Back to Journals » ImmunoTargets and Therapy » Volume 15
Kasugamycin Inhibits Melanoma Lung Metastasis and CHI3L1-Driven M2-Like Tumor-Associated Macrophage Differentiation
Authors Sadanaga T ![]() , Jeong HS
, Jeong HS ![]() , Cortez R, Lee JH
, Cortez R, Lee JH ![]() , Kamle S, Ma B, Shin SJ, Elias JA, Lee CG
, Kamle S, Ma B, Shin SJ, Elias JA, Lee CG
Received 7 October 2025
Accepted for publication 30 January 2026
Published 19 March 2026 Volume 2026:15 563951
DOI https://doi.org/10.2147/ITT.S563951
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Michael Shurin
Takayuki Sadanaga,1,* Han-Seok Jeong,1,* Roberto Cortez,2 Joyce H Lee,3 Suchitra Kamle,1 Bing Ma,1 Sung Jae Shin,4 Jack A Elias,1 Chun Geun Lee1,5
1Department of Molecular Microbiology and Immunology, Brown University, Providence, RI, USA; 2Department of Acute Care Surgery, Morehouse School of Medicine, Grady Memorial Hospital, Atlanta, GA, USA; 3Johns Hopkins School of Medicine, Baltimore, MD, USA; 4Department of Microbiology, Institute for Immunology and Immunological Disease, Graduate School of Medical Science, Brain Korea 21 Project, Yonsei University College of Medicine, Seoul, South Korea; 5Department of Translational Medicine, Intercollege, Hanyang University, Seoul, South Korea
*These authors contributed equally to this work
Correspondence: Chun Geun Lee, Department of Molecular Microbiology and Immunology, Brown University, Providence, RI, USA, Email [email protected]; [email protected]
Purpose: Chitinase-3-like-1 (CHI3L1) is a potent immune modulator implicated in tumor progression and immune suppression, including melanoma lung metastasis. Kasugamycin (KSM) has been reported as a pan-chitinase inhibitor with antifibrotic activity, but its effects on CHI3L1-driven immune regulation remain poorly defined. This study aimed to determine whether KSM suppresses CHI3L1-mediated tumor progression by modulating tumor-associated macrophage (TAM) differentiation and to elucidate the underlying molecular mechanisms.
Methods: The anti-tumor effects of KSM were evaluated using a B16/F10 melanoma lung metastasis model. CHI3L1 gain-of-function approaches were used to assess specificity. Lung immune populations were analyzed by flow cytometry. Human THP-1 monocytes were used to examine CHI3L1-induced macrophage differentiation in vitro. Bulk RNA sequencing was performed on differentiated macrophages to identify downstream signaling pathways. Pharmacologic inhibition studies were conducted using the epidermal growth factor receptor (EGFR) inhibitor gefitinib to validate mechanistic links.
Results: KSM treatment significantly reduced melanoma lung metastasis in a dose-dependent manner. CHI3L1 overexpression enhanced melanoma lung colony formation, which was effectively abrogated by KSM, indicating CHI3L1-specific anti-tumor activity. In melanoma-challenged lungs, KSM markedly decreased M2-like macrophages expressing CD206, CD163, and PD-L1. In vitro, CHI3L1 promoted M2 macrophage differentiation in THP-1 cells, which was strongly suppressed by KSM. Transcriptomic analysis revealed that EGFR expression was robustly induced by CHI3L1 and counter-regulated by KSM. Inhibition of EGFR signaling with gefitinib significantly attenuated CHI3L1-driven STAT3 activation and M2 macrophage polarization.
Conclusion: These findings identify a previously unrecognized anti-tumor mechanism of KSM through inhibition of CHI3L1-EGFR-STAT3 signaling and suppression of M2-like TAM differentiation. KSM may therefore represent a promising immunomodulatory strategy for treating melanoma lung metastasis and other CHI3L1-driven malignancies.
Keywords: kasugamycin, chitinase 3-like 1, anti-CHI3L1, tumor associated macrophages
Introduction
Lung cancer is the leading cause of cancer-related mortality worldwide for both men and women, accounting for approximately 1 in 5 cancer deaths in the United States.1 Non-small cell lung cancer (NSCLC) comprises 80–85% of all lung cancer cases, with over half of patients being diagnosed at an advanced stage of the disease.1,2 Additionally, the lung is the second most frequent site for metastatic spread, with 20–54% of malignant tumors originating elsewhere in the body leading to pulmonary metastases.3 Despite significant advances in lung cancer management, including the use of immune checkpoint inhibitors, the 5-year survival rate for advanced-stage cases remains under 10%. Disease progression during or after treatment continues to be the primary cause of mortality.3 Therefore, developing effective therapeutic strategies to block disease progression and improve patient outcomes remains a critical unmet need.
