")
Back to Journals » OncoTargets and Therapy » Volume 12
Knockdown of cyclooxygenase-2 leads to growth inhibition and cell cycle arrest in hepatocellular carcinoma cells
Authors Lv X, Chen Z, Li S, Xie H
Received 3 December 2018
Accepted for publication 21 February 2019
Published 31 May 2019 Volume 2019:12 Pages 4341—4349
DOI https://doi.org/10.2147/OTT.S196822
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr William C. Cho
Xiuhe Lv,* Zhen Chen,* Shaohua Li, Huahong Xie
State Key Laboratory of Cancer Biology, National Clinical Research Center for Digestive Diseases and Xijing Hospital of Digestive Diseases, Air Force Military Medical University/Fourth Military Medical University, Xi’an 710032, Shaanxi Province, People’s Republic of China
*These authors contributed equally to this work
Background & aims: Cyclooxygenase-2 (COX-2) is proved to play important roles in the development and progression of various human tumors, including hepatocellular carcinoma (HCC). However, the antitumor effect of RNA interference (RNAi) technology targeting COX-2 in HCC has not yet been verified.
Methods: We silenced COX-2 expression using a lentivirus-mediated RNAi and further investigated the effects of COX-2 knockdown on cell growth and cell cycle in Huh7 and SMMC-7721 cells. COX-2 mRNA was detected by RT-PCR while COX-2 protein was detected by Western blotting. The cell proliferation was measured by MTT assay. The cell cycle was measured by flow cytometry. The tumorigenicity of HCC cells was evaluated using soft-agar clonogenic assay in vitro and nude mouse xenograft model in vivo.
Results: The down-regulation of COX-2 expression significantly inhibited cell proliferation and colony formation, and led to cell cycle arrest in vitro, and reduced the potential of tumorigenicity in vivo in both Huh7 and SMMC-7721 cells. Furthermore, PGE2 production was also decreased after COX-2 expression was suppressed. Finally, knockdown of COX-2 also induced the down-regulation of cell cycle-related protein, cyclinD1.
Conclusions: The abrogation of COX-2 expression can lead to potent antitumor activity and knockdown of COX-2 may be served as a prospective therapeutic strategy against HCC.
Keywords: COX-2, RNA interference, hepatocellular carcinoma
Introduction
Hepatocellular carcinoma (HCC) is one of the most frequent solid tumors, which remains the third leading cause of cancer-related death throughout the world. Statistic showed that the incidence of HCC is particularly high in eastern/south-eastern Asia and Africa.1,2 Chronic infection with hepatitis B virus or hepatitis C virus is the major cause of HCC, which may account for 80% of HCC cases globally. Other risk factors, including alcohol consumption, tobacco smoking, diabetes, and obesity, may also play important roles in the occurrence and development of HCC.3 Besides surgical resection, radioembolization, percutaneous ethanol injection, radiofrequency ablation, and transarterial chemoembolization have been used to treat HCC.4 Despite remarkable progress achieved in diagnosis and treatment of HCC, the prognosis of HCC remains unsatisfactory, partially due to the fact that cancer has exhibited vascular invasion and intrahepatic metastasis by the time symptoms have developed.5 Therefore, it is important to find new treatment options for HCC patients.
Cyclooxygenases (COXs) are the rate-limiting enzymes which play important roles in the synthesis of prostaglandin from arachidonic acid. The COXs consist of two isoforms, COX-1 and COX-2. COX-1 is ubiquitously expressed in tissues, while COX-2 is selectively expressed.6 High-level COX-2 expression has been found in colorectal, gastric, ovarian, and other cancers, and COX-2 expression has been found to be well correlated with invasiveness, prognosis, and survival in cancer cells.7–9 Elevated production of the COX-2 metabolite, PGE2, can promote epithelial cancer cell growth, survival, and invasion. Long-term use of NSAIDs or COX-2 selective inhibitors can greatly reduce the incidence of colon cancer partially by blocking the function of COX-2.10
Overexpression of COX-2 has also been observed in HCC recently. Clinical studies demonstrated that increased COX-2 expression was associated with decreased overall and disease-free survival in HCC patients.11 Furthermore, experimental studies demonstrated that inhibition of COX-2 could suppress the growth of human HCC in vitro and reduce the incidence of HCC in rodents.12 These studies strongly indicated that COX-2 might be a critical factor involved in HCC. However, COX-2 inhibitors have been proved to be associated with potential cardiovascular side effects, which limited their use in clinical practice. In the present study, we tried to silence COX-2 expression in HCC cells through lentivirus-mediated RNA interference (RNAi) technology, and further investigated the effects of COX-2 silencing on HCC cell proliferation, cell cycle, and tumor-forming potential, hoping to provide new clues for HCC treatment.
