In: Tulving E, Donaldson W, editors. Organization of Memory. New York: Academic Press; 1972:381–403. | ||
Squire LR. Memory and brain systems: 1969–2009. J Neurosci. 2009;29(41):12711–12716. | ||
Squire LR, Dede AJO. Conscious and unconscious memory systems. Perspect Biol. 2015;7:a021667. | ||
Amaral DG, Rempel-Clower NL, Zola SM, Squire LR. Three cases of enduring memory impairment after bilateral damage limited to the hippocampal formation. J Neurosci. 1996;16(16):5233–5255. | ||
Butters N, Stuss D. Diencephalic amnesia. In: Boller F, Grafman J, editors. Handbook of Neuropsychology Vol.3 Vpl. 4. Amsterdam: Elsevier; 1989:107–148. | ||
Wheeler MA, Stuss DT, Tulving E. Toward a theory of episodic memory: the frontal lobes and autonoetic consciousness. Psychol Bull. 1997;121(3):331–354. | ||
El Haj M, Allain P. What do we know about the relationship between source monitoring deficits and executive dysfunction? Neuropsychol Rehabil. 2012;22(3):449–472. | ||
Kessels RPC, Kopelman MD. Context memory in Korsakoff’s syndrome. Neuropsychol Rev. 2012;22(2):117–131. | ||
Kopelman MD. Remote and autobiographical memory, temporal context memory and frontal atrophy in Korsakoff and Alzheimer patients. Neuropsychologia. 1989;27(4):437–460. | ||
Mayes AR, Meudell PR, Pickering A. Is organic amnesia caused by a selective deficit in remembering contextual information? Cortex. 1985;21(2):167–202. | ||
McKone E, French B. In what sense is implicit memory “episodic”? The effect of reinstating environmental context. Psychon Bull Rev. 2001;8(4):806–811. | ||
Kopelman MD, Stanhope N, Kingsley D. Temporal and spatial context memory in patients with focal frontal, temporal lobe, and diencephalic lesions. Neuropsychologia. 1997;35:1533–1545. | ||
Shoqeirat MA, Mayes AR. Disproportionate incidental spatial-memory and recall deficits in amnesia. Neuropsychologia. 1991;29(8):749–769. | ||
Mayes AR, Meudell PR, MacDonald C. Disproportionate intentional spatial-memory impairments in amnesia. Neuropsychologia. 1991;29(8):771–784. | ||
El Haj M, Kessels RPC, Allain P. Source memory rehabilitation: a review toward recommendations for setting up a strategy training aimed at the “what, where, and when” of episodic retrieval. Appl Neuropsychol Adult. 2016;23(1):53–60. | ||
El Haj M, Kessels RPC, Matton C, et al. Destination memory in Korsakoff’s syndrome. Alcohol Clin Exp Res. 2016;40(6):1321–1327. | ||
Albert MS, Butters N, Brandt J. Patterns of remote memory in amnesic and demented patients. Arch Neurol. 1981;38(8):495–500. | ||
Kopelman MD, Stanhope N, Kingsley D. Retrograde amnesia in patients with diencephalic, temporal lobe or frontal lesions. Neuropsychologia. 1999;37(8):939–958. | ||
Moscovitch M, Nadel L, Winocur G, Gilboa A, Rosenbaum RS. The cognitive neuroscience of remote episodic, semantic and spatial memory. Curr Opin Neurobiol. 2006;16(2):179–190. | ||
Race E, Verfaellie M. Remote memory function and dysfunction in Korsakoff’s syndrome. Neuropsychol Rev. 2012;22(2):105–116. | ||
Rensen YCM, Kessels RPC, Migo EM, Wester AJ, Eling PATM, Kopelman MD. Personal semantic and episodic autobiographical memories in Korsakoff syndrome: a comparison of interview methods. J Clin Exp Neuropsychol. 2016;39(6):1–13. | ||
Zola-Morgan S, Cohen NJ, Squire LR. Recall of remote episodic memory in amnesia. Neuropsychologia. 1983;21(5):487–500. | ||
Weingartner H, Grafman J, Boutelle W, Kayne W, Martin PR. Forms of memory failure. Science. 1983;221(17):380–382. | ||
Perani D, Bressi S, Cappa SF, et al. Evidence of multiple memory systems in the human brain. A [18F]FDG PET metabolic study. Brain. 1993;116(4):903–919. | ||
Smith ME, Oscar-Berman M. Repetition priming of words and pseudowords in divided attention and in amnesia. J Exp Psychol Learn Mem Cogn. 1990;16(6):1033–1042. | ||
Verfaellie M, Cermak LS, Blackford SP, Weiss S. Strategic and automatic priming of semantic memory in alcoholic Korsakoff patients. Brain Cogn. 1990;13(2):178–192. | ||