The glycosyl hydrolase family 18 (GH18) includes true chitinases, such as Chitinase 1 (CHIT1), and several chitinase-like proteins (CLPs) that lack enzymatic activity. Among these, Chitinase 3-like 1 (CHI3L1, also known as YKL-40) is a prototypic CLP that is overexpressed in the serum and tissues of lung cancer patients. Elevated serum levels of CHI3L1 have been identified as an independent prognostic biomarker for poor survival in lung cancer patients.4 CHI3L1 has been shown to drive a pro-tumoral microenvironment through modulation of innate and adaptive immune responses in various cancers, including lung cancer.5–11 Recent studies from our laboratory revealed that CHI3L1 stimulates the expression of immune checkpoint molecules and their receptors including PD-1/PD-L1 and CTLA-4 in macrophages and T cells in the lung, contributing to enhanced melanoma lung metastases.10,11 Among immune cells, macrophages, particularly M2-like tumor-associated macrophages (TAMs) are a major component of the tumor microenvironment. These cells, which highly express CHI3L1, play a crucial role in tumor progression, and CHI3L1’s involvement in promoting M2 macrophage differentiation has been proposed.5,12 However, the direct role and mechanism of CHI3L1 in specific macrophage differentiation driving its pro-tumoral effects and potential therapeutic interventions have not been sufficiently explored.
Kasugamycin (KSM) is a naturally occurring aminoglycoside antibiotic characterized by two sugar moieties and low animal toxicity.13 Recently, KSM was identified as a specific inhibitor of chitinase, demonstrating strong antifibrotic activity by blocking CHIT1-stimulated TGF-β signaling in the lung.14,15 KSM has also been shown to enhance host resistance to viral infections by upregulating interferon-stimulated genes (ISGs).16 Further studies have revealed KSM’s broad anti-chitinase activity across various chitinases from humans and other organisms,17 highlighting its potential use as an inhibitor of chitinase-like proteins, which share significant structural homology with chitinases. However, its activity against CHI3L1 and other GH18 family members that share structural and functional similarities has not been determined.
To evaluate the effects of kasugamycin (KSM) in metastatic lung cancer, we employed a melanoma lung metastasis model, as it is a validated, lung-focused, immune-competent, and mechanistically informative platform for evaluating metastatic cancer to the lung and for testing therapeutic strategies targeting tumor-lung microenvironment interactions.10,11,17
In this study, we demonstrate that KSM significantly suppresses melanoma lung metastasis. Our findings reveal that CHI3L1 drives M2 macrophage differentiation, a process that is counter-regulated by KSM treatment. Specifically, CHI3L1 increased the number of macrophages expressing CD206, CD163, and PD-L1, hallmarks of M2-like tumor-associated macrophages (TAMs) in melanoma lung metastases. This effect was more pronounced in CHI3L1-overexpressing transgenic mice than in wild-type controls. Using bulk RNA sequencing of differentiated macrophages and signaling pathway analyses, we further identified that epidermal growth factor receptor (EGFR) expression was strongly induced by CHI3L1 and counter-regulated by KSM. Treatment with an EGFR inhibitor significantly reduced CHI3L1-driven STAT3signaling as well as M2 macrophage activation, supporting EGFR contribution to CHI3L1-driven M2 macrophage activation. Collectively, these findings reveal a novel anti-tumor mechanism of KSM, in which it inhibits M2-like tumor-associated macrophage differentiation, at least in part through disruption of the CHI3L1-EGFR axis. They also suggest that KSM may be effectively repurposed as an anti-tumor agent for melanoma lung metastasis, in which CHI3L1 plays a significant role.
Materials and Methods
Genetically Modified Mice
Lung-specific CHI3L1-overexpressing transgenic mice (CHI3L1 Tg; Chil1 Tg), in which CHI3L1 is targeted to the lung using the Clara cell 10-kD protein (CC10) promoter to achieve lung epithelial-specific expression. Transgenic founder lines were established and backcrossed to the C57BL/6 background for more than 10 generations. Transgene expression was confirmed by PCR genotyping. All experiments were conducted using 8–10-week-old wild-type (WT) and Chil1 Tg mice. All animal experiments were approved by the Brown University Institutional Animal Care and Use Committee (IACUC, protocol number: 22–11-004), and were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals and the American Veterinary Medical Association (AVMA) Guidelines for the Euthanasia of Animals (2020 Edition).
