Back to Journals » OncoTargets and Therapy » Volume 11
KDM2B overexpression correlates with poor prognosis and regulates glioma cell growth
Authors Wang Y, Zang J, Zhang D ![]() , Sun Z
, Sun Z ![]() , Qiu B, Wang X
, Qiu B, Wang X
Received 23 August 2017
Accepted for publication 28 November 2017
Published 8 January 2018 Volume 2018:11 Pages 201—209
DOI https://doi.org/10.2147/OTT.S149833
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Samir Farghaly
Yiwei Wang,1 Jin Zang,1 Dongyong Zhang,2 Zhenxiang Sun,1 Bo Qiu,2 Xiaojie Wang1
1Department of Human Anatomy, Shenyang Medical College, Huanggu District, Shenyang City, 2Department of Neurosurgery, First Affiliated Hospital of China Medical University, Heping District, Shenyang City, Liaoning Province, China
Background: Gliomas are one of the most lethal cancers in the human central nervous system. Despite clinical treatment advancements, the prognosis of patients with glioma remains poor. KDM2B is a histone lysine demethylase, which has been observed in multiple tumors. But the concrete role of KDM2B in gliomas remains to be further illustrated.
Methods: The KDM2B expression in gliomas was detected with immunohistochemistry and Western blot assay. Furthermore, knockdown of KDM2B in U87 and U251 glioma cell lines, the proliferation capacity was evaluated by cell viability assay, colon formation assay and flow cytometry in vitro. Western blot assay was used to analyze the p21, EZH2 and cyclinD1 changes followed by knockdown of KDM2B.
Results: KDM2B was upregulated in tissues of glioma patients, and the expression was correlated to cancer progression. Downregulation of KDM2B in U87 and U251 glioma cell lines inhibited cell proliferation and arrested cell cycle in G0/G1 phase. In addition, silencing KDM2B promoted the upregulation of p21 while reduced the expression of EZH2 and cyclinD1.
Conclusion: Taken together, our results revealed that KDM2B might influence gliomas growth and act as a novel therapeutic target for glioma patients.
Keywords: EZH2, glioma, KDM2B, P21
Introduction
Gliomas are one of the most lethal cancers of the human central nervous system, exhibiting aggressive behavior.1,2 Despite using several methods in the treatment of gliomas, including microsurgery, chemotherapy, and radiotherapy, the prognosis of patients who suffered from glioma remains poor, with a survival rate of only 2 years. The World Health Organization (WHO) classified glioma tumor into four grades (I–IV) according to the histopathological characteristics that correlate with prognosis. Grade IV glioblastoma (GBM) is most fatal, and the results of current therapies show that the 5-year survival rate for GBM patients is approximately 10%, accounting for the difficulties in resection and the poor sensitivity to radiation and chemical therapies.3–5 Indeed, a comprehensive understanding of glioma pathogenesis is extremely necessary, which provides a new therapy target, thus improving the survival rates. Despite current problems in clinical therapy for gliomas, the molecular underpinnings of the pathogenesis of glioma tumors are rapidly advancing, and several genetic and epigenetic factors have been described in different grades of gliomas. Histone methylation is a novel factor that has been demonstrated to be correlated with transcriptional activation, and some studies have shown that modification in histone methylation contributes to the development of tumor.6,7 The changes in epigenetic genes can be reversed by multiple factors; therefore, targeting these types of genes might be a novel therapy for neoplastic processes.
KDM2B, also known as NDY1, JHDM1B, or FBXL10, is a histone lysine demethylase with specificity to H3K36me1, H3K36me2, and H3K36me3. KDM2B has multiple domains, including a JmjC domain at the N-terminal, a CXXC zinc finger, a plant homeodomain (PHD), an F-box motif, and leucine-rich repeat (LRR).8 Recent research has identified KDM2B as an oncogene in many types of tumors, such as breast cancer,9 leukemia,10–12 pancreatic cancer,13 and gastric cancer.14 In addition, the mutation of KDM2B has been found in human large B-cell lymphoma.15 In mouse embryonic fibroblasts (MEFs), KDM2B inhibits Ink4/Arf by the enhancer of zeste homolog 2 (EZH2), which is a highly conserved histone methyltransferase that targets lysine-27 of histone H3, which influences aging and cell proliferation.16,17 EZH2 is the core catalytic subunit of the polycomb repressor complex-2 (PRC2), which can deactivate the expression of tumor suppressor genes by methylating lysine 27 of histone 2 (H2K27).18 In addition, EZH2 was found to be associated with the proliferation and differentiation of tumors.19 Many studies have demonstrated that EZH2 is upregulated in multiple types of malignant tumors, including prostate cancer20 and breast cancer.21 Moreover, several reports have shown that EZH2 plays an important role in accelerating growth and migration in several tumors.22–24 A study has found that EZH2 is overexpressed in gliomas by targeting miR-101 and inhibiting the proliferation and migration of gliomas.25 Moreover, a defect in EZH2 repairs the self-renewal of GBM cancer stem cell in vitro.26 In addition, a recent study showed that the inhibition of EZH2 in GBM led to more than 80% reduction in the clonogenic factor.26 Thus, EZH2 might be used as a new therapeutic target for the treatment of aggressive tumors, especially gliomas. A previous study demonstrated that KDM2B inhibits miR-let-7b, thus altering the expression of EZH2 in human bladder cancer cell lines.27 These findings indicate that KDM2B plays an important role in the development of malignant cancers by targeting EZH2.
