")
Back to Journals » Cancer Management and Research » Volume 13
Knockdown of SLC39A4 Expression Inhibits the Proliferation and Motility of Gallbladder Cancer Cells and Tumor Formation in Nude Mice
Authors Li M , Fan K , Zheng B , Zekria D, Suo T, Liu H, Shen S, Liu H, Ni X
Received 6 October 2020
Accepted for publication 1 February 2021
Published 8 March 2021 Volume 2021:13 Pages 2235—2246
DOI https://doi.org/10.2147/CMAR.S282269
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Eileen O'Reilly
Min Li,1,* Kun Fan,1,* Bohao Zheng,1,* David Zekria,2 Tao Suo,1 Han Liu,1 Sheng Shen,1 Houbao Liu,1 Xiaoling Ni1
1Department of General Surgery, Zhongshan Hospital, Fudan University, Shanghai, 200032, People’s Republic of China; 2Department of Radiology, Leicester Royal Infirmary, Leicester, LE1 5WW, UK
*These authors contributed equally to this work
Correspondence: Xiaoling Ni; Houbao Liu
Department of General Surgery, Zhongshan Hospital, Fudan University, Shanghai, 200032, People’s Republic of China
Tel +86 21 64041998; +86 21 64041997
Email [email protected]; [email protected]
Purpose: Gallbladder cancer (GBC) is a common malignancy of the biliary tract and is characterized by rapid progression and early metastasis. Elucidating the molecular mechanisms of GBC could help to develop better treatment strategies.
Materials and Methods: Human GBC cell lines (GBC-SD and NOZ) were applied to determine the capacity of the proliferation and migration of cells using the MTT assay, colony formation, wound-healing assay as well as the Transwell™ assay. A nude xenograft was used to evaluate tumor growth in vivo.
Results: Using two types of GBC cell lines, we found that absence of solute carrier family (SLC) 39A4 (which encodes the zinc transporter ZRT/IRT-like protein [ZIP]4), could suppress the proliferation and migration of cells. Additionally, absence of ZIP4 could impair growth of xenografts in nude mice. While, over-expression of SLC39A4 could promote the GBC cell proliferation and migration, and inhibit apoptosis. We revealed that SLC39A4 might affect GBC progression by modulating the signaling pathways responsible for the survival, energy supply and metastasis of cells, and indicated that SLC39A4 could serve as a novel therapeutic target for GBC.
Conclusion: SLC39A4 promoted the viability and motility of GBC cells, and tumor formation in nude mice. We demonstrated an oncogenic potential for SLC39A4.
Keywords: gallbladder cancer, SLC39A4 (ZIP4), proliferation, migration, potential therapeutic target
Introduction
Gallbladder cancer (GBC) is aggressive, is more common in females, and is characterized by rapid progression and early metastasis. Treatment options are surgery, chemotherapy and radiotherapy.1,2 However, GBC is usually diagnosed late due to a lack of early signs and clinical symptoms, which limits therapy choices and undermines a better prognosis. Thus, identification of specific diagnostic markers and elucidation of the underlying molecular mechanisms of GBC are very important. Indeed, the molecular pathology of GBC is poorly understood despite extensive research efforts.3,4
Zinc is a vital element in the human body. It is not only a catalytic cofactor for several enzymes, it also has key roles in the signal transduction involved in cell differentiation, tissue development, and metabolism.5,6 Zinc deficiency is associated with several diseases,7–10 but zinc levels in most tumor cells are increased due to abnormal overexpression of zinc importers, which allow them to survive.11
Solute carrier family (SLC) 39 (also known as ZRT/IRT-like protein [ZIP]) is responsible for transferring zinc from the extracellular space and organelles into the cytosol.12 Among them, ZIP4 (which is encoded by SLC39A4) is the major transporter for zinc uptake. Its aberrant expression has been found in different types of cancers. In hepatocellular carcinoma (HCC), SLC39A4 suppresses the apoptosis and promotes the migration of cells. In addition, SLC39A4 mediates drug resistance in non-small-cell lung cancer (NSCLC). Moreover, it serves as a prognostic marker in multiple cancer types.13–17 Nevertheless, the role of SLC39A4 in GC is not clear.
We wished to assess the impact of SLC39A4 on the proliferation and migration of GBC cells. We also wished to clarify the changes of signaling pathways in response to SLC39A4 deficiency.
Materials and Methods
Cell Culture and Construction of a Stable Cell Line
Two human GBC cell lines, GBC-SD and NOZ, were obtained from the Japanese Collection of Research Bioresources Cell Bank (Tokyo, Japan). Both cell types were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) at 37°C in an atmosphere of 5% CO2. HEK293T cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS at 37°C in an atmosphere of 5% CO2.
GBC-SD cells and NOZ cells were infected with lentivirus encoding SLC39A4 short hairpin (sh) RNA or control lentivirus co-expressed with green fluorescent protein (GFP) according to manufacturer instructions. Briefly, HEK293T cells were used for lentivirus production. Stable cell lines expressing shSLC39A4 or control shRNA were sorted against GFP fluorescence 48 h after infection using fluorescence activated cell sorting and maintained in growth medium supplemented with puromycin (2 µg/mL) for one week.
Cell Proliferation
Cells were plated in 96-well plates at 2×103/well and cultured in complete medium for 5 days. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; catalog number, JT343; Geneview) was added and incubation allowed for 30 min. Then, the absorbance of the formazan solution was read using a spectrophotometer at 490 nm. All experiments were carried out in triplicate.
Colony Formation
Cells were seeded in six-well plates at 1×103/well and then incubated for 13 days (GBC-SD) or 8 days (NOZ). Incubation was followed by staining with 0.1% Crystal Violet for 20 min at room temperature. Colonies containing ≥50 cells were counted manually under a microscope. Each assay was undertaken in triplicate.
Wound Healing
Cells were plated in 96-well plates and cultured overnight. Wounds were made in confluent monolayer cells using the 96 Wounding Replicator (V&P Scientific, San Diego, CA, USA). Cells were cultured in medium supplemented with 0.5% FBS. Wound healing was detected at 0 h and 48 h within scraped lines. Representative fields at different time points were photographed (×100 magnification). Migration area was analyzed by Celigo™ (Nexcelom, Lawrence, MA, USA).
Transwell™ Migration
Cells were plated in the upper chamber of the apparatus with serum-free medium in the Transwell with inserts of pore size 8-μm (3422; Millipore, Billerica, MA, USA). The lower chambers were filled with complete culture medium. After incubation for 24 h, cells on the upper-membrane surface were removed by cotton tips. Then, membranes were washed with phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde, washed twice with PBS, and stained with 0.2% Crystal Violet. The migrated cells from nine fields were counted under a light microscope (×200 magnification). All experiments were done in triplicate.