Mice were anesthetized with 2–3% isoflurane inhalation for all surgical or invasive procedures, and euthanized by gradual CO2 inhalation followed by cervical dislocation to ensure death, in compliance with the institutional and federal regulations.
No additional animal license was required for this study, as all procedures were performed under the approval of the IACUC protocol.
Melanoma Lung Metastasis and KSM Treatment
The B16-F10 mouse melanoma cell line (#CRL-6475, ATCC, Manassas, VA) was cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. Cells were collected upon reaching 80% confluence, adjusted to a concentration of 1×106 cells/mL, and injected into mice via the lateral tail vein (2×105 cells/mouse in 200 μL DMEM; low-dose challenge in a setting including CHI3L1 transgenic mice, 0.2×105 cells/mouse).11 Intraperitoneal PBS or KSM (100–200 mg/kg) was administered every other day for two weeks after the tumor cell challenge. Lung metastases were quantified by counting melanoma colonies (black nodules) on the pleural surface as described previously.7,9 In vivo KSM doses were selected based on the prior PK/PD data used in animal toxicology studies that demonstrated no toxicity up to 400 mg/kg18 and reported animal studies.14
FACS Analysis
Single-cell suspensions from whole mouse lungs were prepared using the Lung Dissociation Kit (Miltenyi Biotec) per the manufacturer’s instructions. Cells were stained with fluorescently labeled antibodies directed against CD45 (30-F11, #103126, Thermo Fisher Scientific), PD-L1 (MIH5, #12-5982-8, Thermo Fisher Scientific), CD11b (M1/70, #101210, Biolegend), CD206 (MR5D3, #MA5-16870, Thermo Fisher Scientific), CD163 (TNKUPJ, #17-1631-82, Thermo Fisher Scientific). Flow cytometry data were collected using the BD FACSAria IIIu and analyzed with FlowJo (v10) software. The gating strategy of the macrophage has been illustrated in Figure S7.
In vitro Macrophage Differentiation Using CHI3L1 and Tumor Cell Conditioned Media
Human monocytic THP-1 cells (# TIB-202, ATCC) were differentiated into TAMs using recombinant (r) M-CSF, CHI3L1 along with tumor cell-conditioned media (from A549 cells (#CRM-CCL-185, ATCC)), following established procedures with modification.19 In this protocol, we replaced the traditional IL-4/IL-10 cytokines with CHI3L1. KSM effects were evaluated by adding 3 µM KSM to the TAM differentiation medium. Differences in TAM differentiation (measured by CD206, CD163, and PD-L1 co-expression) were assessed by real-time qRT-PCR, FACS, and immunoblot evaluations. The expression of other markers of macrophage activation such as CX3CR1, CD36 and iNOS, was also evaluated by real-time RT-PCR evaluations. The specific role of CHI3L1 in macrophage differentiation was also investigated using a CHI3L1-neutralizing monoclonal antibody (FRG antibody).
RNA Extraction and Semi-Quantitative Real-Time qPCR
Total RNA was isolated using TRIzol reagent (ThermoFisher Scientific) followed by RNA purification with the RNeasy Mini Kit (Qiagen, Germantown, MD) per the manufacturer’s instructions. Reverse transcription and real-time PCR were performed as previously described.8,20 Ct values for target genes were normalized to internal housekeeping genes GAPDH or Rpl13a. Primer sequences are listed in Table S5.
Bulk RNA Sequencing and Data Analysis
For mechanistic understanding, we used unbiased bulk RNA-seq analysis, similar to approaches used in other studies.21,22 Macrophages differentiated from THP-1 cells under various treatments were subjected to bulk RNA sequencing. Total RNA isolated from the pooled duplicated samples was subjected to evaluation. Differential transcriptomic gene expression profiling and data analysis were conducted to identify genes regulated by CHI3L1 and KSM using Illumina’s next-generation sequencing, which detects coding and long non-coding RNAs (Standard RNA-Seq Service provided by Genewiz/Azenta Life Science, Waltham, MA).