However, the possible upregulation and oncogenic role of KDM2B in glioma is still incompletely defined. To examine the role of KDM2B in glioma, we analyzed the expression of KDM2B in various grades of human glioma tissues using immunohistochemistry (IHC) and Western blotting. The correlation between KDM2B expression with clinical pathological and prognostic characteristics was also investigated. We found that the expression of KDM2B in human glioma tissues was significantly higher than that in normal brain tissues, and it was correlated with pathological grades. Then, we used RNA targeting KDM2B to silence the functional expression in the two GBM cell lines, U87 and U251, and its effects on cell proliferation and clone formation were evaluated to identify the function of KDM2B in glioma cells and the biological behavior of the tumor cells. Our findings indicated that KMD2B might be a novel biomarker in the clinical diagnosis and prognosis in glioma by targeting EZH2.
Patients and methods
Patients and specimens
Surgical specimens were collected from 62 glioma patients from the First Affiliated Hospital of China Medical University from 2008 to 2011. Clinicopathological information about the glioma patients is presented in Table 1. Six normal brain tissue samples were used for the control group. Normal tissues were obtained from patients suffering from cerebral injury who underwent internal decompression. None of these patients had received radiotherapy or chemotherapy prior to surgery. All tumor samples were classified as grade I, grade II, grade III, or grade IV based on the WHO classification guidelines. This study was approved by the Ethical Committee of the First Affiliated Hospital of China Medical University at Shenyang. Written informed consent was obtained from all participants.
 | Table 1 Clinicopathological features of 62 glioma patients |
Cell lines, production of lentiviral particles, and establishment of stable cell lines
U87 and U251 human GBM cells were purchased from the Chinese Academy of Sciences (Shanghai, China). Cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Thermo Fisher Scientific, Waltham, MA, USA), 10% fetal bovine serum (FBS; HyClone, South Logan, UT, USA), 100 U/mL penicillin G, and 100 μg/mL streptomycin. The cells were incubated in a sterile incubator maintained at 37°C with 5% CO2. KDM2B-RNAi-lentiviral vectors were purchased from Shanghai Genechem Company (Shanghai, China). The KDM2B #1 sequence was 5′-GATGAGCATGTCCCAGTTT-3′, the KDM2B #2 sequence was 5′-TGAGCGTGAAAGGTTGTTT-3′, and the shRNA control sequence was 5′-TTCTCCGAACGTGTCACGTtt-3′. The U87 and U251 cells were cultured in a 12-well plate and transfected with lentivirus with 3 mg/mL polybrene.
IHC
Immunostaining was performed according to the manufacturer’s protocol. Briefly, all specimens were fixed with 4% paraformaldehyde to prepare the paraffin-embedded specimens in 4 μm sections. Rabbit polyclonal anti-KDM2B (1:150; Abcam, Cambridge, UK) and biotinylated goat anti-rabbit immunoglobulin G were used as primary and secondary antibodies. After counterstaining with diaminobenzidine, the sections were inspected under an optical microscope. For the control sections, the primary antibody was replaced by phosphate-buffered saline (PBS).
Three independent pathologists were invited to evaluate the IHC analysis, without knowledge of the clinical and pathological characteristics of the patients. The intensity of the staining was scored as follows: 0 (negative), 1 (weakly positive), 2 (moderately positive), and 3 (strongly positive). The percentage of KDM2B-positive cells was scored as follows: 0 (0% glioma cells stained), 1 (1%–25% glioma cells stained), 2 (26%–50% glioma cells stained), and 3 (50%–100% glioma cells stained). The intensity and percentage scores were multiplied and classified into two groups: low expression (0–4.5) and high expression (4.5–9).
