")
Back to Journals » Cancer Management and Research » Volume 11
Knockdown Of CCDC132 Attenuates Gastric Cancer Cells Proliferation And Tumorigenesis By Facilitating DNA Damage Signaling
Authors Xu X, Cao W, Sun W, Wang Z , Chen H, Zheng Z, Yang X
Received 14 May 2019
Accepted for publication 15 October 2019
Published 12 November 2019 Volume 2019:11 Pages 9585—9597
DOI https://doi.org/10.2147/CMAR.S215631
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Antonella D'Anneo
Xiaowu Xu,1 Weilang Cao,1 Wei Sun,2 Zhaohong Wang,1 Hui Chen,1 Zhiqiang Zheng,1 Xiaomin Yang3
1Department of General Surgery, The Second Affiliated Hospital and Children’s Hospital of Wenzhou Medical University, Wenzhou, Zhejiang 325027, People’s Republic of China; 2Department of Pharmacy, The Second Affiliated Hospital and Children’s Hospital of Wenzhou Medical University, Wenzhou, Zhejiang 325027, People’s Republic of China; 3Department of Pathology, Wenzhou People’s Hospital, Wenzhou, Zhejiang 325000, People’s Republic of China
Correspondence: Xiaomin Yang
Department of Pathology, Wenzhou People’s Hospital, Wenzhou, Zhejiang 325000, People’s Republic of China
Tel/Fax +86-577-057788256807
Email [email protected]
Background: Aberrant endocytic recycling has fundamental functions on plasma membrane component turnover. Recent studies have identified an uncharacterized protein, CCDC132, in the endosome-associated recycling protein complex. Besides, our preliminary data first showed that CCDC132 was elevated in malignant neoplasms, especially in esophagus/stomach cancers. However, the functions and the underlying mechanisms of CCDC132 in gastric cancer (GC) biology remain unclear.
Methods: The CCDC132 mRNA expression in 4 GC cell lines and normal gastric epithelial cell lines was detected by qRT-PCR. Then, CCDC132 was downregulated in AGS and MGC-803 cells by lentivirus-induced RNA interfere, and cell viability assay, clone formation assay and apoptosis assay were carried out. The mechanism of CCDC132 on cell proliferation and apoptosis activation was explored using PathScan® Stress, apoptosis signaling arrays and Western blot. We further investigated the pro-oncogenesis of CCDC132 in vivo. Meanwhile, immunohistochemistry was utilized to analyze the association between CCDC132 expression and clinicopathological features and prognosis. Finally, the correlation between CCDC132 and p53 was analyzed by Spearman’s rank correlation analysis.
Results: In this study, knockdown of CCDC132 significantly decreased cell proliferation and clone formation ability and facilitated apoptosis, and increased phosphorylation of p53 and Chk2 and protein levels of γ-H2AX, 53BP1, cleaved Caspase 3 and cleaved PARP. Additionally, knockdown of CCDC132 attenuated tumorigenesis and tumor growth of MGC-803 cell xenografts. CCDC132 expression was significantly higher in GC tissues compared with that in adjacent normal tissues and was positively correlated with nodal metastasis and TNM stage and negatively associated with prognosis. The survival rate of CCDC132 positive patients was lower than that of CCDC132-negative patients. Furthermore, CCDC132 expression was negatively related to p53.
Conclusion: This study unravels that knockdown of CCDC132 attenuates GC cell proliferation and tumorigenesis by facilitating DNA damage signaling, indicating that CCDC132 may serve as a potential target for GC therapy.
Keywords: CCDC132, gastric cancer, proliferation, DNA damage, tumorigenesis
Introduction
Gastric cancer (GC) remains as one of the most prevalent diseases worldwide, which leads to high mortality, especially in East Asia and Eastern Europe.1,2 The risk factors of GC include Helicobacter pylori infection, inherited mutation of CDH1 gene and obesity.3–7 On the molecular events, human epidermal growth factor receptor 2 (HER2) and vascular endothelial growth factor (VEGF) were significantly amplified to promote angiogenesis in GC.8 A previous study showed that sialylation of HER2 activated AKT and ERK pathways to contribute to proliferation, suppress apoptosis and promote chemoresistance.9 Thus, HER2 has a pivotal role in oncogenesis, chemotherapy and poor prognosis.8–10 Besides, Eph receptor A3 (EphA3) was elevated in GC to activate STAT3/VEGF signaling, which could promote tumor growth and angiogenesis.11 Upregulation of EphB3 also activated the MAPK pathway to counteract the therapeutic effects of FGFR inhibitor.12 Yes-associated protein (YAP) activated EGFR/AKT and EGFR/ERK signals to attenuate the sensitivity of cisplatin treatment in GC cells.13 Upregulation of transducin (β)-like 1 X-linked receptor 1 (TBL1XR1) activated β-catenin/MMP7/EGFR/ERK signaling to promote metastasis and lead to poor prognosis of GC.14 Taken together, these findings indicated that various receptors were aberrantly activated to involve in tumorigenesis and progression of GC by triggering pro-oncogenic signals.
The endocytic cycle, including endocytosis and exocytosis, plays a pivotal role in cellular material exchange and plasma membrane content turnover, which is crucial for cell homeostasis and signaling transduction.15–17 Previous studies suggested that endocytic cargos were sorted and recycled in a specific motif-dependent manner, rather than a relatively passive process.18 Particularly, endocytic cargoes sorted in the Golgi-associated retrograde protein (GARP) complex (composed of ANG2, VPS52, VPS53 and VPS54 subunits) were retrograde transported to trans-Golgi network (TGN) for further degradation, while those cargoes sorted in the endosome-associated recycling protein (EARP) complex (composed of ANG2, VPS52, VPS53 and syndetin subunits) were recycled back to the plasma membrane for further reuse.19–22 Therefore, the EARP complex might serve as a potentially important way for upregulating pro-oncogenic cargo turnover in the plasma membrane. Moreover, syndetin was unique in the EARP complex and might serve as a very important candidate target for cancer treatments.
Aberrant endocytic recycling of plasma membrane contents, especially the receptors involved in cell migration/invasion, angiogenesis, oncogenesis and chemotherapy,23–28 was done to facilitate pro-oncogenic pathways such as growth factor receptor signaling and GTPase signaling.23,29,30 On the molecular events, a systematic analysis study unraveled that endocytosis dysregulation was correlated with cell onco-transformation.28 PKCs promoted erythroblastic oncogene B-2 (ErbB2) to enter the Arf6-dependent endocytic recycling complex for further reuse, but not lysosome for further degradation.23 Thus, targeting endocytic recycling exhibited promising prospects in cancer treatments. For example, phosphatidic acid phosphohydrolase (PAP) inhibition could suppress EGFR activation and defected its endocytic recycling, which in turn to impair cell proliferation and induce apoptosis.27 Unfortunately, although more candidates were identified in the endocytic recycling complex, the underlying mechanisms remain largely elusive. Moreover, the functions and the underlying mechanisms within the EARP complex in cancer biology remain largely unknown.
CCDC132 configured at least two aliases, VPS50 or syndetin, which are required to join and tether the EARP complex to recycling endosomes.20 So far as we know, only several studies had shown that CCDC132 was elevated in pancreatic cancer, which might involve in proliferation and immuno-escape of pancreatic cancer cells via regulating EGFR and RCAS1 turnover in plasma membrane.31–33 However, the role and mechanism of CCDC132 in cancers, especially in the pancreatic cancer, remain largely unknown. Nevertheless, in our preliminary study, we first investigated the expression of CCDC132 in a wide variety of cancers from multiple independent TCGA databases, and then we found that CCDC132 of esophagus/stomach cancers was tremendously higher than the others. This promising result strongly drove us to further investigate the function and mechanism of CCDC132 in GC.
