Back to Journals » Infection and Drug Resistance » Volume 19
Klebsiella pneumoniae and Disseminated Intravascular Coagulation: A Comprehensive Review of Pathogenesis, Clinical Manifestations, and Management Strategies
Authors Namaki M, Ghoreyshizadeh E, Shokouhi B, Naeimi Mazraeh F, Mahmoudpour A, Taghizadeh S, Aghazadeh M, Samadi Kafil H ![]()
Received 8 February 2026
Accepted for publication 29 June 2026
Published 8 July 2026 Volume 2026:19 602149
DOI https://doi.org/10.2147/IDR.S602149
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Hemant Joshi
Malek Namaki1,2, Erfan Ghoreyshizadeh2, Behrooz Shokouhi2, Fariba Naeimi Mazraeh3, Ata Mahmoudpour4, Sepehr Taghizadeh5, Mohammad Aghazadeh2, Hossein Samadi Kafil5
1Student Research Committee, Faculty of Medicine, Tabriz University of Medical Sciences, Tabriz, Iran; 2Pharmaceutical Nanotechnology Research Center, Faculty of Medicine, Tabriz University of Medical Sciences, Tabriz, Iran; 3Immunology Research Center, Faculty of Medicine, Tabriz University of Medical Sciences, Tabriz, Iran; 4Resuscitation and Critical-Care Research Center, Department of Anesthesiology, Faculty of Medicine, Tabriz University of Medical Sciences, Tabriz, Iran; 5Drug Applied Research Center, Faculty of Medicine, Tabriz University of Medical Sciences, Tabriz, Iran
Correspondence: Hossein Samadi Kafil, Drug Applied Research Center, Faculty of Medicine, Tabriz University of Medical Sciences, Tabriz, Iran, Email [email protected]; Mohammad Aghazadeh, Drug Applied Research Center, Faculty of Medicine, Tabriz University of Medical Sciences, Tabriz, Iran, Email [email protected]
Abstract: K. pneumoniae represents a significant opportunistic pathogen capable of causing life-threatening infections, particularly in immunocompromised and critically ill patients. Despite increasing reports of severe coagulopathy in K. pneumoniae sepsis, pathogen-specific mechanisms and tailored management strategies remain insufficiently characterized. This review aims to systematically examine the intricate relationship between K. pneumoniae infections and the development of DIC, a critical complication with high mortality rates, by integrating microbial virulence factors with the host’s thrombo-inflammatory response. We explore the microbial virulence factors that contribute to disease pathogenesis, including capsular polysaccharides, lipopolysaccharides, siderophores, and various genetic determinants that enable immune evasion and systemic dissemination. The pathophysiological mechanisms linking K. pneumoniae sepsis to DIC are detailed, emphasizing the role of inflammatory cytokines, coagulation cascade activation, and endothelial dysfunction. Clinical manifestations, diagnostic challenges including a thorough overview of global diagnostic criteria, and current management strategies are discussed, highlighting the importance of early recognition and targeted intervention. In conclusion, this review underscores that DIC in K. pneumoniae sepsis results from a synergistic interaction between specific bacterial virulence factors (eg, LPS, capsule, siderophores) and a dysregulated host thrombo-inflammatory response, advocating for a preemptive, multimodal management strategy that combines rapid antimicrobial therapy with early recognition of coagulopathy to improve patient outcomes.
Keywords: Klebsiella pneumoniae, intensive care units, disseminated intravascular coagulation
Introduction
K. pneumoniae is a formidable Gram-negative pathogen whose pathogenicity is driven by key virulence factors including a polysaccharide capsule essential for immune evasion and high-affinity siderophore systems for iron acquisition.1 Hypermucoviscous strains, especially K1/K2 capsular serotypes, exhibit heightened invasiveness.2 Furthermore, biofilm formation widely implicated in 65–80% of bacterial infections and a Type VI secretion system (T6SS) significantly enhance the persistence and virulence of K. pneumoniae.3,4
In intensive care units (ICUs), K. pneumoniae, particularly carbapenem-resistant (CRKP) strains, poses a critical threat. ICU admission is an independent risk factor for CRKP bloodstream infection.5–7 The pooled prevalence of CRKP among nosocomial K. pneumoniae infections in ICUs is alarmingly high at 62%.8 Associated mortality is substantial, with a global 30-day mortality rate of 34% for CRKP bacteremia and a pooled mortality of 37.2% for CRKP infections.9,10 ICU-acquired K. pneumoniae pneumonia carries mortality exceeding 50% in several series.11,12
In septic patients, K. pneumoniae can trigger disseminated intravascular coagulation (DIC), a life-threatening coagulopathy characterized by systemic thrombosis and bleeding. The pathogenesis involves lipopolysaccharide (LPS)-driven tissue factor expression and a cytokine storm, initiating coagulation.13 This is amplified by neutrophil extracellular traps (NETs), which provide a prothrombotic scaffold,14 and bacterial outer membrane vesicles (OMVs) that systemically disseminate procoagulant stimuli.15 Concurrent endothelial damage and depletion of natural anticoagulants culminate in DIC.16
Diagnosis relies on scoring systems due to the absence of a single definitive test.13 The International Society on Thrombosis and Haemostasis (ISTH) overt DIC criteria are widely used.16 For sepsis-associated DIC, the Sepsis-Induced Coagulopathy (SIC) score, endorsed by the ISTH, is valuable for early identification using platelet count, prothrombin time (PT), and Sequential Organ Failure Assessment (SOFA) score.17,18 The Japanese Association for Acute Medicine (JAAM) criteria offer an alternative with high sensitivity.16,19 To our knowledge, this is the first review specifically integrating microbiological virulence, immunothrombosis, and clinical management of DIC triggered by K. pneumoniae.
Epidemiology and Risk Factors
The epidemiology of K. pneumoniae infections varies significantly by healthcare setting, with ICUs representing a major burden. The global pooled prevalence of CRKP among nosocomial infections is 28%, rising sharply to 62% specifically within ICUs.8 Colonization is a key risk factor for subsequent infection; rates are markedly higher in hospitalized patients, particularly those exposed to broad-spectrum antibiotics.3,20 Major risk factors for ICU-acquired CRKP infection include mechanical ventilation, central venous catheterization, and prior antibiotic exposure.21 Hypervirulent and resistant strains, such as ST11 in China and ST258 in Europe, are frequently implicated in severe outbreaks within these settings.22,23 K. pneumoniae-associated DIC occurs most frequently in patients with specific risk factors that predispose them to severe infection and coagulation dysfunction. Diabetes mellitus emerges as a particularly significant risk factor, as demonstrated in a case report of a 72-year-old woman with newly diagnosed diabetes who developed fatal K. pneumoniae meningitis with concomitant DIC.24 Other important predisposing conditions include alcoholism, malignant tumors, renal failure, liver disease, and chronic lung disease.25 Immunosuppression, whether from disease processes or therapeutic interventions, dramatically increases susceptibility to severe K. pneumoniae infections and subsequent DIC.26 About 50% to 70% of patients with sepsis develop coagulation dysfunction, and about 35% of patients have significant DIC.27 The development of DIC in K. pneumoniae infections significantly worsens prognosis. Mortality rates for K. pneumoniae pneumonia requiring ICU admission reach 58%, while CRKP bloodstream infections are associated with a 30-day mortality rate of 34%.11 This substantial risk underscores the critical importance of preventing hematogenous dissemination and recognizing coagulation abnormalities early in the course of infection (Table 1).