Western Blotting (Immunoblotting)
Protein lysates (25 µg) from lung tissues or cells were subjected to SDS-PAGE, transferred to membranes, and immunoblotted with primary antibodies against CD163 (#PA5-109327, Thermo Fisher Scientific), CD206 (E6T5J, #24595S, Cell Signaling Technology), phosphorylated AKT (p-Akt) (193H12, #4058S, Cell Signaling Technology), and total Akt (11E7, #4685S, Cell Signaling Technology), phosphorylated EGFR (Tyr1068) (p-EGFR) (#44-788G, Thermo Fisher Scientific), EGFR (D38B1, #4267S, Cell Signaling Technology), phosphorylated Erk (p-Erk) (#9101S, Cell Signaling Technology), total Erk (#9102S, Cell Signaling Technology), phosphorylated STAT3 (Tyr705) (p-STAT3) (D3A7, #9145, Cell Signaling Technology), total STAT3 (79D7, #4904, Cell Signaling Technology) and b-actin (C4, #sc-47778 HRP, Santa Cruz Biotechnology). HRP-conjugated anti-rabbit IgG secondary antibody (#7074S, Cell Signaling Technology) was used for detection. Immunoblotting was performed following standard protocols as described previously.23
Measurement of Oxidative Stress-Induced Apoptosis
AMJ2-C11 cells (#CRL-2456, ATCC) were treated with hydrogen peroxide (#216763, Sigma-Aldrich, 1 mM, 24 hours) to induce oxidative stress. Apoptotic cells were labeled with Annexin V and propidium iodide (#556547, BD Biosciences), followed by flow cytometry analysis.
Double-Label Fluorescent Immunohistochemistry
Formalin-fixed, paraffin-embedded (FFPE) lung tissue blocks were sectioned into 5 µm-thick slices and mounted onto glass slides. Sections were deparaffinized, rehydrated, and subjected to heat-induced epitope retrieval using a steamer and antigen unmasking solution (Abcam, citrate buffer, pH 6.0) for 30 minutes. Blocking was performed with serum-free protein blocking solution (Dako/Agilent, Santa Clara, CA) for 60 minutes at room temperature. Slides were incubated overnight at 4°C with primary antibodies against CD163 (EPR19518, #ab182422, abcam), CD206 (#AF2535, R&D Systems). After washing, fluorescent-labeled secondary antibodies of Donkey anti-Rabbit IgG (H+L) Alexa Fluor™ 488 (#A-21206, Thermo Fisher Scientific) or Donkey anti-Goat IgG (H+L) Alexa Fluor™ 594 (#A-11058, Thermo Fisher Scientific) were applied for 1 hour at room temperature. Sections were counterstained with DAPI and mounted with coverslips.
Quantification and Statistical Analysis
All statistical analyses were conducted using GraphPad Prism software. Comparisons between groups were made using two-tailed Student’s t-tests. One-way ANOVA followed by appropriate post-hoc tests was performed for multiple group comparisons. Data are presented as mean ± SEM, and statistical significance was defined as P<0.05.
Results
Kasugamycin Inhibits Melanoma Lung Metastasis
To evaluate the effects of KSM on pulmonary melanoma metastasis, wild-type (WT) mice were injected via the tail vein with either B16-F10 melanoma cells (B16) or vehicle control (PBS), as illustrated in Figure 1A. Fourteen days post-challenge, metastatic melanoma colonies (pleural colonies) were prominent in mouse lungs and were counted and compared between PBS-treated mice and those treated with varying doses of KSM (Figure 1B). These studies revealed that KSM significantly attenuated melanoma lung metastasis in a dose-dependent manner. Hematoxylin and eosin (H&E) staining further demonstrated reduced parenchymal tumor growth in the lungs of KSM-treated mice compared to PBS-treated controls (Figure 1C). Additionally, the degree of inhibition in melanoma lung metastasis correlated positively with the duration of KSM treatment (Figure S1).