Distribution of the Cell Cycle
Cells were treated as indicated. Approximately 48 h later, cells were harvested, washed with ice-cold PBS, and fixed in 70% ethanol overnight at −20°C. Subsequently, fixed cells were washed thrice with PBS and stained with propidium iodide (PI) solution (50 µg/mL PI and 100 µg/mL RNase A in PBS) in the dark for 30 min. DNA content was measured on a FACScan flow cytometer (BD Biosciences, San Jose, CA, USA) with FlowJo 7.6.1 (Tree Star, Ashland, OR, USA). Experiments were repeated thrice.
Apoptosis
Pellets of GBC-SD cells and NOZ cells were washed with ice-cold PBS and resuspended in 200 μL of 1× binding buffer. Subsequently, cells were incubated with 10 μL of Annexin V-allophycocyanin (Beyotime Institute of Technology, Beijing, China) for 15 min in the dark at room temperature. Samples were detected using a BD Accuri™ C6 Plus flow cytometer (BD Biosciences). Data were quantified and analyzed using a Guava easyCyte™ flow cytometer (EMD Millipore, Burlington, MA, USA).
Xenograft Model in Nude Mice
Animal studies were conducted in compliance with the regulations on management of animal welfare set by the Chinese Association for Laboratory Animal Sciences (Beijing, China). The protocol for animal experiments was approved by the Ethics Committee of Zhongshan Hospital (Shanghai, China).
Briefly, 4 × 106 stably transfected NOZ cells were suspended in 200 μL of PBS and injected (s.c.) into female BALB/c nude mice (4 weeks). Nude mice were sacrificed after 18 days. Tumor masses were removed surgically and weighed. Tumor volume was calculated as: tumor volume = (length × width2) × 0.5.
RNA Extraction and Real-Time Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
RNA was extracted using TRIzol™ Reagent (Invitrogen, Carlsbad, CA, USA) and used for complementary (c) DNA synthesis with PrimeScript™ RT Reagent Kit with gDNA Eraser (TaKaRa Biotechnology, Shiga, Japan). RT-qPCR was conducted with SYBR™ Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) using the ABI Prism 7500 Real-time PCR System (Applied Biosystems).
The following primer sequences (forward and reverse, respectively) were used: ZIP4, 5ʹ-CGGCGATGTTGAAAGTACG-3ʹ and 5ʹ-ACAGCAGCAGCAGGACGGT-3ʹ; glyceraldehyde 3-phosphate dehydrogenase (GAPDH), 5ʹ-TGACTTCAACAGCGACACCCA-3ʹ and 5ʹ-CACCCTGTTGCTGTAGCCAAA-3ʹ; BMP4, 5ʹ-CCTGGGCACCTCATCACA-3ʹ and 5ʹ-CATAGTTTGGCTGCTTCTC-3ʹ; GJA1, 5ʹ-CTGCCTTTCGTTGTAACACT-3ʹ and 5ʹ-TCTCTTCCTTTCGCATCAC-3ʹ; SYK, 5ʹ-GTGTCATTCAATCCGTATGAGCC-3ʹ and 5ʹ-TTTCGGTCCAGGTAAACCTCC-3ʹ; RUNX3, 5ʹ-GAACTGAACCCATTCTCCGACC-3ʹ and 5ʹ-ACGCTGAGGCTGCTGATGCT-3ʹ; SGK1, 5ʹ-GAAGCAGAGGAGGATGGGTCT-3ʹ and 5ʹ-GTTTAGCATGAGGATTGGACG-3ʹ; CDK4, 5ʹ-AGTGGTGGAACAGTCAAG-3ʹ and 5ʹ-AGCCCAATCAGGTCAAAG-3ʹ; EGFR, 5ʹ-AGGCACGAGTAACAAGCTCAC-3ʹ and 5ʹ-ATGAGGACATAACCAGCCACC-3ʹ; MET, 5ʹ-TGTTGTACCACTCCTTCCCTG-3ʹ and 5ʹ-ACGGCTTCAGAATGTAAGTGTAT-3ʹ. Analyses of melting curves were done to confirm specificity. The 2−ΔΔCt method was used for quantification. Each sample was run in triplicate, and GAPDH was used as an internal control.
Western Blotting
Cells were washed with cold PBS. Proteins were extracted with lysis buffer supplemented with a proteinase inhibitor. Cell lysates were centrifuged at 12,000 × g for 10 min at room temperature. Supernatants were collected and protein concentrations were quantified using a bicinchoninic acid kit (P0010S; Beyotime Institute of Technology). Then, proteins (35 mg) with loading buffer were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Western blots were developed using anti-ZIP4 (orb395779; Biorbyt, Cambridge, UK), anti-GAPDH (sc-32,233; Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-GJA1 (ab51067; Abcam, Cambridge, UK), anti-SGK1 (12,103; Cell Signaling Technology, Danvers, MA, USA), anti-MET (ab51067; Abcam), anti-VEGFC (ab83905; Abcam), mouse anti-rabbit IgG (7074; Cell Signaling Technology) and rabbit anti-mouse IgG (7076; Cell Signaling Technology).
Statistical Analysis
Each experiment was carried out at least three times independently. Data are the mean ± SD. Comparisons between two groups were carried out using two-tailed unpaired t-tests. P < 0.05 was considered significant.
Results
Establishment of SLC39A4 Stably Knocked-Down GBC Cells
Studies have uncovered a tumor-promoting role of ZIP4 (SLC39A4) in various cancer types.14,15,17,18 We wished to explore its potential relationship to GBC. First, we knocked-down SLC39A4 expression in a GBC cell line (GBC-SD) with lentivirus expressing three SLC39A4-specific shRNA sequences. shSLC39A4-2 had relatively higher efficiency in downregulating its expression of mRNA and protein (Figure 1A and B). Thus, lentivirus harboring shSLC39A4-2 was chosen to infect another GBC cell line: NOZ. Real-time RT-qPCR and Western blotting showed that expression of ZIP4 (SLC39A4) had been inhibited significantly (Figure 1C and D).