Materials and methods
Cell culture
Human HCC cell line Huh7 and SMMC-7721 were obtained from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). All cell lines were grown in RPMI 1640 (Invitrogen, Carlsbad, CA, USA) culture medium supplemented with 10% heat-inactivated fetal calf serum (FCS) and 2 mM L-glutamine (GIBCO/BRL) at 37°C with 5% CO2 in a humidified incubator (Forma Scientific, Marietta, OH, USA).
Construction of lentivirus vectors
In order to generate lentivirus expressing RNAi specific for COX-2 gene, the RNAi sequence for human COX-2 was 5ʹ-AACTG CTCAACACCGGAATTTTT-3ʹ, targeting human COX-2 at the 291–313 position in the sequence (Gene Bank Accession: NM000963.1), which was proved to be effective in the recent paper. The negative control constructs having no homology with the human genome was 5ʹ-AATTCTCCGAACGTGTCACGT-3ʹ.13 The sequences were cloned into the pGCSIL-Green Fluorescent Protein (GeneChem, Shanghai, China) to generate the lentiviral vectors. Lentiviral vectors and packaging vectors were then transfected into 293T cells. Supernatants containing lentiviruses were attained by filtering through 0.45 μm cellulose acetate filters and using ultracentrifugation 72 hrs later transfection, and the titer of lentiviruses was determined.
Infection of lentivirus
The target cells were plated at 40–50% confluence and incubated overnight. On the day of infection, the culture medium was replaced by the appropriately titered viral supernatant (1.5 mL/well) and incubated at 37°C for 10 hrs. Then, the viral supernatant was replaced with fresh media. After 5 days of infection, the knockdown efficiency was determined by RT-PCR and western blot.14 Huh7 and SMMC-7721 parental cells, control transfectants, and COX-2 transfectants were named Huh7-P, Huh7-C, Huh7-S, SMMC-7721-P, SMMC-7721-C, and SMMC-7721-S cells, respectively.
Cell proliferation assays
MTT assay was applied to investigate the effect of COX-2 inhibition on cell proliferation. Briefly, cells were plated onto 96-well plates at a density of 2×103 cells/well. After 1, 2, 3, 4, 5, 6, 7 days of incubation, 15 μL MTT (5 mg/mL; Sigma, St. Louis, MO, USA) was added and incubated for 6 hrs in each day. The supernatant was removed and replaced with 150 mL dimethyl sulfoxide. After shaking for 15 mins at room temperature to dissolve MTT crystals in culture plates, absorbance values were quantified by an ELISA reader (Bio-Rad Laboratories, Richmond, CA, USA) at a wavelength of 490 nm.
Cell cycle analysis
For cell cycle analysis, 1×104 cells were seeded and incubated in six-well plates until they reach 80–85% confluence. Then, cells were harvested by trypsinization and fixed with 70% ethanol overnight. After treated with RNase A (2 mg/mL) for 30 mins, cells were reacted with propidium iodide (50 mg/mL) for 30 mins at room temperature. DNA content was determined by FACScan analysis. The proliferous index (PI) was calculated: PI = (S + G2)/(S + G2 + G1).
Cell clonogenic assay
Soft-agar clonogenic assay was done to detect the clone formation potential of HCC cell after COX-2 expression was decreased. Briefly, HCC cells at a density of 1×102 cells/well were suspended in 0.3% agar in RPMI 1640 culture medium containing 10% FCS, and plated on solidified agar (0.6%) in six-well dishes. The plates were incubated for 2 weeks at 37°C in 5% CO2. Colonies identified by crystal violet staining of over 50 cells per colony were precisely counted to determine the efficacy. The results were presented as a percentage of the control.
Tumorigenicity test in nude mice
To further detect the effect of COX-2 RNAi on HCC cell clone formation potential, tumorigenicity test in nude mice was also done. Transfected cells were trypsinized and resuspended in PBS after washing twice with a serum-free medium. About 5×106 cells in 0.2 mL were injected subcutaneously in the right lateral aspect of the thoracic wall of one 4-week-old female BALB/c nude mice. Each group had three mice. Mice were observed daily, and tumor size was inspected once a week. After 4 weeks of observation, the mice were sacrificed and the xenograft tumors were removed for further analysis. Tumor volume (TV) was calculated: TV = (length ×width2)×0.5. All animal experiments were approved by the Ethics Committee of the Fourth Military Medical University, and all procedures conformed to the National Institutes of Health guidelines for the care and use of laboratory animals.
RT-PCR
Total RNA was isolated from cells by using the TRIZOL Reagent (GIBCO BRL, Grand Island, NY, USA). According to the manufacturer’s protocol, RT reaction was performed using the First-Strand cDNA Synthesis Kit (MBI Fermentas, Vilnius, Lithuania). The PCR primers were as follows: COX-2, forward: 5ʹ-TTCAAATGAGATTGTGGGAAAATTGCT-3ʹ and reverse: 5ʹ-AGATCATCTCT GCCTGAGTA TCTT-3ʹ; β-actin, forward: 5ʹ-AGCGGGAAATCGTGCGTG-3ʹ and reverse: 5ʹ-CAGGGTACATGGTGGTGCC-3ʹ.