Verfaellie M, Cermak LS, Letourneau L, Zuffante P. Repetition effects on a lexical decision task: the role of episodic memory in the performance of alcoholic Korsakoff patients. Neuropsychologia. 1991;29(7):641–657. | ||
Pitel AL, Beaunieux H, Guillery-Girard B, et al. How do Korsakoff patients learn new concepts? Neuropsychologia. 2009;47(3):879–886. | ||
Cohen NJ, Squire LR. Preserved learning and retention of pattern-analyzing skill in amnesia: dissociation of knowing how and knowing that. Science. 1980;210(4466):207–210. | ||
Cermak LS, Lewis R, Butters N, Goodglass H. Role of verbal mediation in performance of motor tasks by Korsakoff patients. Psychol Sci. 1973;37(1):259–262. | ||
Charness N, Milberg W, Alexander MP. Teaching an amnesic a complex cognitive skill. Brain Cogn. 1988;8(2):253–272. | ||
Beaunieux H, Desgranges B, Lalevée C, de la Sayette V, Lechevalier B, Eustache F. Preservation of cognitive procedural memory in a case of Korsakoff’s syndrome: methodological and theoretical insights. Percept Mot Skills. 1998;86(3 Pt 2):1267–1287. | ||
Kessels RPC, van Loon E, Wester AJ. Route learning in amnesia: a comparison of trial-and-error and errorless learning in patients with the Korsakoff syndrome. Clin Rehabil. 2007;21(10):905–911. | ||
Swinnen SP, Puttemans V, Lamote S. Procedural memory in Korsakoff’s disease under different movement feedback conditions. Behav Brain Res. 2005;159(1):127–133. | ||
Heyselaar E, Segaert K, Walvoort SJW, Kessels RPC, Hagoort P. The role of procedural memory in the skill for language: evidence from syntactic priming in patients with amnesia. Neuropsychologia. 2017;101:97–105. | ||
Butters N. Alcoholic Korsakoff’s syndrome: some unresolved issues concerning etiology, neuropathology, and cognitive deficits. J Clin Exp Neuropsychol. 1985;7(2):181–210. | ||
Nissen MJ, Willingham D, Hartman M. Explicit and implicit remembering: when is learning preserved in amnesia? Neuropsychologia. 1989;27(3):341–352. | ||
Schmidtke K, Handschu R, Vollmer H. Cognitive procedural learning in amnesia. Brain Cogn. 1996;32(3):441–467. | ||
Xu Y, Corkin S. HM revisits the Tower of Hanoi Puzzle. Neuropsychology. 2001;15(1):69. | ||
Beaunieux H, Pitel AL, Witkowski T, Vabret F, Viader F, Eustache F. Dynamics of the cognitive procedural learning in alcoholics with korsakoff’s syndrome. Alcohol Clin Exp Res. 2013;37(6):1025–1032. | ||
Hayes SM, Fortier CB, Levine A, Milberg WP, McGlinchey R. Implicit memory in Korsakoff’s syndrome: a review of procedural learning and priming studies. Neuropsychol Rev. 2012;22(2):132–153. | ||
Oudman E, Nijboer TCW, Postma A, Wijnia JW, Van der Stigchel S. Procedural learning and memory rehabilitation in Korsakoff’s syndrome – a review of the Literature. Neuropsychol Rev. 2015;25(2):134–148. | ||
Lezak M. Neuropsychological Assessment. 2nd ed. Oxford: Oxford University Press; 1983. | ||
Miyake A, Friedman NP, Emerson MJ, Witzki AH, Howerter A, Wager TD. The unity and diversity of executive functions and their contributions to complex “frontal lobe” tasks: a latent variable analysis. Cogn Psychol. 2000;41(1):49–100. | ||
Bouman L, Grünbaum A. Eine Störung der Chronognosie und ihre Bedeutung im betreffenden Symptomenbild. Mschr Psychiat Neurol. 1929;73:1–19. | ||
Van Oort R, Kessels RPC. Executive dysfunction in Korsakoff’s syndrome: time to revise the DSM criteria for alcohol-induced persisting amnestic disorder? Int J Psychiatry Clin Pract. 2009;13(1):78–81. | ||
Brion M, Pitel A-L, Beaunieux H, Maurage P. Revisiting the continuum hypothesis: toward an in-depth exploration of executive functions in Korsakoff syndrome. Front Hum Neurosci. 2014;8(July):1–7. | ||
Walvoort SJW, van der Heijden PT, Wester AJ, Kessels RPC, Egger JIM. Self-awareness of cognitive dysfunction: self-reported complaints and cognitive performance in patients with alcohol-induced mild or major neurocognitive disorder. Psychiatry Res. 2016;245:291–296. | ||
