Back to Journals » Cancer Management and Research » Volume 12
Knockdown of Long Non-Coding RNA HOTAIR Suppresses Cisplatin Resistance, Cell Proliferation, Migration and Invasion of DDP-Resistant NSCLC Cells by Targeting miR-149-5p/Doublecortin-Like Kinase 1 Axis
Authors Zhan Y, Abuduwaili K, Wang X, Shen Y, Nuerlan S, Liu C
Received 16 January 2020
Accepted for publication 5 July 2020
Published 24 August 2020 Volume 2020:12 Pages 7725—7737
DOI https://doi.org/10.2147/CMAR.S246299
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lu-Zhe Sun
Yiyi Zhan, Kahaerjiang Abuduwaili, Xiuli Wang, Yanli Shen, Saiteer Nuerlan, Chunling Liu
The Second Department of Pulmonary Medicine, The Affiliated Tumor Hospital of Xinjiang Medical University, Urumqi, Xinjiang Uygur Autonomous Region, People’s Republic of China
Correspondence: Chunling Liu
The Second Department of Pulmonary Medicine, The Affiliated Tumor Hospital of Xinjiang Medical University, No. 789 Suzhou East Road, Urumqi 830011, Xinjiang Uygur Autonomous Region, People’s Republic of China
Tel +86-991-7819352
Email [email protected]
Background: Long non-coding RNA (lncRNA) HOTAIR has been reported to be associated with cisplatin (DDP) resistance in different human cancers including non-small cell lung cancer (NSCLC). However, the mechanism of HOTAIR in cisplatin resistance of NSCLC remains largely undefined.
Materials and Methods: Expression of HOTAIR, miR-149-5p and doublecortin-like kinase 1 (DCLK1) was detected using real-time quantitative PCR (RT-qPCR) and Western blotting. Cisplatin resistance was determined with cell counting kit (CCK)-8 assay and transwell assays in vitro, and xenograft tumor models in vivo. The target binding between miR-149-5p and either HOTAIR or DCLK1 was predicted on Diana Tools website, and confirmed by dual-luciferase reporter assay and RNA immunoprecipitation.
Results: Expression of HOTAIR was upregulated in DDP-resistant NSCLC tumor tissues and cell lines (A549/DDP and H1299/DDP). Knockdown of HOTAIR decreased the acquired cisplatin resistance of A549/DDP and H1299/DDP cells, as evidenced by attenuated 50% inhibitory concentration (IC50) of DDP, cell proliferation, migration and invasion in vitro, as well as tumor growth inhibition in vivo. Mechanically, HOTAIR negatively regulated miR-149-5p expression via targeting, and DCLK1 was a downstream target for miR-149-5p. DCLK1 was indirectly regulated by HOTAIR in DDP-resistant NSCLC cells as well. Functionally, miR-149-5p deletion could counteract the inhibitory effect of HOTAIR knockdown on cisplatin resistance; contrarily, restoring miR-149-5p exhibited the similar inhibition on cisplatin resistance in DDP-resistant cells in vitro, which was then abated by DCLK1 upregulation.
Conclusion: Knockdown of HOTAIR enhances DDP-resistant NSCLC cells to overcome cisplatin resistance partially via regulating miR-149-5p/DCLK1 axis.
Keywords: HOTAIR, NSCLC, cisplatin resistance
Introduction
Lung cancer is the leading cause of cancer-related death among males, and non-small-cell lung cancer (NSCLC) accounts for approximately 85% in all primary lung cancers.1 NSCLC is a type of heterogeneous tumor and classified into three different subtypes: squamous cell carcinoma, adenocarcinoma and large cell carcinoma.2 In terms of the treatment of NSCLC, platinum-based chemotherapy following surgical resection has been a standard strategy.3 Cisplatin (DDP), a platinum-containing compound, remains a reference standard for the first-line chemotherapy of multiple cancers including NSCLC. However, the clinical outcome remains disappointing in NSCLC patients, largely due to the acquired clinical resistance.4 Therefore, reducing drug resistance may be a promising approach for the treatment of DDP-resistant NSCLC patients.
Recently, accumulating evidence has reported the link between the dysregulation of long non-coding RNAs (lncRNAs) and drug resistance in cancers.5 LncRNAs are a type of transcripts with more than 200 nucleotides with little protein-coding capacity. Dysregulation of lncRNAs plays an essential role in the initiation and development of tumors.6 HOTAIR is one of the key lncRNAs that is found to be upregulated in various human cancers. Acting as one oncogene, HOTAIR is closely related to the resistance of chemotherapy drugs.7 In lung cancer, HOTAIR correlates with metastasis and poor prognosis in these patients, and acts as an oncogene in cell proliferation, metastasis and drug resistance.8 Nevertheless, the complete biological roles of HOTAIR, especially on drug resistance, in NSCLC are undisclosed.
HOTAIR can potentially regulate lung cancer through multiple mechanisms such as crosstalk with microRNAs (miRNAs), which has become an emerging light-spot in the non-coding world.9 MiRNAs are another group of noncoding transcripts with approximately 22 nucleotides. It has been well recognized about miRNAs as pivotal regulators of cisplatin resistance in lung cancers10 including NSCLC.11,12 MiRNA (miR)-149-5p was predicted as a novel potential target gene for HOTAIR according to DianaTools database in the present study. The linking between miR-149-5p and cisplatin resistance has already reported in several types of cancers13,14 including NSCLC.15
Doublecortin-like kinase 1 (DCLK1) is often overexpressed in human cancers including NSCLC, and takes part in tumorigenesis, metastasis and drug resistance.16–18 In this study, we detected the expression of HOTAIR, miR-149-5p and DCLK1 in DDP-resistant and -sensitive NSCLC tissues and cells. Then, 50% inhibitory concentration (IC50), cell proliferation, migration and invasion in vitro and tumor growth in vivo were analyzed to determine the effect of HOTAIR dysregulation on cisplatin resistance. Furthermore, the relationship among HOTAIR, miR-149-5p and DCLK1 in cisplatin resistance in NSCLC was confirmed.
Materials and Methods
Clinical Samples and Tissue Acquirement
Seventy patients with NSCLC were recruited from the Affiliated Tumor Hospital of Xinjiang Medical University, and the clinicopathological parameters of these NSCLC patients were presented in Table 1. All patients were received definitive chemotherapy with cisplatin after surgery. The NSCLC tumor tissues were acquired and directly preserved in liquid nitrogen during surgery. No enrolled patients in our study received anti-cancer therapy prior to surgery. According to the tracking survey, NSCLC tissue samples divided into two groups: DDP resistant (n=35) and DDP sensitive (n=35). DDP-resistant NSCLC was defined as tumor progression or recurrence within 6 months after the last DDP treatment, while those recurrence or progression more than 6 months were identified as DDP-sensitive NSCLC. This study was approved by the Ethics Committee of the Affiliated Tumor Hospital of Xinjiang Medical University and participators in the form of written informed consents.