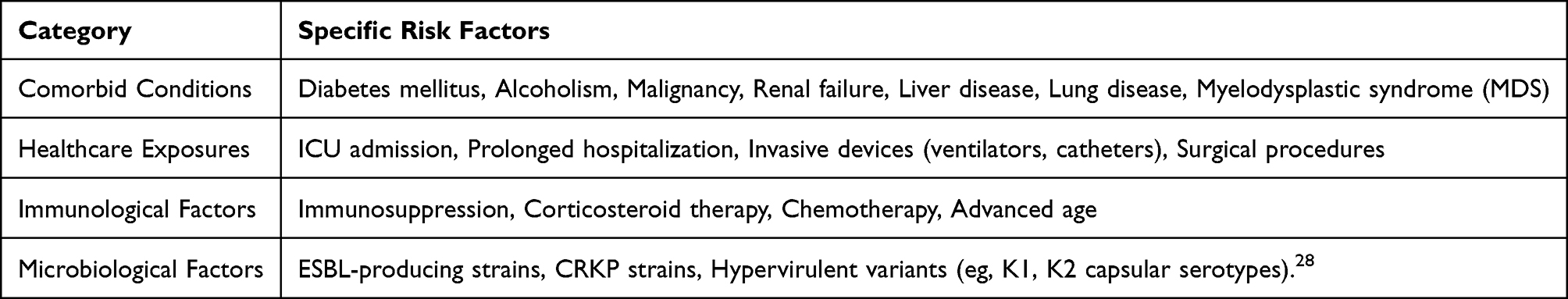 |
Table 1 Risk Factors for K. pneumoniae Infection and DIC |
Virulence Mechanisms of K. pneumoniae
The pathogenesis of K. pneumoniae infections and their ability to trigger DIC relies on an arsenal of virulence factors that enable immune evasion, tissue invasion, and systemic dissemination. Understanding these mechanisms is essential for comprehending the link between infection and coagulopathy.29
LPS
The LPS component of K. pneumoniae’s outer membrane functions as a potent endotoxin that triggers intense inflammatory responses through activation of pattern recognition receptors such as TLR4 on immune cells, contributing to the cytokine storm associated with severe sepsis and DIC.30 Furthermore, LPS within OMVs is pivotal in driving DIC during Gram-negative sepsis by synergistically amplifying host procoagulant responses via TLR4 activation, suggesting OMV production as a novel therapeutic target.15 The outermost O-antigen subunit of LPS represents the initial interface with the innate immune system, with O1 being the most common serotype among clinical K. pneumoniae isolates. Specifically, the O1 antigen binds complement component C3b, weakening macrophage activation and promoting bacteremia and liver abscess formation. Genes involved in LPS core synthesis (eg, waaG, waaF, waaC) are crucial for maintaining membrane integrity and resistance to hydrophobic agents and host defenses.31 LPS is a powerful Pathogen-Associated Molecular Pattern (PAMP) that binds to TLR4 on macrophages and endothelial cells, leading to the release of pro-inflammatory cytokines (TNF-α, IL-1, IL-6, HMGB-1).32 This cytokine storm is a primary cause of DIC, as these mediators induce tissue factor expression on monocytes and endothelium, a critical mechanism linking bacteremia to septic coagulopathy.33 This activation of the coagulation cascade leads to thrombin generation and a thrombotic phenotype characterized by significant fibrin deposition, particularly in renal glomeruli, contributing to organ dysfunction.32,34 Simultaneously, LPS-mediated pathways suppress natural anticoagulant pathways (eg, antithrombin, protein C) and impair fibrinolysis via a sustained increase in Plasminogen Activator Inhibitor-1 (PAI-1).32,35,36 Furthermore, LPS directly activates the contact pathway of coagulation cascade by binding to and autoactivating Factor XII (FXII or Hageman factor).37 In conclusion, compelling evidence delineates LPS as a central mediator in the pathogenesis of K. pneumoniae sepsis-associated DIC. Therefore, we conclude that LPS is a potential biomarker for DIC in patients with K. pneumoniae sepsis, which could inform preventive strategies and improve prognostic assessment after further clinical validation.
Capsular Polysaccharides
The capsular polysaccharide represents the most important virulence factor of K. pneumoniae, serving as the primary determinant of serotype classification and providing protection against phagocytosis and serum bactericidal activity.38 To date, 77 distinct capsular types have been identified, with specific capsular serotypes (K1, K2, K5, K20, K54, and K57) consistently associated with invasive pyogenic infections including liver abscess, septicemia, and pneumonia.39,40 The K1/K2 capsular serotypes demonstrate particularly high virulence, attributed to their chemical composition which lacks mannose and rhamnose, sugars normally recognized by macrophage lectin receptors for phagocytosis induction. Capsular synthesis is encoded by the chromosomal cps gene cluster, which includes genes such as wzi, wza, wzb, wzc, gnd, wca, cpsB, cpsG, and galF. The wzy gene serves as a serotype-specific target often used for differentiating various cps types. Interestingly, the chromosomally encoded mucoviscosity-associated gene A (magA), discovered in 2004, was subsequently identified as the wzy gene specific to the K1 capsular serotype (K1_wzy).40,41 The magA gene is essential for capsular polysaccharide synthesis and appears to mediate crucial role in hypermucoviscosity and complement-mediated resistance, while magA mutants lose these characteristics. The hypermucoviscous phenotype, identifiable by a positive string test (>5 mm filament), is a key feature of hypervirulent strains and is strongly associated with the K1/K2 capsular serotypes and the presence of the rmpA/rmpA2 regulators.42 This thick capsule is paramount for resisting phagocytosis by neutrophils and macrophages, allowing the bacterium to achieve high bacterial loads in the blood, a potent trigger for systemic inflammatory response and DIC.26,43 The capsule is an anionic polysaccharide layer with negatively charged surface, due to carboxyl and phosphate groups, and can promote the binding and autoactivation of FXII, the initiator of the contact pathway of coagulation. This process drives systemic thrombin generation, microvascular thrombosis, consumptive coagulopathy, and can progress to DIC and multi-organ failure.37,44 Also, during sepsis, capsule-mediated intense inflammation and endothelial activation may promote coagulopathy by contributing to endothelial injury, suppression of anticoagulant pathways (eg, the protein C system), and the creation of a procoagulant state through increased tissue factor and elevated PAI-1 levels.45 Consequently, the bacterial capsule may contribute to a procoagulant microenvironment that exacerbates SIC, underscoring its potential as a therapeutic target for reducing infection-associated thrombosis.
Siderophores and Iron Acquisition Systems
Iron acquisition represents a critical determinant of K. pneumoniae pathogenicity, as iron is essential for bacterial proliferation within the host environment. K. pneumoniae employs several siderophore systems including enterobactin, yersiniabactin, salmochelin, and aerobactin to scavenge iron from host proteins.46,47 Hypervirulent K. pneumoniae (hvKP) strains demonstrate a distinctive 6- to 10-fold increase in siderophore production compared to classical K. pneumoniae strains. The genes required for enterobactin biosynthesis are located on the chromosomal ent cluster (eg, entB), while its receptor is encoded by fepA. To circumvent host defense mechanisms like lipocalin-2 (which binds enterobactin), K. pneumoniae produces modified siderophores including glycosylated enterobactin (salmochelin, iro genes) and the non-catecholate siderophore yersiniabactin (ybt genes). Aerobactin (encoded by the iucABCD-iutA operon) appears particularly important in hvKP strains, accounting for approximately 90% of total siderophore production and serving as a reliable biomarker for hvKP identification.2,39 The genes for aerobactin are often located on virulence plasmids (eg, pLVPK), frequently co-localizing with rmpA. Other iron acquisition systems include the kfu ABC transporter (associated with liver abscess and meningitis) and the yersiniabactin system (ybtS).42 By efficiently scavenging iron, these systems support rapid bacterial growth in vivo, leading to high-grade bacteremia and sustained inflammatory stimulus, thereby exacerbating the risk of DIC.43 In addition, Siderophores, such as enterobactin and yersiniabactin, can modulate host immune responses. They induce proinflammatory cytokines, increase vascular permeability, and promote endothelial damage. This inflammatory milieu activates the coagulation system, leading to microthrombosis and consumption of clotting factors hallmarks of DIC.48
Adhesion Factors and Biofilm
Fimbriae (pili) represent proteinaceous structural appendages that facilitate bacterial attachment to host surfaces. K. pneumoniae expresses type 1 and type 3 fimbriae, encoded by fim and mrk gene clusters, respectively.47 Type 1 fimbriae (adhesin FimH) are expressed in the urinary tract and enable bladder cell invasion and biofilm formation, while type 3 fimbriae (adhesin MrkD) facilitate attachment to both biotic (eg, endothelial cells, extracellular matrix) and abiotic surfaces (eg, catheters).46 The allS gene, often associated with the K1 capsular serotype, is involved in allantoin metabolism and also contributes to virulence. Adhesion is the first step towards colonization and subsequent invasion. While the capsule can sometimes mask fimbriae, their role in establishing infection foci is critical. Biofilm formation, aided by fimbriae, creates a protected niche for persistent infection, which can serve as a continuous source of bacteremia and ongoing activation of the coagulation and inflammatory systems.35,43,49,50 It is hypothesized that fimbriae in Gram-negative bacteremia facilitate initial tissue invasion and systemic dissemination. Through promoting endothelial attachment and activation, they may bind directly to coagulation FXII, potentially triggering its autoactivation and contributing to DIC. Activated FXII (FXIIa or Activated Hageman factor) initiates the intrinsic coagulation cascade, promoting thrombin generation and a hypercoagulable state. Simultaneously, FXIIa activates the kallikrein-kinin system, releasing bradykinin, which enhances vascular permeability and triggers a potent pro-inflammatory host response. This combined dysregulation of coagulation and inflammation drives the consumption of clotting factors and microvascular thrombosis, thereby serving as a critical early pathogenic mechanism in the progression from bacteremia to septic coagulopathy and potential DIC.37,44,48
T6SS and OMPs
The T6SS is a contact-dependent mechanism used to inject effector proteins into eukaryotic host cells or competing bacteria in K. pneumoniae. Presence of T6SS has been associated with hvKP and the K1 capsular serotype, enhancing survival and pathogenicity in the host.51 Outer Membrane Proteins (OMPs) in K. pneumoniae like OmpK35 and OmpK36 are involved in nutrient uptake and can contribute to antibiotic resistance and serum resistance. Lipoproteins are important for maintaining outer membrane integrity and resistance to phagocytosis.2 The T6SS functions as a molecular syringe, delivering effector proteins directly into host endothelial cells. This action disrupts endothelial integrity, causing vascular leakage and exposing subendothelial tissue factor a potent initiator of the extrinsic coagulation pathway. Concurrently, T6SS effectors can activate pro-inflammatory pathways (eg, NF-κB), contributing to a cytokine storm that further upregulates tissue factor on monocytes and endothelial cells, thereby amplifying thrombin generation.52,53 OMPs, exacerbate this process through distinct mechanisms. They facilitate bacterial adhesion to and invasion of endothelial cells, causing direct injury. Furthermore, OMPs engage TLR2/TLR4, intensifying the inflammatory response that promotes a hypercoagulable state. They also aid in immune evasion by binding host regulators, perpetuating the infection that fuels coagulation activation.52,54 In concert, T6SS-mediated direct damage and OMP-driven hyperinflammation create a vicious cycle. This leads to excessive fibrin deposition, platelet consumption, and secondary fibrinolysis, the hallmark features of DIC. Consequently, microvascular thrombosis contributes to multi-organ failure, underscoring the pivotal role of these virulence factors in sepsis-associated coagulopathy.