 |
Figure 1 Kasugamycin inhibits melanoma lung metastasis. (A) Schematic illustration of melanoma lung metastasis evaluation. (B) Dose-dependent kasugamycin (KSM) inhibition of melanoma lung metastasis. B16-F10 (B16) melanoma cells were delivered to the mice through tail vein injection with and without KSM treatment (100 mg/kg and 200 mg/kg mouse, i.p.). Representative lung photos with melanoma lung metastasis (left panel) and their pleural colony counts (right panel). (C) Representative H&E stains of B16-F10 (B16) melanoma cell-challenged lungs with and without KSM treatment. The values in (B) for the pleural colony count are the mean ± SEM. *P<0.05, ****P<0.0001 (One-Way ANOVA, multiple comparisons). Scale bar in (C) = 250 µm. |
Kasugamycin Inhibits CHI3L1-Mediated Signaling and Anti-Apoptotic Responses of Lung Macrophages
Given that KSM is a potent inhibitor of CHIT1 and other enzymatically active chitinases,24,25 we hypothesized that KSM may similarly influence the signaling and biological activity of CHI3L1. To test this, we first investigated the impact of KSM on well-characterized CHI3L1 signaling pathways.26 As shown in Figure 2A, recombinant CHI3L1 (rCHI3L1) enhanced the phosphorylation of Erk and Akt in AMJ2-C11 mouse lung macrophages. We also observed that CHI3L1 increased the expression of M2 polarization markers (Cd206, Cd163), whereas there were no significant changes in the expression of M1 markers, such as Nos2 (Figure S2). These CHI3L1-stimulated signaling activations were effectively abrogated by KSM treatment, similar to anti-CHI3L1 antibody (FRG) treatment (Figure 2A), and M2 polarizations were also effectively abrogated by KSM treatment (Figure S2). CHI3L1 is also known for its anti-apoptotic effects, particularly in reducing oxidative stress-induced apoptosis.25 To evaluate KSM’s impact, macrophages were treated with hydrogen peroxide (H2O2) to induce oxidative stress, and apoptosis was assessed by fluorescence-activated cell sorting (FACS) analysis. CHI3L1 treatment significantly decreased the number of Annexin V (+) apoptotic lung macrophages. However, this anti-apoptotic effect was significantly reduced upon KSM treatment to levels comparable to anti-CHI3L1 antibody (FRG) treatment (Figure 2B), suggesting that KSM effectively blocks CHI3L1-mediated anti-apoptotic signaling in macrophages. Together, these findings demonstrate that KSM effectively inhibits CHI3L1-mediated signaling and cellular responses in macrophages.
 |
Figure 2 KSM inhibits CHI3L1-stimulated Erk and Akt activation and the antiapoptotic effect of CHI3L1 in macrophages. (A) AMJ-C11 mouse lung macrophages were stimulated with recombinant (r) CHI3L1 overnight, and the activation of Erk and Akt was detected by Western blot evaluations. pErk, phosphorylated Erk; T-Erk, total Erk; pAkt, phosphorylated Akt, T-Akt, total Akt. Right panel, densitometric quantitation on the blots of pErk and pAkt. (B) After exposure of AMJ-C11 cells with H2O2 (1 mM, 24 hours) with and without rCHI3L1 and KSM treatment, cellular apoptosis (% of Annexin V+ cells) was measured by FACS evaluations. (A and B) are representatives of 3 independent experiments. Right panel, the values of Annexin 5 (+) cells (%) are the mean ± SEM. *P<0.05, **P<0.01, ***P<0.001 (One-Way ANOVA, multiple comparisons). |
KSM Inhibits M2-Like TAM Accumulation in Lungs with Melanoma Metastasis
To investigate the roles of CHI3L1 and KSM in macrophage differentiation during melanoma lung metastasis, we characterized macrophages accumulated in the lungs of WT and CHI3L1 transgenic mice following B16-F10 challenge and KSM treatment. Immunohistochemistry and FACS analyses revealed increased numbers of macrophages expressing CD206, CD163, and PD-L1 (markers of M2-like TAMs,27,28) in the lungs of B16-challenged mice (Figure 3A and B). Notably, KSM treatment significantly reduced CD206(+)/CD163(+) macrophages in peritumoral regions, indicating that KSM inhibits M2-like TAM accumulation in the lung (Figure 3A). PD-L1 expression was similarly elevated in CD206(+) and CD163(+) macrophages within the lungs of melanoma-challenged mice. However, it was markedly reduced following KSM treatment (Figure 3B).
 |
Figure 3 KSM inhibits M2-like TAMs accumulation in lungs of melanoma metastasis. (A) Co-immunostaining of CD206 (red) and CD163 (green) in lungs challenged with B16-F10 (B16) melanoma cells with vehicle (PBS) or KSM treatment (100 mg/kg/mouse). Arrows indicate CD206/CD163 double (+) cells (yellow) or CD206 or CD163 single positive cells (white). (B) Representative FACS evaluations on CD206(+)/CD163(+), CD206(+)/ PD-L1(+), and CD163(+)/PD-L1(+) macrophages in lungs with PBS or melanoma cell challenge and KSM treatment. Scale bar in (A) = 25 µm. |
CHI3L1 Enhances the Melanoma Lung Metastasis and M2 Macrophage Accumulation in the Lung
To see the impact of CHI3L1 in melanoma lung metastasis and tumor-associated macrophage activation, we evaluated the WT and CHI3L1 Tg mice after low-dose B16-F10 melanoma cell challenge (0.2x105 cells/mouse). The number of melanoma lung colonies was significantly increased in CHI3L1 Tg mice compared to WT colonies, and the increases were abrogated with KSM treatment (Figure 4A and B). Immunohistochemical evaluations further revealed a significant accumulation of CD206(+)/CD163(+) and CD206(+)/PD-L1(+) cells in the lungs of CHI3L1 Tg mice and those increases were abrogated with KSM treatment (Figure 4C and D). These findings suggest that CHI3L1 promotes M2 or M2-like TAM accumulation in melanoma lung metastases and that KSM effectively counteracts this process.