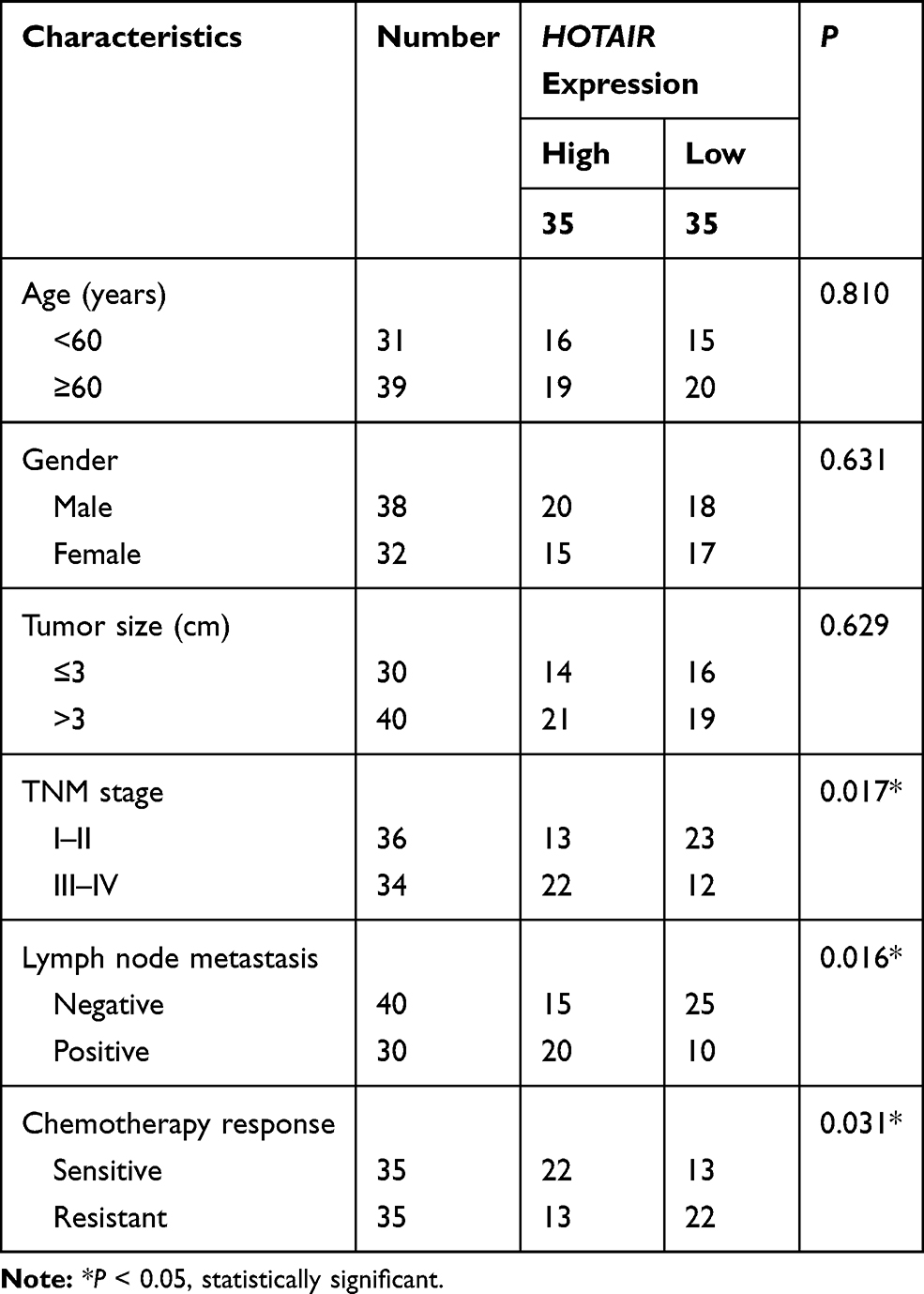 |
Table 1 Correlation Between HOTAIR Expression and Clinicopathological Parameters of Patients with NSCLC |
Cells and Cell Culture
Human NSCLC cell lines A549 (CCL-185) and H1299 (CRL-5803) were obtained from American Type Culture Collection (Manassas, VA, USA). All cells were grown up in Roswell Park Memorial Institute 1640 medium (RPMI-1640; Gibco, Grand Island, NY, USA) supplemented with 10% (v/v) fetal bovine serum (FBS; Gibco) at 37°C in 5% CO2.
Construction of DDP-Resistant NSCLC Cells in vitro
A549 and H1299 cells were prepared to forcedly acquired DDP resistance. The cells were pre-treated with stepwise increasing concentrations of DDP (Sigma-Aldrich Co., St Louis, MO, USA). To maintain the resistance phenotype of DDP-resistant A549 and H1299 cells, 5 μM DDP was additionally added into the RPMI medium for long-time culture. After the determination for 50% inhibitory concentration (IC50), these cells were named as A549/DDP and H1299/DDP.
Cell Transfection
For overexpression, miR-149-5p mimic and miR-NC mimic were purchased from GenePharma (Shanghai, China); the sequence of HOTAIR and coding domain sequence of DCLK1 were cloned into pcDNA3.1 (Invitrogen, Carlsbad, CA, USA), respectively. For knockdown, miR-149-5p inhibitor (in-miR-149-5p), siRNA against HOTAIR (si-HOTAIR), and their controls were obtained from GenePharma. Cell transfection was carried out with Lipofectamine™ 2000 (Invitrogen) according to the manufacturers’ instruction. After transfection for 24 h, A549/DDP and H1299/DDP cells were collected for further experiments. Sequences of siRNAs were as follows: si-HOTAIR: 5ʹ- GAACGGGAGUACAGAGAGAUU-3ʹ; si-NC: 5ʹ-GAACGGAGCGAGCAGACCUUU-3ʹ.
Cell Counting Kit (CCK)-8 Assay
For IC50 analysis, the parental cells and A549/DDP and H1299/DDP cells were seeded into 96-well plate (Corning, NY, USA) for overnight and then exposed to DDP (10, 20, 40, 60, 80, and 100 μM) for 48 h. 10 μL CCK-8 solution was added to each well and the cultures were incubated for another 4 h at 37°C. After Mixing on an orbital shaker for 5 min, optical density at 450 nm was recorded using a microplate reader. The experiments were conducted at least 3 times. The IC50 values were calculated by the relative dose–response survival curve. For cell viability assay, transfected A549/DDP and H1299/DDP cells were exposed to 60 μM DDP for 0, 24, 48 and 72 h. All the other operations were the same with IC50 analysis.