Regulatory Genes and Virulence Plasmids
Regulatory genes play crucial roles in modulating K. pneumoniae virulence expression. The regulator of mucoid phenotype A and A2 (rmpA and rmpA2) genes are known positive regulators of capsule production and the hypermucoviscosity phenotype.41,55 Additionally, the Klebsiella virulence regulator genes kvrA and kvrB influence virulence in K1/K2 hvKP strains by activating capsule gene expression, an effect not observed in classical strains. The ferric uptake regulator (Fur) modulates iron acquisition and also suppresses rmpA, rmpA2, and rcsA genes responsible for capsule production. Many virulence genes (eg, iucABCD-iutA [aerobactin], iroBCDN [salmochelin], rmpA/rmpA2) are carried on large, conjugative plasmids (eg, pLVPK) that enable horizontal gene transfer between bacteria.55 This co-localization of virulence and sometimes resistance determinants on mobile genetic elements facilitates the emergence of increasingly pathogenic and difficult-to-treat strains, such as carbapenem-resistant hypervirulent K. pneumoniae (CR-hvKP).2 Regulatory genes and virulence plasmids in K. pneumoniae critically exacerbate sepsis-induced DIC by amplifying key virulence determinants. These elements upregulate capsular polysaccharide production and siderophore systems, enhancing immune evasion, iron acquisition, and high-grade bacteremia. The resulting sustained inflammatory response and endothelial injury promote tissue factor expression, thrombin generation, and consumptive coagulopathy. Consequently, this dysregulation of host coagulation and inflammation drives the progression to DIC.
Summary
The pathogenesis of DIC in K. pneumoniae sepsis is driven by a hierarchical synergy of key bacterial virulence factors that sequentially ignite and amplify a thrombo-inflammatory cascade. The primary instigator is LPS, which binds host TLR4 to trigger a cytokine storm; this upregulates tissue factor on monocytes and endothelium to initiate coagulation, directly injures the endothelium to create a procoagulant state, stimulates PAI-1 to cause fibrinolytic shutdown, and primes neutrophils for prothrombotic NETosis. This process is critically enabled by the Capsular Polysaccharide, which facilitates immune evasion and persistent high-grade bacteremia, thereby ensuring a continuous, overwhelming source of LPS to sustain the inflammatory drive. The severity of the septic insult is further potentiated by Siderophores (eg, aerobactin), which sequester host iron to enhance bacterial proliferation and tissue invasion, thereby increasing the pathogen load and magnifying the release of all pro-DIC mediators. Contributing factors like fimbriae, promoting endothelial adherence and biofilm persistence and OMVs, delivering concentrated virulence packages, reinforce localized inflammation and systemic signaling. Ultimately, this coordinated virulence strategy creates a self-reinforcing cycle where bacterial factors initiate a massive inflammatory response, which in turn unleashes host-derived effectors like extracellular histones and NETs that directly execute the widespread microvascular thrombosis and consumptive coagulopathy defining DIC (Figure 1).
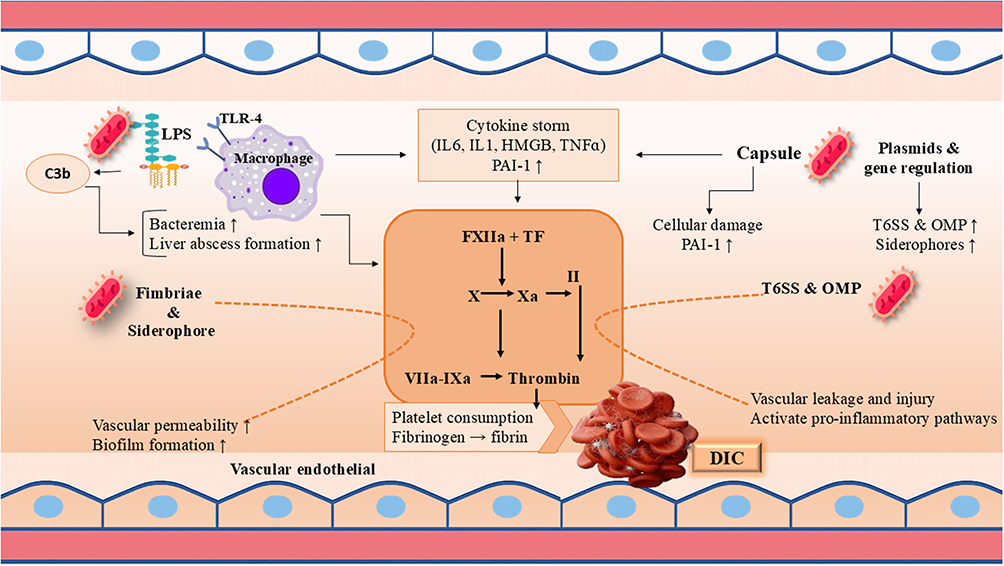 |
Figure 1 Virulence Mechanisms of K. pneumoniae and DIC. This figure illustrates how different virulence factors can participate in pathogenesis. |
Pathophysiology of DIC in K. pneumoniae Infections
DIC represents a systemic thrombo-hemorrhagic disorder characterized by widespread activation of coagulation pathways, leading to simultaneous thrombosis and hemorrhage. The pathophysiology of DIC in K. pneumoniae infections involves complex interactions between microbial components, host inflammatory responses, and the coagulation system.