Western blot assay
Cells and tissue proteins were lysed in radioimmunoprecipitation (RIPA) buffer, and the total protein concentration was determined with a bicinchoninic acid (BCA) assay (Beyotime Institute of Biotechnology, Shanghai, China). Twenty micrograms of total protein were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to polyvinylidene fluoride (PVDF) membranes. The membranes were washed, blocked, and incubated sequentially with specific primary antibodies, namely, rabbit polyclonal anti-Cyclin D1 (1:1,500; Abcam), rabbit polyclonal anti-EZH2 (1:1,500; Abcam), and rabbit monoclonal anti-P21 (1:1,500; Abcam). Incubation with the primary antibodies was followed by incubation with goat antirabbit secondary antibody (1:5,000; OriGene Co. Ltd, Beijing, China). The reactions were detected by an enhanced chemiluminescence assay. The ImageJ software was used to quantify the protein expression levels. Each experiment was performed in triplicate.
Cell viability assay
Cell proliferation was measured using an 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) Assay Kit (BioDev-Tech, Beijing, China). Briefly, the cells were seeded in 96-well plates at a density of 2×103 cells/well (200 μL/well). The plate was incubated for 1, 2, 3, 4, 5, and 6 days. MTT (20 μL, 5 mg/mL) was added to each well. After 4 hours of incubation at 37°C, the culture medium was removed, and the intracellular formazan crystals were solubilized with 150 μL DMSO. After shaking for 10 minutes, the absorbance of each well at 490 nm was measured by an enzyme immunoassay instrument. All experiments were repeated in triplicate.
Clone formation assay
The cellular proliferation potential was assessed by colony formation, which involved seeding 400–1,000 cells in six-well plates and incubated at 37°C in a 5% CO2 incubator. After 14 days of culture, the cells were stained with crystal violet and the colonies were counted. The data are represented as the mean ± SD from three independent experiments.
Cell cycle analysis
Cells were harvested and fixed in 70% ethanol and stored at 4°C overnight. The fixed cells were centrifuged at 1,000× g for 5 minutes and washed with cold PBS three times. Finally, the cells were incubated with 50 μg/mL propidium iodide (PI) containing 8 μg/mL RNase in the dark at 37°C for 30 minutes. The DNA content of the cells was quantified by flow cytometry (FCM).
Statistical analysis
Differences between the groups were evaluated by one-way ANOVA with Tukey’s post hoc test. The Kaplan–Meier method was used for the survival analysis, and univariate and multivariate Cox regressions were used to explore the association between the biomarkers with the outcomes and patient characteristics. All statistical analyses were performed using SPSS 19.0 software (IBM Corporation, Armonk, NY, USA). The results were considered significant if P<0.05, and the data are presented as the mean ± SD from at least three independent experiments.
Results
KDM2B overexpressed in human glioma tissues
We first measured the expression of KDM2B by IHC analysis in 62 glioma tissues (grades II–IV) and six normal brain tissues. As shown in Figure 1A, KDM2B staining was mainly located at the nucleus and cytoplasm of glioma cells, and the noncancerous brain tissue specimens were not stained. Of the 62 glioma samples, 38 (61.29%) and 24 (38.71%) samples displayed high and low expression of KDM2B (Table 1), respectively, and further analysis showed that patients with high KDM2B expression more frequently had advanced WHO grade than those with low KDM2B expression (P<0.05, Table 1). There was no statistical significance between the KDM2B expression and other clinicopathological features, including the patients’ age, gender, tumor location, tumor size, and Karnofsky Performance Scale (KPS) score (P>0.05, Table 1).