Western blotting
Protein lysate was extracted from cells and tumor xenograft tissues in lysis buffer (50 mM Tris, pH 7.2, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 500 mM NaCl, 10 mM MgCl2, 10 μg/mL leupeptin, 10 μg/mL aprotinin, and 1 mM PMSF) and quantified by the Bradford method. 50 micrograms of cytosolic proteins were electrophoresed on 10% SDS polyacrylamide gels and then transferred to nitrocellulose membranes. After blocked with 5% fat-free milk in Tween-TBS (20 mmol/L Tris, 0.15 mol/L NaCl [pH 7.0], 0.1% Tween 200), the membranes were incubated at 4°C overnight with anti-human COX-2 antibody (1:400, Cayman Chemical, MI, USA), anti-cyclinD1 antibody (1:100, Santa Cruz Biotech, CA,USA) and anti-β-actin antibody (1:5,000, Sigma Chemical Co., MO, USA). After washed in Tween-TBS for three times, membranes were incubated with the corresponding horseradish peroxidase-conjugated secondary antibody (1:2,000, Cell Signaling Technology, MA, USA). Blots were developed with the enhanced chemiluminescence system (Amersham, Freiburg, Germany). Western blot for β-actin was performed as the internal control.
Prostaglandin E2 analysis
PGE2 production was detected through PGE2 high sensitivity ELISA-Kit (Cayman Chemical, MI, USA). Firstly, cells were plated onto 24-well plates at a density of 5×104 cells/well. After 24 hrs, the supernatant was removed and centrifuged. Then, the amount of PGE2 in the supernatant was determined by using the PGE2 ELISA-Kit according to the manufacturer's introduction. Parallel cells were harvested and counted simultaneously. The amount of PGE2 in the supernatants was evaluated according to the number of cells in the dishes.
Statistical analysis
All statistical analyses were performed using the SPSS17.0 statistical software. Student’s t-test and one-way analysis of variance test were adopted. Results are expressed as means ± standard deviation (SD). Values of P<0.05 were considered as significant.
Results
COX-2 RNAi led to decrease of COX-2 expression and PGE2 production
After infection of the constructed lentiviruses, both Huh7-S cells and SMMC-7721-S cells showed a significant reduction in COX-2 expression when compared with control cells or parental cells (P<0.05, respectively), while no significant difference of COX-2 expression between control cells and parental cells was discovered (Figure 1A and B). Similarly, both Huh7-S cells and SMMC-7721-S cells showed significantly decreased PGE2 expression when compared with control cells or parental cells (P<0.05, respectively), while no significant difference of PGE2 expression between control cells and parental cells was discovered (Figure 1C and D).
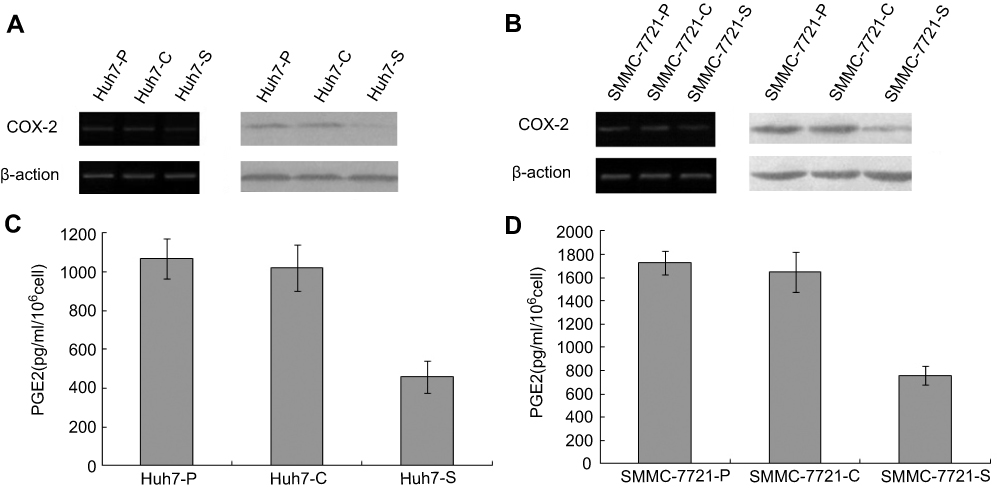 | Figure 1 COX-2 siRNA led to a decrease of COX-2 and PGE2 expression in Huh7 and SMMC-7721 cells. (A) Western blot and RT-PCR analysis of the Huh7-P, Huh7-C, and Huh7-S cells; (B) Western blot and RT-PCR analysis of the SMMC-7721-P, SMMC-7721-C, and SMMC-7721-S cells; (C) PGE2 production was detected in Huh7 transfected cells and control cells; (D) PGE2 production was detected in SMMC-7721 transfected cells and control cells. |
COX-2 knockdown inhibited cell proliferation
MTT assay was performed to examine the effects of COX-2 RNAi on cell growth of HCC cells. From the fourth day, significant growth inhibition of Huh7-S cells was observed when compared with Huh7-C cells or Huh7-P cells (P<0.05, respectively, Figure 2A). SMMC-7721-S cells also showed significant growth inhibition on the fourth day when compared with SMMC-7721-C cells or SMMC-7721-P cells (P<0.05, respectively, Figure 2B). However, no significant difference between control cells and parental cells was discovered, either for Huh7 or SMMC-7721 cells.