Real-Time Quantitative PCR (RT-qPCR)
Total RNA in tissues and cells was isolated with TRIzol (Invitrogen). The first-strand cDNA with SuperScript II reverse transcriptase (Invitrogen) with special stem-loop primer for miRNA and the quantitative PCR was performed with SYBR® Premix Ex Taq™ (Takara, Shiga, Japan) on Bio-Rad iQ5 real-time PCR detection system (Bio-Rad, Hercules, CA, USA). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and U6 small nuclear RNA (U6) were used as an internal control to HOTAIR, DCLK1 and miR-149-5p. The expression was calculated according to the comparative threshold (Ct) method as normalized to that of U6 or GAPDH (2−ΔCt) and the fold changes were calculated by the equation 2−ΔΔCt. The reactions were performed in triplicate for each sample and the primers involved were listed as follows: HOTAIR, 5ʹ-TGCTACTTGTGTAGACCCAG-3ʹ (sense) and 5ʹ-AGCAAAGGCTGGACCTTTGCT-3ʹ (anti-sense); miR-149-5p, 5ʹ-TCGGCAGGUCUGGCUCCGUGUC-3ʹ (sense) and 5ʹ-CCGAGGACGGGAGTG −3ʹ (anti-sense); DCLK1, 5ʹ-GGAGTGGTGAAACGCCTGTAC-3ʹ (sense) and 5ʹ-GGTTCCATTAACTGAGCTGG-3ʹ (anti-sense); GAPDH, 5ʹ-CCCCTTCATTGACCTCAACTACAT-3ʹ (sense) and 5ʹ-CGCTCCTGGAAGATGGTGA-3ʹ (anti-sense); U6, 5ʹ-TTCACGAATTTGCGTGTCAT-3ʹ (sense) and 5ʹ-CGCTCGGCAGCACATATAC-3ʹ (anti-sense).
Transwell Assay
The ability of cell migration and invasion was measured using Transwell chamber (8 μm pore size, Corning) with matrigel-free (for migration) or matrigel-coated (Bection Dickinson, Franklin Lakes, NJ, USA) (for invasion). Transfected A549/DDP and H1299/DDP cells (2 × 104 cells) in 200 μL serum-free medium were implanted in the upper chambers. The 500 μL medium containing 10% FBS was used as a chemo-attractant and loaded in the low chamber. Transwell system was stayed at 37°C for 24 h. After removing the cells on the top surface with a cotton swab, cells on the lower surface were stained with 0.1% crystal violet for 15 min at room temperature, followed with being photographed and counted under a microscope in five predetermined fields (×200). Three independent experiments were carried out.
Western Blotting
Total protein from cultured A549/DDP and H1299/DDP cells was isolated in RIPA lysis buffer (Beyotime, Shanghai, China). According to Bradford protein assay reagent (Bio-Rad), equal amounts of protein (20 μg) from each sample were loaded for the standard procedures of Western blot assay. β-actin was used to normalize the DCLK1 protein level. The primary antibodies including DCLK1 (#62,257, 1:1000) and β-actin (#4967, 1:1000) were purchased from Cell Signaling Technology (CST; Danvers, MA, USA), and Ki-67 (#ab197547, 1:500), cleaved caspase-3 (#ab49822, 1:500) were from abcam (Cambridge, UK).
Luciferase Reporter Assay and RNA Immunoprecipitation (RIP)
According to in cilico data, there were potential complementary binding sites of miR-149-5p in human HOTAIR and 3ʹUTR of DCLK1 (NM_004734.5). Then, the wild types of HOTAIR and 3ʹUTR of DCLK1 (HOTAIR-WT and DCLK1 3ʹUTR-WT) were separately cloned into psi-CHECK-2 vector (Invitrogen) using PCR methods, as well as their mutants HOTAIR-MUT and DCLK1 3ʹUTR-MUT. A549/DDP and H1299/DDP cells were co-transfected with miR-149-5p/NC mimic and either HOTAIR-WT/MUT or DCLK1 3ʹUTR-WT/MUT. After 24 h incubation, cells were harvested to measure Firefly and Renilla luciferase activities using the dual-luciferase reporter assay system (Promega, Madison, WI, USA). The transfections were repeated at least three times.
The RIP assay was performed in A549/DDP and H1299/DDP cells after the transfection of miR-149-5p/NC mimic. Magna RIPTM RNA-binding protein immunoprecipitation kit (Millipore-Sigma, Billerica, MA, USA) was chosen to enrich HOTAIR from the samples bound to the Ago2 or IgG antibody obeyed the standard instructions. The co-precipitated RNAs were detected by RT-qPCR.
Xenograft Mouse Model
Six-week athymic BALB/c mice were obtained from Shanghai SLAC Laboratory Animal Co. Ltd. The animal experiments were approved by The Institutional Review Board of the Affiliated Tumor Hospital of Xinjiang Medical University and were untaken in accordance with National Institutions of Health Guide for Care and Use of Laboratory Animals. H1299/DDP cells were stably transfected lentiviral particles encoding shRNA against HOTAIR (sh-HOTAIR; Neuron Biotech, Shanghai, China) or the negative control (sh-NC) using Polybrene reagent (Sigma). Equal numbers (106) of transfected H1299/DDP cells/0.2 mL were subcutaneously injected in subcutaneous area of flanks (5 mice per group) for 35 days. One week later after transplantation, xenograft mice were subjected to intra-peritoneal injection of DDP at a dose of 3.0 mg/kg body weight or phosphate buffer solution (PBS; pH 7.4) every 7 days from the 7th day. Xenograft experiments were divided into three groups: sh-NC+PBS, sh-HOTAIR+PBS, sh-NC+DDP, and sh-HOTAIR+DDP. The tumors were measured with a caliper once 7 days, and the mice were practiced with euthanasia on day 35. The tumor volume was calculated using the formula: 0.5 × l × w2 (l is the length of tumor and w is the width of tumor). And the weight of tumors was evaluated with electronic balance. Immediately, the tumors were frozen in −80°C for further isolation of total RNA.
Statistical Analysis
Statistics were analyzed by SPSS 21.0 (SPSS Inc, Chicago, IL, USA) and presented as the mean ± standard deviation. Unpaired Student’s t-test method was utilized for comparison between two groups, while one-way analysis of variance was used for data comparison in multiple groups. P < 0.05 was considered as statistically significant.