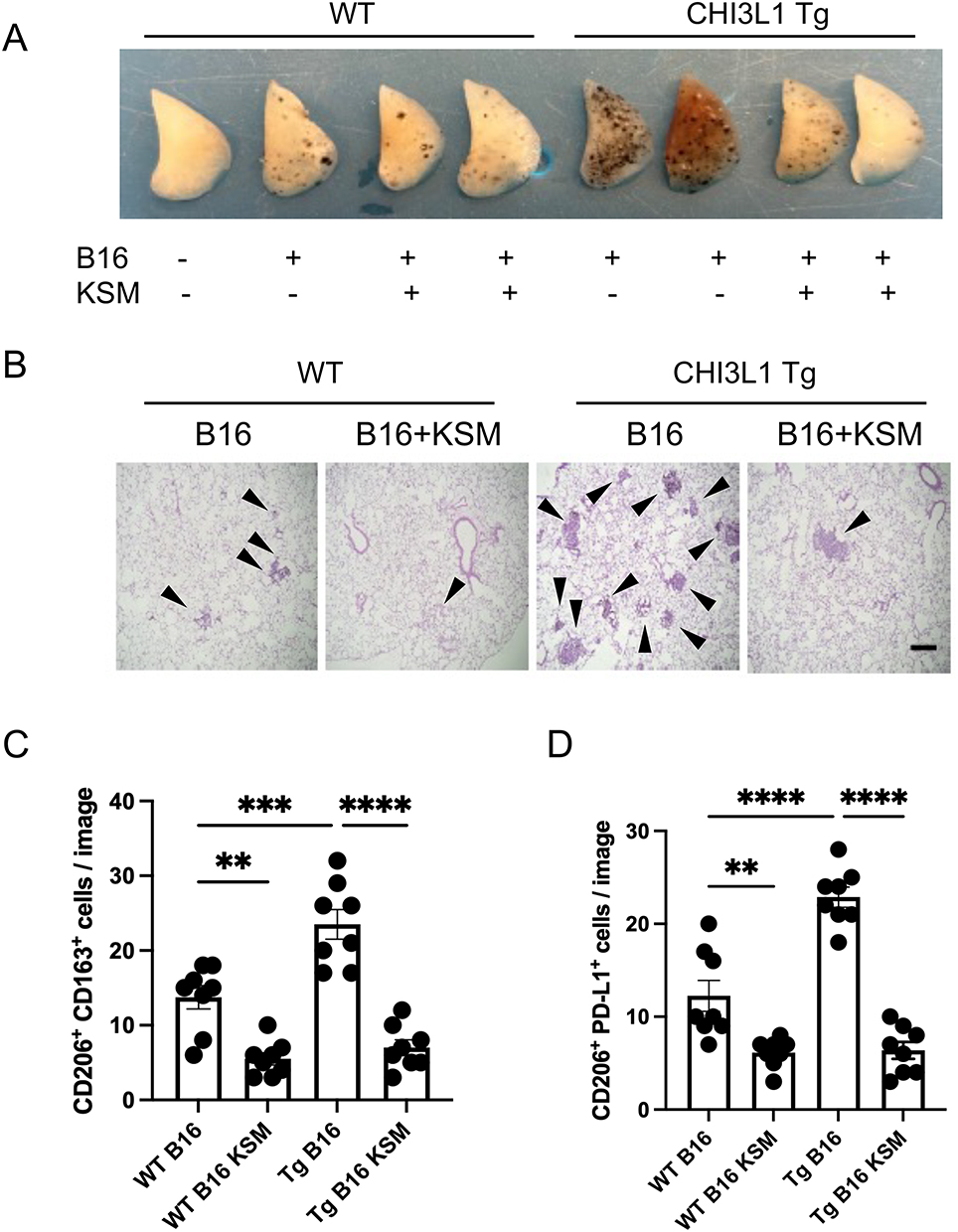 |
Figure 4 KSM inhibits CHI3L1-induced M2 macrophage accumulation in melanoma lung metastasis. (A) Representative photographs of WT and CHI3L1 transgenic lungs with and without KSM treatment after B16-F10 melanoma cell challenge. (B) Representative H&E histology of WT and Transgenic lungs with and without KSM treatment after B16-F10 melanoma cell challenge (x4 original magnification). Black arrowheads indicate metastatic tumors. (C and D) The number of CD206(+)/CD163(+) and CD206(+)/PD-L1(+) macrophages detected by immunohistochemical staining in lungs of WT and CHI3L1 Tg mice with and without KSM treatment. The number of macrophages co-expressing CD206 and CD163 or PD-L1 was counted using microscopic images under 20x magnification (n=8 each). The values in (C) are the mean ± SEM. **P<0.01 ***P<0.001 ****P<0.0001 (One-Way ANOVA, multiple comparisons). |
KSM Inhibits CHI3L1-Driven M2 Macrophage Differentiation in vitro
To further investigate CHI3L1’s capacity to promote M2 or M2-like TAM differentiation, we conducted in vitro studies using THP-1 human monocytic cells. After overnight stimulation with PMA, THP-1 cells were exposed to rCHI3L1/M-CSF for 48 hours in the presence and absence of tumor-conditioned media (TCM; supernatant of A549 cells) (Figure 5A). The expression of M2 or M2-like TAM activation markers, including CD206, CD163, CD274 (PD-L1), CX3CR1, and CD36, was assessed by real-time qRT-PCR. These experiments revealed that rCHI3L1 upregulated expression of typical markers of M2 or M2-like TAM differentiation with no significant effect on the expressions of NOS2 and HLA-DRA, markers of M1 macrophage differentiation (Figure 