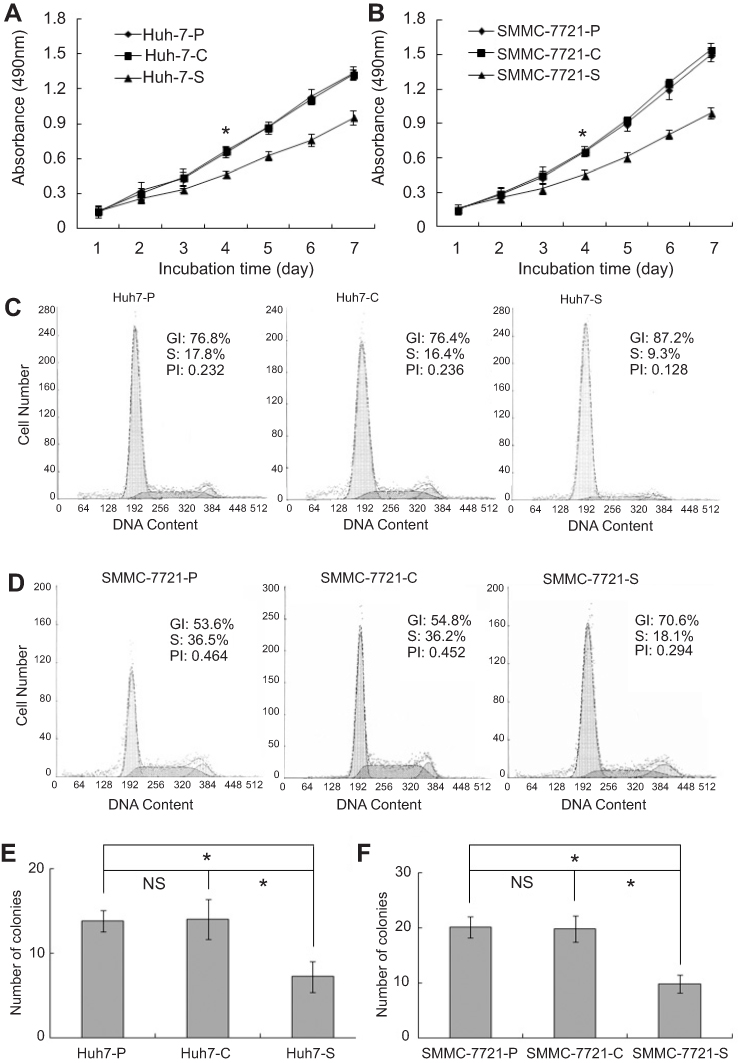 | Figure 2 COX-2 down-regulation induced HCC cell growth inhibition and cell cycle arrest in vitro. (A) Cell’s growth rate of the Huh7-P, Huh7-C, and Huh7-S cells (*P<0.05); (B) Cell’s growth rate of SMMC-7721-P, SMMC-7721-C, and SMMC-7721-S (*P<0.05); (C) The cell cycle distribution and the proliferous indexes of Huh7-P, Huh7-C, and Huh7-S cells; (D) The cell cycle distribution and the proliferous indexes of SMMC-7721-P, SMMC-7721-C, and SMMC-7721-S cells; (E) Colony numbers of Huh7-P, Huh7-C, and Huh7-S cells (*P<0.05; NS, no significant difference); (F) Colony numbers of SMMC-7721-P, SMMC-7721-C, and SMMC-7721-S cells (*P<0.05; NS, no significant difference). |
Decreased COX-2 expression induced cell cycle arrest
The effects of COX-2 RNAi on the cell cycle distribution of HCC cells were examined with flowcytometry analysis. As shown in Table 1, when compared with Huh7-C cells or Huh7-P cells, the percentage of Huh7-S cells was significantly increased in the G1 phase and decreased in the S phase (P<0.05, respectively, Figure 2C). As shown in Table 2, similar changes were also found for SMMC-7721 cells. When compared with SMMC-7721-C cells or SMMC-7721-P cells, the percentage of SMMC-7721-S cells was significantly increased in the G1 phase and decreased in the S phase (P<0.05, respectively, Figure 2D). Again, no significant difference between control cells and parental cells was discovered, either for Huh7 or SMMC-7721 cells.