Initiation of Coagulation Activation
The primary trigger for DIC in K. pneumoniae sepsis is the systemic inflammatory response to infection. Bacterial components, particularly LPS, activate monocytes, macrophages, and endothelial cells to express tissue factor, the principal initiator of the extrinsic coagulation cascade.35 Tissue factor expression leads to thrombin generation and fibrin formation. Simultaneously, physiological anticoagulant pathways are downregulated: antithrombin levels are consumed and suppressed by inflammation, the protein C system is impaired due to endothelial thrombomodulin downregulation, and tissue factor pathway inhibitor is overwhelmed.16,35 The extensive capsular polysaccharide of K. pneumoniae provides resistance against complement-mediated killing and phagocytosis, allowing sustained bacteremia and ongoing activation of these pathways.56 Furthermore, NETs, released in response to infection (NETosis), contain DNA, histones, and antimicrobial proteins. While aimed at trapping bacteria, these NET components are highly prothrombotic. They can activate platelets, provide a scaffold for thrombus formation, and directly activate the contact pathway (FXII), further amplifying thrombin generation.30,35 Activated FXII by LPS, initiates the intrinsic coagulation cascade, leading to thrombin generation, fibrin formation, and microvascular thrombosis. Concurrently, FXIIa activates plasma kallikrein, triggering the kallikrein-kinin system and resulting in bradykinin release, which amplifies vascular permeability and inflammatory responses.37 Extracellular vesicles or microparticles derived from activated platelets, monocytes, and endothelial cells also contribute significantly. These extracellular vesicles expose phosphatidylserine and often carry tissue factor, providing catalytic surfaces that greatly amplify coagulation reactions.35
Cytokine-Mediated Coagulation Dysregulation
A “cytokine storm” featuring elevated levels of TNF-α, IL-1, IL-6, and IL-8 plays a central role in the pathogenesis of sepsis-induced DIC.30,36 These proinflammatory cytokines promote tissue factor expression, downregulate thrombomodulin expression, inhibit fibrinolysis through increased production of PAI-1, and activate endothelial cells. The NET effect is enhanced thrombin generation, impaired anticoagulation, and suppressed fibrin removal, leading to a severe hemostatic imbalance that manifests as both thrombosis and hemorrhage. Damage-Associated Molecular Patterns (DAMPs), such as extracellular histones, HMGB1, and cell-free DNA released from damaged host cells, further exacerbate this process by amplifying inflammation and directly activating platelets and coagulation factors.16,35
Endothelial Dysfunction
Endothelial injury represents a critical event in the pathogenesis of DIC. K. pneumoniae components and host inflammatory mediators disrupt the antithrombotic properties of the endothelium. The protective glycocalyx layer is shed (“shedding”), exposing adhesion molecules like E-selectin and promoting platelet adhesion, leukocyte recruitment, and procoagulant activity. Damaged endothelial cells release ultralarge von Willebrand factor (vWF) multimers that enhance platelet aggregation and microvascular thrombosis. The simultaneous exposure of subendothelial collagen further activates platelets and the intrinsic coagulation pathway. This endothelial dysfunction also leads to increased vascular permeability, contributing to shock and organ edema.35,36
Impairment of Fibrinolytic System
Dysregulated fibrinolysis is a hallmark of sepsis-induced DIC. Initially, the fibrinolytic system is activated in response to fibrin deposition, evidenced by elevated D-dimer and FDP levels. However, as DIC progresses, there is progressive inhibition of fibrinolysis mediated primarily by a dramatic increase in PAI-1 levels, leading to “fibrinolytic shutdown”.35 This transition from hyperfibrinolysis to hypofibrinolysis results in accumulated fibrin deposition in the microvasculature, contributing to end-organ ischemia and dysfunction. Thrombin-Activatable Fibrinolysis Inhibitor may also contribute to inhibiting fibrinolysis in sepsis.16,35
Clinical Manifestations and Diagnosis of K. pneumoniae Associated-DIC
Clinical Presentation of DIC
The clinical manifestations of DIC in patients with K. pneumoniae sepsis often reflect a thrombotic-dominant phenotype, driven by intense systemic inflammation and endothelial injury.26 Microvascular thrombosis is common, leading to organ dysfunction such as acute kidney injury, acute respiratory distress syndrome (ARDS), and neurological deterioration. In severe invasive syndromes eg, liver abscess or metastatic infections macrovascular thrombosis like portal vein thrombosis (pylephlebitis) may occur.26,57 Hemorrhagic features, including petechiae, mucosal oozing, or catheter-site bleeding, are observed but are typically less severe than in obstetric or malignancy-associated DIC.58 The progression is usually acute, evolving over hours to days, aligning with the rapid clinical course of CR-hvKP or CRKP sepsis.59 Early recognition of DIC in this setting is critical, as it correlates with higher mortality and complicated metastatic infections such as endogenous endophthalmitis or disseminated abscesses.60
Diagnostic Approach and Global Criteria
The diagnosis of DIC requires integration of clinical and laboratory findings, as no single test is pathognomonic. It is a clinicopathological syndrome whose diagnosis relies on scoring systems based on readily available coagulation tests. The dynamic nature of DIC necessitates repeated evaluations over time for accurate diagnosis and monitoring.16
Key Laboratory Tests for DIC
The diagnosis of sepsis-induced DIC relies on a constellation of laboratory abnormalities that reflect the underlying pathophysiological processes of systemic coagulation activation, consumption of clotting factors and platelets, and secondary fibrinolysis. Thrombocytopenia, typically defined as a platelet count below 100–150 × 109/L, is one of the earliest and most consistent hematological findings, resulting from consumption, sequestration, and adherence to damaged endothelium.61,62 Global coagulation screening tests often reveal a prolonged PT or international normalized ratio (INR) due to the consumption of coagulation factors, particularly factors II, V, VII, and X.63 The activated partial thromboplastin time (aPTT) may also be prolonged, though it can be variable. Markers of fibrin formation and degradation are crucially elevated. Fibrin degradation products (FDP) and, more specifically, D-dimer levels are significantly increased, indicating intense thrombin generation, fibrin deposition, and subsequent fibrinolysis.63,64 In contrast, fibrinogen levels, being an acute-phase reactant, may remain normal or even elevated in the early stages of sepsis, masking ongoing consumption. A low or rapidly declining fibrinogen level is therefore considered a late sign of severe coagulopathy and carries a poor prognosis.65 Supportive laboratory findings include decreased antithrombin activity, resulting from consumption by thrombin and inactivation by neutrophil elastase.66 While a peripheral blood smear may show schistocytes suggestive of microangiopathic hemolysis, this finding is less characteristic of septic DIC than of primary thrombotic microangiopathies.61 Viscoelastic point-of-care tests, such as thromboelastography or rotational thromboelastometry, provide a dynamic assessment of the clotting process and serve as adjunctive tools that do not replace established diagnostic scoring systems. They can detect a spectrum of states, from a hypercoagulable profile with shortened clotting time and increased clot strength in early sepsis to a hypocoagulable pattern with prolonged clotting time and reduced clot firmness in overt, consumptive DIC.67
Global Diagnostic Scoring Systems for DIC
The diagnosis of DIC is challenging due to its variable presentation and the lack of a single pathognomonic test. To standardize the diagnostic process, several validated scoring systems have been developed, each with specific objectives, parameters, and clinical contexts.61 These systems integrate readily available laboratory tests to provide an objective assessment of coagulopathy, facilitating earlier recognition and consistent diagnosis across different clinical settings.
The most widely recognized international standard is the overt-DIC diagnostic score established by the ISTH. This system is designed to identify overt, decompensated DIC by evaluating four key parameters: platelet count, PT or INR, fibrinogen level, and a fibrin-related marker such as D-dimer or fibrin/fibrinogen degradation products. A cumulative score of 5 or higher is considered compatible with a diagnosis of overt DIC.68 While this score has high specificity for advanced DIC, its major limitation is lower sensitivity for detecting the early, non-overt stages of the syndrome, potentially delaying intervention.61