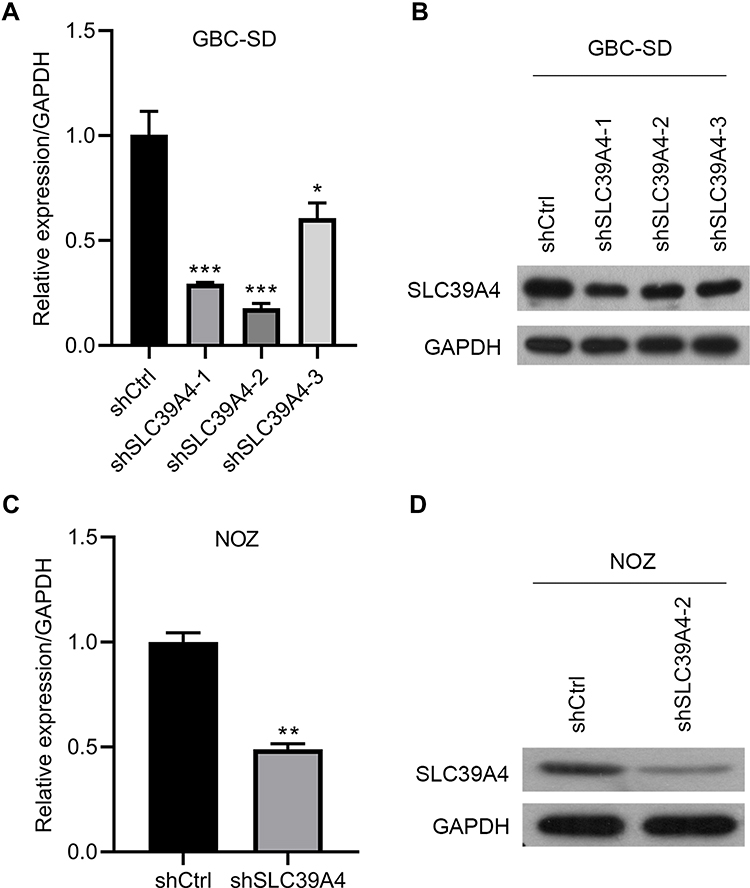 |
Figure 1 Establishment of SLC39A4 stably knocked-down cell lines in GBC-SD cells and NOZ cells. (A and B) Knockdown efficiency of three shSLC39A4s was evaluated in GBC-SD cells by real-time RT-qPCR (A) and Western blotting (B) and shSLC39A4-2 was selected to carry out subsequent experiments. (C and D) Confirmation of SLC39A4 expression in NOZ shSLC39A4-2 (NOZ/shSLC39A4-2) and control (NOZ/shCtrl) cells by real-time RT-qPCR (C) and Western blotting (D). The results of real-time RT-qPCR are the mean ± SD (unpaired t-test). *p < 0.05, **p < 0.01, ***p < 0.001, compared with NOZ/shCtrl or GBC-SD/shCtrl. |
Knockdown of SLC39A4 Expression Suppressed Cell Proliferation and Colony Formation
ZIP4 has been shown to repress apoptosis and promote the cell cycle in pancreatic cancer cells and HCC cells.13,17 We wondered if this was also the case in GBC cells. Cell proliferation was inhibited significantly by silencing of SLC39A4 expression in NOZ cells (Figure 2A) and in GBC-SD cells (Figure 2B). The ability to form colonies was also decreased dramatically in SLC39A4 knocked-down cells (Figure 2C and D), which suggested that ZIP4 was required for the proliferation of GBC cells.
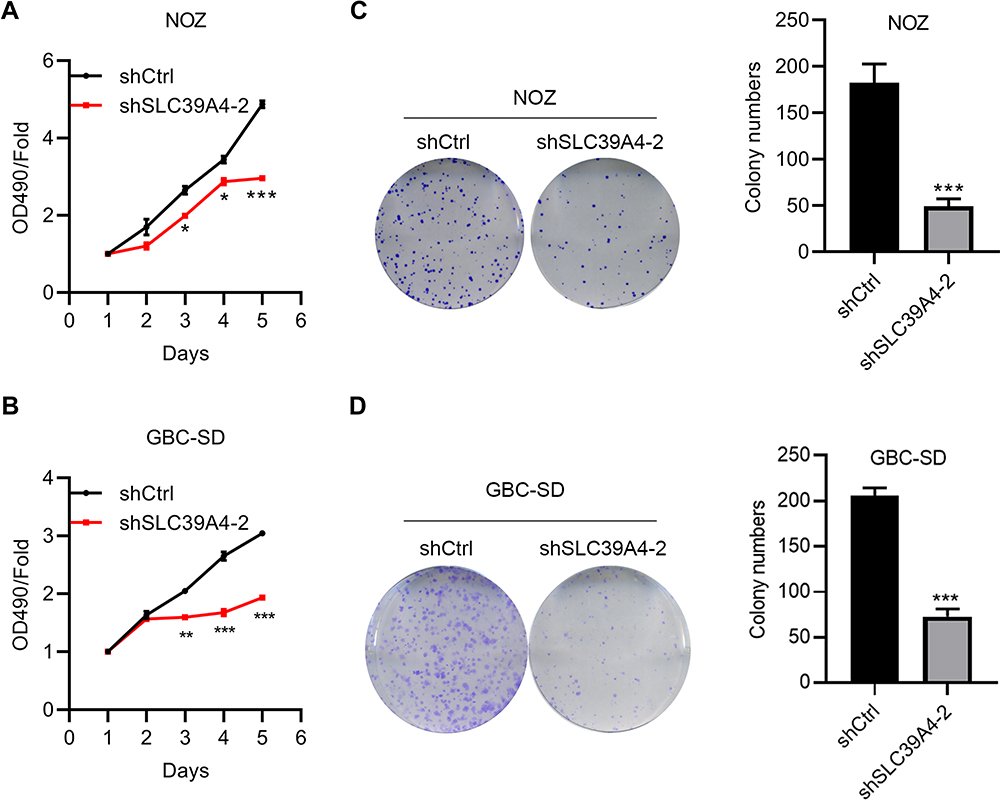 |
Figure 2 Knockdown of SLC39A4 expression suppressed cell proliferation and colony formation. (A and B) MTT assays were undertaken to evaluate proliferation of NOZ/shSLC39A4-2 cells (A) and GBC-SD/shSLC39A4-2 cells (B). (C and D) Colony-formation assays of NOZ/shSLC39A4-2 cells (C) and GBC-SD/shSLC39A4-2 cells (D). Images and quantitation are shown. Data are the mean ± SD (unpaired t-test) *p<0.05, **p<0.01, ***p < 0.001, compared with NOZ/shCtrl or GBC-SD/shCtrl. |
Knockdown of SLC39A4 Expression Impeded Cell Migration
It has been reported that ZIP4 is overexpressed in NSCLC, and is associated with enhanced migration of cells.14 In HCC, ZIP4 expression is also activated and cell migration increased.17 Next, we sought to discover what would happen after SLC39A4 expression was silenced. In the wound-healing assay, NOZ/shSLC39A4 cells and GBC-SD/shSLC39A4 cells migrated much more slowly than control cells (Figure 3A and B). Also, in the Transwell migration assay, silencing of SLC39A4 expression inhibited transmigration of NOZ cells considerably. An identical result was obtained in GBC-SD/shSLC39A4 cells (Figure 3C and D). Taken together, these data indicated that ZIP4 facilitated migration of GBC cells.