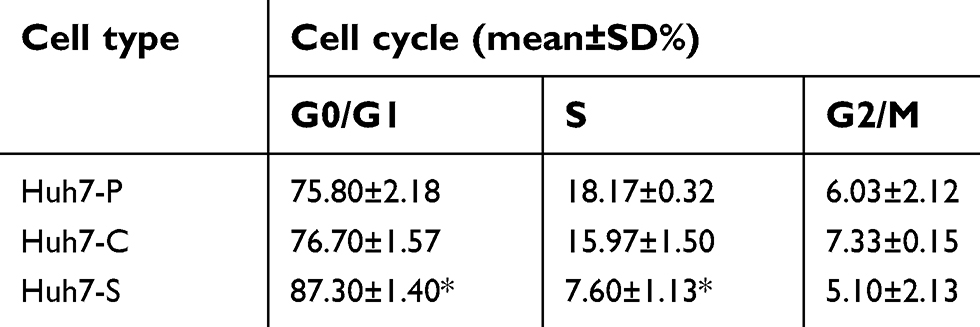 | Table 1 Effects of COX-2 RNAi on the cell cycle of Huh7-P, Huh7-C, and Huh7-S cells |
 | Table 2 Effects of COX-2 RNAi on the cell cycle of SMMC-7721-P, SMMC-7721-C, and SMMC-7721-S cells |
COX-2 knockdown inhibited tumor formation potential in vivo
After each cell was cultured in soft agar for 4 weeks, Huh7-S cells yielded fewer colonies (7±1.4 colonies) than those from Huh7-C cells (14±2.3 colonies) or Huh7-P cells (14.8±1.9 colonies) (P<0.05, respectively, Figure 2E). Colonies formed by SMMC-7721-S cells (9.6±1.3 colonies) were also fewer than those from SMMC-7721-C cells (19.4±1.5 colonies) or SMMC-7721-P cells (20±2.2 colonies) (P<0.05, respectively, Figure 2F). No significant difference between control cells and parental cells was discovered, either for Huh7 or SMMC-7721 cells.
COX-2 knockdown inhibited tumor formation potential in vitro
In nude mice, tumors developed from Huh7-S cells were significantly smaller compared with those developed from Huh7-P cells or Huh7-C cells in the third week (P<0.05, respectively, Figure 3E). After the xenograft tumors were excised, tumors derived from Huh7-S cells were smaller compared with those derived from Huh7-P cells or Huh7-C cells (Figure 3A). The average weight of Huh7-S cell xenograft tumor was less than that in control groups (P<0.05, respectively, Figure 3C). On the other side, tumors developed from SMMC-7721-S cells grew slower than those from SMMC-7721-P cells or SMMC-7721-C cells after two weeks (P<0.05, respectively, Figure 3F). Again, after the xenograft tumors were excised, tumors derived from SMMC-7721-S cells were smaller compared with those derived from SMMC-7721-P cells or SMMC-7721-C cells (Figure 3B). The average weight of SMMC-7721-S cell xenograft tumor was less than that in control groups (P<0.05, respectively, Figure 3D).
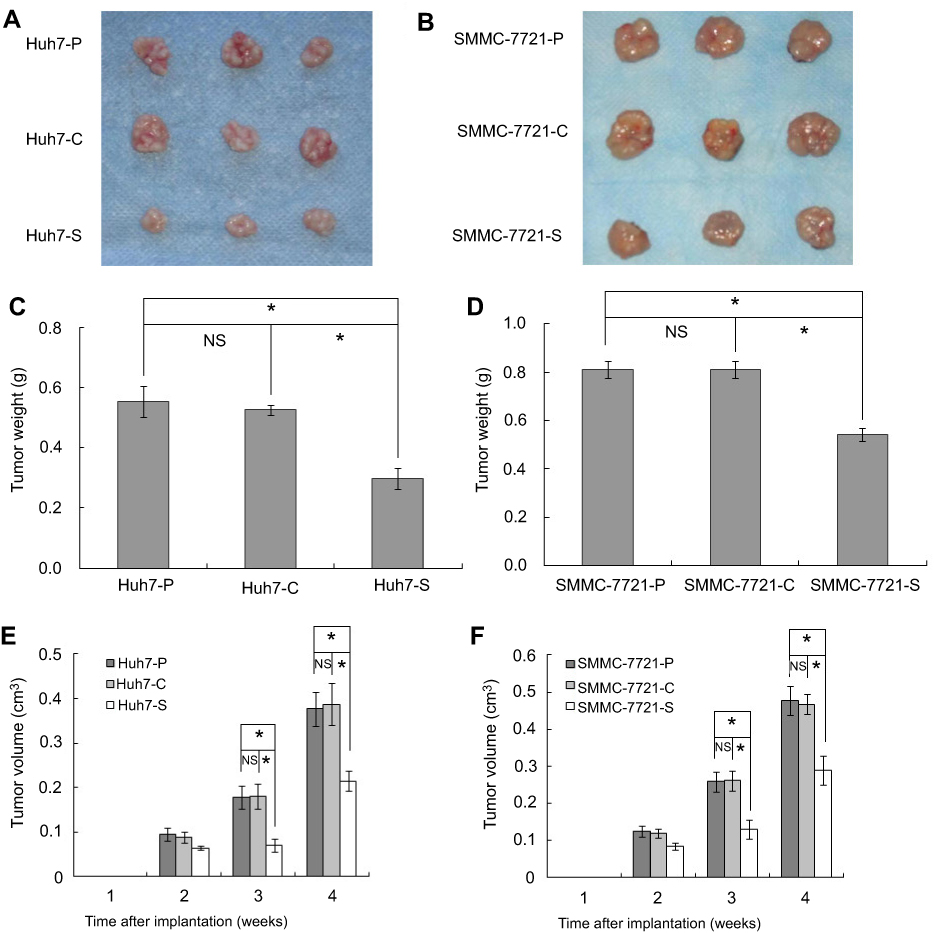 | Figure 3 COX-2 down-regulation inhibited tumor formation potential in vivo. (A) Xenograft tumors of Huh7-P, Huh7-C, and Huh7-S cells in BALB/c nu/nu mice; (B) Xenograft tumors of SMMC-7721-P, SMMC-7721-C, and SMMC-7721-S cells in BALB/c nu/nu mice; (C) Tumor weights of xenograft tumors of Huh7-P, Huh7-C, and Huh7-S cells (*P<0.05; NS, no significant difference); (D) Tumor weights of xenograft tumors of SMMC-7721-P, SMMC-7721-C, and SMMC-7721-S cells (*P<0.05; NS, no significant difference); (E) Tumorigenicity of Huh7-P, Huh7-C, and Huh7-S cells in BALB/c nu/nu mice (*P<0.05; NS, no significant difference); (F) Tumorigenicity of SMMC-7721-P, SMMC-7721-C, and SMMC-7721-S cells in BALB/c nu/nu mice (*P<0.05; NS, no significant difference). |
Possible mechanism of COX-2 involved in the regulation of cell cycle
We detected the expressions of cell cycle-related protein cyclinD1 in HCC parental and transfected cells and xenograft tumor cells. The results from RT-PCR and western blotting showed that knockdown of COX-2 could significantly down-regulate the expressions of cyclinD1 in both Huh7 and SMMC-7721 cell lines and xenograft tumors (Figure 4A and B).