5B, C, and Figure S3). Importantly, KSM treatment significantly suppressed CHI3L1-stimulated M2 activation marker expression regardless of TCM treatment (Figure 5B and C). FACS analysis and immunoblot evaluation further confirmed that CHI3L1 drives CD206(+)/CD163(+) M2 macrophage differentiation, which is strongly inhibited by KSM treatment (Figure 5D).
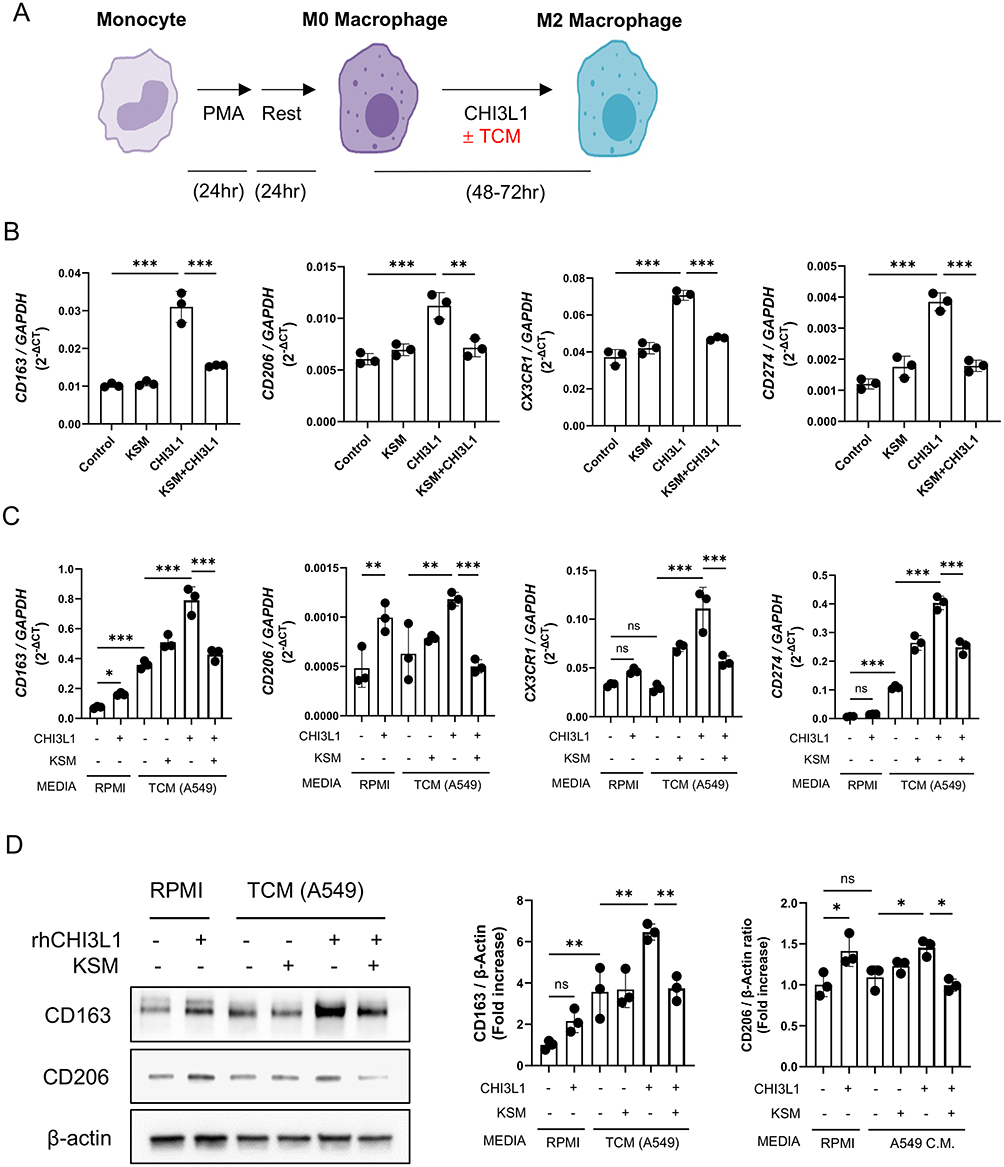 |
Figure 5 KSM inhibits CHI3L1-stimulated differentiation of monocytic macrophages to M2 or M2-like TAM macrophages. (A) Schematic illustration of macrophage differentiation using human THP-1 cells. (B) Realtime PT-PCR evaluations on the M2 macrophage activation markers in the macrophages with and without CHI3L1 (500 ng/mL) and KSM (250 ng/mL) treatment. (C) Real-time qRT-PCR evaluations on the M2 macrophage activation markers in the macrophages cultured in the presence and absence of tumor cell conditioned medium (TCM; A549 cell supernatant) together with CHI3L1 and KSM treatment. (D) Representatives of immunoblot evaluations on the expression of CD206/CD163 in differentiated macrophages stimulated in the presence and absence of tumor cell conditioned medium (TCM; A549 cell supernatant) together with CHI3L1 and or KSM treatment. Right panel, densitometric quantitation on the blots of CD163 and CD206. The values in (B–D) are the mean ± SEM. *P<0.05, **P<0.01, ***P<0.001 (One-Way ANOVA, multiple comparisons). Abbreviation: ns, not significant. |
Identification of KSM-Regulated Genes in CHI3L1-Driven M2 Macrophage