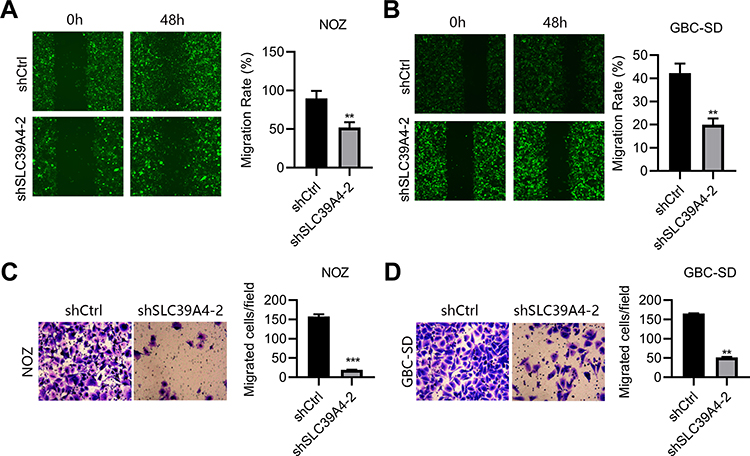 |
Figure 3 Knockdown of SLC39A4 expression impeded cell migration. (A and B) Wound-healing assays were undertaken to evaluate migration of NOZ/shSLC39A4-2 cells (A) and GBC-SD/shSLC39A4-2 cells (B). Representative images (magnification, ×100) and quantitation are shown. (C and D) Transwell-migration assays were conducted to observe the transmigration capability of NOZ/shSLC39A4-2 cells (C) and GBC-SD/shSLC39A4-2 cells (D). Representative images (magnification, ×200) and quantitation are shown. Data are the mean ± SD (unpaired t-test). **p < 0.01, ***p < 0.001, compared with NOZ/shCtrl or GBC-SD/shCtrl. |
Knockdown of SLC39A4 Expression Induced the Apoptosis and Cycle Arrest of Cells
To reveal the mechanisms underlying repression of cell proliferation, we analyzed the cell-cycle distribution of GBC cells after knockdown of SLC39A4 expression. After knockdown of SLC39A4 expression, the number of apoptotic GBC-SD cells and NOZ cells increased (Figure 4A–D). We observed accumulation of GBC-SD/shSLC39A4-2 cells in the G0/G1 phase compared with that in control cells, and an obvious reduction in the number of GBC-SD/shSLC39A4-2 cells in the S phase (Figure 4E and F). Knockdown of shSLC39A4 expression also led to increased proportions of NOZ cells in the G0/G1 phase and decreased proportions in the S phase (Figure 4G and H), suggesting that cell-cycle arrest and apoptosis contributed considerably to inhibition of cell proliferation.
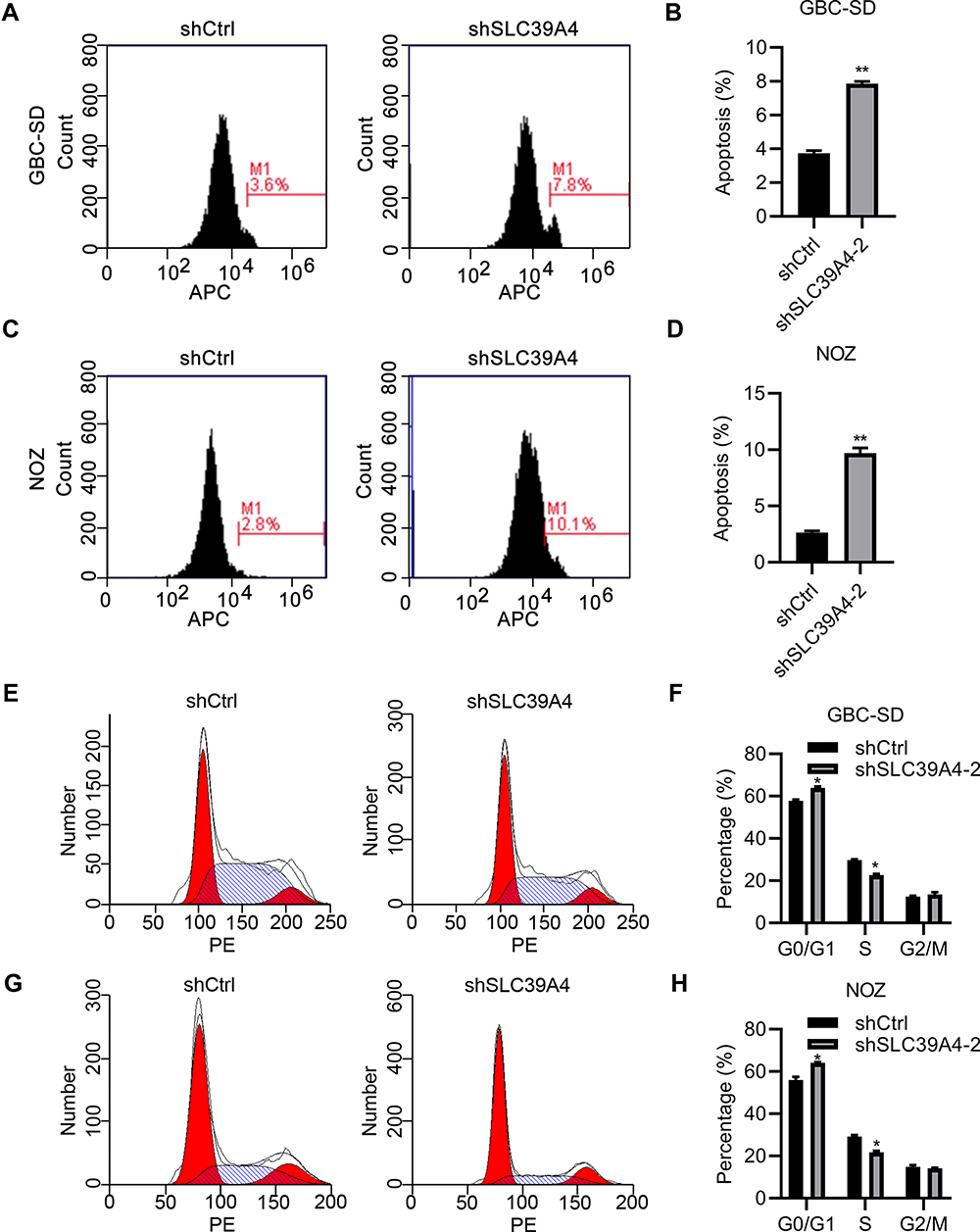 |
Figure 4 Knockdown of SLC39A4 expression induced apoptosis and cell-cycle arrest. (A–D) Apoptosis analyses were done in GBC-SD cells and NOZ cells using FACS (A and C), and apoptotic percentages were calculated by normalizing values to the total number of cells (B and D). Data are the mean ± SD (unpaired t-test). **p<0.01, compared with NOZ/shCtrl or GBC-SD/shCtrl. (E–H) Cell-cycle distributions were analyzed by FACS (E and G) and quantitation are shown in (F and H), after normalization to the total cell number. Data are the mean ± SD (unpaired t-test). *p < 0.05, compared with control cells. |
Overexpression of SLC39A4 Promoted the Proliferation, Cycle Progression, and Migration of Cells
To exclude the off-target effects of shRNA knockdown, we overexpressed SLC39A4 in GBC-SD cells and NOZ cells to observe the growth and migration of cells. SLC39A4 was efficiently overexpressed in GBC-SD and NOZ cells (Figure 5A). As expected, GBC-SD cells and NOZ cells transfected with exogenous SLC39A4 exhibited enhanced viabilities (Figure 5B). The proportion of GBC-SD cells in the G0/G1 phase showed an obvious decline, but an increase in the proportion of GBC-SD cells in the S and G2/M phases after SLC39A4 overexpression was documented (Figure 5C and D). In SLC39A4-overexpressed NOZ cells, the number of G0/G1-phase cells also showed a decline but their number in the G2/M phase increased (Figure 5E and F). These results indicated that the division activity of GBC-SD cells was motivated considerably by increased expression of SLC39A4. Besides, more GBC-SD cells and NOZ cells with SLC39A4 overexpression transmigrated than cells with the control vector (Figure 5G–J).