Materials And Methods
Cell Culture
GC cell lines (AGS, SGC-7901, MGC-803 and BGC-823) and normal gastric epithelial cell line (GES-1) were purchased from the Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). All cells were maintained in Roswell Park Memorial Institute (RPMI) −1640 medium (Gibco, Scotland, UK) containing 10% fetal bovine serum (Sigma, St. Louis, MO, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin. All cell lines were incubated at 37°C with 5% CO2.
Establishment Of Scramble- And CCDC132-shRNA-Expressing GC Cell Lines
Stable knockdown of CCDC132 cell lines was established as described in a previous study.46 Lentiviruses-related plasmids were purchased and constructed by Shanghai Genechem CO., LTD (Shanghai, China). The inserted sequences were as follows: 5ʹ-TTCTCCGAACGTGTCACGT-3ʹ (scramble), 5ʹ-TTCCACCTGTTCTCAATTT-3ʹ (CCDC132-shRNA). Scramble- and CCDC132-shRNA expressing lentiviruses were also prepared by this company, and lentiviruses with titer more than 1×108 PFU/mL were used in the experiments. Stably scramble- and CCDC132-shRNA-expressing AGS cells and MGC-803 cells were established by infecting with the indicated lentivirus supernatants, respectively. Highly GFP-expressing cells were sorted and collected by FACS. After that, the highly GFP-expressing clones were passaged and expanded for further experiments.
Cell Viability Assay
GFP-expressing cells were seeded into 96-well plates at 1000 cells/well and cultured for 5 days; the growth medium was changed every 2 days. Cellular green fluorescence of each well was captured and calculated every day by using the Celigo® Image Cytometer (Nexcelom Bioscience, Lawrence, MA, USA) as per manufacturer’s instructions.
Apoptosis Assay
The indicated cells were resuspended at 1×106 cells/mL and then proceeded with eBioscience™ Annexin V Apoptosis Detection Kit APC (eBioscience, USA) as per manufacturer’s instructions. After washed with D-Hanks buffer twice, cells were resuspended in 1×binding buffer and loaded immediately by Guava® easyCyte HT Systems (Millipore, USA).
Clone Formation
Clone formation assay was performed as described in a previous study.46 In brief, cells were harvested and washed twice, after resuspending in growth medium and then passed through a 40 μm cell strainer (FALCON, Corning, USA); single cells were plated in 6-well plates at a density of 500. The growth medium of each well was carefully changed every 2 days for 2 weeks, and then cells were washed with Dx-PDFRestrictionsRemover Hanks buffer twice and stained with crystal violet. Clones of each well were captured and subsequently analyzed by Clone-Counter software.47
Western Blot
Western Blot was performed as described in a previous study.9 In brief, protein extracts from the indicated cells were prepared by RIPA lysis buffer (cat. no. P0013B; Beyotime, China). Protein concentrations were determined using the BCA Protein Assay Kit (cat. no. P0010; Beyotime, China), and total protein samples were loaded to SDS-PAGE, then followed by a transfer onto PVDF membranes (Millipore, Bedford, MA, USA). Membranes were incubated overnight at 4°C with primary antibodies. The antibodies to CCDC132 (dilution at 1:1000; cat. no. ab117763; Abcam, Cambridge, MA, USA), and GAPDH (dilution at 1:2000; cat. no. ab9484; Abcam) were used in knockdown CCDC132 AGS cells. Besides, the antibodies to the apoptosis relative proteins and the DNA damage marks were used in knockdown CCDC132 MGC-803 cells, GAPDH served as the loading control. The antibodies to the cell proliferation pathway proteins were used in knockdown CCDC132 MGC-803 cells, JNK, AKT and ERK served as loading control. After washing with TBST three times, membranes were then incubated with the matching anti-rabbit (dilution at 1:3000; cat. no. A0208; Beyotime) or anti-mouse secondary antibody (dilution at 1:3000; cat. no. A0216; Beyotime) or for 1 hr at room temperature. Proteins were visualized by using an enhanced Pierce™ ECL Western Blotting Substrate (Thermo, USA), and then captured by Gel Logic 1500 imaging system (Eastman Kodak, USA).
RNA Isolation And Quantitative RT-PCR
RNA isolation and quantitative RT-PCR were performed as described in a previous study.9 In brief, total RNA was isolated by using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.), and cDNA was synthesized by using the TransScript One-Step gDNA Removal and cDNA Synthesis SuperMix (cat. no. AT311-02; Transgen Biotech, China) according to the manufacturer’s protocol. All cDNAs were amplified by using UltraSYBR Mixture (cat. no. CW2602; CWBIO, China). The PCR condition was as follows: 95°C for 10 mins, followed by 40 cycles of 95°C for 15 s, 60°C for 40 s. The sequences of primers were used as follows: CCDC132: forward, 5ʹ‑CATCTGGGGATACGCTGTATG‑3ʹ and reverse, 5ʹ‑GTAGTTCACTGGCGGTTGAG‑3ʹ; GAPDH: forward, 5ʹ‑AGCCTCAAGATCATCAGC‑3ʹ and reverse, 5ʹ‑GAGTCCTTCCACGATACC‑3ʹ.
Cellular Stress And Apoptosis Signaling Array Assay
Cellular stress and apoptosis signaling array were mainly referred to the manufacturer’s instructions. In brief, protein extracts from the indicated cells were prepared by PathScan® Sandwich ELISA Lysis Buffer (cat. no. 7018; Cell Signaling, USA). Protein concentrations were determined using the BCA Protein Assay Kit (cat. no. P0010; Beyotime). Affix the multi-well gasket to the glass slide; 100 μL array blocking buffer was added to each well and incubated for 15 mins at room temperature on an orbital shaker. After decanting the blocking buffer, 60 μL lysate or positive/negative control was added to the appropriate well and covered with a sealing tape. After incubating it overnight at 4°C on an orbital shaker, all liquid were gently flicking out and then washed with 100 μL 1× array wash buffer 4 times. 75 μL 1× detection antibody cocktail was added to each well and incubated for 1 hr at room temperature on an orbital shaker, followed by washing with 100 μL 1× array wash buffer four times. 75 μL 1×HRP-linked streptavidin was added and incubated for 30 mins at room temperature on an orbital shaker. After washing with 100 μL 1× array wash buffer 4 times, the slide was incubated with LumiGLO®/Peroxide reagent, and then immediately captured by Gel Logic 1500 imaging system (Eastman Kodak).
Animal Xenograft Study
Mouse models bearing Scramble-MGC-803 cells and CCDC132-shRNA-MGC-803 cells were generated as follows. 4×106 indicated cells were subcutaneously inoculated into the left armpit of 4-week female nude mice (Shanghai Lingchang Biotechnology co. LTD, Shanghai, China); each group had 10 mice repeats. 8 days post-inoculation, xenografts started to emerge, and the body weights were recorded every 3 days. The volume of xenograft was calculated by the formula (V) = (a + b) × (a) × (b) × (0.2618), where V is the xenograft volume; a is the short diameter; and b is the long diameter. Three weeks after inoculation, the Perkin Elmer (Lumina LT, Boston, MA, USA) in vivo imaging system was used to measure the average fluorescence intensity and distribution using a region of interest centered on the xenograft tumors using Living Image 4.4 software. Mice were sacrificed on day 21, and the xenografts were harvested to weigh its weight. All mice were housed at Wenzhou Medical University Animal Care Facility according to the institutional guidelines for laboratory animals, and the protocol was approved by the Wenzhou Medical University Institutional Animal Care and Use Committee.