For earlier detection, particularly in critically ill and septic patients, the JAAM score was introduced for DIC. This system incorporates systemic inflammatory response syndrome (SIRS) criteria alongside laboratory markers platelet count, PT ratio, and FDP levels to enhance sensitivity. A score of 4 or greater indicates DIC. The JAAM score is considered more sensitive than the ISTH overt-DIC criteria and is useful for identifying DIC at an earlier phase, though it may be less specific.69 Of note, the updated JAAM-2 DIC score replaces SIRS with antithrombin activity as a parameter.70
To further refine early risk stratification specifically in sepsis, the ISTH subcommittee proposed the SIC score in 2017. This simplified, two-step tool utilizes only three parameters: platelet count, INR, and the SOFA score as a marker of organ dysfunction. A SIC score of 4 or higher identifies septic patients with early coagulopathy who are at significant risk of progressing to overt DIC, thereby aiding in timely decisions regarding supportive management and potential anticoagulant therapy.17,61
In Japan, other simplified criteria are also in use. The Japanese Society on Thrombosis and Hemostasis (JSTH) has proposed diagnostic criteria that can incorporate molecular markers.71 A further simplified version, sometimes referred to as the modified JAAM (m-JAAM) score, replaces the SIRS component with antithrombin activity, aiming to improve specificity. These systems typically use a combination of platelet count, PT-INR, FDP, and antithrombin activity, with a threshold score of 4 for diagnosing DIC.14,71
Selecting the appropriate scoring system depends on the clinical context. The ISTH overt-DIC score remains the international benchmark for diagnosing established DIC (Table 2). For early intervention in sepsis, the SIC score is a valuable screening tool, while the JAAM and its modifications are extensively applied in Japan for sensitive early detection.61,69 It is crucial to note that, given the dynamic nature of DIC, sequential measurements and the trend of the score over time often provide more meaningful diagnostic and prognostic information than a single assessment (Table 3).61
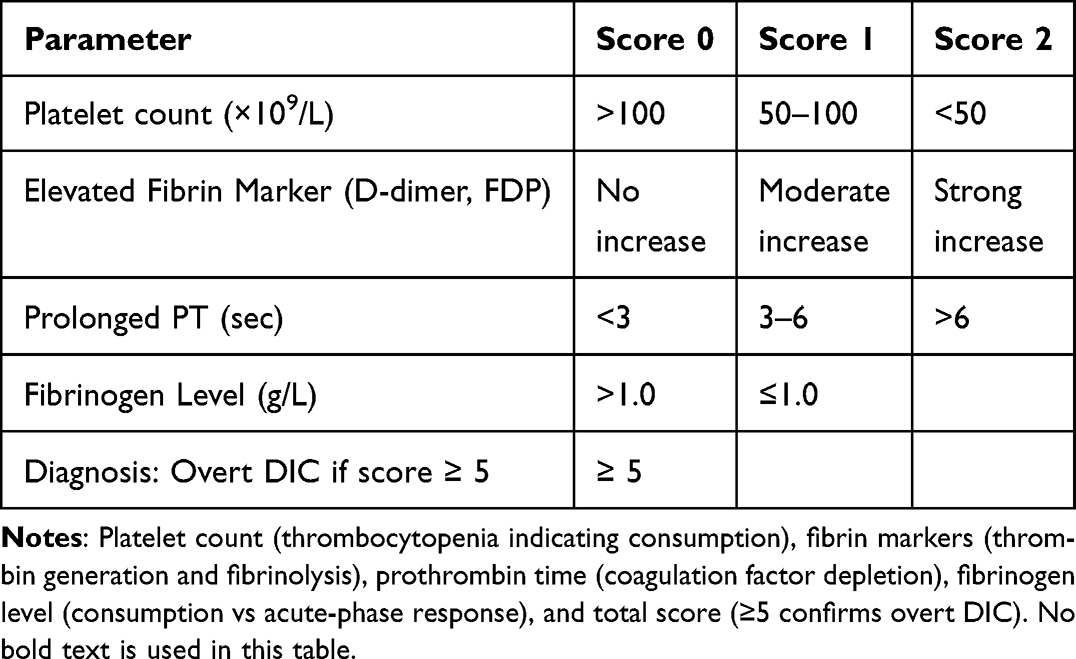 |
Table 2 ISTH Scoring System for Overt DIC |
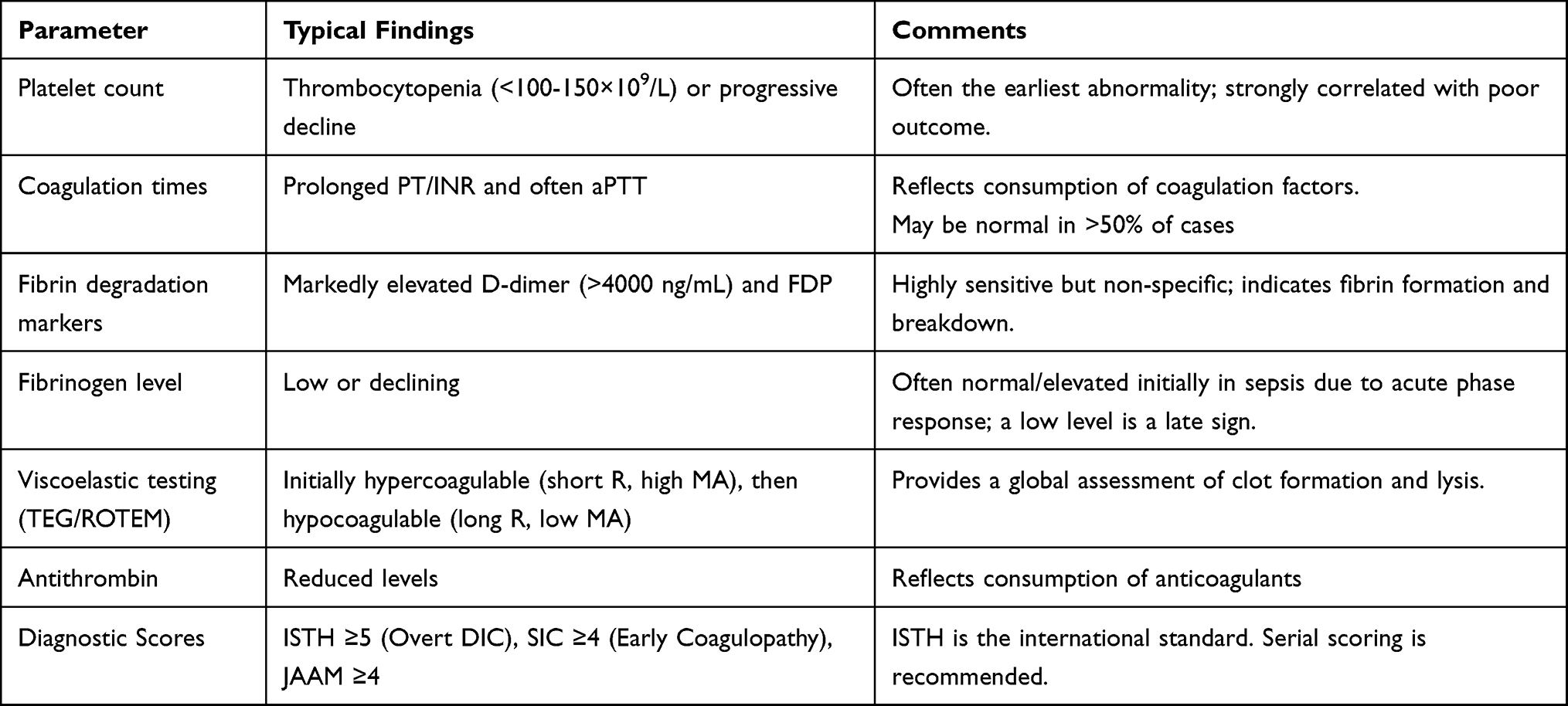 |
Table 3 Laboratory Features and Diagnostic Criteria for DIC |
Management Strategies
Antimicrobial Therapy and Source Control
Appropriate antibiotic therapy represents the cornerstone of management for K. pneumoniae infections complicated by DIC. Initial empirical regimens for community-acquired K. pneumoniae pneumonia typically include third- or fourth-generation cephalosporins or respiratory fluoroquinolones, with possible addition of an aminoglycoside. For penicillin-allergic patients, aztreonam or respiratory fluoroquinolones represent alternatives. The increasing prevalence of antibiotic-resistant strains necessitates careful antimicrobial selection based on local susceptibility patterns and individual risk factors. ESBL-producing strains require treatment with carbapenems (eg, meropenem, imipenem); however, emerging therapeutic options such as cefiderocol or the combination of ceftazidime-avibactam plus aztreonam are increasingly highlighted in recent literature.72 For carbapenem-resistant Enterobacteriaceae infections, combination therapy with polymyxins (colistin), tigecycline, fosfomycin, aminoglycosides, or newer β-lactam/β-lactamase inhibitor combinations (eg, ceftazidime-avibactam, meropenem-vaborbactam) is often necessary.46 Infectious disease consultation is recommended for carbapenem-resistant Enterobacteriaceae infections. Source control measures, such as percutaneous or surgical drainage of abscesses (eg, liver abscess) and removal of infected devices, are essential adjuncts to antimicrobial therapy. The duration of antibiotic therapy typically spans 14 days, though longer courses may be required for complicated infections.25,57
Supportive Care and Coagulation Management
Supportive care within the ICU is fundamental for managing patients with K. pneumoniae sepsis complicated by DIC, focusing on stabilizing hemodynamics, respiratory function, and failing organs. Alongside treating the underlying infection, meticulous coagulation management is essential to address the concurrent risks of bleeding and thrombosis inherent to DIC. Guideline-directed blood component therapy should be administered based on clinical evidence of bleeding, the need for invasive procedures, and the overall trajectory of coagulopathy, rather than on laboratory abnormalities alone. The ISTH guidelines recommend platelet transfusion for actively bleeding patients with a platelet count below 50 × 109/L, or below 100 × 109/L in those at high risk of bleeding, such as prior to an invasive procedure.58,73 For patients with significant bleeding and prolonged PT or aPTT, infusion of fresh frozen plasma (FFP) at an initial dose of 15–30 mL/kg is indicated to replenish coagulation factors.73,74 Specific fibrinogen replacement with cryoprecipitate or fibrinogen concentrate is warranted when levels fall below 1.5 g/L in bleeding patients, with a higher threshold (eg, >2.0 g/L) recommended in obstetric contexts or cases of severe hemorrhage. This targeted, supportive approach aims to correct hemostatic imbalances while minimizing the risks of volume overload and thrombotic complications.73,75
Anticoagulation Therapy
Historical randomized controlled trials (RCTs) of agents like antithrombin, activated protein C, tissue factor pathway inhibitor, and thrombomodulin in broad sepsis populations have largely failed to show consistent mortality benefit. This failure stems from several factors: heterogeneous pathophysiology, enrollment of patients without significant coagulopathy, inappropriate timing of intervention often beyond the window of maximal thromboinflammation, the limitations of 28-day all-cause mortality as a sole endpoint, and confounding from concomitant heparin use which increased bleeding risk.73,76 The 2013 ISTH guidelines suggest considering unfractionated heparin for prophylaxis or treatment of thrombosis in DIC, but not for routine use.73 Consequently, robust evidence from large RCTs supporting routine anticoagulant use remains lacking, leading international guidelines outside Japan to typically not recommend their use. However, in Japan, based on observational data and pathophysiological rationale, antithrombin and recombinant thrombomodulin are approved and used for sepsis-induced DIC. Meta-analyses of observational studies on combination therapy suggest a non-significant trend toward reduced mortality without significantly increased bleeding, though high heterogeneity and absence of RCT data preclude definitive conclusions.77,78
For future trial success, a paradigm shift is required. Firstly, patients with clear SIC (eg, meeting SIC or overt DIC criteria) must be precisely selected, potentially using machine learning to identify high-risk phenotypes. Secondly, therapy should be initiated early upon coagulopathy detection to avoid diluted effects. Thirdly, composite endpoints like DIC resolution combined with improved organ function (eg, SOFA score) should be incorporated rather than relying solely on 28-day mortality; tools like a Composite Prognostic Index show promise for early prediction. Fourthly, bleeding risk must be meticulously monitored, especially with combination therapies or concomitant heparin, while emphasizing individualized dosing.79 Pending results from such improved RCTs, a pragmatic clinical approach involves several steps: considering anticoagulants only for septic patients with clear, progressive coagulopathy; initiating therapy early while avoiding concomitant heparin; assessing treatment response early (eg, by day 3) using dynamic DIC scores and organ function to guide decisions; and individualizing the choice between monotherapy or combination therapy, acknowledging the latter’s stronger pathophysiological rationale but less certain risk-benefit profile.78,79
Advanced Supportive Measures
Novel therapeutic approaches targeting specific pathological pathways in sepsis and DIC continue to be investigated. These include monoclonal antibodies against bacterial components, immunomodulatory agents (eg, IL-6 inhibitors), and complement inhibitors (eg, anti-C5a), all of which aim to attenuate the thromboinflammatory cascade.35,43 In terms of supportive management, viscoelastic testing may help identify specific coagulation abnormalities and guide transfusion strategies, though direct evidence in K. pneumoniae-associated DIC remains limited.25 Furthermore, dynamic prognostic tools such as the CPI which incorporates both coagulation resolution and organ dysfunction trends may help clinicians evaluate early treatment responses and guide therapeutic adjustments in septic DIC [A]. The integration of such indices into clinical practice could provide a more nuanced and timely assessment of patient trajectory, complementing traditional outcome measures such as 28-day mortality and facilitating personalized anticoagulant management in high-risk sepsis populations.