Results
HOTAIR Was Upregulated in DDP-Resistant NSCLC Tissues and Cells
To investigate the association of HOTAIR expression with NSCLC chemoresistance against DDP, RT-qPCR was carried out to confirm its expression in patients with NSCLC. As shown in Figure 1A, HOTAIR levels were significantly higher in DDP-resistant tumors (n=35) than DDP-sensitive tumors (n=35). In clinic, high HOTAIR level was significantly associated with TNM stage, lymph node metastasis and DDP response (Table 1). The DDP-resistant NSCLC cells (A549/DDP and H1299/DDP) in vitro were developed based on their parental cell lines (A549 and H1299). CCK-8 assay further confirmed this acquired cisplatin resistance, as depicted by the increased IC50 values of DDP in A549/DDP and H1299/DDP cells than that in parental cells (Figure 1B and C). To further testify whether HOTAIR plays a critical role in the acquired DDP resistance of NSCLC cells, the expression of HOTAIR was also detected utilizing RT-qPCR. Expectedly, HOTAIR was upregulated in A549/DDP and H1299/DDP cells (Figure 1D). These data showed an increase of HOTAIR in DDP-resistant NSCLC tissues and cells.
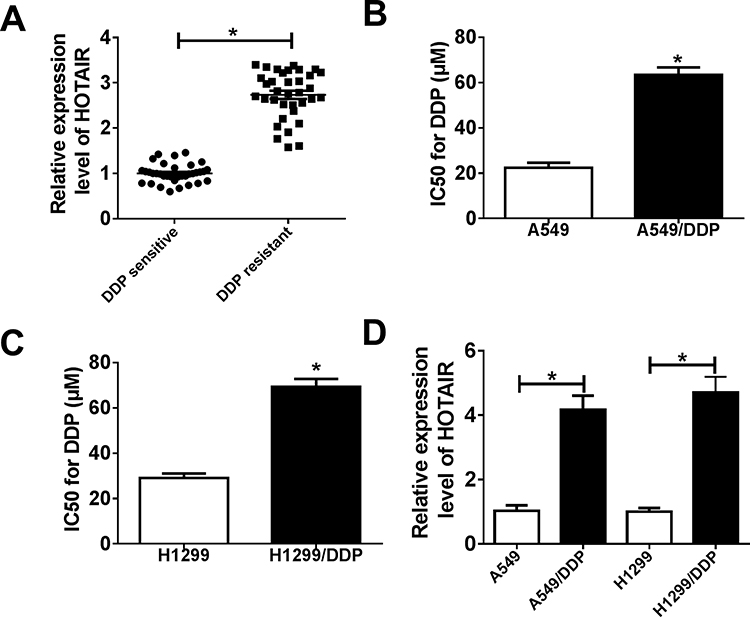 |
Figure 1 Expression of lncRNA HOTAIR (HOTAIR) in cisplatin (DDP)-resistant non-small cell lung cancer (NSCLC) tissues and cells. (A) Real-time quantitative PCR (RT-qPCR) assay showed the expression levels of HOTAIR in DDP-resistant and -sensitive NSCLC tissues. (B, C) The 50% inhibitory concentration (IC50) values of DDP were determined by Cell Counting Kit (CCK)-8 assay in A549/DDP and H1299/DDP cells with their parental cells. The cells were exposed to different concentrations (10, 20, 40, 60, 80, and 100 μM) of DDP for 48 h. (D) RT-qPCR assay showed HOTAIR levels in A549/DDP and H1299/DDP cells with their parental cells. *P < 0.05. |
HOTAIR Knockdown Inhibited Cisplatin Resistance in DDP-Resistant NSCLC Cells in vitro
The role of HOTAIR in cisplatin resistance was determined in loss-of-function experiments. The siRNA against HOTAIR was used in A549/DDP and H1299/DDP, and RT-qPCR confirmed the transfection efficiency (Figure 2A). Secondly, drug resistance was assessed by IC50 value, cell proliferation, migration and invasion. The CCK-8 results showed that si-HOTAIR1 could significantly decrease the IC50 of DDP in A549/DDP and H1299/DDP cells (Figure 2B), as well as cell proliferation (Figure 2C and D); the ability of cell migration and invasion analyzed by transwell assays was lowered by si-HOTAIR transfection for 24 h (Figure 2E and F). Thus, knockdown of HOTAIR could reverse the high IC50 of DDP, cell proliferation, migration and invasion in A549/DDP and H1299/DDP cells. On the contrary, overexpression of HOTAIR via pcDNA vector transfection led to increased malignant behaviors of the parental cells (A549 and H1299), as evidenced by increased IC50 value of DDP, cell proliferation, migration and invasion (Supplementary Figure 1A–F).
 |
Figure 2 Effects of HOTAIR knockdown on cisplatin resistance in NSCLC cells. A549/DDP and H1299/DDP cells were transfected with siRNA against HOTAIR (si-HOTAIR) or its negative control (si-NC). (A) RT-qPCR assay showed HOTAIR expression levels after transfection for 24 h. (B) IC50 values of DDP were determined by CCK-8 assay after treated with different concentrations (10, 20, 40, 60, 80, and 100 μM) of DDP for 48 h. (C, D) CCK-8 assay detected cell viability after transfection for 0, 24, 48 and 72 h. (E, F) Transwell assays measured cell migration and invasion after transfection for 24 h. The cell migration/invasion ability was calculated as % of total cells. *P < 0.05. |
miR-149-5p Sponged by HOTAIR Was Downregulated in DDP-Resistant NSCLC Tissues and Cells
A possible target miRNAs of HOTAIR were retrieved on DianaTools website, and miR-149-5p was further identified to be highly expressed in HOTAIR-silenced A549/DDP and H1299/DDP cells (Figure 3A). The sequences of the putative binding site of miR-149-5p in HOTAIR-WT were mutated as the complementary sequences (Figure 3B). Next, a dual-luciferase reporter assay was performed to show that miR-149-5p mimic significantly diminished luciferase activity of HOTAIR-WT in both A549/DDP and H1299/DDP cells (Figure 3C and D); whereas there was little influence on the luciferase activity of HOTAIR-MUT whenever transfected with miR-149-5p mimic or miR-NC mimic. In addition, RIP assay was carried out to further verify this target binding. As a result, HOTAIR was abundantly enriched by Ago2 in A549/DDP and H1299/DDP cells with miR-149-5p overexpression (Figure 3E). These results indicated that miR-149-5p was sponged by HOTAIR via targeting. Moreover, expression of miR-149-5p was lower in DDP-resistant NSCLC tissues and cells (Figure 3F and G); its expression was downregulated in A549/DDP and H1299/DDP cells transfected with pcDNA-HOTAIR and upregulated with si-HOTAIR transfection (Figure 3H).