Patients And Specimens
In this study, we used a study cohort of 90 patients who underwent radical gastrotomy for gastric adenocarcinoma from May 2007 to February 2008 at The Second Affiliated Hospital of Wenzhou Medical University (Wenzhou, China) and were followed‑up from July 2015, with an observation time of 7.4–8.1 years. No patient was treated with radiotherapy or chemotherapy before surgery. The clinical information, including age, sex, tumor size, histological differentiation, recurrence, metastasis, p53 and Ki67, was obtained from the patients’ medical records. The pathological TNM status was assessed according to the criteria of the TNM Classification of Malignant Tumors and American Joint Committee on Cancer (edition 8, 2017). This study was approved by the Ethics Committee of The Second Affiliated Hospital of Wenzhou Medical University, and the need for informed consent was waived. However, written informed consent for radical gastrotomy and the use of data for research purposes were obtained from the patient prior to treatment.
Tissue Microarray (TMA) And Immunohistochemistry
The TMA was constructed as described in the reference.48 IHC detection in TMA was performed manually. Briefly, the sections were dewaxed by incubating in dimethyl benzene at 45°C for 60 mins, followed by immersion in distilled water. The slides were immersed in citrate buffer (0.01 M pH 6.0), boiling bathing for 15 mins. Block endogenous peroxidase by 3% hydrogen peroxide bath for 15 mins. After blocking in 1% bovine serum albumin for 30 mins at 24°C, the slides were incubated with rabbit anti-CCDC132 polyclonal antibody (cat. no. 10816A; Celplor LLC-A Molecular Biology CRO Company, Carolina, NC USA; diluted 1:200) in a humidified chamber overnight at 24°C, followed by conjugation to the secondary antibody and DAB staining (cat. no. ZLI-9018; Beijing Zhongshan Golden Bridge Biotechnology Co, Ltd, Beijing, China) for 5 mins; slides were then counterstained using hematoxylin, dehydrated, and sealed with neutral gum.
CCDC132 staining was scored for the percentage of positive cells and the intensity of staining in the cytoplasm. The scoring system for intensity was: 0, no staining; 1, weak staining; 2, moderate staining; and 3, strong staining. The scoring system for the percentage of stained tumor cells was: 0, <5% stained cells; 1, 5–25% stained cells; 2, 26–50% stained cells; 3, 51–75% stained cells; and 4, >75% stained cells. A final score was the product of the staining intensity and the percentage of stained cells. A score of 0 to 4 was considered CCDC132 negative and a score >4 was considered CCDC132 positive.
Statistical Analysis
The quantitative data were compared using the Wilcoxon–Mann–Whitney two-group (two-tailed) test. Student’s t-test was used to analyze any two groups. The correlation between CCDC132 expression and the clinicopathological features of the patients was performed with the Chi‑square test or Fisher’s exact test. The correlation between CCDC132 expression and p53 expression was analyzed by Spearman’s rank correlation analysis. The overall survival (OS) curve was analyzed with the Kaplan–Meier method and was compared with a log-rank test. P values <0.05 were considered statistically significant.
Results
The Expression Of CCDC132 In GC
To investigate the pro-oncogenic potentiality of CCDC132, cancer genomics data were extracted from multiple independent Cancer Genomic Atlas (TCGA) databases, and then analyzed by the cBioPortal online analyzing tool (the cBioPortal for Cancer Genomics). We found that CCDC132 of esophagus/stomach cancers was tremendously higher than the others (Figure 1A). Besides, compared with GES-1, GC cell lines all showed a high expression of CCDC132, and there was a significant difference between different GC cell lines (Figure 1B). Then, we successfully downregulated the expression of CCDC132 in AGS cells and MGC-803 cells by lentivirus-induced RNAi; the knockdown efficiency of CCDC132 of two cell lines was more than 70% (Figure 1C and D). This stable scramble cells and CCDC132 knockdown cells were passaged and expanded for further experiments.
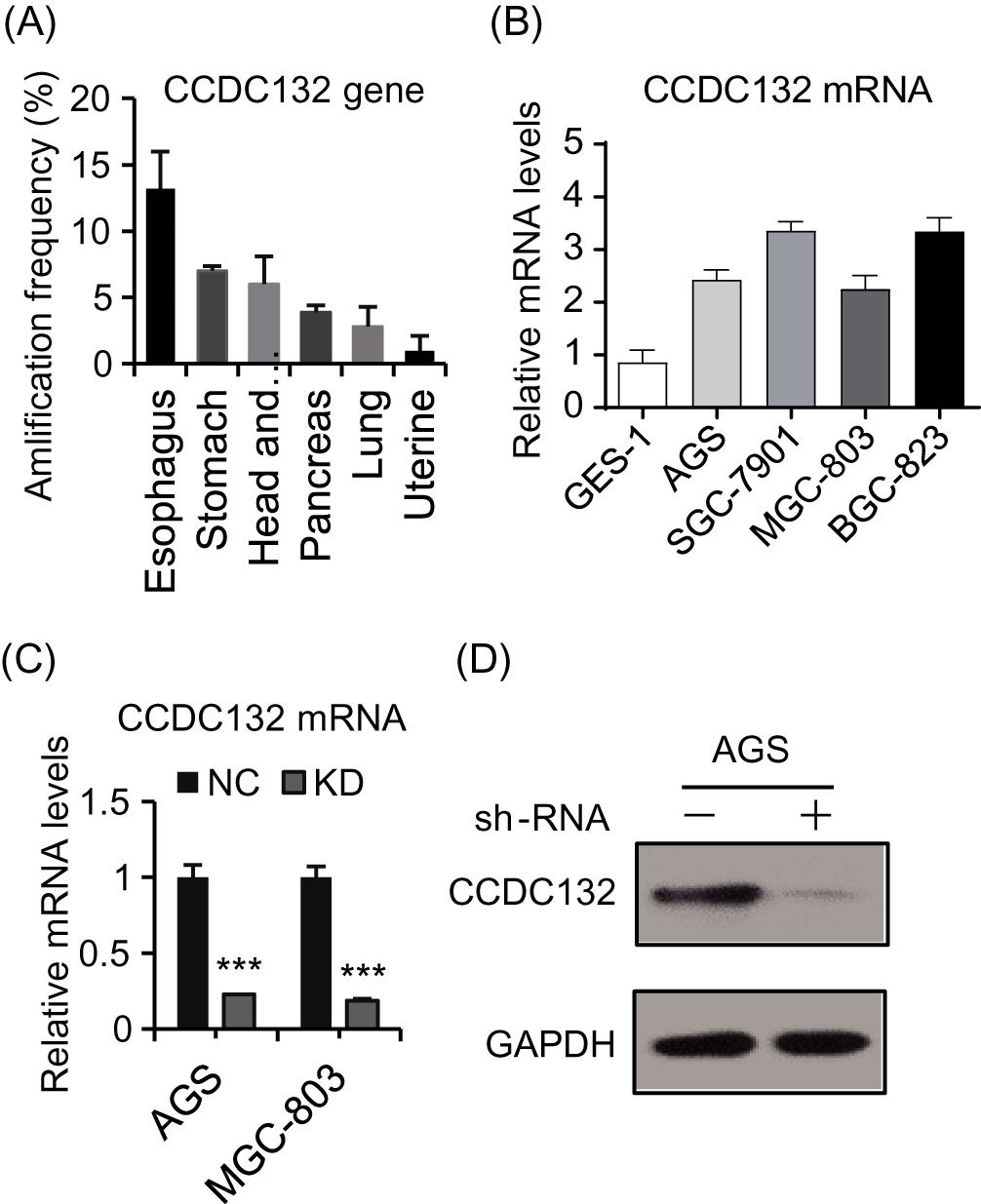 |
Figure 1 The expression of CCDC132 in GC. (A) The top amplification frequency of CCDC132 gene in various cancers. (B) Quantitative RT-PCR for the CCDC132 mRNA expression in the four GC cell lines and normal gastric epithelial cell line, GAPDH was loaded as the housekeeping gene. (C) Parent AGS cells and MGC-803 cells were infected by scramble-shRNA (NC) and CCDC132-shRNA (KD) expressing lentiviruses, respectively; then, quantitative RT-PCR was performed to detect the knockdown efficiency of CCDC132, GAPDH was loaded as the housekeeping gene. (D) The knockdown efficiency of CCDC132 of AGS cells was further confirmed by Western blot, GAPDH served as the loading control. Results are presented as the mean ± SD. ***p<0.001. |
CCDC132 Is Essential For Proliferation And Survival Of GC Cells
To investigate the primary function of CCDC132 on cellular biology of GC, we constructed stable knockdown GC cell line as described above (Figure 1C and D); our results showed that knockdown of CCDC132 significantly inhibited proliferation of AGS cells and MGC-803 cells (Figure 2A–D). Knockdown of CCDC132 almost decreased 70% clone formation abilities in AGS cells and 50% clone formation abilities in MGC-803 cells (Figure 2E–G). Otherwise, knockdown of CCDC132 slightly but significantly facilitated apoptosis of AGS cells and MGC-803 cells (Figure 2H and I). Thus, these data showed that CCDC132 is essential for proliferation and survival of GC cells.