Future Directions and Research Priorities
Several research priorities emerge from our current understanding of K. pneumoniae-associated DIC. Furthermore, emerging evidence implicates the contact activation pathway as a critical contributor to thrombosis in sepsis. Targeting this pathway, for instance with FXII or FXI inhibitors, represents a novel antithrombotic strategy that could potentially uncouple coagulation from inflammation, reducing microvascular thrombosis without increasing bleeding risk. Preclinical and clinical evaluation of such inhibitors is therefore a priority for sepsis-associated DIC, including that triggered by K. pneumoniae. First, improved rapid diagnostic methods are needed to identify high-risk patients and initiate preemptive therapy. Molecular techniques for detecting virulence genes (eg, iucA, rmpA for hvKP) and resistance determinants may guide targeted antimicrobial therapy. Second, further elucidation of the pathophysiological mechanisms linking specific virulence factors (eg, LPS, capsule) to coagulation activation may identify novel therapeutic targets (eg, anti- tissue factor agents, PAI-1 inhibitors). Third, clinical trials focusing on antithrombotic therapies (eg, rTM, AT) in sepsis-induced DIC should stratify patients by pathogen type to determine whether specific approaches are more effective for K. pneumoniae infections. Finally, infection prevention strategies, including vaccine development (eg, targeting common capsular serotypes) and enhanced infection control measures to curb the spread of multidrug-resistant and hypervirulent strains, contribute crucial approaches for reducing the burden of K. pneumoniae infections and their complications.
Conclusion
In conclusion, K. pneumoniae sepsis presents a significant risk for the development of fatal DIC. This review underscores the critical importance of a proactive, two-step clinical approach. First, patients with suspected or confirmed K. pneumoniae bacteremia, particularly those infected with hypervirulent or multidrug-resistant strains, must be promptly and systematically evaluated for early signs of coagulopathy using established DIC scoring systems (eg, SIC, ISTH). Second, once a patient is stratified as high-risk, a preemptive and multimodal management strategy must be urgently instituted. This strategy synergizes aggressive, targeted antimicrobial therapy and source control with vigilant supportive care and consideration of adjuvant anticoagulation, as discussed. Implementing this paradigm of early risk assessment followed by rapid, multifaceted intervention is essential to disrupt the lethal synergy between bacterial virulence and the host’s dysregulated thrombo-inflammatory response, thereby improving survival outcomes in this devastating complication.
Data Sharing Statement
Due to the nature of the study, there is no supporting data for availability.
Acknowledgment
This study was supported by Drug applied Research Center and Faculty of Medicine, Tabriz University of Medical Sciences with grant number 76601 (Ph.D dissertation of Malek Namaki with supervision of Dr. Mohammad Aghazadeh and Dr. Hossein Samadi Kafil). We thank all staff of DARC and Department of Microbiology and Imam Reza Hospital (Ms. Shabnam Kalani) for their helps and collaborations.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Chen T, Wang Y, Zhou Y, et al. Recombination drives evolution of carbapenem-resistant Klebsiella pneumoniae sequence type 11 KL47 to KL64 in China. Microbiol Spectr. 2023;11(1):e01107–14. doi:10.1128/spectrum.01107-22
2. Zhu J, Wang T, Chen L, et al. Virulence factors in hypervirulent Klebsiella pneumoniae. Front Microbiol. 2021;12:642484. doi:10.3389/fmicb.2021.642484
3. Chen T-A, Chuang Y-T, Lin C-H. A decade-long review of the virulence, resistance, and epidemiological risks of Klebsiella pneumoniae in ICUs. Microorganisms. 2024;12(12):2548. doi:10.3390/microorganisms12122548
4. Zeng L, Yang C, Zhang J, et al. An outbreak of carbapenem-resistant Klebsiella pneumoniae in an intensive care unit of a major teaching hospital in Chongqing, China. Front Cell Infect Microbiol. 2021;11:656070. doi:10.3389/fcimb.2021.656070
5. Giannella M, Trecarichi EM, De Rosa FG, et al. Risk factors for carbapenem‐resistant Klebsiella pneumoniae bloodstream infection among rectal carriers: a prospective observational multicentre study. Clin Microbiol Infect. 2014;20(12):1357–1362. doi:10.1111/1469-0691.12747
6. Bialvaei AZ, Samadi Kafil H, Ebrahimzadeh Leylabadlo H, et al. Dissemination of carbapenemases producing Gram negative bacteria in the Middle East. Iran J Microbiol. 2015;7(5):226. doi:10.1973/947815Y0000000063
7. Bialvaei AZ, Kafil HS, Asgharzadeh M, et al. Current methods for the identification of carbapenemases. J Chemother. 2016;28(1):1–19. doi:10.1179/1973947815Y.0000000063
8. Lin X-C, Li C-L, Zhang S-Y, et al. The global and regional prevalence of hospital-acquired carbapenem-resistant Klebsiella pneumoniae infection: a systematic review and meta-analysis. Open Forum Infect Dis. 2024;11. doi:10.1093/ofid/ofad649
9. Semet C, Efe K, Akalın H, et al. Outcome of carbapenem or colistin resistant Klebsiella pneumoniae bacteremia in the intensive care unit. Sci Rep. 2024;14(1):25805. doi:10.1038/s41598-024-73786-x
10. Agyeman AA, Bergen PJ, Rao GG, et al. A systematic review and meta-analysis of treatment outcomes following antibiotic therapy among patients with carbapenem-resistant Klebsiella pneumoniae infections. Int J Antimicrob Agents. 2020;55(1):105833. doi:10.1016/j.ijantimicag.2019.10.014
11. Grosjean V, Gressens SB, Pham T, et al. Community-acquired Klebsiella pneumoniae pneumonia in ICU: a multicenter retrospective study. Ann Intens Care. 2024;14(1):69. doi:10.1186/s13613-024-01269-3
12. Rafat C, Messika J, Barnaud G, et al. Hypervirulent Klebsiella pneumoniae, a 5-year study in a French ICU. J Med Microbiol. 2018;67(8):1083–1089. doi:10.1099/jmm.0.000788
13. Levi M, Schultz M, van der Poll T. Disseminated intravascular coagulation in infectious disease. Semin Thromb Hemost. 2010;36(4):367–377. doi:10.1055/s-0030-1254046
14. Iba T, Ito T, Maruyama I, et al. Potential diagnostic markers for disseminated intravascular coagulation of sepsis. Blood Rev. 2016;30(2):149–155. doi:10.1016/j.blre.2015.10.002