 |
Figure 3 Identification of the negative regulatory relationship between HOTAIR and miR-149-5p. (A) RT-qPCR assay measured expression levels of miRNAs in A549/DDP and H1299/DDP cells transfected with si-HOTAIR or si-NC. (B) The predicted hsa-miR-149-5p binding sites in HOTAIR according to DianaTools. The corresponding sequence in the mutated version was shown as well. (C, D) Luciferase activity of HOTAIR wild type (HOTAIR-WT) and mutant (HOTAIR-MUT) was examined by dual-luciferase reporter assay in A549/DDP and H1299/DDP cells when transfected with miR-149-5p mimic (miR-149-5p) or miR-NC mimic (miR-NC). (E) Expression of HOTAIR was detected with RNA immunoprecipitation (RIP) assay in A549/DDP and H1299/DDP cells transfected with miR-149-5p/NC. The enrichment of HOTAIR level was showed as % of input. (F, G) RT-qPCR assay showed the expression levels of miR-149-5p in DDP-resistant NSCLC tissues and cells (A549/DDP and H1299/DDP), compared to DDP-sensitive NSCLC tissues and parental cells. (H) RT-qPCR assay measured miR-149-5p expression levels in A549/DDP and H1299/DDP cells transfected with pcDNA-HOTAIR (HOTAIR), si-HOTAIR or their controls. *P < 0.05. |
Reduced miR-149-5p Counteracted the Inhibitory Effect of HOTAIR Knockdown on Cisplatin Resistance
Then, a series of rescue experiments were performed to testify the occurrence of HOTAIR/miR-149-5p axis. A549/DDP and H1299/DDP cells were transfected with si-HOTAIR or si-NC and co-transfected with si-HOTAIR and in-miR-149-5p or in-miR-NC. As depicted in Figure 4A, si-HOTAIR transfection led to increased expression of miR-149-5p, whereas this upregulation was then impaired by in-miR-149-5p transfection. CCK-8 assay demonstrated that si-HOTAIR-mediated the decrease of IC50 of DDP (Figure 4B) and cell proliferation (Figure 4C and D in A549/DDP and H1299/DDP cells) were apparently improved by introducing in-miR-149-5p. Consistently, si-HOTAIR-induced inhibition on cell migration and invasion was attenuated after in-miR-149-5p transfection (Figure 4E and F). These outcomes showed that blocking miR-149-5p could counteract the inhibitory effect of HOTAIR knockdown on acquired cisplatin resistance in A549/DDP and H1299/DDP cells, suggesting that HOTAIR modulated cisplatin resistance in DDP-resistant NSCLC cells via at least partially targeting miR-149-5p.
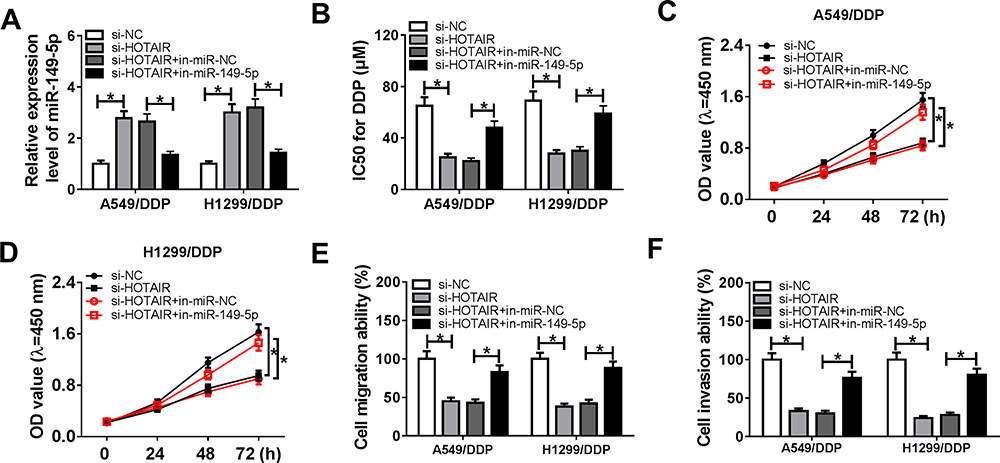 |
Figure 4 Influence of miR-149-5p reduction on the inhibitory effect of HOTAIR knockdown on cisplatin resistance. A549/DDP and H1299/DDP cells were transfected with si-HOTAIR or si-NC, and co-transfected with si-HOTAIR and miR-149-5p/NC inhibitor (in-miR-149-5p/NC). (A) Expression levels of miR-149-5p were detected by RT-qPCR after transfection. CCK-8 assay detected (B) IC50 values of DDP after transfection for 48 h and (C, D) cell viability after transfection for 0, 24, 48 and 72 h. (E, F) Transwell assays measured cell migration and invasion after transfection for 24 h. *P < 0.05. |
DCLK1 Was Positively Regulated by HOTAIR via miR-149-5p
We suspected DCLK1 as one target gene of miR-149-5p according to DianaTools website, and the putative binding sites between DCLK1 and miR-149-5p were presented (Figure 5A). Dual-luciferase reporter assay further identified this potential target binding, as evidenced by the decline of luciferase activity in A549/DDP and H1299/DDP cells co-transfected with DCLK1 3ʹUTR-WT and miR-149-5p mimic (Figure 5B and C). Expression of DCLK1 was measured by RT-qPCR and DCLK1 mRNA levels were higher in DDP-resistant NSCLC tissues and cells (Figure 5D and E); moreover, its protein level analyzed by Western blotting was downregulated in A549/DDP and H1299/DDP cells transfected with miR-149-5p mimic and upregulated with in-miR-149-5p transfection or pcDNA-HOTAIR transfection (Figure 5F and G); meanwhile, HOTAIR overexpression-mediated upregulation of DCLK1 was further attenuated with the presence of miR-149-5p mimic. These results indicated that HOTAIR sponging miR-149-5p indirectly regulated DCLK1 expression in DDP-resistant NSCLC.
 |
Figure 5 Verification of the target relationship between miR-149-5p and doublecortin-like kinase 1 (DCLK1). (A) The predicted hsa-miR-149-5p binding sites in DCLK1 3ʹ untranslated regions (3ʹUTR) according to DianaTools. The corresponding sequence in the mutated version was shown as well. (B, C) Luciferase activity of DCLK1 3ʹUTR wild type (DCLK1 3ʹUTR-WT) or mutant (DCLK1 3ʹUTR-MUT) in A549/DDP and H1299/DDP cells transfected with miR-149-5p or miR-NC. (D, E) RT-qPCR assay showed the expression levels of DCLK1 mRNA in DDP-resistant NSCLC tissues and cells (A549/DDP and H1299/DDP), compared to DDP-sensitive NSCLC tissues and parental cells. (F, G) Western blotting measured DCLK1 protein expression levels in A549/DDP and H1299/DDP cells when transfected with miR-149-5p, in-miR-149-5p, pcDNA-HOTAIR, si-HOTAIR, or their controls. β-actin was detected as the internal reference. *P < 0.05. |
Elevated DCLK1 Abated the Suppressive Effect of miR-149-5p Overexpression on Cisplatin Resistance in DDP-Resistant NSCLC Cells in vitro
The role of miR-149-5p in cisplatin resistance in NSCLC cells was researched, as well as the presence of miR-149-5p/DCLK1 axis. A549/DDP and H1299/DDP cells were transfected with miR-149-5p/NC mimic and co-transfected with miR-149-5p mimic and pcDNA-DCLK1 or pcDNA empty vector. As depicted in Figure 6A and B, IC50 of DDP was decreased with miR-149-5p mimic transfection accompanied with downregulated DCLK1 protein. Cell proliferation of A549/DDP and H1299/DDP cells was diminished by miR-149-5p mimic transfection (Figure 6C and D), as well as the ability of cell migration and invasion (Figure 6E and F). More importantly, the inhibition of miR-149-5p overexpression on DCLK1 expression, IC50 of DDP, cell proliferation, migration and invasion was overall significantly abated in A549/DDP and H1299/DDP cells co-transfected with pcDNA-DCLK1 (Figure 6A–F). These data showed a suppressive role of miR-149-5p overexpression in cisplatin resistance in DDP-resistant NSCLC cells in vitro partially through downregulating DCLK1.