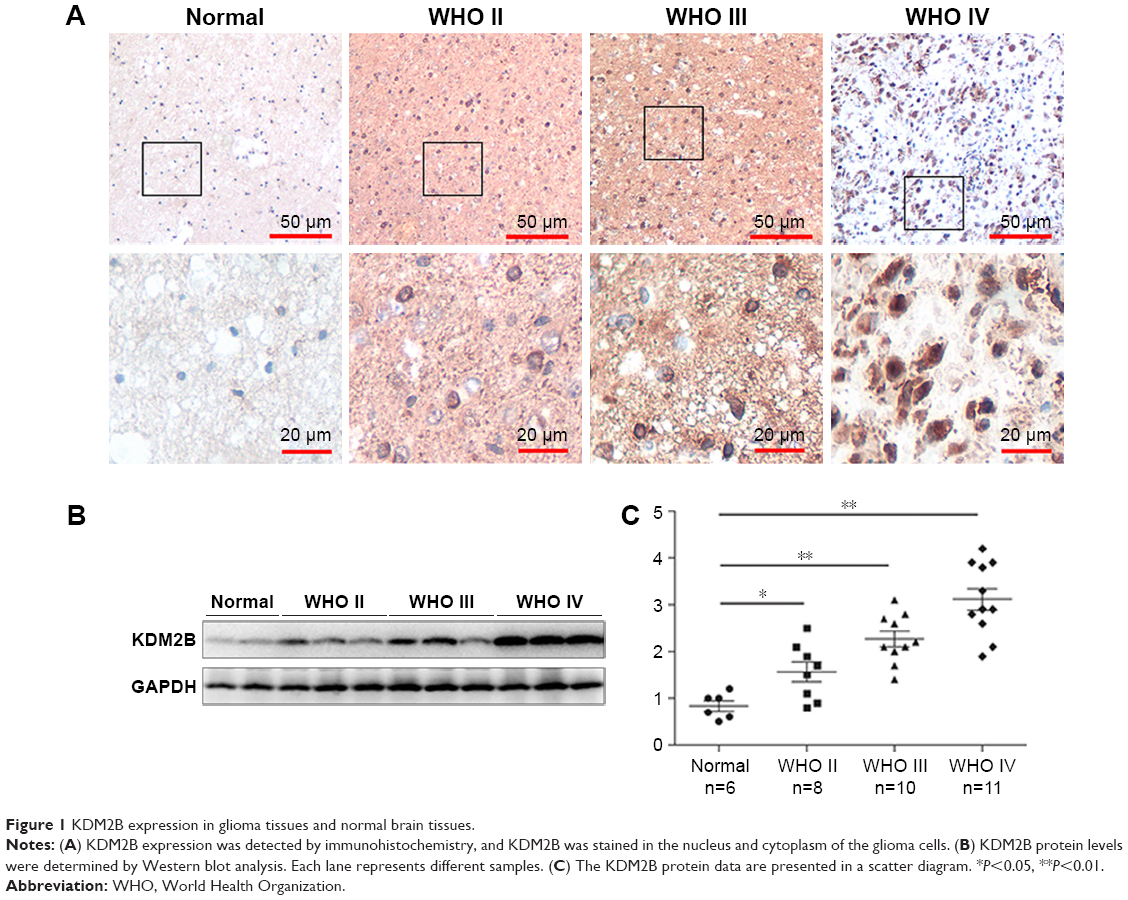 | Figure 1 KDM2B expression in glioma tissues and normal brain tissues. |
Furthermore, we measured the protein expression in the glioma samples and normal brain tissues by Western blot. We found that the expression of the KDM2B protein in glioma samples was higher than that in the normal brain tissues, and quantitative results showed that the KDM2B protein expression levels were highest in the high-grade glioma tissues (grades III and IV; Figure 1B and C). These results suggested that KDM2B was overexpressed in the human glioma tissues and its expression was correlated with glioma grade.
KDM2B expression level correlated with poor prognosis of glioma patients
To evaluate the effect of the KDM2B expression level on the prognosis of glioma patients, we divided the patients into two groups: the high expression group and the low expression group by IHC, and were followed up for 60 months. The survival time of the high KDM2B expression patients was shorter than that of the low KDM2B expression patients according to Kaplan–Meier survival analysis (P<0.05, Figure 2). Furthermore, we used univariate and multivariate analyses to examine the relative risk of the prognostic parameters (Table 2). The Cox regression model showed that the KDM2B expression level (hazard ratio 2.224; 95% confidence interval [CI] 1.171–4.227, P=0.015) and glioma grade (hazard ratio 9.894; 95% CI 3.435–28.500, P=0.000) were independent prognostic factors for the poor overall survival of glioma patients.