15. Wang E, Liu Y, Qiu X, et al. Bacteria-released outer membrane vesicles promote disseminated intravascular coagulation. Thromb Res. 2019;178:26–33. doi:10.1016/j.thromres.2019.03.019
16. Iba T, Helms J, Connors JM, et al. The pathophysiology, diagnosis, and management of sepsis-associated disseminated intravascular coagulation. J Intensive Care. 2023;11(1):24. doi:10.1186/s40560-023-00672-5
17. Iba T, Nisio MD, Levy JH, et al. New criteria for sepsis-induced coagulopathy (SIC) following the revised sepsis definition: a retrospective analysis of a nationwide survey. BMJ Open. 2017;7(9):e017046. doi:10.1136/bmjopen-2017-017046
18. Iba T, Levy JH, Wada H, et al. Differential diagnoses for sepsis‐induced disseminated intravascular coagulation: communication from the SSC of the ISTH. J Thromb Haemost. 2019;17(2):415–419. doi:10.1111/jth.14354
19. Ha SO, Park SH, Hong S-B, et al. Performance evaluation of five different disseminated intravascular coagulation (DIC) diagnostic criteria for predicting mortality in patients with complicated sepsis. J Korean Med Sci. 2016;31(11):1838–1845. doi:10.3346/jkms.2016.31.11.1838
20. Itani R, Khojah HMJ, Kibrit R, et al. Risk factors associated with multidrug-resistant Klebsiella pneumoniae infections: a multicenter observational study in Lebanese hospitals. BMC Public Health. 2024;24(1):2958. doi:10.1186/s12889-024-20474-0
21. Mouloudi E, Massa E, Papadopoulos S, et al. Bloodstream infections caused by carbapenemase-producing Klebsiella pneumoniae among intensive care unit patients after orthotopic liver transplantation: risk factors for infection and impact of resistance on outcomes. Transplant Proc. 2014;46(9):3216–3218. doi:10.1016/j.transproceed.2014.09.159
22. Gu D, Dong N, Zheng Z, et al. A fatal outbreak of ST11 carbapenem-resistant hypervirulent Klebsiella pneumoniae in a Chinese hospital: a molecular epidemiological study. Lancet Infect Dis. 2018;18(1):37–46. doi:10.1016/S1473-3099(17)30489-9
23. Unlu O, Ersoz BR, Istanbullu Tosun A, et al. Epidemic Klebsiella pneumoniae ST258 incidence in ICU patients admitted to a university hospital in Istanbul. J Infect Developing Countries. 2021;15(05):665–671. doi:10.3855/jidc.13430
24. Lee B, Yeroushalmi K, Me HM, et al. Community acquired Klebsiella pneumoniae meningitis: a case report. Germs. 2018;8(2):92. doi:10.18683/germs.2018.1136
25. Chung GC, Lee JE, Byon I, et al. Endogenous endophthalmitis due to Klebsiella pneumoniae liver abscess: a retrospective study of clinical course, treatment pattern, and prognosis. Int J Ophthalmol. 2025;18(8):1553. doi:10.18240/ijo.2025.08.18
26. Yamamoto K, Hasegawa I, Suga Y, Kano Y. A case of pylephlebitis without intra-abdominal infection secondary to pneumonia caused by hypermucoviscous Klebsiella pneumoniae. Cureus. 2024;16(9):e69428. doi:10.7759/cureus.69428
27. Qi W, Liu J, Li A. Effect of anticoagulant versus non-anticoagulant therapy on mortality of sepsis-induced disseminated intravascular coagulation: a systematic review and meta-analysis. Clin App Thrombosis/Hemostasis. 2023;29:10760296231157766. doi:10.1177/10760296231157766
28. Desai D, Misra RN, Gandham NR, et al. First report of virulence factors in carbapenem-resistant Klebsiella pneumoniae from Maharashtra, India. J Datta Meghe Inst Med Sci Univ. 2024;19(4):729–735. doi:10.4103/jdmimsu.jdmimsu_374_24
29. Unar A, Bertolino L, Patauner F, et al. Pathophysiology of disseminated intravascular coagulation in sepsis: a clinically focused overview. Cells. 2023;12(17):2120. doi:10.3390/cells12172120
30. Yamasaki O, Sugihara S, Kajita A, et al. Staphylococcal enterotoxin B‐and lipopolysaccharide‐induced toxic shock syndrome in a burn patient. J Dermatol. 2021;48(4):547–550. doi:10.1111/1346-8138.15729
31. Li Y, Li X, Wu W, et al. Insights into the roles of macrophages in Klebsiella pneumoniae infections: a comprehensive review. Cell Mol Biol Lett. 2025;30(1):34. doi:10.1186/s11658-025-00717-7
32. Suga Y, Kubo A, Katsura H, et al. Detailed exploration of pathophysiology involving inflammatory status and bleeding symptoms between lipopolysaccharide-and tissue factor-induced disseminated intravascular coagulation in rats. Int J Hematol. 2021;114(2):172–178. doi:10.1007/s12185-021-03158-y
33. Sachetto AT, Mackman N. Monocyte tissue factor expression: lipopolysaccharide induction and roles in pathological activation of coagulation. Thromb Haemost. 2023;123(11):1017–1033. doi:10.1055/a-2091-7006
34. Minomo H, Inoue K, Sakaki S, et al. Establishment of disseminated intravascular coagulation (DIC) model by a single iv administration of Escherichia coli-derived lipopolysaccharide (LPS) to cynomolgus monkeys and evaluation of its pathophysiological status. J Pharmacol Sci. 2017;133(2):88–95. doi:10.1016/j.jphs.2017.01.005
35. Iba T, Connors JM, Nagaoka I, et al. Recent advances in the research and management of sepsis-associated DIC. Int J Hematol. 2021;113(1):24–33. doi:10.1007/s12185-020-03053-y
36. Liufu R, Chen Y, Wan X-X, et al. Sepsis-induced coagulopathy: the different prognosis in severe pneumonia and bacteremia infection patients. Clin App Thrombosis/Hemostasis. 2023;29:10760296231219249. doi:10.1177/10760296231219249
37. Lira AL, Puy C, Shatzel JJ, et al. Bacterial infection and activation of the contact pathway of coagulation. Blood Vessels Thromb Hemost. 2025;2(4):100091. doi:10.1016/j.bvth.2025.100091
38. Hasani A, Soltani E, Ahangarzadeh Rezaee M, et al. Serotyping of Klebsiella pneumoniae and its relation with capsule-associated virulence genes, antimicrobial resistance pattern, and clinical infections: a descriptive study in medical practice. Infect Drug Resist. 2020;13:1971–1980. doi:10.2147/IDR.S243984
39. Jiang S, Ma Z, Cao H, et al. Genomic study substantiates the intensive care unit as a reservoir for carbapenem-resistant Klebsiella pneumoniae in a teaching hospital in China. Microb Genom. 2024;10(9):001299. doi:10.1099/mgen.0.001299
40. Ssekatawa K, Byarugaba DK, Nakavuma JL, et al. Prevalence of pathogenic Klebsiella pneumoniae based on PCR capsular typing harbouring carbapenemases encoding genes in Uganda tertiary hospitals. Antimicrob Resist Infect Control. 2021;10(1):57. doi:10.1186/s13756-021-00923-w
41. Sikarwar AS, Omasanggar RAP. A review on advanced serotyping methods for identification of Klebsiella pneumoniae capsular serotypes. Indian J Med Res Pharm Sci. 2014;1(7):27–33.