Moscovitch M. Confabulation and the frontal systems: strategic versus associative retrieval in neuropsychological theories of memory. In: Roediger HL, Craik F, editors. Varieties of Memory and Consciousness: Essays in Honor of Endel Tulving. Hillsdale: Lawrence Erlbaum Associates; 1989:133–160. | ||
Berlyne N. Confabulation. Br J Psychiatry. 1972;120(554):31–39. | ||
Glowinski R, Payman V, Frencham K. Confabulation: a spontaneous and fantastic review. Aust N Z J Psychiatry. 2008;42(11):932–940. | ||
Kopelman MD. Varieties of confabulation and delusion. Cogn Neuropsychiatry. 2010;15(1–3):14–37. | ||
Schnider A, Gutbrod K, Hess CW, Schroth G. Memory without context: amnesia with confabulations after infarction of the right capsular genu. J Neurol Neurosurg Psychiatry. 1996;61(2):186–193. | ||
Schnider A, von Däniken C, Gutbrod K. The mechanisms of spontaneous and provoked confabulations. Brain. 1996;119(4):1365–1375. | ||
Kopelman MD. Two types of confabulation. J Neurol Neurosurg Psychiatry. 1987;50(11):1482–1487. | ||
Schnider A. The Confabulating Mind; How the Brain Creates Reality. Oxford: Oxford University Press; 2008. | ||
Rensen YCM, Oosterman JM, Walvoort SJW, Eling PATM, Kessels RPC. Intrusions and provoked and spontaneous confabulations on memory tests in Korsakoff’s syndrome. J Clin Exp Neuropsychol. 2016;39(2):1–11. | ||
Dalla Barba GF, Nedjam Z, Dubois B. Confabulation, executive functions and source memory in Alzheimer’s disease. Cogn Neuropsychol. 1999;16:385–398. | ||
Schnider A, Ptak R. Spontaneous confabulators fail to suppress currently irrelevant memory traces. Nat Neurosci. 1999;2(7):677–681. | ||
Stuss DT, Alexander MP, Lieberman A, Levine H. An extraordinary form of confabulation. Neurology. 1978;28(11):1166–1172. | ||
Kessels RPC, Kortrijk HE, Wester AJ, Nys GMS. Confabulation behavior and false memories in Korsakoff’s syndrome: role of source memory and executive functioning. Psychiatry Clin Neurosci. 2008;62(2):220–225. | ||
Gilboa A, Alain C, Stuss DT, Melo B, Miller S, Moscovitch M. Mechanisms of spontaneous confabulations: a strategic retrieval account. Brain. 2006;129(6):1399–1414. | ||
Moscovitch M, Winocur G. The frontal cortex and working with memory. In: Stuss DT, Knight R, editors. Principles of Frontal Lobe Function. New York: Oxford University Press; 2002:188–209. | ||
Stuss DT, van Reekum R, Murphy K. Differentiation of states and causes of apathy. In: Borod J, editor. Neuropsychology of Emotion. New York: Oxford University Press; 2000:340–363. | ||
Marin R. Apathy: a neuropsychiatric syndrome. J Neuropsychiatry Clin Neurosci. 1991;3(3):243–254. | ||
Robert P, Onyike CU, Leentjens AFG, et al. Proposed diagnostic criteria for apathy in Alzheimer’s disease and other neuropsychiatric disorders. Eur Psychiatry. 2009;24(2):98–104. | ||
Arts N. Abulie, apathie en avolitie: stoornissen van “de wil”? In: Eling P, Krabbendam L, Aleman A, editors. Cognitieve Neuropsychiatrie. Amsterdam: Boom; 2013:359–395. | ||
Talland G. Psychological studies of Korsakoff’s psychosis: V. Spontaneity and activity rate. J Nerv Ment Dis. 1960;130:16–25. | ||
Levy R, Dubois B. Apathy and the functional anatomy of the prefrontal cortex-basal ganglia circuits. Cereb Cortex. 2006;16(7):916–928. | ||
Levy R, Czernecki V. Apathy and the basal ganglia. J Neurol. 2006;253(Suppl 7:VIII):54–61. | ||
Jorge RE, Starkstein SE, Robinson RG. Apathy following stroke. Can J Psychiatry. 2010;55(6):350–354. | ||
Stella F, Radanovic M, Aprahamian I, Canineu PR, De Andrade LP, Forlenza OV. Neurobiological correlates of apathy in alzheimer’s disease and mild cognitive impairment: a critical review. J Alzheimer’s Dis. 2014;39(3):633–648. | ||
Douglas JJ, Wilkinson DA. Evidence of normal emotional responsiveness in alcoholic Korsakoff’s syndrome in the presence of profound memory impairment. Addiction. 1993;88(12):1637–1645. | ||
Labudda K, Todorovski S, Markowitsch HJ, Brand M. Judgment and memory performance for emotional stimuli in patients with alcoholic Korsakoff syndrome. J Clin Exp Neuropsychol. 2008;30(2):224–235. | ||