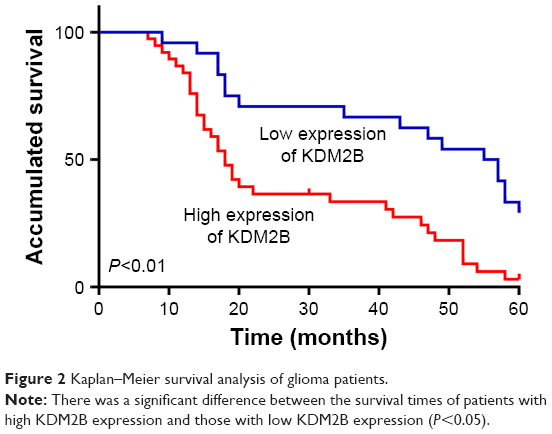 | Figure 2 Kaplan–Meier survival analysis of glioma patients. |
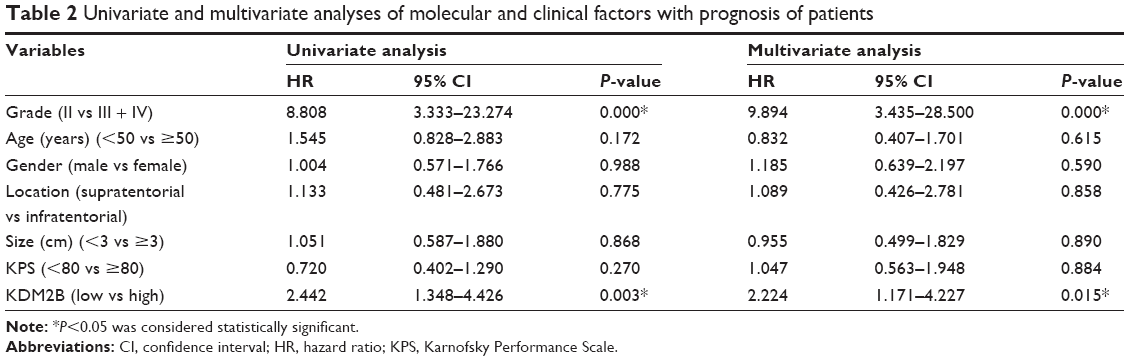 | Table 2 Univariate and multivariate analyses of molecular and clinical factors with prognosis of patients |
Knockdown of KDM2B in glioma cell lines inhibited cell growth
To determine the function of KDM2B in glioma cells, basal KDM2B levels were knocked down by shRNA in the two glioma cell lines, U251 and U87. The transfection efficiency was determined by Western blot (Figure 3A). Compared to the shNC (nonspecific control) group, the protein levels of KDM2B in U251 and U87 glioma cell lines were significantly decreased in the shKDM2B groups. Thus, shKDM2B#1 and shKDM2B#2 were used for the next experiments.
 | Figure 3 KDM2B knockdown in the GBM cell lines reduced cellular proliferation and induced G1/G0 cell cycle arrest. |
We examined the effects of KDM2B on glioma growth by MTT assay and colony-forming assay. The cell viability of the glioma was observed for 6 days in the NC and shKDM2B groups by MTT assay. The growth of shKDM2B groups was obviously decreased from the fourth day in U251 and U87 glioma cell lines (Figure 3B). We also found that the downregulation of KDM2B remarkably inhibited the colony-forming ability of U251 and U87 cells (Figure 3C). The colony-forming numbers of the two glioma cell lines in the shKDM2B groups were significantly decreased compared with those in the NC group. These results showed that shKDM2B could inhibit the growth of the glioma cells in vitro.
Knockdown of KDM2B induced cell cycle arrest of glioma cell lines
Because KDM2B knockdown can reduce cell proliferation, we further examined the effect of KDM2B knockdown on the cell cycle by FCM. The results showed that knockdown of the KDM2B gene can reduce the proportion of cells in the S and G2/M phases while increasing the proportion of cells in the G0/G1 phase in both glioma cell lines (Figure 3D). The results of the cell cycle assay showed that the knockdown of KDM2B induced cell cycle arrest at the G0/G1 phase.
Silencing of KDM2B regulated the expression of cyclin D1 EZH2 and P21 in glioma cell lines
Following the knockdown of KDM2B in the U87 and U251 cell lines, cyclin D1, a cell cycle-related protein, was examined by Western blot assay. We found that the downregulation of KDM2B significantly decreased the protein levels of cyclin D1 (Figure 3E). Moreover, silencing of KDM2B induced the downregulation of EZH2 and the expression of P21 was increased (Figure 3F). These results indicated that the KDM2B/EZH2 pathway plays an important role in glioma cell lines.