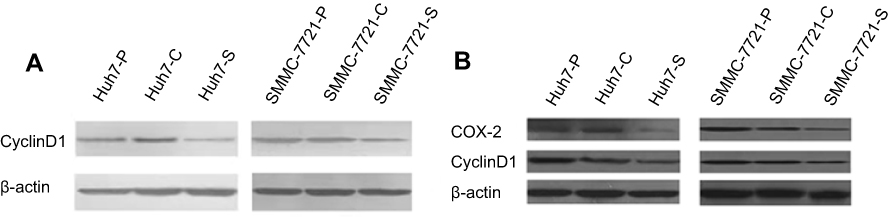 | Figure 4 COX-2 siRNA downregulated the expression of cyclinD1. (A) Western blot analysis of cyclinD1 in HCC parental cells and transfectants; (B) Western blot analysis of cyclinD1 in xenograft tumors derived from HCC parental cells and transfectants. |
Discussion
Recently, lentivirus-mediated RNAi, which is mediated by 21–25 nucleotide short interfering RNA (siRNA) duplexes homologous to the interested gene, has rapidly become an innovative and elective tool for studying the regulation of gene expression and function in vitro.15 Furthermore, based on its high selectivity and specialty, this powerful tool is gradually become a novel anti-cancer therapeutic method by silencing some key oncogenic proteins.16 Compared with COX-2 inhibitors, silencing of COX-2 by lentivirus-mediated RNAi can avoid the side effects induced by NSAIDs. COX-2 RNAi has shown high efficacy and specialty in COX-2 blocking in endometrial cancer cell, pancreatic cancer cell, and other cancer cells.17–19 Besides inhibition of tumor cell proliferation, colony formation, and metastasis potential, down-regulation of COX-2 expression by RNAi can enhance the efficiency of chemotherapy drugs.20
In this study, we used lentivirus-mediated RNAi targeting COX-2, and further investigated its efficacy on the suppression of COX-2 expression and the consequent anticancer potential in HCC. The results showed that after COX-2 expression was silenced with COX-2 RNAi, HCC cell proliferation was inhibited, and HCC cell cycle progression was suppressed in G1-S phase. Furthermore, knockdown of COX-2 enzyme in HCC cells strongly impaired cell anchorage-independent growth in soft agar, and inhibited tumor growth in nude mice. At the same time, PGE2, a kind of COX-2 enzymatic product, was down-regulated after COX-2 silenced. PGE2 was discovered to be overproduced in many tumors, which could promote cancer cell growth by promoting angiogenesis and metastasis and by influencing the immune response.21 Through impairing PGE2 production, COX-2 RNAi affected HCC cell proliferation, cell cycle distribution, and tumor formation ability. Moreover, to study the possible molecular mechanism of COX-2 involved in the development of HCC, we detected the expression of cell cycle-related protein cyclin D1 in both groups. It is well known that cyclin D1 controls cell cycle progression through the G1 phase and G1-S phase.22 Our results showed that knockdown COX-2 significantly down-regulated the expression of cyclinD1. The probable mechanism of this may because a positive feedback loop is established between COX-2/PGE2 axis and STAT3 signaling, and STAT3 regulates a range of genes contributing to cell growth, differentiation, and angiogenesis, including Cyclin D1.23 These findings further strengthened the results that inhibiting COX-2 could repress HCC cell growth in vivo and in vitro.