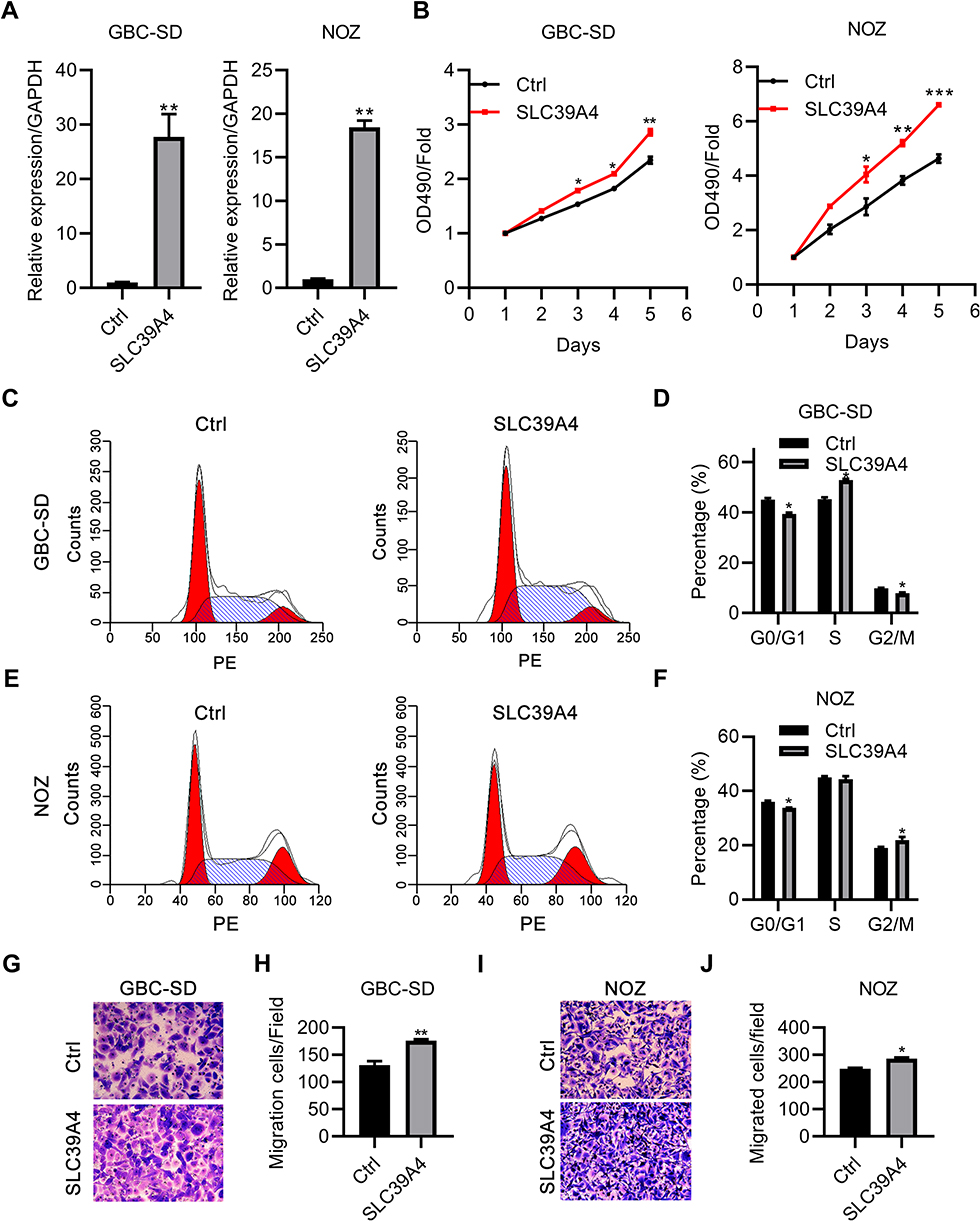 |
Figure 5 Overexpression of SLC39A4 promoted the growth and migration of cells. (A) Real-time RT-qPCR was done to confirm expression of exogenous SCL39A4 in GBC-SD cells and NOZ cells. mRNA expression was normalized to that of GAPDH. (B) MTT assays were undertaken to evaluate cell proliferation. (C–F) Cycle distributions were analyzed in GBC-SD cells and NOZ cells overexpressing SLC39A4 by FACS (C and E) and data are presented in (D and F) normalized to the total cell number. (G–J) Transwell-migration assays were conducted to observe the transmigration capability of NOZ/SLC39A4 cells and GBC-SD/SLC39A4 cells. (G and I) Representative images (magnification, ×200) and (H and J) quantitation are shown. Data are the mean ± SD (unpaired t-test). *p < 0.05, **p < 0.01, ***p < 0.001, compared with control cells. |
Knockdown of SLC39A4 Expression Repressed Tumor Formation in Nude Mice
SLC39A4 expression was related positively to the proliferation and colony formation of GBC cells in vitro. Hence, we proceeded to discover if SLC39A4 expression could affect tumor formation in vivo. After subcutaneous injection of control cells or NOZ/shSLC39A4 cells, xenografts were allowed to grow freely up to 18 days. As expected, tumors derived from NOZ/shSLC39A4 cells were restrained obviously compared with those in control groups (Figure 6A and B), and the average weight of tumors was also lighter (Figure 6C). In accordance with these findings, expression of c-Met and Ki-67 was downregulated in tumor tissues of mice with knockdown of SLC39A4 expression (Figure 6D). These results suggested that SLC39A4 might serve to potentiate tumorigenesis.