42. Jung S, Chae HJ, Park YJ, et al. Microbiological and clinical characteristics of bacteraemia caused by the hypermucoviscosity phenotype of Klebsiella pneumoniae in Korea. Epidemiol Infect. 2013;141(2):334–340. doi:10.1017/S0950268812000933
43. Adelborg K, Larsen JB, Hvas AM. Disseminated intravascular coagulation: epidemiology, biomarkers, and management. Br J Haematol. 2021;192(5):803–818. doi:10.1111/bjh.17172
44. Minasyan H. Sepsis: mechanisms of bacterial injury to the patient. Scand J Trauma Resusc Emerg Med. 2019;27(1):19. doi:10.1186/s13049-019-0596-4
45. Nadel S, Ninis N. Invasive meningococcal disease in the vaccine era. Front Pediatr. 2018;6:321. doi:10.3389/fped.2018.00321
46. Ahmadi M, Ranjbar R, Behzadi P, Mohammadian T. Virulence factors, antibiotic resistance patterns, and molecular types of clinical isolates of Klebsiella pneumoniae. Expert Rev Anti Infect Ther. 2022;20(3):463–472. doi:10.1080/14787210.2022.1990040
47. Riwu KHP, Effendi MH, Rantam FA, et al. A review: virulence factors of Klebsiella pneumonia as emerging infection on the food chain. Vet World. 2022;15(9):2172. doi:10.14202/vetworld.2022.2172-2179
48. Holmes CL, Anderson MT, Mobley HLT, et al. Pathogenesis of gram-negative bacteremia. Clin Microbiol Rev. 2021;34(2). doi:10.1128/cmr.00234-20
49. Hemmati F, Rezaee MA, Ebrahimzadeh S, et al. Novel strategies to combat bacterial biofilms. Mol Biotechnol. 2021;63(7):569–586. doi:10.1007/s12033-021-00325-8
50. Hajiagha MN, Samadi Kafil H. Efflux pumps and microbial biofilm formation. Infect Genet Evol. 2023;112:105459. doi:10.1016/j.meegid.2023.105459
51. Zhou M, Lan Y, Wang S, et al. Epidemiology and molecular characteristics of the type VI secretion system in Klebsiella pneumoniae isolated from bloodstream infections. J Clin Lab Anal. 2020;34(11):e23459. doi:10.1002/jcla.23459
52. Selim MI, El‑banna T, Sonbol F, et al. Arthrospira maxima and biosynthesized zinc oxide nanoparticles as antibacterials against carbapenem-resistant Klebsiella pneumoniae and Acinetobacter baumannii: a review article. Microb Cell Fact. 2024;23(1):311. doi:10.1186/s12934-024-02584-x
53. Maitra R, Saxena D, Singh S, et al. Multidrug-Resistant Acinetobacter baumannii: a Wily, Existential Threat to Modern Healthcare. ACS Infect Dis. 2025;11:2951–2978. doi:10.1021/acsinfecdis.5c00570
54. EL-Hakeem RA, Saleh S, Aboulwafa M, et al. Acinetobacter baumannii virulence factors, resistance mechanisms, and new insights on infection treatment. Arch Pharm Sci Ain Shams Univ. 2023;7(1):208–230. doi:10.21608/aps.2023.212124.1125
55. Li L, Ma J, Cheng P, et al. Roles of two-component regulatory systems in Klebsiella pneumoniae: regulation of virulence, antibiotic resistance, and stress responses. Microbiol Res. 2023;272:127374. doi:10.1016/j.micres.2023.127374
56. Asgari F, Supino D, Parente R, et al. The long pentraxin PTX3 controls klebsiella pneumoniae severe infection. Front Immunol. 2021;12:666198. doi:10.3389/fimmu.2021.666198
57. Sato K, Takahashi K, Kusuta T. A case of hypermucoviscosity phenotype of Klebsiella pneumoniae liver abscess saved by damage control strategy. Case Rep Surg. 2022;2022(1):6019866. doi:10.1155/2022/6019866
58. Levi M, Sivapalaratnam S. Disseminated intravascular coagulation: an update on pathogenesis and diagnosis. Exp Rev Hematol. 2018;11(8):663–672. doi:10.1080/17474086.2018.1500173
59. Saeed GT, Al Smady MN, Awatramani G, et al. A unique case of hypervirulent Klebsiella pneumoniae invasive syndrome with endogenous endophthalmitis and left renal vein thrombosis without liver abscess. Eur J Case Rep Intern Med. 2024;11(11):004927. doi:10.12890/2024_004927
60. Kim E, Byon I, Lee JJ, et al. Endogenous endophthalmitis from a Klebsiella pneumoniae liver abscess: the incidence, risk factors, and utility of imaging. Am J Ophthalmol. 2023;252:69–76. doi:10.1016/j.ajo.2023.03.009
61. Iba T, Watanabe E, Umemura Y, et al. Sepsis-associated disseminated intravascular coagulation and its differential diagnoses. J Intensive Care. 2019;7(1):32. doi:10.1186/s40560-019-0387-z
62. Levi M. Inflammation and coagulation. Inflammation. 2017:833–860.
63. Zaidi SRH, Rout P. Interpretation of blood clotting studies and values (PT, PTT, aPTT, INR, anti-factor Xa, D-dimer). In: StatPearls. StatPearls Publishing; 2024.
64. Morelli B, Montaruli B, Steffan A, et al. Recommendations for harmonization of the coagulation screening tests laboratory report. Biochim Clin. 2023;47(4):377.
65. Tsantes AG, Parastatidou S, Tsantes EA, et al. Sepsis-induced coagulopathy: an update on pathophysiology, biomarkers, and current guidelines. Life. 2023;13(2):350. doi:10.3390/life13020350
66. Iba T, Levi M, Levy JH. Sepsis-induced coagulopathy and disseminated intravascular coagulation. Semin Thromb Hemost. 2020;46(1):89–95. doi:10.1055/s-0039-1694995
67. Nath SS, Pandey CK, Kumar S. Clinical application of viscoelastic point-of-care tests of coagulation-shifting paradigms. Ann Card Anaesth. 2022;25(1):1–10. doi:10.4103/aca.aca_319_20
68. Taylor FB; Scientific Subcommittee on Disseminated Intravascular Coagulation (DIC) of the International Society on Thrombosis and Haemostasis (ISTH). Towards definition, clinical and laboratory criteria, and a scoring system for disseminated intravascular coagulation. Thromb Haemost. 2001;86:1327–1330. doi:10.1055/s-0037-1616068
69. Gando S, Saitoh D, Ogura H, et al. A multicenter, prospective validation study of the Japanese Association for Acute Medicine disseminated intravascular coagulation scoring system in patients with severe sepsis. Crit Care. 2013;17(3):R111. doi:10.1186/cc12783
70. Matsuoka T, Yamakawa K, Umemura Y, et al. The modified Japanese Association for Acute Medicine disseminated intravascular coagulation diagnostic criteria in sepsis is useful for an indicator of initiating treatment for disseminated intravascular coagulation. Thromb Res. 2025;253:109408. doi:10.1016/j.thromres.2025.109408
71. Mori H, Harada-Shirado K, Kawano N, et al. Net reclassification index in comparison of prognostic value of disseminated intravascular coagulation diagnostic criteria by Japanese Society on Thrombosis and Hemostasis and International Society on Thrombosis and Haemostasis: a multicenter prospective cohort study. Thromb J. 2023;21(1):84. doi:10.1186/s12959-023-00523-1
72. Bush K. Past, present, and future perspectives on aztreonam and avibactam. Exp Rev Anti-Infective Ther. 2025;23(5):277–290. doi:10.1080/14787210.2025.2473047
73. Wada H, Yamamoto A, Tomida M, et al. Proposal of quick diagnostic criteria for disseminated intravascular coagulation. J Clin Med. 2022;11(4):1028. doi:10.3390/jcm11041028
74. Squizzato A, Gallo A, Levi M, et al. Underlying disorders of disseminated intravascular coagulation: communication from the ISTH SSC subcommittees on disseminated intravascular coagulation and perioperative and critical care thrombosis and hemostasis. J Thromb Haemost. 2020;18(9):2400–2407. doi:10.1111/jth.14946
75. Iba T, Helms J, Neal MD, et al. Mechanisms and management of the coagulopathy of trauma and sepsis: trauma-induced coagulopathy, sepsis-induced coagulopathy, and disseminated intravascular coagulation. J Thromb Haemost. 2023;21(12):3360–3370. doi:10.1016/j.jtha.2023.05.028
76. Totoki T, Makino Y, Yamakawa K, et al. Effects of combination therapy of antithrombin and thrombomodulin for sepsis-associated disseminated intravascular coagulation: a systematic review and meta-analysis. Thromb J. 2024;22(1):10. doi:10.1186/s12959-023-00579-z
77. Egi M, Ogura H, Yatabe T, et al. The Japanese Clinical Practice Guidelines for management of sepsis and septic shock 2020 (J-SSCG 2020). J Intensive Care. 2021;9(1):53. doi:10.1186/s40560-021-00555-7
78. Iba T, Yamakawa K, Shiko Y, et al. Determining prognostic indicator for anticoagulant therapy in sepsis-induced disseminated intravascular coagulation. J Intensive Care. 2024;12(1):24. doi:10.1186/s40560-024-00739-x
79. Iba T, Helms J, Maier CL, Ferrer R, Levy JH. Evolution of clinical trials in anticoagulation for sepsis: bridging past to future. Semin Thromb Hemost. 2025;52(1):126–138. doi:10.1055/a-2657-6380
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
A Rare Case of Acute Infectious Purpura Fulminans Caused by Klebsiella Pneumoniae and Human Herpesvirus Type 5
Li XL, Luan CY, Fan YJ, Lin XY, Jiang D, Su MX, Wang G, Yang X
Journal of Inflammation Research 2022, 15:4251-4260
Published Date: 26 July 2022