Brion M, D’Hondt F, Davidoff DA, Maurage P. Beyond cognition: understanding affective impairments in Korsakoff syndrome. Emot Rev. 2016;8(4):376–384. | ||
Johnson MK, Kim JK, Risse G. Do alcoholic Korsakoff’s syndrome patients acquire affective reactions? J Exp Psychol Learn Mem Cogn. 1985;11(1):22–36. | ||
Snitz BE, Hellinger A, Daum I. Impaired processing of affective prosody in Korsakoff’s syndrome. Cortex. 2002;38(5):797–803. | ||
Oosterman JM, De Goede M, Wester AJ, Van Zandvoort MJE, Kessels RPC. Perspective taking in Korsakoff’s syndrome: the role of executive functioning and task complexity. Acta Neuropsychiatr. 2011;23(6):302–308. | ||
Montagne B, Kessels RPC, Wester AJ, de Haan EHF. Processing of emotional facial expressions in Korsakoff’s syndrome. Cortex. 2006;42(5):705–710. | ||
Clark US, Oscar-Berman M, Shagrin B, Pencina M. Alcoholism and judgments of affective stimuli. Neuropsychology. 2007;21(3):346–362. | ||
Arciniegas D, Silver J. Pharmacotherapy of cognitive impairment. In: Zasler N, Katz D, Zafonte R, editors. Brain Injury Medicine; Principles and Practice, 2nd ed. New York: Demos Medical; 2013. | ||
Arts N, Wiersma J. Psychofarmaca bij de behandeling van langetermijngevolgen van chronisch alcoholmisbruik. Psyfar. 2011;(3):54–57. | ||
de Joode EA, van Boxtel MPJ, Hartjes P, Verhey FRJ, van Heugten CM. Use of an electronic cognitive aid by a person with Korsakoff syndrome. Scand J Occup Ther. 2013;20(6):446–453. | ||
Svanberg J, Evans JJ. Neuropsychological rehabilitation in alcohol-related brain damage: a systematic review. Alcohol Alcohol. 2013;48(6):704–711. | ||
Clare L, Jones RSP. Errorless learning in the rehabilitation of memory impairment: a critical review. Neuropsychol Rev. 2008;18(1):1–23. | ||
Haslam C, Kessels RPC. Errorless Learning in Neuropsychological Rehabilitation: Mechanisms, Efficacy and Application. Oxon, UK: Routledge; 2018. | ||
Oudman E, Nijboer TCW, Postma A, et al. Acquisition of an instrumental activity of daily living in patients with Korsakoff’s syndrome: a comparison of trial and error and errorless learning. Neuropsychol Rehabil. 2013;23(6):888–913. | ||
Wilson BA, Baddeley A, Evans J, Shiel A. Errorless learning in the rehabilitation of memory impaired people. Neuropsychol Rehabil. 1994;4:307–326. | ||
Komatsu SI, Mimura M, Kato M, Wakamatsu N, Kashima H. Errorless and effortful processes involved in the learning of face–name associations by patients with alcoholic Korsakoff’s syndrome. Neuropsychol Rehabil. 2000;10:113–132. | ||
Aggleton JP, Brown MW. Episodic memory, amnesia, and the hippocampal–anterior thalamic axis. Behav Brain Sci. 1999;22(3):425–489. | ||
Aggleton JP, Dumont JR, Warburton EC. Unraveling the contributions of the diencephalon to recognition memory: a review. Learn Mem. 2011;18(6):384–400. | ||
Aggleton JP. Looking beyond the hippocampus: old and new neurological targets for understanding memory disorders. Proc R Soc B Biol Sci. 2014;281(1786):20140565. | ||
Ward LM. The thalamic dynamic core theory of conscious experience. Conscious Cogn. 2011;20(2):464–486. | ||
Dalrymple-Alford JC, Harland B, Loukavenko EA, et al. Anterior thalamic nuclei lesions and recovery of function: relevance to cognitive thalamus. Neurosci Biobehav Rev. 2015;54:145–160. | ||
Saalmann YB. Intralaminar and medial thalamic influence on cortical synchrony, information transmission and cognition. Front Syst Neurosci. 2014;8:1–8. | ||
Saalmann YB, Kastner S. Cognitive and perceptual functions of the visual thalamus. Neuron. 2011;71(2):209–223. | ||
Mitchell AS, Chakraborty S. What does the mediodorsal thalamus do? Front Syst Neurosci. 2013;7(August):1–19. | ||
Baxter MG. Mediodorsal thalamus and cognition in non-human primates. Front Syst Neurosci. 2013;7(August):1–5. | ||
Jankowski MM, Ronnqvist KC, Tsanov M, et al. The anterior thalamus provides a subcortical circuit supporting memory and spatial navigation. Front Syst Neurosci. 2013;7(August):1–12. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.