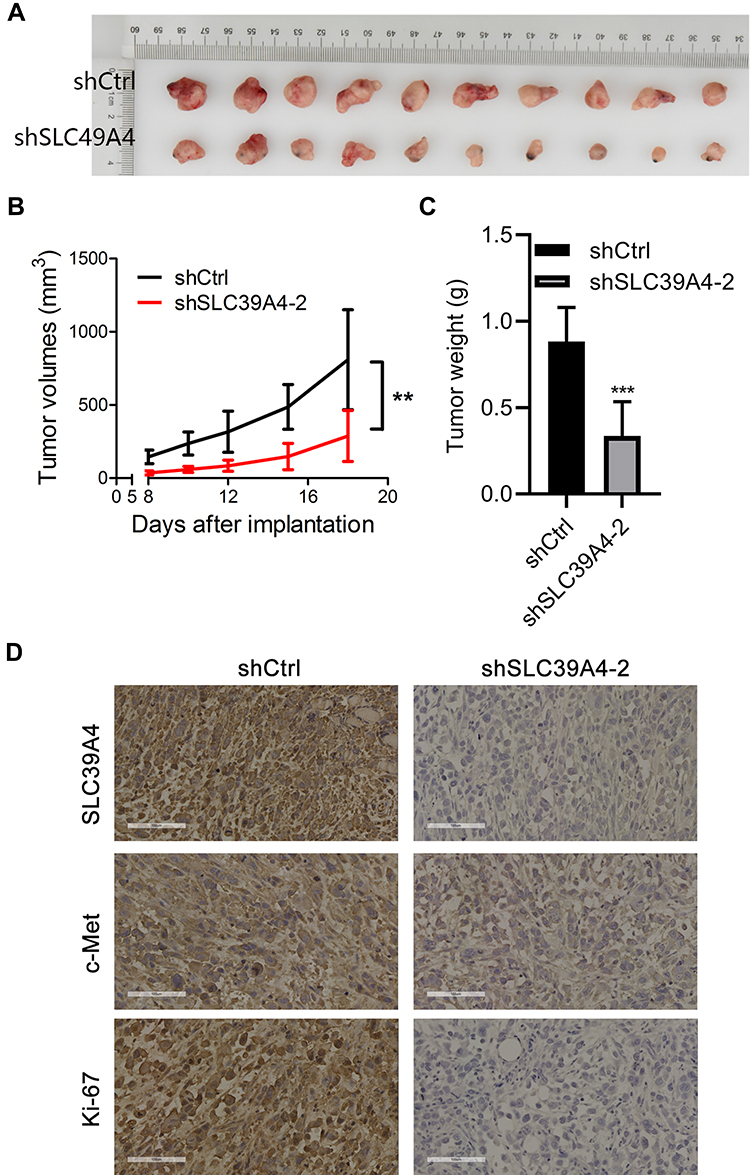 |
Figure 6 Knockdown of SLC39A4 expression repressed tumor formation in nude mice. (A) Surgically excised tumor tissue 18 days after initial implantation. (B) Growth curves of subcutaneously implanted tumors. Tumor volumes were measured at the indicated timepoints. (C) Average weight of implanted tumors in NOZ/shSLC39A4-2 xenograft-bearing mice. Data are the mean ± SD (unpaired t-test). **p < 0.01, ***p < 0.001, compared with NOZ/shCtrl. (D) IHC analyses of SLC39A4, c-Met and Ki-67 in tumors of control and shSLC39A4-2 groups. Scale bar, 100 μm. |
Downregulation of SLC39A4 Expression Influenced the Signaling Pathways Involved in Tumor Progression
Based on the results detailed above, we tried to identify the signaling pathways that were affected by SLC39A4 insufficiency. As expected, expression of genes that regulated cell proliferation, such as EGFR, CDK4 and c-MET, was decreased (Figure 7A and B).19,20 Besides, expression of genes that promote metastasis and vascular formation, such as GJA1 and SYK, was repressed (Figure 7A and B).21–23 In particular, expression of VEGFC, a positive regulator of lymph-node metastasis and lymphangiogenesis,24,25 was downregulated in response to SLC39A4 absence (Figure 7B). Inflammation and modulation of the immune microenvironment have key roles in influencing tumor progression.26,27 RUNX3 aids in the control of immunity and inflammation.28 After silencing of SLC39A4 expression, RUNX3 expression was reduced (Figure 7A). Also, RUNX3 has been found to correlate with anchorage-independent growth in pancreatic cancer cells.29 Moreover, expression of BMP4 and SGK1 was downregulated (Figure 7A and B), both of which have been found to favor tumor-cell survival and whose inhibition could lead to apoptosis.30–32 All the above data illustrated that ZIP4 deficiency could affect the different signaling pathways involved in tumor progression.
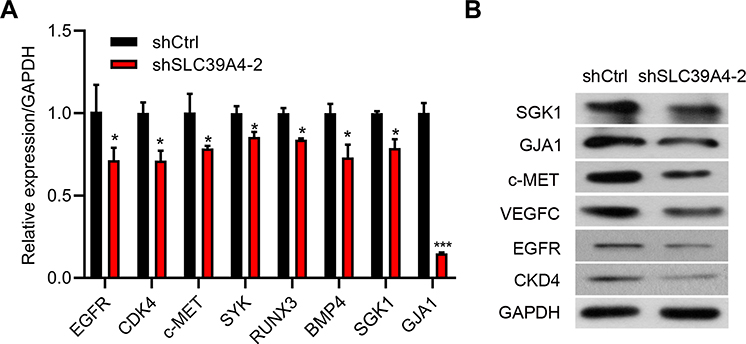 |
Figure 7 Downregulation of SLC39A4 expression influenced the malignant-development potential of GBC-SD cells. (A) mRNA expression of target genes in GBC-SD/shSLC39A4-2 cells by real-time RT-qPCR. Results are representative of three independent experiments, and are shown as the mean ± SD (unpaired t-test). *p < 0.05, ***p < 0.001, compared with GBC-SD/shCtrl. (B) Protein expression of indicated target genes in GBC-SD/shSLC39A4-2 cells by Western blotting. |
Discussion
The gallbladder stores bile and is located under the liver. GBC is the most common malignancy of the biliary tract, and often occurs in women. There are limited treatment options for GBC because it is often detected late and specific targets for therapy have not been identified.2 Therefore, obtaining specific diagnostic markers for GBC is very important to better understand and treat this disease. However, little is known about the mechanisms of GBC.4
Zinc is an essential mineral for life. It is indispensable for most enzymes to carry out catalytic activities and for nucleic acids to be synthesized. Zinc inadequacy leads to growth retardation, impaired immune function, and delayed healing of wounds.5–7,9 Conversely, excess zinc can cause disorders. For instance, a large intake of zinc results in low copper status and reductions in the levels of high-density lipoproteins.8,10 Moreover, the zinc level has been found to be increased in some tumor cells.33 As a result, zinc concentration must be controlled tightly.