The effects of COX-2 RNAi on cancer cell proliferation and cell cycle distribution were of great dispute in recent studies. In Capan-2 pancreatic cancer cells, COX-2 RNAi could inhibit cell growth significantly by decreasing the cell proliferation, increasing cell apoptosis and regulating the cell cycle.17 And a decrease of COX-2 expression through lentivirus mediated-RNAi significantly inhibited the growth, decreased the invasion and migration ability of SaOS2 osteosarcoma cells.16 Furthermore, silencing COX-2 mediated by RNAi in human laryngeal carcinoma Hep-2 cells could induce a reduction of proliferation, invasiveness, and tumorigenesis potential, coupled with increased apoptosis.24 On the other side, COX-2 RNAi induced a notable reduction of PGE2 levels but had no effects on proliferation in OVCAR-3 human ovarian carcinoma cells.25 And in HT-29 colon cancer cells, the invasiveness, as well as their ability to form colonies in soft-agar, was reduced after COX-2 expression was silenced, while the cell proliferation was not affected.13 To our knowledge, there were few studies using lentivirus-mediated RNAi targeting COX-2 for determining the therapeutic significance of COX-2 in HCC. Liu et al, firstly explored the effects of silencing COX-2 expression via RNAi on the tumorigenicity of SMMC-7721 cell line.26 However, their study only provided primary results of the animal experiment rather than investigation of the detailed mechanism at the cellular level. Our results combined with recent findings, strongly suggested that COX-2 directed siRNAs, which could exert antitumor effects both in vitro and in vivo, might be a promising genetic therapeutic approach for HCC treatment.
Conclusion
We demonstrated that the abrogation of COX-2 expression could lead to potent antitumor activity and knockdown of COX-2 might be served as a prospective therapeutic strategy against HCC.
Acknowledgments
We greatly thank Jiugang Song, and GeneChem engineers for their excellent technical assistance. This work was supported by the Science and Technology Support Program of National Clinical Research Center for Digestive Diseases (2015BAI13B07), the Natural Science Foundation Research Project of Shaanxi Province (Grant No. 2016JM8148), and the New Clinical Technique Project of Xijing Hospital to Dr. Huahong Xie.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Kulik L, El-Serag HB. Epidemiology and management of hepatocellular carcinoma. Gastroenterology. 2019;156:477-491.e1. doi:10.1053/j.gastro.2018.08.065
2. Njei B, Rotman Y, Ditah I, Lim JK. Emerging trends in hepatocellular carcinoma incidence and mortality. Hepatology. 2015;61:191–199. doi:10.1002/hep.27388
3. Jerrells TR. Commentary with regard to the role of alcohol and cofactors in the development of hepatocellular carcinoma. Alcohol Clin Exp Res. 2012;36:64–65. doi:10.1111/j.1530-0277.2012.01780.x
4. Tian G, Yang S, Yuan J, et al. Comparative efficacy of treatment strategies for hepatocellular carcinoma: systematic review and network meta-analysis. BMJ Open. 2018;8:e021269. doi:10.1136/bmjopen-2017-021269
5. Xie H, Song J, Liu K, et al. The expression of hypoxia-inducible factor-1alpha in hepatitis B virus-related hepatocellular carcinoma: correlation with patients‘ prognosis and hepatitis B virus X protein. Dig Dis Sci. 2008;53:3225–3233.
6. Ghosh N, Chaki R, Mandal V, Mandal SC. COX-2 as a target for cancer chemotherapy. Pharmacol Rep. 2010;62:233–244.
7. Bengi G, Keles D, Topalak Ö, Yalçin M, Kiyak R, Oktay G. Expressions of TIMP-1, COX-2 and MMP-7 in colon polyp and colon cancer. Euroasian J Hepatogastroenterol. 2015;5:74–79. doi:10.5005/jp-journals-10018-1138
8. Ren J, Liu J, Sui X. Correlation of COX-2 and MMP-13 expressions with gastric cancer and their effects on prognosis. J BUON. 2018;23:665–671.