 |
Figure 2 CCDC132 is essential for GC cell proliferation and survival. (A) and (C) the scramble (NC) and CCDC132-shRNA (KD)-expressing lentiviruses were also simultaneously expressing green fluorescent protein (GFP); these cells were seeded and then captured the fluorescent signal every day; (B) and (D) then the cell number was autocalculated by manipulating those fluorescent signals in Celigo® Image Cytometer and its software. (E) and (F) Colony formation analysis for the indicated cells. (G) Quantification of colony formation from (E) and (F). (H) FACS-based apoptosis analysis for the indicated cells. (I) Quantification of apoptosis ratio from (H). Results are presented as the mean ± SD. **p < 0.01; ***p < 0.001. |
The Underlying Mechanism Of CCDC132 On Proliferation And Survival Of MGC-803 Cells
We next applied cellular stress and apoptosis signaling arrays and Western blot analysis to explore the potential mechanism of CCDC132 on cell proliferation and survival. The results of cellular stress and apoptosis signaling arrays found that knockdown of CCDC132 significantly increased the phosphorylation of Bad, HSP27, p53, p38MAPK, SAPK/JNK and Chk2 and promoted the protein level of cleaved PARP, cleaved Caspase 3 and lκBα, while there was no significant change in p-ERK1/2, p-AKT, p-Smad2, cleaved Caspase-7, p-Chk1, p-lκBα, p-eIF2α, p-TAK1, Survivin, or α-Tubulin with or without knockdown of CCDC132. Knockdown of CCDC132 significantly increased the total protein level of lκBα with not significant changing in phosphorylated form of lκBα; thus, knockdown of CCDC132 actually decreased the phosphorylation of lκBα (Figure 3A and B). Western blot analysis also showed that knockdown of CCDC132 increased the protein expression level of p-p53, cleaved PARP and cleaved Caspase 3, while it had no significant effect on the protein expression level of p-JNK, p-AKT and p-ERK, which was consistent with the results of cellular stress and apoptosis signaling arrays. Besides, the protein expression levels of γ-H2AX and 53BP1 were significantly improved after knocking down CCDC132 in MGC-803 cells, which suggested that knockdown of CCDC132 could cause DNA damage (Figure 3C–H). Thus, these results suggested that knockdown of CCDC132 might induce DNA damage to facilitate cell apoptosis and attenuate cell division and proliferation.
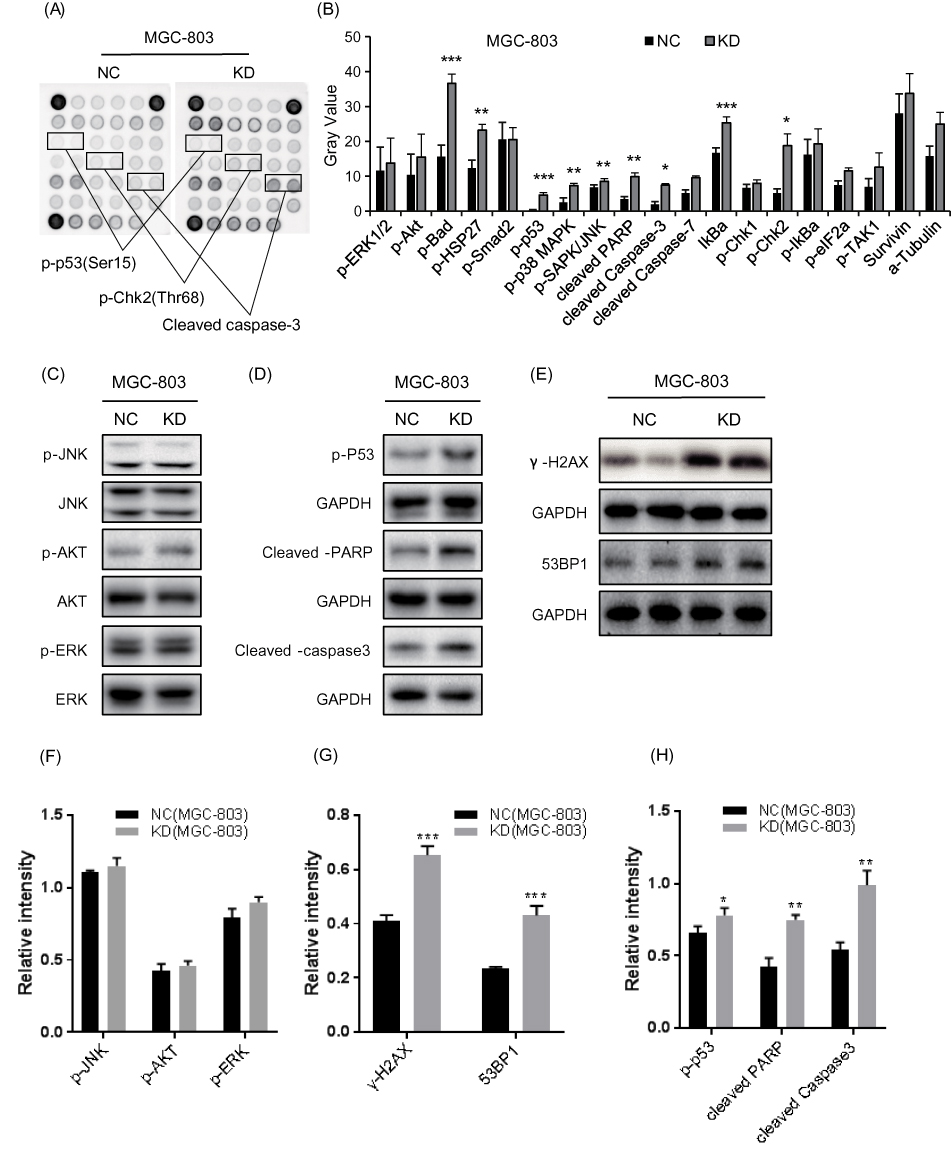 |
Figure 3 Knockdown of CCDC132 led to activate the pro-apoptotic signals in MGC-803 cells. (A) The PathScan® Stress and Apoptosis Signaling Antibody Array Kit was applied to detect the signaling molecules of stress response and apoptosis that responded to knockdown of CCDC132 in MGC-803 cells, and the interest candidates were present as described above. (B) Quantitative the array data by array analysis software. Results are presented as the mean ± SD. (C) Cell proliferation pathway proteins were confirmed by Western blot in knockdown CCDC132MGC-803 cells, JNK, AKT and ERK served as the loading control. (D) The apoptosis relative proteins were confirmed by Western blot in knockdown CCDC132 MGC-803 cells, GAPDH served as the loading control. (E) The DNA damage marks were detected by western bolt in knockdown CCDC132 MGC-803cells, GAPDH served as the loading control. (F) Cell proliferation pathway proteins were confirmed by Western blot in knockdown CCDC132MGC-803 cells, JNK, AKT and ERK served as the loading control. (G) The apoptosis relative proteins were confirmed by Western blot in knockdown CCDC132 MGC-803 cells, GAPDH served as the loading control. (H) The DNA damage marks were detected by western bolt in knockdown CCDC132 MGC-803cells, GAPDH served as the loading control. *p < 0.05; **p < 0.01; ***p < 0.001. |