 |
Figure 6 Influence of DCLK1 elevation on the role of miR-149-5p in cisplatin resistance. A549/DDP and H1299/DDP cells were transfected with miR-149-5p or miR-NC, and co-transfected with miR-149-5p and pcDNA-DCLK1 (DCLK1) or pcDNA. (A) Expression levels of DCLK1 were detected by Western blotting after transfection. CCK-8 assay detected (B) IC50 values of DDP after transfection for 48 h and (C, D) cell viability after transfection for 0, 24, 48 and 72 h. (E, F) Transwell assays measured cell migration and invasion after transfection for 24 h. *P < 0.05. |
Knockdown of HOTAIR Inhibited Tumor Growth and Cisplatin Resistance of DDP-Resistant NSCLC Cells in vivo
To confirm the effects of HOTAIR on cisplatin resistance of NSCLC cells in vivo, H1299/DDP cells stably infected with sh-HOTAIR or sh-NC was subcutaneously injected into BALB/c nude mice (n=5), followed with DDP (3.0 mg/kg body weight) administration or PBS treatment. As shown in Figure 7A and B, xenograft tumor was generated after implantation for 7 days; sh-HOTAIR extremely decreased tumor volume and tumor weight in both groups treated with DDP or PBS. RT-qPCR analysis clarified that sh-HOTAIR transfection resulted in lower HOTAIR and DCLK1 expression, and higher miR-149-5p expression in xenograft tumor tissues, accompanied with downregulated Ki-67 and upregulated cleaved caspase-3 (Figure 7C and D). Collectively, these results implicated that HOTAIR knockdown could suppress cisplatin resistance of H1299/DDP cells in vivo partially through upregulating miR-149-5p and downregulating DCLK1.
 |
Figure 7 Knockdown of HOTAIR inhibited tumor growth of H1299/DDP cells in vivo. H1299/DDP cells were lentiviral infected with short hairpin RNA against HOTAIR (sh-HOTAIR) or its negative control sh-NC prior to injection into BALB/c nude mice (n=5). Xenograft tumors were exposed to DDP (3.0 mg/kg body weight) or phosphate buffer solution (PBS; pH 7.4) every 7 days from 7th day after transplantation. (A) The volumes were calculated every week and the growth curve was drawn. (B) Tumor weight was recorded on day 35 after transplantation. (C) Expression of HOTAIR, miR-149-5p and DCLK1 was confirmed in xenograft tumors using RT-qPCR. (D) Western blotting evaluated protein levels of DCLK1, Ki-67 and cleaved caspase-3. *P < 0.05. |
Discussion
Previous studies have shown dysregulation of lncRNAs participate in tumor progression and chemoresistance.19 HOTAIR contributes to cisplatin resistance of NSCLC through several mechanisms such as downregulating p21,20 upregulating Kruppel-like factor 4,21 targeting miR-326/specificity protein 1 axis,22 and activating Wnt signaling pathway.23 In the present study, we observed the upregulation of HOTAIR in DDP-resistant NSCLC tissues and investigated the promoting effect of HOTAIR on acquired cisplatin resistance in NSCLC cells through targeting miR-149-5p/DCLK1 axis.
HOTAIR takes part in different drug resistance of NSCLC cells. For instance, silencing of HOTAIR decreased crizotinib resistance by suppressing the phosphorylation of ULK1 in inactivating autophagy.24 Liu et al25 indicated that elevated HOTAIR upregulated expression of KLF4, tumor stem cell-related biomarkers, which might lead to cisplatin resistance in A549/DDP cells. Liu et al26 reported that the HOTAIR downregulation could restore gefitinib sensitivity through activating Bax/Caspase3 pathway and suppressing TGFα/EGFR signaling. HOTAIR targeting miRNAs has been a well-documented mechanism in regulating cisplatin resistance of human cancers. For example, HOTAIR targeted miRNA-12627 or miRNA-34a28 to promote cisplatin resistance in gastric cancer cells. It was depicted that miRNA-326 targeting SP1 was declared to reverse chemoresistance of A549/DDP cells both in vitro and in vivo, which was mediated by HOTAIR repression.22 Here, we noticed a decrease in cell viability, migration and invasion, as well as attenuated IC50 values and tumor growth in A549/DDP and H1299/DDP cells after HOTAIR was knocked down. Besides, overexpressing HOTAIR could induce A549 and H1299 cells to acquire cisplatin resistance as well. These outcomes suggested a promoting role of HOTAIR in cisplatin resistance in NSCLC. Besides, the anti-growth and anti-metastasis properties of HOTAIR deficiency in A549 and H1299 cells both in vitro and in vivo as well.24,29,30 Notably, luciferase reporter assay and RIP assay testified miR-149-5p was a novel target for HOTAIR. Thus, our results implied that inhibiting HOTAIR/miR-149-5p axis contributed to reverse chemoresistance in DDP-resistant NSCLC cells.
In NSCLC, six miRNAs including miR-149-5p and nine target genes were validated to distinguish squamous cell carcinoma and adenocarcinoma,31 which are two different subtypes of NSCLC. According to the miRNA expression profile, expression of miR-149-5p was observed to be downregulated in NSCLC tissues and cells than normal controls.32 Functionally, Zhao et al33 demonstrated that miR-149-5p inhibited tumor growth, epithelial–mesenchymal transition (EMT) phenotype, invasion and metastasis in NSCLC by negatively modulating Forkhead box M1/cyclin D1/MMP2 axis. In terms of the relationship between miR-149-5p and chemoresistance, miR-149-5p downregulation could effectively attenuate gefitinib resistance, as described by decreased cell viability and colony formation ability, and increased apoptosis rate and caspase 3 expression.34 MiR-149-5p was also declared to be linked to cisplatin-vinorelbine response and progression-free survival in NSCLC patients.35 However, the part of miR-149-5p in chemoresistance of many chemotherapy drugs including cisplatin in NSCLC remains largely unknown. Herein, we wondered whether miR-149-5p was complicated in cisplatin resistance in DDP-resistant NSCLC cells. Expression level of miR-149-5p was lower in drug-resistant NSCLC tissues and cells, which is in consistent with the previous study.32,33,36 Its forced high expression could decrease IC50 of DDP, cell proliferation, migration and invasion of A549/DDP and H1299/DDP cells. Our data implied that miR-149-5p upregulation could descend the acquired DDP resistance in NSCLC cells in vitro, which supports the announcement of miR-149-5p role in cisplatin resistance in ovarian cancer,13 esophageal cancer14 and gastric cancer.37 Furthermore, we testified that the suppressive effect of miR-149-5p in cisplatin resistance in NSCLC cells probably via directly interacting with upstream HOTAIR and downstream DCLK1. This study established a new evidence of HOTAIR/miR-149-5p axis in functioning in cisplatin resistance.