9. Sun H, Zhang X, Sun D, et al. COX-2 expression in ovarian cancer: an updated meta-analysis. Oncotarget. 2017;8:88152–88162. doi:10.18632/oncotarget.21538
10. Xie H, Gao L, Chai N, et al. Potent cell growth inhibitory effects in hepatitis B virus X protein positive hepatocellular carcinoma cells by the selective cyclooxygenase-2 inhibitor celecoxib. Mol Carcinog. 2009;48:56–65. doi:10.1002/mc.20455
11. Chen G, Li X, Yang J, et al. Prognostic significance of cyclooxygenase-2 expression in patients with hepatocellular carcinoma: a meta-analysis. Arch Med Sci. 2016;12:1110–1117. doi:10.5114/aoms.2016.61916
12. Li T, Zhong J, Dong X, et al. Meloxicam suppresses hepatocellular carcinoma cell proliferation and migration by targeting COX-2/PGE2-regulated activation of the β-catenin signaling pathway. Oncol Rep. 2016;35:3614–3622. doi:10.3892/or.2016.4764
13. Strillacci A, Griffoni C, Spisni E, Manara MC, Tomasi V. RNA interference as a key to knockdown overexpressed cyclooxygenase-2 gene in tumour cells. Br J Cancer. 2006;94:1300–1310.
14. Guo X, Wang W, Hu J, et al. Lentivirus-mediated RNAi knockdown of NUPR1 inhibits human nonsmall cell lung cancer growth in vitro and in vivo. Anat Rec (Hoboken). 2012;295:2114–2121. doi:10.1002/ar.22571
15. Cong P, Yi C, Wang XY. Expression of Smo in pancreatic cancer CD44+CD24+cells and construction of a lentiviral expression vector to silence Smo. Oncol Lett. 2018;16:4855–4862. doi:10.3892/ol.2018.9315
16. Kang X, Wang F, Lan X, et al. Lentivirus-mediated shRNA targeting CNN2 inhibits hepatocarcinoma in vitro and in vivo. Int J Med Sci. 2018;15:69–76. doi:10.7150/ijms.21113
17. Zhong Y, Xia Z, Liu J, Lin Y, Zan H. The effects of cyclooxygenase-2 gene silencing by siRNA on cell proliferation, cell apoptosis, cell cycle and tumorigenicity of Capan-2 human pancreatic cancer cells. Oncol Rep. 2012;27:1003–1010. doi:10.3892/or.2011.1595
18. Xiao YT, Luo LM, Zhang R. Effect of lentivirus mediated cyclooxygenase-2 gene shorthairpinRNA on invasiveness of endometrial carcinoma. Eur J Gynaecol Oncol. 2015;36:44–48.
19. Strillacci A, Griffoni C, Valerii MC, Lazzarini G, Tomasi V, Spisni E. RNAi-based strategies for cyclooxygenase-2 inhibition in cancer. J Biomed Biotechnol. 2010;828045.
20. Du Y, Shi A, Han B, et al. COX-2 silencing enhances tamoxifen antitumor activity in breast cancer in vivo and in vitro. Int J Oncol. 2014;44:1385–1393. doi:10.3892/ijo.2014.2299
21. Peng WT, Sun WY, Li XR, et al. Emerging roles of G protein-coupled receptors in hepatocellular carcinoma. Int J Mol Sci. 2018;19:E1366. doi:10.3390/ijms19051366
22. Mukhopadhyay S, Saqcena M, Foster DA. Synthetic lethality in KRas-driven cancer cells created by glutamine deprivation. Oncoscience. 2015;2:807–808. doi:10.18632/oncoscience.253
23. Lin A, Wang G, Zhao H, et al. TLR4 signaling promotes a COX-2/PGE2/STAT3 positive feedback loop in hepatocellular carcinoma cells. Oncoimmunology. 2015;5:e1074376. doi:10.1080/2162402X.2015.1074376
24. Zhao Q, Wang C, Zhu J, et al. RNAi-mediated knockdown of cyclooxygenase2 inhibits the growth, invasion and migration of SaOS2 human osteosarcoma cells: a case control study. J Exp Clin Cancer Res. 2011;30:26. doi:10.1186/1756-9966-30-24
25. Denkert C, Fürstenberg A, Daniel PT, et al. Hauptmann S: induction of G0/G1 cell cycle arrest in ovarian carcinoma cells by the anti-inflammatory drug NS-398, but not by COX-2-specific RNA interference. Oncogene. 2003;22:8653–8661. doi:10.1038/sj.onc.1206920
26. Liu LL, Lin LR, Tong ML, et al. Effects of silencing cyclooxygenase-2 expression via rna interference on the tumorigenicity of the smmc-7721 human hepatocarcinoma cell line. Oncol Rep. 2012;27:1829–1834. doi:10.3892/or.2012.1702
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.