Knockdown Of CCDC132 Attenuated Tumorigenesis And Tumor Growth Of MGC-803 Cells In Vivo
We investigated the pro-oncogenesis of CCDC132 in vivo, and NC-MGC-803 cells (infected by scramble-shRNA expressing lentiviruses) and KD-MGC-803 cells (infected by CCDC132-shRNA expressing lentiviruses) were seeded into 4-week female nude mice and the tumor volume was calculated every 3 days. Our results showed that the volume of the xenografts of the two group grew in a time-dependent manner, but the xenografts from NC-MGC-803 cells grew significantly faster than the xenografts from KD-MGC-803 cells (Figure 4B). At the ending point, all of xenografts were collected. The xenografts from NC-MGC-803 cells were much bigger than the xenografts from KD-MGC-803 cells (Figure 4A). Knockdown of CCDC132 led to a significant decrease of over 70% weight of xenografts (Figure 4C). Nude mouse was imaged under a small animal imaging system in vivo 3 weeks after inoculation (Figure 4D). Quantitatively, the fluorescent intensity was diminished in the xenograft of the KD group compared with that in the NC group (Figure 4E). Thus, these results show that CCDC132 plays a pivotal role in tumorigenesis and tumor growth of MGC-803 cells.
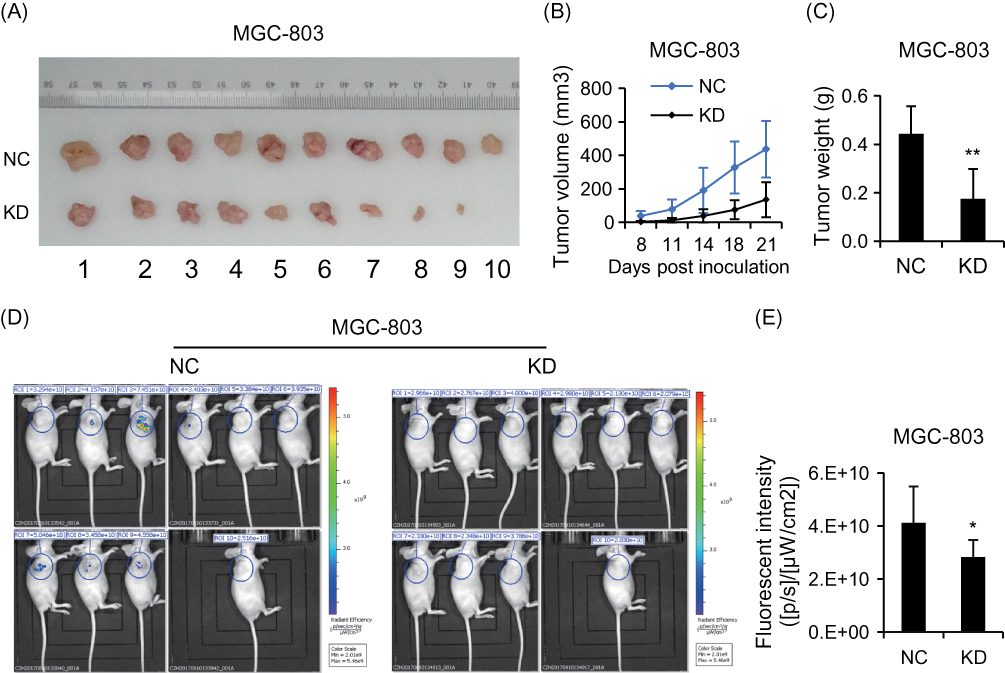 |
Figure 4 CCDC132 plays an important role in tumorigenesis and tumor growth of MGC-803 cells. (A) Xenografts from NC-MGC-803 cells and KD-MGC-803 cells were present at the ending point. (B) The tumor growth curves of xenografts from NC-MGC-803 cells and KD-MGC-803 cells, respectively. (C) Xenografts from NC-MGC-803 cells and KD-MGC-803 cells were weighted at the ending point. (D) Fluorescent intensity of the xenograft model 3 weeks after inoculation. (E) Quantitative analysis shows that of the fluorescent intensity of the KD group was obviously lower than that of the NC group. Results are presented as the mean ± SD. *p<0.05; **p<0.01. |
Overexpression Of CCDC132 Protein In GC Tissues Associated With A Poor Prognosis
We used tissue microarray to analyze the expression of the CCDC132 protein in 90 primary GC tissues and the corresponding adjacent normal tissues. Immunohistochemical staining revealed that CCDC132 protein was localized dominantly in the cytoplasm in GC cell, some expression was detected in the nucleus (Figure 5A and B). CCDC132 expression was significantly higher in the cancer tissues (56.7%) than that in the adjacent normal tissues (41.1%) (Figure 5C). We also analyzed the associations between CCDC132 expression and pathological features. Results showed that the expression of CCDC132 was significantly and positively correlated with nodal metastasis and TNM stage, yet there was no significant correlation on age, gender, tumor size, pathological grade, invasion depth and distant metastasis (Table 1). Besides, the expression of CCDC132 was negatively associated with prognosis. The survival rate for patients with GC who were CCDC132 positive was lower than that of patients who were CCDC132 negative [29 months (median), 95% confidence interval (CI): 22.37‑35.63 vs. 44 months, 95% CI: 16.93‑71.07] (Figure 5D).