In humans, intracellular zinc homeostasis is regulated by two major zinc transporter families: the SLC30 (ZnT) family and SLC39 (ZIP) family.12 The latter is responsible for zinc influx and could play a crucial part in malignancies in humans.33,34 For instance, SLC39A6 promotes the proliferation and invasion, and inhibits apoptosis, in esophageal squamous cell carcinoma (ESCC) cells.35 Suppression of SLC39A7 can abrogate survival of colorectal cancer cells.36 Also, in metastatic breast cancer, SLC39A10 expression is correlated positively with lymph-node metastasis.37 Expression of SLC39A4 (which encodes ZIP4) has been reported to be correlated positively with progression of pancreatic cancer.13,38 Also, activated ZIP4 inhibits apoptosis of HCC cells and enhances their cell cycle and migration.17 In NSCLC, SLC39A4 expression has been shown to be associated with strengthened cell migration, cisplatin resistance, and poor survival.14 SLC39A4 could even serve as a prognostic marker in ESCC.15 All the observations mentioned above indicate that SLC39A4 could be used as a therapeutic target for human cancers. We wished to determine the effects of SLC39A4 on proliferation of GBC cells in vitro and in vivo, as well as the molecular signaling mechanisms involved. We discovered that downregulation of SLC39A4 expression could repress the growth and migration of cells significantly, as well as tumor formation in nude mice. Taken together, these results implied an oncogenic character for SLC39A4 in GBC cells.
Gene expression analysis showed that SLC39A4 regulated the expression of VEGFC and SYK. The latter is downstream of SRC kinases and VEGFC is the ligand of VEGFR3. SYK is involved in vascular development, and VEGFC has been shown to promote lymphangiogenesis and lymph-node metastasis.24,25,39 These phenomena are consistent with the effects of SLC39A10 mentioned above.37 c-MET and CDK4 are important oncogenes which regulate GBC cell survival, metastasis and cell cycle.40–42 Combination of CDK4/6 and c-MET inhibitor have been applied for the treatment of glioblastoma.43 Here, we showed that CDK4 and c-MET were regulated by SLC39A4. Both mRNA and protein abundance of CDK4 and c-MET was reduced in GBC cells with silenced SLC39A4. Thus, we proposed that SLC39A4 knockdown suppressed GBC growth and migration partly through downregulation of CDK4 and c-MET.
Owing to inhibition of ZIP4 expression, zinc concentrations may change in the cytosol of tumor cells. We found that various aspects of the behaviors and signaling pathways of GBC cells were influenced if SLC39A4 expression was downregulated. Considering zinc as a vital element for numerous enzymes to function normally, we wondered if these outcomes were caused by changes in the zinc concentration, or if ZIP4 participated in these pathways directly. Additional investigations are needed to clarify the exact roles of ZIP4 in GBC.
Conclusions
We demonstrated an oncogenic potential for SLC39A4. Suppression of SLC39A4 expression in GBC cells weakened the viability and motility of GBC cells, and tumor formation in nude mice. Conversely, high expression of SLC39A4 could promote the growth and migration of tumor cells. Based on these findings, we speculate that SLC39A4 is very likely to be a novel target for developing new strategies for treating GBC. Importantly, this study may help us gain some novel insights of the molecular events triggering GBC progression.
Acknowledgments
The authors were supported by the National Natural Science Foundation of China (81600630; 81872352), Most Important Clinical Medical Center and Key Discipline Construction of Shanghai (2017ZZ02007); and Shanghai Sailing Program (20YF1407300). The funders had no role in study design, data collection/analyses, decision to publish, or manuscript preparation.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hickman L, Contreras C. Gallbladder cancer: diagnosis, surgical management, and adjuvant therapies. Surg Clin North Am. 2019;99(2):337–355. doi:10.1016/j.suc.2018.12.008
2. Javle M, Zhao H, Abou-Alfa GK. Systemic therapy for gallbladder cancer. Chin Clin Oncol. 2019;8(4):44. doi:10.21037/cco.2019.08.14
3. Sharma A, Sharma KL, Gupta A, Yadav A, Kumar A. Gallbladder cancer epidemiology, pathogenesis and molecular genetics: recent update. World J Gastroenterol. 2017;23(22):3978–3998. doi:10.3748/wjg.v23.i22.3978
4. Schmidt MA, Marcano-Bonilla L, Roberts LR. Gallbladder cancer: epidemiology and genetic risk associations. Chin Clin Oncol. 2019;8(4):31. doi:10.21037/cco.2019.08.13
5. Hojyo S, Fukada T. Roles of zinc signaling in the immune system. J Immunol Res. 2016;2016:6762343. doi:10.1155/2016/6762343
6. John E, Laskow TC, Buchser WJ, et al. Zinc in innate and adaptive tumor immunity. J Transl Med. 2010;8:118. doi:10.1186/1479-5876-8-118
7. Fukunaka A, Fujitani Y. Role of zinc homeostasis in the pathogenesis of diabetes and obesity. Int J Mol Sci. 2018;19:2. doi:10.3390/ijms19020476
8. Hooper PL, Visconti L, Garry PJ, Johnson GE. Zinc lowers high-density lipoprotein-cholesterol levels. JAMA. 1980;244(17):1960–1961. doi:10.1001/jama.1980.03310170058030
9. Prasad AS, Beck FW, Grabowski SM, Kaplan J, Mathog RH. Zinc deficiency: changes in cytokine production and T-cell subpopulations in patients with head and neck cancer and in noncancer subjects. Proc Assoc Am Physicians. 1997;109(1):68–77.