Discussion
Glioma is one of the most aggressive tumors, and the prognosis for glioma remains very poor. Despite multiple reports have demonstrated new treatment for this specific tumor type, the destruction caused by glioma did not improve dramatically. Thus, the pathogenesis of glioma remains to be further elucidated. The development of glioma is a multistep progress, which involves multiple epigenetic changes. As reversible biomarkers, epigenetic markers might be used as a potential therapeutic target for glioma.
In this study, we first investigated the expression of KDM2B in human glioma tissues and normal brain tissues. We found that KDM2B was upregulated in glioma tissue compared with normal tissues. In addition, we analyzed the association between KDM2B expression and the pathological features of glioma. In our results, the expression of KDM2B was highly correlated with the glioma grade. The survival analysis showed that the high expression of KDM2B was associated with low survival rates. These results demonstrated that KDM2B might play a vital role in the development of glioma. These results were consistent with the previous study on the upregulation of KDM2B, which was associated with the progression and low survival rate of gastric cancer. Moreover, a previous study has shown that KDM2B mediates the apoptotic response in GBM cells by activating the apoptotic machinery.28
In the present study, knockdown of KDM2B dramatically reduced the viability of both the U87 and U251 glioma cells in vitro. In addition, inhibition of KDM2B contributed to cell cycle progression in the two glioma cell lines and resulted in an increase in the percentage of G0/G1 phase cells while decreasing the percentage of S phase cells. These results showed that KDM2B might play a vital role in the proliferation of glioma cells.
It has previously been reported that the knockdown of KDM2B inhibits cell proliferation and affects cell cycle progression in tumor cells.29 Moreover, genetic knockdown of KDM2B can also induce G0/G1 arrest in cancer cells.9 These results were consistent with the findings in the present study. Previous results showed that downregulation of the cell cycle plays a vital role in glioma cells.30,31 In addition, silencing KDM2B can enhance the protein expression of P21 and reduce the expression of cyclin D1. Accumulating expression of P21 represses RB phosphorylation, which is necessary for the cell cycle.32 In multiple cancer cells, cyclin D1, a pro-oncogene protein, regulates the G1-to-S phase transition in cell cycle.33 These results indicated that, in glioma cells, KDM2B regulates the cell cycle through coordinated regulation of a variety of cell cycle-related factors.
The experiments in this study showed that silencing KDM2B reduced the protein expression of EZH2 in two glioma cell lines. EZH2 is an oncogenic gene with multiple aspects, which promotes the migration and proliferation of tumors by the methylation of tumor inhibitor proteins.34 The prognostic and biological role of EZH2 has been fully investigated in gliomas.35–40 In addition, deactivation of EZH2 might alleviate many vital signals relating to glioma self-renewal, which suggests that EZH2 can act as a novel therapeutic factor for GBM.41 A previous study identified EZH2 as a prognostic biomarker of survival rates in glioma patients undergoing resection treatment. Previous evidence showed that KDM2B regulates the expression of EZH2 by transcriptionally repressing microRNAs such as let-7.16,42 It has been reported that EZH2 expression in myelodysplastic syndrome cells was downregulated when KDM2B was silenced.41 These results indicated that EZH2 may be regulated by KDM2B. Furthermore, EZH2 overexpression was found to be related to poor prognosis of gliomas and to promote the tumorigenicity of GBM by targeting STAT3.43,44 These results indicated that KDM2B can mediate the expression of EZH2 and participate in the pathogenesis of gliomas.
Conclusion
The present study showed that KDM2B was overexpressed in glioma tissues and might reflect the prognosis of glioma patients. Moreover, the expression of KDM2B is associated with glioma degree in a grade-dependent manner. In addition, genetic silencing of KDM2B has been found to have growth-inhibiting effects in the U87 and U251 human glioma cell lines and is correlated with G0/G1 phase cell cycle arrest by targeting the EZH2 genes. Further studies are needed to explore the potential role of KDM2B in gliomas in animal models.
Acknowledgments
This study was supported by the Doctoral Startup Foundation of Liaoning Province (grant number 201501004 to DZ), the Science and Technology Project of Liaoning Province (grant number 201005036), the Natural Science Foundation of Liaoning Province (grant number 20170520040), and the Science and Technology Project of the Technology Division of Shenyang City (grant number F13-221-9-33).
Disclosure
The authors report no conflicts of interest in this work.