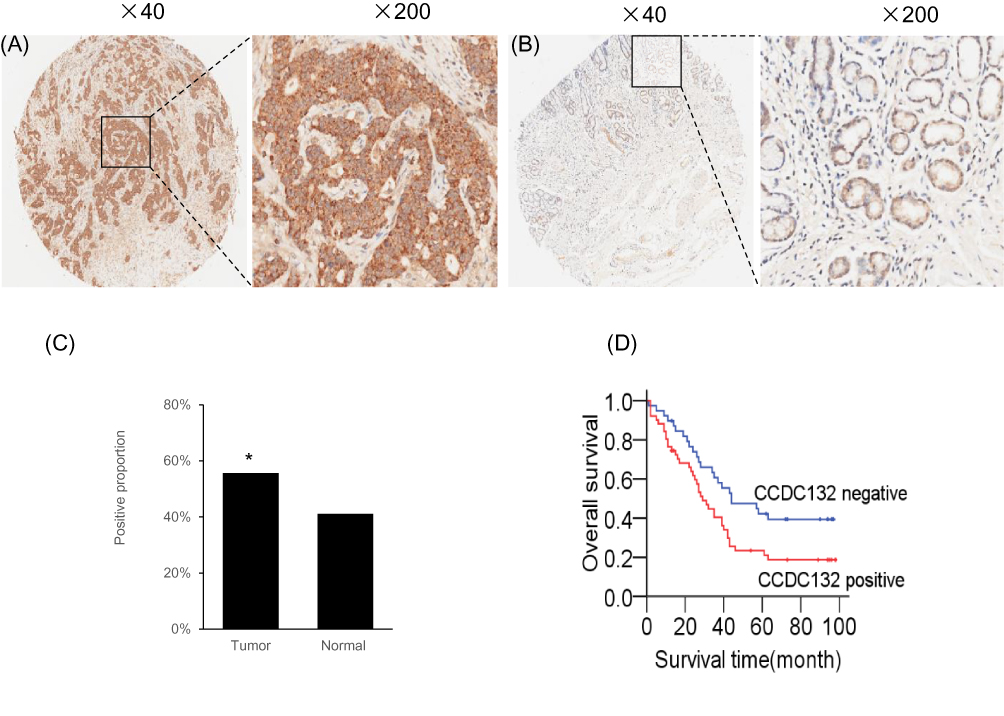 |
Figure 5 CCDC132 confers poor prognosis in patients with GC. (A) Representative Immunohistochemical staining for CCDC132 in cancer tissues. (B) Representative Immunohistochemical staining for CCDC132 in adjacent normal tissues. (C) Frequency of CCDC132 expression in cancer and adjacent normal tissues (n=90), (D) Kaplan–Meier curves of the overall survival of patients negative or positive for CCDC132 expression (log rank = 5.240, p=0.022). *p < 0.05. |
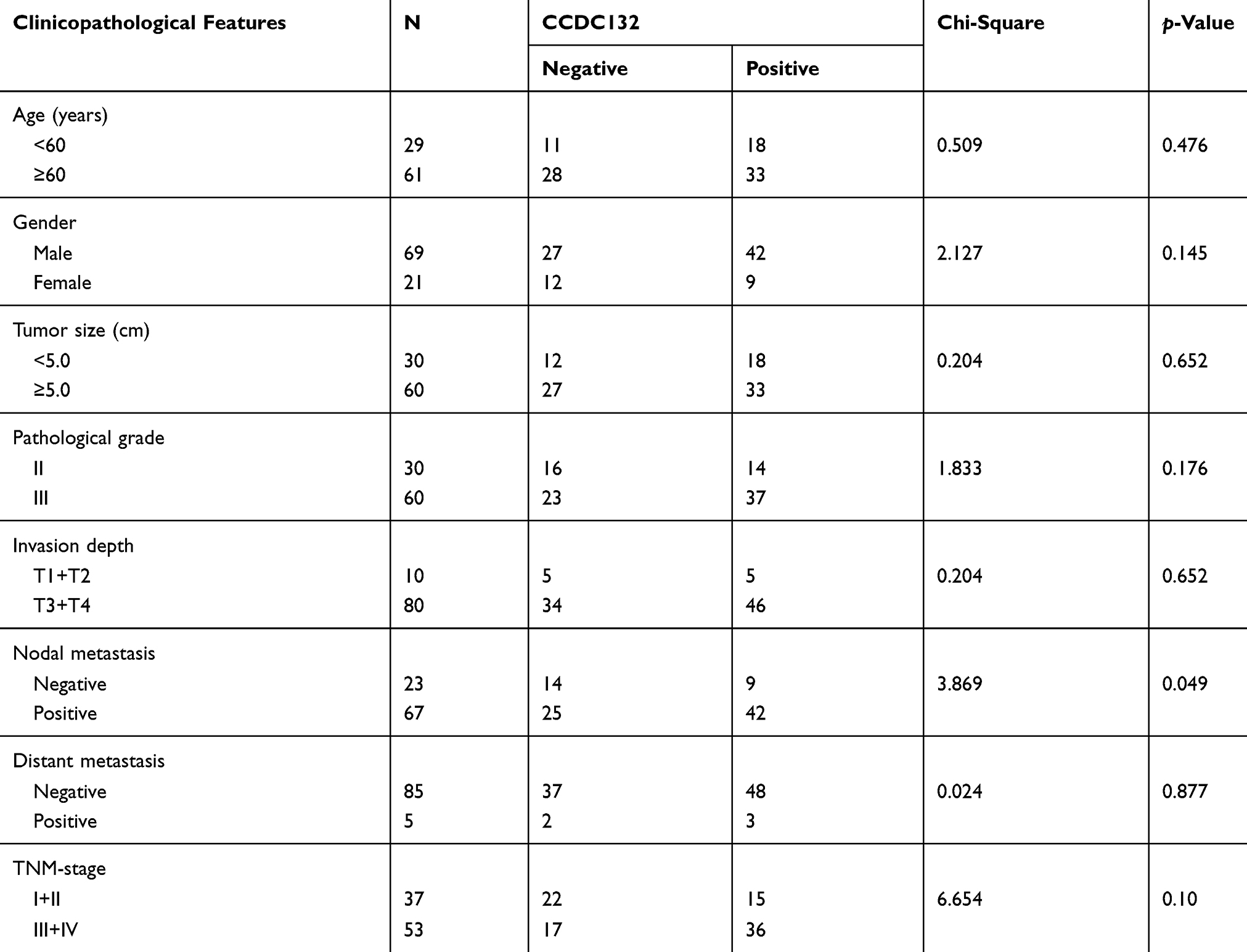 |
Table 1 The Associations Of CCDC132 Expression With Clinicopathological Features In Patients With Resectable GC. |
Correlation Between CCDC132 Expression And P53 Expression In GC
We further analyzed the correlation between CCDC132 expression and p53 expression using Spearman’s rank correlation analysis. The results revealed that the expression of CCDC132 was negatively correlated with p53 (R=−0.53, p<0.01) expression in GC.