10. Willis MS, Monaghan SA, Miller ML, et al. Zinc-induced copper deficiency: a report of three cases initially recognized on bone marrow examination. Am J Clin Pathol. 2005;123(1):125–131. doi:10.1309/V6GVYW2QTYD5C5PJ
11. Hara T, Takeda TA, Takagishi T, Fukue K, Kambe T, Fukada T. Physiological roles of zinc transporters: molecular and genetic importance in zinc homeostasis. J Physiol Sci. 2017;67(2):283–301. doi:10.1007/s12576-017-0521-4
12. Baltaci AK, Yuce K. Zinc transporter proteins. Neurochem Res. 2018;43(3):517–530. doi:10.1007/s11064-017-2454-y
13. Cui X, Zhang Y, Yang J, et al. ZIP4 confers resistance to zinc deficiency-induced apoptosis in pancreatic cancer. Cell Cycle. 2014;13(7):1180–1186. doi:10.4161/cc.28111
14. Wu DM, Liu T, Deng SH, Han R, Xu Y. SLC39A4 expression is associated with enhanced cell migration, cisplatin resistance, and poor survival in non-small cell lung cancer. Sci Rep. 2017;7(1):7211. doi:10.1038/s41598-017-07830-4
15. Xia C, Chen X, Li J, Chen P. SLC39A4 as a novel prognosis marker promotes tumor progression in esophageal squamous cell carcinoma. Onco Targets Ther. 2020;13:3999–4008. doi:10.2147/OTT.S245094
16. Liu M, Yang J, Zhang Y, et al. ZIP4 promotes pancreatic cancer progression by repressing ZO-1 and claudin-1 through a ZEB1-dependent transcriptional mechanism. Clin Cancer Res. 2018;24(13):3186–3196. doi:10.1158/1078-0432.CCR-18-0263
17. Weaver BP, Zhang Y, Hiscox S, et al. Zip4 (Slc39a4) expression is activated in hepatocellular carcinomas and functions to repress apoptosis, enhance cell cycle and increase migration. PLoS One. 2010;5:10. doi:10.1371/journal.pone.0013158
18. Jin H, Liu P, Wu Y, et al. Exosomal zinc transporter ZIP4 promotes cancer growth and is a novel diagnostic biomarker for pancreatic cancer. Cancer Sci. 2018;109(9):2946–2956. doi:10.1111/cas.13737
19. O’Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. 2016;13(7):417–430. doi:10.1038/nrclinonc.2016.26
20. M V, S J, Vo E, et al. Dusp3 deletion in mice promotes experimental lung tumour metastasis in a macrophage dependent manner. PLoS One. 2017;12(10):e0185786. doi:10.1371/journal.pone.0185786
21. Moncayo G, Grzmil M, Smirnova T, et al. SYK inhibition blocks proliferation and migration of glioma cells and modifies the tumor microenvironment. Neuro Oncol. 2018;20(5):621–631. doi:10.1093/neuonc/noy008
22. Bonacquisti EE, Nguyen J. Connexin 43 (Cx43) in cancer: implications for therapeutic approaches via gap junctions. Cancer Lett. 2019;442:439–444. doi:10.1016/j.canlet.2018.10.043
23. Torres-Hernandez A, Wang W, Nikiforov Y, et al. Targeting SYK signaling in myeloid cells protects against liver fibrosis and hepatocarcinogenesis. Oncogene. 2019;38(23):4512–4526. doi:10.1038/s41388-019-0734-5
24. Chaudary N, Milosevic M, Hill RP. Suppression of vascular endothelial growth factor receptor 3 (VEGFR3) and vascular endothelial growth factor C (VEGFC) inhibits hypoxia-induced lymph node metastases in cervix cancer. Gynecol Oncol. 2011;123(2):393–400. doi:10.1016/j.ygyno.2011.07.006
25. Wang CA, Tsai SJ. The non-canonical role of vascular endothelial growth factor-C axis in cancer progression. Exp Biol Med. 2015;240(6):718–724. doi:10.1177/1535370215583802
26. Gupta SC, Kunnumakkara AB, Aggarwal S, Aggarwal BB. Inflammation, a double-edge sword for cancer and other age-related diseases. Front Immunol. 2018;9:2160. doi:10.3389/fimmu.2018.02160
27. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–867. doi:10.1038/nature01322
28. Lotem J, Levanon D, Negreanu V, et al. Runx3 in immunity, inflammation and cancer. Adv Exp Med Biol. 2017;962:369–393.
29. Rossi E, Bagala C, Inzani F, et al. RUNX3 as a potential predictor of metastasis in human pancreatic cancer. In Vivo. 2017;31(5):833–840. doi:10.21873/invivo.11136
30. Deng G, Zeng S, Qu Y, et al. BMP4 promotes hepatocellular carcinoma proliferation by autophagy activation through JNK1-mediated Bcl-2 phosphorylation. J Exp Clin Cancer Res. 2018;37(1):156. doi:10.1186/s13046-018-0828-x
31. Liu W, Wang X, Liu Z, et al. SGK1 inhibition induces autophagy-dependent apoptosis via the mTOR-Foxo3a pathway. Br J Cancer. 2017;117(8):1139–1153. doi:10.1038/bjc.2017.293
32. Ma J, Zeng S, Zhang Y, et al. BMP4 enhances hepatocellular carcinoma proliferation by promoting cell cycle progression via ID2/CDKN1B signaling. Mol Carcinog. 2017;56(10):2279–2289. doi:10.1002/mc.22681
33. Bafaro E, Liu Y, Xu Y, Dempski RE. The emerging role of zinc transporters in cellular homeostasis and cancer. Signal Transduct Target Ther. 2017;2. doi:10.1038/sigtrans.2017.29
34. Bogdan AR, Miyazawa M, Hashimoto K, Tsuji Y. Regulators of iron homeostasis: new players in metabolism, cell death, and disease. Trends Biochem Sci. 2016;41(3):274–286. doi:10.1016/j.tibs.2015.11.012
35. Cui XB, Shen YY, Jin TT, et al. SLC39A6: a potential target for diagnosis and therapy of esophageal carcinoma. J Transl Med. 2015;13:321. doi:10.1186/s12967-015-0681-z
36. Sheng N, Yan L, You W, et al. Knockdown of SLC39A7 inhibits cell growth and induces apoptosis in human colorectal cancer cells. Acta Biochim Biophys Sin. 2017;49(10):926–934. doi:10.1093/abbs/gmx094
37. Kagara N, Tanaka N, Noguchi S, Hirano T. Zinc and its transporter ZIP10 are involved in invasive behavior of breast cancer cells. Cancer Sci. 2007;98(5):692–697. doi:10.1111/j.1349-7006.2007.00446.x
38. Li M, Zhang Y, Bharadwaj U, et al. Down-regulation of ZIP4 by RNA interference inhibits pancreatic cancer growth and increases the survival of nude mice with pancreatic cancer xenografts. Clin Cancer Res. 2009;15(19):5993–6001.
39. Sun J, Zhang Z, S Y. Circ_RUSC2 upregulates the expression of miR-661 target gene SYK and regulates the function of vascular smooth muscle cells. Biochem Cell Biol. 2019;97(6):709–714. doi:10.1139/bcb-2019-0031
40. Qian L, Su H, Wang G, Li B, Shen G, Gao Q. Anti-tumor activity of bufalin by inhibiting c-MET mediated MEK/ERK and PI3K/AKT signaling pathways in gallbladder cancer. J Cancer. 2020;11(11):3114–3123. doi:10.7150/jca.38393
41. Moon W, Park H, Lee H, et al. Co-expression of cox-2, C-met and beta-catenin in cells forming invasive front of gallbladder cancer. Cancer Res Treatment. 2005;37(3):171–176. doi:10.4143/crt.2005.37.3.171
42. Feng Z, Chen J, Wei H, et al. The risk factor of gallbladder cancer: hyperplasia of mucous epithelium caused by gallstones associates with p16/CyclinD1/CDK4 pathway. Exp Mol Pathol. 2011;91(2):569–577. doi:10.1016/j.yexmp.2011.06.004
43. Olmez I, Zhang Y, Manigat L, et al. Combined c-Met/Trk inhibition overcomes resistance to CDK4/6 inhibitors in glioblastoma. Cancer Res. 2018;78(15):4360–4369. doi:10.1158/0008-5472.CAN-17-3124
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.