References
Giese A, Bjerkvig R, Berens ME, Westphal M. Cost of migration: invasion of malignant gliomas and implications for treatment. J Clin Oncol. 2003;21(8):1624–1636. | ||
Lowenstein PR, Castro MG. The long and winding road: from the high-affinity choline uptake site to clinical trials for malignant brain tumors. Adv Pharmacol. 2016;76:147–173. | ||
Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. | ||
Mirimanoff RO. High-grade gliomas: reality and hopes. Chin J Cancer. 2014;33(1):1–3. | ||
Qiu ZK, Shen D, Chen YS, et al. Enhanced MGMT expression contributes to temozolomide resistance in glioma stem-like cells. Chin J Cancer. 2014;33(2):115–122. | ||
Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6(11):838–849. | ||
Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. | ||
Tsukada Y, Fang J, Erdjument-Bromage H, et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439(7078):811–816. | ||
Kottakis F, Foltopoulou P, Sanidas I, et al. NDY1/KDM2B functions as a master regulator of polycomb complexes and controls self-renewal of breast cancer stem cells. Cancer Res. 2014;74(14):3935–3946. | ||
He J, Nguyen AT, Zhang Y. KDM2b/JHDM1b, an H3K36me2-specific demethylase, is required for initiation and maintenance of acute myeloid leukemia. Blood. 2011;117(14):3869–3880. | ||
Pfau R, Tzatsos A, Kampranis SC, Serebrennikova OB, Bear SE, Tsichlis PN. Members of a family of JmjC domain-containing oncoproteins immortalize embryonic fibroblasts via a JmjC domain-dependent process. Proc Natl Acad Sci U S A. 2008;105(6):1907–1912. | ||
Ueda T, Nagamachi A, Takubo K, et al. Fbxl10 overexpression in murine hematopoietic stem cells induces leukemia involving metabolic activation and upregulation of Nsg2. Blood. 2015;125(22):3437–3446. | ||
Tzatsos A, Paskaleva P, Ferrari F, et al. KDM2B promotes pancreatic cancer via Polycomb-dependent and -independent transcriptional programs. J Clin Invest. 2013;123(2):727–739. | ||
Zhao E, Tang C, Jiang X, et al. Inhibition of cell proliferation and induction of autophagy by KDM2B/FBXL10 knockdown in gastric cancer cells. Cell Signal. 2017;36:222–229. | ||
Pasqualucci L, Trifonov V, Fabbri G, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet. 2011;43(9):830–837. | ||
Tzatsos A, Paskaleva P, Lymperi S, et al. Lysine-specific demethylase 2B (KDM2B)-let-7-enhancer of zester homolog 2 (EZH2) pathway regulates cell cycle progression and senescence in primary cells. J Biol Chem. 2011;286(38):33061–33069. | ||
Tzatsos A, Pfau R, Kampranis SC, Tsichlis PN. Ndy1/KDM2B immortalizes mouse embryonic fibroblasts by repressing the Ink4a/Arf locus. Proc Natl Acad Sci U S A. 2009;106(8):2641–2646. | ||
Tsang DP, Cheng AS. Epigenetic regulation of signaling pathways in cancer: role of the histone methyltransferase EZH2. J Gastroenterol Hepatol. 2011;26(1):19–27. | ||
Huang BY, Pan XY, Li ZX, Wang ZC, Yu YS, Dou ZH. Polycomb group proteins and their roles in regulating stem cell development. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2012;34(3):281–285. | ||
Melling N, Thomsen E, Tsourlakis MC, et al. Overexpression of enhancer of zeste homolog 2 (EZH2) characterizes an aggressive subset of prostate cancers and predicts patient prognosis independently from pre- and postoperatively assessed clinicopathological parameters. Carcinogenesis. 2015;36(11):1333–1340. | ||
Guo S, Li X, Rohr J, et al. EZH2 overexpression in different immunophenotypes of breast carcinoma and association with clinicopathologic features. Diagn Pathol. 2016;11:41. | ||
Yang F, Lv LZ, Cai QC, Jiang Y. Potential roles of EZH2, Bmi-1 and miR-203 in cell proliferation and invasion in hepatocellular carcinoma cell line Hep3B. World J Gastroenterol. 2015;21(47):13268–13276. | ||