Discussion
Aberrant activation of the receptor in cellular surface including mutation, upregulation of transcription level, aberrant degradation and recycling remains as a complicated context. Previous studies have shown that receptors acquired mutation or were amplified via various pathways in GC.10–14 However, the mechanisms of aberrant degradation or recycling of receptor in GC remain largely unknown. The CCDC132 was merely present in the EARP complex, but not the GARP complex.20 In the present study, we first investigated the relationship between endocytic recycling and oncology. After we analyzed cancer genomics data from TCGA databases, our results showed that the CCDC132 expression of esophagus/stomach cancers was tremendously higher than the others. CCDC132 was also highly expressed in GC cell lines compared with the normal gastric epithelial cell line. However, the functions of CCDC132 in GC remain unclear. Therefore, we investigated the expression and biological functions of CCDC132 in GC specimens and GC cells. We found that CCDC132 was markedly upregulated in human GC tissues relative to noncancerous tissues. CCDC132 expression was associated with the lymph node metastasis and TNM-stage. Increased CCDC132 expression was associated with a poor prognosis in GC patients. Based on these data, CCDC132 could be an attractive target for cancer therapy and warrants further exploration; thus we randomly used AGS cells and MGC-803 cells for further study. So far, as we know, only these following studies were related to CCDC132 functions. Previous studies had shown that CCDC132 contributed to dense-core vesicle maturation and acidification, which promoted the formation of the EARP complex.20,34 The elevation of CCDC132 also might contribute to pancreatic cancer and atopic dermatitis.31,35 Besides, CCDC132 significantly correlated with certain clinical pathological features of IgA nephropathy (IgAN), including urine protein, human serum albumin, total cholesterol and Lee’s pathological grades, but the relationship between CCDC132 and the risk of IgAN required further confirmation.36 However, the biological functions of CCDC132 still remain largely unclear. In the present study, we first focused on the effect of CCDC132 on GC biology. To explore the oncogenesis of CCDC132, we knocked down the CCDC132 expression of AGS cells and MGC-803 cells by lentivirus-induced RNAi. Our results first unraveled that knockdown of CCDC132 attenuated the proliferation and survival of GC cells. We next further investigated these underlying mechanisms. We screened multiple molecules involved in stress response and apoptosis that responded to knockdown of CCDC132 in MGC-803 cells. Our results show that knockdown of CCDC132 promoted phosphorylation of p53 and Chk2, and then further activated Bad, caspase 3 and PARP, which in turn triggers pro-apoptotic signaling. The knockdown of CCDC132 also increased the protein levels of γ-H2AX and 53BP1 which were the DNA damage mark. According to previous studies,37,38 these results suggested that knockdown of CCDC132 played a far-reaching effect on DNA damage to facilitate apoptosis. Otherwise, previous studies showed that HSP27 activated cytochrome c/Apaf-1/dATP complex signaling to suppress the activation of procaspase-9,39 and phosphorylation of HSP27-induced TAK1/ERK signaling to antiapoptosis.40 Our results show that knockdown of CCDC132 significantly induced phosphorylation of HSP27, yet there was no significant difference in activation of ERK, AKT, TAK1 or survivin, which indicated that the downstream antiapoptotic signaling of HSP27 was blocked under certain circumstances. Therefore, the underlying mechanism was required for further investigations. Moreover, our results also showed that knockdown of CCDC132 significantly induced phosphorylation of p38, which might respond to genotoxic stress of knockdown of CCDC132. Previous studies showed that phosphorylation of HSP27 activated TAK1/p38 to trigger pro-survival signaling,40,41 but our results found that knockdown of CCDC132 had no effect on TAK1 activity. Otherwise, the other studies had well-documented p38-mediated apoptosis in various conditions.42–44 Our results suggested that knockdown of CCDC132 activated p38-mediated apoptosis, which might be independent of HSP27. HSP27 also activated proteasome to phosphorylate and degrade lκBα, which in turn to activate NF-κB signaling.45 In the present study, our results showed that knockdown of CCDC132 significantly activated HSP27 and decreased phosphorylation of lκBα. It suggested that the effect of CCDC132 on regulating NF-κB signaling was a complicated context, and the function of this regulation was required for further investigations. Taken these together, our data suggested that knockdown of CCDC132 attenuated proliferation and survival mainly via induced apoptosis by inducing DNA damage signaling. Previous studies had shown that multiple pro-oncogenic receptors involved in tumorigenesis and progression of GC.10–14 Dysfunction of HER2/EFGR2 suppressed proliferation and metastasis as well as facilitated apoptosis in GC cells.9 The EARP complex plays a pivotal role in regulating receptor reuse.18 Thus, knockdown of CCDC132 might also serve as a potential treatment for GC via disrupting the EARP complex. In line with this hypothesis, our results showed that knockdown of CCDC132 significantly inhibited tumorigenesis and tumor growth of MGC-803 cells. Thus, CCDC132 may serve as a potential target for GC therapy.
Conclusions
In the present study, so far as we know, it was first found that CCDC132 was highly elevated in GC and first reported that CCDC132 served as a pro-oncogene to play a pivotal role in GC biology. Knockdown of CCDC132 significantly suppressed proliferation, clone formation and tumorigenesis via activating DNA damage signaling to induced apoptosis. Thus, CCDC132 serves as a potential target for GC therapy.
Acknowledgment
This work was supported by grants from Medical and Health Research Fund Project of Zhejiang Province (2017KY468) and Basic Scientific Research Project of Wenzhou (Y20180215).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Freddie B, Jian-Song R, Eric M, Jacques F. Global estimates of cancer prevalence for 27 sites in the adult population in 2008. Int J Cancer Suppl. 2013;132(5):1133–1145. doi:10.1002/ijc.27711
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7. doi:10.3322/caac.21387
3. González CA, Sala N, Rokkas T. Gastric cancer: epidemiologic aspects. Helicobacter. 2013;18(s1):34–38. doi:10.1111/hel.2013.18.issue-s1
4. Hansford S, Kaurah P, Li-Chang H, et al. Hereditary diffuse gastric cancer syndrome: CDH1 mutations and beyond. JAMA Oncol. 2015;1(1):23–32. doi:10.1001/jamaoncol.2014.168
5. van der Post RS, Vogelaar IP, Manders P, et al. Accuracy of hereditary diffuse gastric cancer testing criteria and outcomes in patients with a germline mutation in CDH1. Gastroenterology. 2015;149(4):897–906.e819. doi:10.1053/j.gastro.2015.06.003
6. Buckland G, Agudo A, Luján L, et al. Adherence to a mediterranean diet and risk of gastric adenocarcinoma within the European Prospective Investigation into Cancer and Nutrition (EPIC) cohort study. Am J Clin Nutr. 2010;91(2):381–390. doi:10.3945/ajcn.2009.28209
7. Crew KD, Neugut AI. Epidemiology of gastric cancer. World J Gastroenterol. 2006;12(3):354. doi:10.3748/wjg.v12.i3.354
8. Geng Y, Chen X, Qiu J, et al. Human epidermal growth factor receptor-2 expression in primary and metastatic gastric cancer. Int J Clin Oncol. 2014;19(2):303. doi:10.1007/s10147-013-0542-9
9. Liu N, Zhu M, Linhai Y, et al. Increasing HER2 α2,6 sialylation facilitates gastric cancer progression and resistance via the Akt and ERK pathways. Oncol Rep. 2018;40(5):2997–3005.
10. Narikazu B. HER2-positive gastric cancer. Gastric Cancer. 2014;17(1):1–12. doi:10.1007/s10120-013-0252-z
11. Lv XY, Lv XY, Wang J, et al. EphA3 contributes to tumor growth and angiogenesis in human gastric cancer cells. Oncol Rep. 2018;40(4):2408–2416. doi:10.3892/or.2018.6586
12. SY L, YJ N, YA J, JL K, SC O, DH L. Upregulation of EphB3 in gastric cancer with acquired resistance to a FGFR inhibitor. Int J Biochem Cell Biol. 2018;102:128–137. doi:10.1016/j.biocel.2018.07.008
13. Lu T, Sun L, Zhu X. Yes-associated protein enhances proliferation and attenuates sensitivity to cisplatin in human gastric cancer cells. Biomed Pharmacother. 2018;105:1269–1275. doi:10.1016/j.biopha.2018.06.031
14. Zhou Q, Wang X, Yu Z, et al. Transducin (β)-like 1 X-linked receptor 1 promotes gastric cancer progression via the ERK1/2 pathway. Oncogene. 2017;36(13):1873. doi:10.1038/onc.2016.352
15. Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annu Rev Biochem. 2009;78:857–902. doi:10.1146/annurev.biochem.78.081307.110540
16. Watanabe S, Boucrot E. Fast and ultrafast endocytosis. Curr Opin Cell Biol. 2017;47:64–71. doi:10.1016/j.ceb.2017.02.013
17. Liang K, Wei L, Chen L. Exocytosis, endocytosis, and their coupling in excitable cells. Front Mol Neurosci. 2017;10:109. doi:10.3389/fnmol.2017.00109
18. Cullen PJ, Steinberg F. To degrade or not to degrade: mechanisms and significance of endocytic recycling. Nat Rev Mol Cell Biol. 2018;19(11):679–696.