Next, we searched for the potential gene effectors involved in its functions in NSCLC. In plenty of potential gene effectors, DCLK1 was further confirmed due to many reasons. For example, Powrozek et al38 firstly appointed out that it could be detected of DCLK1 gene promoter methylation in the plasma of lung cancers including NSCLC and SCLC, and this phenomenon was associated with lower overall survival. Later, expression of DCLK1 in pathological stage I NSCLC tumors was investigated and suggested DCLK1 as a new target in clinic.39 Functionally, DCLK1 was confirmed to take part in cell proliferation, migration and invasion of human lung squamous cell carcinoma cells in vitro through serving as target gene for miR-448.40 Besides, cisplatin resistance and PI3K/AKT/mTOR signaling pathway were also altered by miR-539/DCLK1 axis.18 That is why we selected DCLK1 as a potential downstream target of HOTAIR/miR-149-5p axis in regulating the acquired cisplatin-resistant NSCLC cells. Here, we observed the upregulation of DCLK1 in DDP-resistant patients with NSCLC and in A549/DDP and H1299/DDP cells. Upregulation of oncogene DCLK1 could contribute to cisplatin resistance of A549/DDP and H1299/DDP cells with miR-149-5p overexpression through increase IC50, cell proliferation, migration and invasion. Taken together, DCLK1 may be a key target of adjuvant chemotherapy to reverse DDP resistance in patients with NSCLC.
In this study, we provided a novel, promising mechanism underlying HOTAIR at least through activating miR-149-5p/DCLK1 axis. However, the influence of HOTAIR/miR-149-5p/DCLK1 on other cell processes including apoptosis and EMT remains to be further uncovered, as well as the involved signaling pathways such as ERK1/2 MAPK signaling pathway.41
Conclusion
In conclusion, HOTAIR is significantly upregulated in human DDP-resistant NSCLC tissues and cells. Knockdown of HOTAIR partially reverses the acquired cisplatin resistance in DDP-resistant NSCLC cells both in vitro and in vivo through miR-149-5p/DCLK1 axis. This work suggests a novel HOTAIR/miR-149-5p/DCLK1 pathway in the occurrence, development and treatment of cisplatin resistance in NSCLC.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359(13):1367–1380. doi:10.1056/NEJMra0802714
3. Rossi A, Di Maio M. Platinum-based chemotherapy in advanced non-small-cell lung cancer: optimal number of treatment cycles. Expert Rev Anticancer Ther. 2016;16(6):653–660. doi:10.1586/14737140.2016.1170596
4. Chang A. Chemotherapy, chemoresistance and the changing treatment landscape for NSCLC. Lung Cancer. 2011;71(1):3–10. doi:10.1016/j.lungcan.2010.08.022
5. Hu Y, Zhu QN, Deng JL, Li ZX, Wang G, Zhu YS. Emerging role of long non-coding RNAs in cisplatin resistance. Onco Targets Ther. 2018;11:3185–3194. doi:10.2147/OTT.S158104
6. Li L, Zhang X, Liu Q, et al. Emerging role of HOX genes and their related long noncoding RNAs in lung cancer. Crit Rev Oncol Hematol. 2019;139:1–6. doi:10.1016/j.critrevonc.2019.04.019
7. Zhou X, Chen J, Tang W. The molecular mechanism of HOTAIR in tumorigenesis, metastasis, and drug resistance. Acta Biochim Biophys Sin (Shanghai). 2014;46(12):1011–1015. doi:10.1093/abbs/gmu104
8. Loewen G, Jayawickramarajah J, Zhuo Y, Shan B. Functions of lncRNA HOTAIR in lung cancer. J Hematol Oncol. 2014;7:
9. Yoon JH, Abdelmohsen K, Gorospe M. Functional interactions among microRNAs and long noncoding RNAs. Semin Cell Dev Biol. 2014;34:9–14. doi:10.1016/j.semcdb.2014.05.015
10. Chen Y, Gao Y, Zhang K, et al. MicroRNAs as regulators of cisplatin resistance in lung cancer. Cell Physiol Biochem. 2015;37(5):1869–1880. doi:10.1159/000438548
11. Fadejeva I, Olschewski H, Hrzenjak A. MicroRNAs as regulators of cisplatin-resistance in non-small cell lung carcinomas. Oncotarget. 2017;8(70):115754–115773. doi:10.18632/oncotarget.22975
12. Zang H, Peng J, Wang W, Fan S. Roles of microRNAs in the resistance to platinum based chemotherapy in the non-small cell lung cancer. J Cancer. 2017;8(18):3856–3861. doi:10.7150/jca.21267
13. Sun L, Zhai R, Zhang L, Zhao S. MicroRNA-149 suppresses the proliferation and increases the sensitivity of ovarian cancer cells to cisplatin by targeting X-linked inhibitor of apoptosis. Oncol Lett. 2018;15(5):7328–7334.
14. Wang Y, Chen J, Zhang M, et al. MiR-149 sensitizes esophageal cancer cell lines to cisplatin by targeting DNA polymerase beta. J Cell Mol Med. 2018;22(8):3857–3865.
15. Lingzi X, Zhihua Y, Xuelian L, et al. Genetic variants in microRNAs predict non-small cell lung cancer prognosis in Chinese female population in a prospective cohort study. Oncotarget. 2016;7(50):83101–83114. doi:10.18632/oncotarget.13072
16. Nguyen CB, Houchen CW, Ali N. APSA awardee submission: tumor/cancer stem cell marker doublecortin-like kinase 1 in liver diseases. Exp Biol Med (Maywood). 2017;242(3):242–249. doi:10.1177/1535370216672746
17. Makino S, Takahashi H, Okuzaki D, et al. DCLK1 integrates induction of TRIB3, EMT, drug resistance, and poor prognosis in colorectal cancer. Carcinogenesis. 2020;41(3):303–12.