Eskander RN, Ji T, Huynh B, Wardeh R, Randall LM, Hoang B. Inhibition of enhancer of zeste homolog 2 (EZH2) expression is associated with decreased tumor cell proliferation, migration, and invasion in endometrial cancer cell lines. Int J Gynecol Cancer. 2013;23(6):997–1005. | ||
Sun M, Liu XH, Lu KH, et al. EZH2-mediated epigenetic suppression of long noncoding RNA SPRY4-IT1 promotes NSCLC cell proliferation and metastasis by affecting the epithelial–mesenchymal transition. Cell Death Dis. 2014;5:e1298. | ||
Smits M, Nilsson J, Mir SE, et al. miR-101 is down-regulated in glioblastoma resulting in EZH2-induced proliferation, migration, and angiogenesis. Oncotarget. 2010;1(8):710–720. | ||
Suva ML, Riggi N, Janiszewska M, et al. EZH2 is essential for glioblastoma cancer stem cell maintenance. Cancer Res. 2009;69(24):9211–9218. | ||
Kottakis F, Polytarchou C, Foltopoulou P, Sanidas I, Kampranis SC, Tsichlis PN. FGF-2 regulates cell proliferation, migration, and angiogenesis through an NDY1/KDM2B-miR-101-EZH2 pathway. Mol Cell. 2011;43(2):285–298. | ||
Kurt IC, Sur I, Kaya E, et al. KDM2B, an H3K36-specific demethylase, regulates apoptotic response of GBM cells to TRAIL. Cell Death Dis. 2017;8(6):e2897. | ||
Yu X, Wang J, Wu J, Shi Y. A systematic study of the cellular metabolic regulation of Jhdm1b in tumor cells. Mol Biosyst. 2015;11(7):1867–1875. | ||
Ouyang Q, Xu L, Cui H, Xu M, Yi L. MicroRNAs and cell cycle of malignant glioma. Int J Neurosci. 2016;126(1):1–9. | ||
Chang YC, Chou FP, Huang HP, Hsu JD, Wang CJ. Inhibition of cell cycle progression by penta-acetyl geniposide in rat C6 glioma cells. Toxicol Appl Pharmacol. 2004;198(1):11–20. | ||
Igata M, Motoshima H, Tsuruzoe K, et al. Adenosine monophosphate-activated protein kinase suppresses vascular smooth muscle cell proliferation through the inhibition of cell cycle progression. Circ Res. 2005;97(8):837–844. | ||
Lamba Saini M, Weynand B, Rahier J, Mourad M, Hamoir M, Marbaix E. Cyclin D1 in well differentiated thyroid tumour of uncertain malignant potential. Diagn Pathol. 2015;10:32. | ||
Chang CJ, Hung MC. The role of EZH2 in tumour progression. Br J Cancer. 2012;106(2):243–247. | ||
Orzan F, Pellegatta S, Poliani PL, et al. Enhancer of Zeste 2 (EZH2) is up-regulated in malignant gliomas and in glioma stem-like cells. Neuropathol Appl Neurobiol. 2011;37(4):381–394. | ||
Ahani N, Shirkoohi R, Rokouei M, Alipour Eskandani M, Nikravesh A. Overexpression of enhancer of zeste human homolog 2 (EZH2) gene in human cytomegalovirus positive glioblastoma multiforme tissues. Med Oncol. 2014;31(11):252. | ||
Kim SH, Joshi K, Ezhilarasan R, et al. EZH2 protects glioma stem cells from radiation-induced cell death in a MELK/FOXM1-dependent manner. Stem Cell Reports. 2015;4(2):226–238. | ||
Li AM, Dunham C, Tabori U, et al. EZH2 expression is a prognostic factor in childhood intracranial ependymoma: a Canadian Pediatric Brain Tumor Consortium study. Cancer. 2015;121(9):1499–1507. | ||
Lin L, Zheng Y, Tu Y, et al. MicroRNA-144 suppresses tumorigenesis and tumor progression of astrocytoma by targeting EZH2. Hum Pathol. 2015;46(7):971–980. | ||
Sharma V, Purkait S, Takkar S, et al. Analysis of EZH2: micro-RNA network in low and high grade astrocytic tumors. Brain Tumor Pathol. 2016;33(2):117–128. | ||
Kim E, Kim M, Woo DH, et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell. 2013;23(6):839–852. | ||
Karoopongse E, Yeung C, Byon J, et al. The KDM2B- let-7b -EZH2 axis in myelodysplastic syndromes as a target for combined epigenetic therapy. PLoS One. 2014;9(9):e107817. | ||
Wen Y, Cai J, Hou Y, Huang Z, Wang Z. Role of EZH2 in cancer stem cells: from biological insight to a therapeutic target. Oncotarget. 2017;8(23):37974–37990. | ||
Zhang Y, Yu X, Chen L, Zhang Z, Feng S. EZH2 overexpression is associated with poor prognosis in patients with glioma. Oncotarget. 2017;8(1):565–573. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.