19. Gershlick DC, Schindler C, Chen Y, Bonifacino JS. TSSC1 is novel component of the endosomal retrieval machinery. Mol Biol Cell. 2016;27(18):2867–2878. doi:10.1091/mbc.e16-04-0209
20. Schindler C, Chen Y, Pu J, Guo X, Bonifacino JS. EARP, a multisubunit tethering complex involved in endocytic recycling. Nat Cell Biol. 2015;17(5):639–650. doi:10.1038/ncb3129
21. Spang A. Membrane tethering complexes in the endosomal system. Front Cell Dev Biol. 2016;4:51. doi:10.3389/fcell.2016.00035
22. Bonifacino JS, Hierro A. Transport according to GARP: receiving retrograde cargo at the trans-Golgi network. Trends Cell Biol. 2011;21(3):159–167. doi:10.1016/j.tcb.2010.11.003
23. Bailey TA, Luan H, Tom E, et al. A kinase inhibitor screen reveals protein kinase C-dependent endocytic recycling of ErbB2 in breast cancer cells. J Biol Chem. 2014;289(44):30443–30458. doi:10.1074/jbc.M114.608992
24. Zobel M, Disanza A, Senic-Matuglia F, et al. A NUMB–EFA6B–ARF6 recycling route controls apically restricted cell protrusions and mesenchymal motility. J Cell Biol. 2018;217(9):3161–3182. doi:10.1083/jcb.201802023
25. Chrifi I, Louzao-Martinez L, Brandt MM, et al. CMTM4 regulates angiogenesis by promoting cell surface recycling of VE-cadherin to endothelial adherens junctions. Angiogenesis. 2019;22(1):75–93. doi:10.1007/s10456-018-9638-1
26. Das L, Gard JM, Prekeris R, et al. Novel regulation of integrin trafficking by Rab11-FIP5 in aggressive prostate cancer. Mol Cancer Res. 2018;16(8):1319–1331. doi:10.1158/1541-7786.MCR-17-0589
27. Shaughnessy R, Retamal C, Oyanadel C, et al. Epidermal growth factor receptor endocytic traffic perturbation by phosphatidate phosphohydrolase inhibition: new strategy against cancer. FEBS J. 2014;281(9):2172–2189. doi:10.1111/febs.12770
28. Elkin SR, Bendris N, Reis CR, et al. A systematic analysis reveals heterogeneous changes in the endocytic activities of cancer cells. Cancer Res. 2015;75(21):4640. doi:10.1158/0008-5472.CAN-15-0939
29. Caldieri G, Malabarba MG, Fiore PPD, Sigismund S. EGFR Trafficking in Physiology and Cancer. Springer, Cham: 2018;235–272.
30. Tebar F, Enrich C, Rentero C, Grewal T. GTPases Rac1 and ras signaling from endosomes. Prog Mol Subcell Biol. 2018;57:65–105.
31. Porterfield M, Zhao P, Han H, et al. Discrimination between adenocarcinoma and normal pancreatic ductal fluid by proteomic and glycomic analysis. J Proteome Res. 2015;13(2):395–407. doi:10.1021/pr400422g
32. Nobuyuki O, Masaki O, Satoshi I, et al. Serine protease inhibitor kazal type 1 and epidermal growth factor receptor are expressed in pancreatic tubular adenocarcinoma, intraductal papillary mucinous neoplasm, and pancreatic intraepithelial neoplasia. J Hepatobiliary Pancreat Sci. 2013;20(6):620–627. doi:10.1007/s00534-012-0587-6
33. Akashi T, Oimomi H, Nishiyama K, et al. Expression and diagnostic evaluation of the human tumor-associated antigen RCAS1 in pancreatic cancer. Pancreas. 2003;26(1):49–55. doi:10.1097/00006676-200301000-00009
34. Paquin N, Murata Y, Froehlich A, et al. The conserved VPS-50 protein functions in dense-core vesicle maturation and acidification and controls animal behavior. Curr Biol. 2016;26(7):862–871. doi:10.1016/j.cub.2016.01.049
35. Matsumoto Y, Imai Y, Sugita Y, et al. CCDC132 is highly expressed in atopic dermatitis T cells. Mol Med Rep. 2010;3(1):83. doi:10.3892/mmr_00000237
36. Niu D, Ren Y, Xie L, et al. Association between CCDC132, FDX1 and TNFSF13 gene polymorphisms and the risk of IgA nephropathy. Nephrology. 2015;20(12):908–915. doi:10.1111/nep.12611
37. Speidel D. The role of DNA damage responses in p53 biology. Arch Toxicol. 2015;89(4):501–517.
38. Cai Z, Chehab NH, Pavletich NP. Structure and activation mechanism of the CHK2 DNA damage checkpoint kinase. Mol Cell. 2009;35(6):818–829. doi:10.1016/j.molcel.2009.09.007
39. Bonniaud P, Ducasse C, Bonniaud P. Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat Cell Biol. 2000;2(9):645–652. doi:10.1038/35023595
40. Qi Z, Shen L, Zhou H, et al. Phosphorylation of heat shock protein 27 antagonizes TNF-α induced HeLa cell apoptosis via regulating TAK1 ubiquitination and activation of p38 and ERK signaling. Cell Signal. 2014;26(7):1616–1625. doi:10.1016/j.cellsig.2014.03.015
41. Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412(6844):346–351. doi:10.1038/35085597
42. Yan Q, Chen X, Gong H, et al. Delivery of a TNF-α–derived peptide by nanoparticles enhances its antitumor activity by inducing cell-cycle arrest and caspase-dependent apoptosis. FASEB J. 2018;32(12):6948–6964. doi:10.1096/fj.201800377R
43. Beccafico S, Morozzi G, Marchetti MC, et al. Artesunate induces ROS-and p38 MAPK-mediated apoptosis and counteracts tumor growth in vivo in embryonal rhabdomyosarcoma cells. Carcinogenesis. 2015;36(9):1071–1083. doi:10.1093/carcin/bgv098
44. Sui X, Kong N, Ye L, et al. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014;344(2):174–179. doi:10.1016/j.canlet.2013.11.019
45. Parcellier A, Schmitt E, Gurbuxani S, et al. HSP27 is a ubiquitin-binding protein involved in I-kappaBalpha proteasomal degradation. Mol Cell Biol. 2003;23(16):5790. doi:10.1128/MCB.23.16.5790-5802.2003
46. Liu N, Mei L, Fan X, et al. Phosphodiesterase 5/protein kinase G signal governs stemness of prostate cancer stem cells through Hippo pathway. Cancer Lett. 2016;378(1):38–50. doi:10.1016/j.canlet.2016.05.010
47. Niyazi M, Niyazi I, Belka C. Counting colonies of clonogenic assays by using densitometric software. Radiat Oncol. 2007;2(1):4. doi:10.1186/1748-717X-2-4
48. Xu J, Zhao ZG, Ye L, et al. Prognostic significance of Daxx NCR (nuclear/cytoplasmic ratio) in gastric cancer. Cancer Med. 2017;6(9):2063–2075. doi:10.1002/cam4.1144
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.