18. Deng H, Qianqian G, Ting J, Aimin Y. miR-539 enhances chemosensitivity to cisplatin in non-small cell lung cancer by targeting DCLK1. Biomed Pharmacother. 2018;106:
19. Hou Z, Xu C, Xie H, et al. Long noncoding RNAs expression patterns associated with chemo response to cisplatin based chemotherapy in lung squamous cell carcinoma patients. PLoS One. 2014;9(9):e108133. doi:10.1371/journal.pone.0108133
20. Liu Z, Sun M, Lu K, et al. The long noncoding RNA HOTAIR contributes to cisplatin resistance of human lung adenocarcinoma cells via downregulation of p21(WAF1/CIP1) expression. PLoS One. 2013;8(10):e77293. doi:10.1371/journal.pone.0077293
21. Liu MY, Li XQ, Gao TH, et al. Elevated HOTAIR expression associated with cisplatin resistance in non-small cell lung cancer patients. J Thorac Dis. 2016;8(11):3314–3322. doi:10.21037/jtd.2016.11.75
22. Li J, Li S, Chen Z, et al. miR-326 reverses chemoresistance in human lung adenocarcinoma cells by targeting specificity protein 1. Tumour Biol. 2016;37(10):13287–13294. doi:10.1007/s13277-016-5244-2
23. Guo F, Cao Z, Guo H, Li S. The action mechanism of lncRNA-HOTAIR on the drug resistance of non-small cell lung cancer by regulating Wnt signaling pathway. Exp Ther Med. 2018;15(6):4885–4889. doi:10.3892/etm.2018.6052
24. Yang Y, Jiang C, Yang Y, et al. Silencing of LncRNA-HOTAIR decreases drug resistance of non-small cell lung cancer cells by inactivating autophagy via suppressing the phosphorylation of ULK1. Biochem Biophys Res Commun. 2018;497(4):1003–1010. doi:10.1016/j.bbrc.2018.02.141
25. Liu MY, Li XQ, Gao TH, et al. Elevated HOTAIR expression associated with cisplatin resistance in non-small cell lung cancer patients. J Thorac Dis. 2016;8(11):3314–3322. doi:10.21037/jtd.2016.11.75
26. Liu Y, Jiang H, Zhou H, et al. Lentivirus-mediated silencing of HOTAIR lncRNA restores gefitinib sensitivity by activating Bax/Caspase-3 and suppressing TGF-alpha/EGFR signaling in lung adenocarcinoma. Oncol Lett. 2018;15(3):2829–2838. doi:10.3892/ol.2017.7656
27. Yan J, Dang Y, Liu S, Zhang Y, Zhang G. LncRNA HOTAIR promotes cisplatin resistance in gastric cancer by targeting miR-126 to activate the PI3K/AKT/MRP1 genes. Tumour Biol. 2016;37(12):16345–16355. doi:10.1007/s13277-016-5448-5
28. Cheng C, Qin Y, Zhi Q, Wang J, Qin C. Knockdown of long non-coding RNA HOTAIR inhibits cisplatin resistance of gastric cancer cells through inhibiting the PI3K/Akt and Wnt/beta-catenin signaling pathways by up-regulating miR-34a. Int J Biol Macromol. 2018;107(Pt B):2620–2629. doi:10.1016/j.ijbiomac.2017.10.154
29. Chen S, Peng M, Zhou G, et al. Long non-coding RNA HOTAIR regulates the development of non-small cell lung cancer through miR-217/DACH1 signaling pathway. Eur Rev Med Pharmacol Sci. 2019;23(2):670–678. doi:10.26355/eurrev_201901_16905
30. Jiang C, Yang Y, Yang Y, et al. Long noncoding RNA (lncRNA) HOTAIR affects tumorigenesis and metastasis of non-small cell lung cancer by upregulating miR-613. Oncol Res. 2018;26(5):725–734. doi:10.3727/096504017X15119467381615
31. Molina-Pinelo S, Gutierrez G, Pastor MD, et al. MicroRNA-dependent regulation of transcription in non-small cell lung cancer. PLoS One. 2014;9(3):e90524. doi:10.1371/journal.pone.0090524
32. Yang C, Sun C, Liang X, Xie S, Huang J, Li D. Integrative analysis of microRNA and mRNA expression profiles in non-small-cell lung cancer. Cancer Gene Ther. 2016;23(4):90–97. doi:10.1038/cgt.2016.5
33. Zhao L, Liu L, Dong Z, Xiong J. miR-149 suppresses human non-small cell lung cancer growth and metastasis by inhibiting the FOXM1/cyclin D1/MMP2 axis. Oncol Rep. 2017;38(6):3522–3530. doi:10.3892/or.2017.6047
34. Hu Y, Qin X, Yan D, et al. Genome-wide profiling of micro-RNA expression in gefitinib-resistant human lung adenocarcinoma using microarray for the identification of miR-149-5p modulation. Tumour Biol. 2017;39(3):1010428317691659. doi:10.1177/1010428317691659
35. Berghmans T, Ameye L, Willems L, et al. Identification of microRNA-based signatures for response and survival for non-small cell lung cancer treated with cisplatin-vinorelbine A ELCWP prospective study. Lung Cancer. 2013;82(2):340–345. doi:10.1016/j.lungcan.2013.07.020
36. Ke Y, Zhao W, Xiong J, Cao R. miR-149 inhibits non-small-cell lung cancer cells EMT by targeting FOXM1. Biochem Res Int. 2013;2013:506731. doi:10.1155/2013/506731
37. Li X, Liang J, Liu YX, et al. miR-149 reverses cisplatin resistance of gastric cancer SGC7901/DDP cells by targeting FoxM1. Pharmazie. 2016;71(11):640–643. doi:10.1691/ph.2016.6696
38. Powrozek T, Krawczyk P, Nicos M, Kuznar-Kaminska B, Batura-Gabryel H, Milanowski J. Methylation of the DCLK1 promoter region in circulating free DNA and its prognostic value in lung cancer patients. Clin Transl Oncol. 2016;18(4):398–404. doi:10.1007/s12094-015-1382-z
39. Tao H, Tanaka T, Okabe K. Doublecortin and CaM kinase-like-1 expression in pathological stage I non-small cell lung cancer. J Cancer Res Clin Oncol. 2017;143(8):1449–1459. doi:10.1007/s00432-017-2405-7
40. Shan C, Fei F, Li F, et al. miR-448 is a novel prognostic factor of lung squamous cell carcinoma and regulates cells growth and metastasis by targeting DCLK1. Biomed Pharmacother. 2017;89:1227–1234. doi:10.1016/j.biopha.2017.02.017
41. Cook SJ, Stuart K, Gilley R, Sale MJ. Control of cell death and mitochondrial fission by ERK1/2 MAP kinase signalling. FEBS J. 2017;284(24):4177–4195. doi:10.1111/febs.14122
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.