To elucidate the molecular mechanisms underlying KSM’s antitumor activity, we performed mRNA expression profiling using bulk RNA sequencing on differentiated macrophages in the presence of CHI3L1 alone or in combination with KSM. Differential gene expression analysis identified 519 upregulated and 494 downregulated genes (≥ 2-fold change) in rCHI3L1-treated macrophages compared to PBS controls. As illustrated by volcano plots (Figure 6A), CHI3L1 stimulated the expression of multiple genes including metalloproteinases (MMP-8, MMP-9), growth factor (EGFR), chemokine (CXCL13), and other signaling molecules, which are associated with pathways involved in anti-apoptosis, proliferation, adhesion, and immune modulation (Figure S4 and Table S1). On the other hand, KSM treatment significantly increased the expression of genes associated with protein translation and mitochondrial function, including ribosomal genes such as RPL41P1, RPL35, PVALB, and ROMO1 (Figure S5 and Table S2). Interestingly, several genes associated with protumor activities, such as EGFR, FP671120.4, ITGB8, TNC, GUCY1A2, GREM1, MACC1, and ABAC13, were upregulated by CHI3L1 but downregulated by KSM (Figure 6B and Table S3). Among these genes, EGFR expression was one of the most prominently regulated genes by CHI3L1 and KSM treatment. Conversely, KSM increased the expression of several genes that were downregulated by CHI3L1, including those with known antitumor activities, such as CCL23, NKD2, AZU1, and PLD4 (Table S4). In particular, we further confirmed increased mRNA and protein expression of EGFR in CHI3L1-stimulated differentiated M2 macrophages (Figure 6C and D). Consistent with these protein-level findings, qPCR analyses demonstrated that CHI3L1-induced changes in M2- and M1-associated gene expression at the mRNA level were also attenuated by EGFR inhibition (Figure S6A and B).
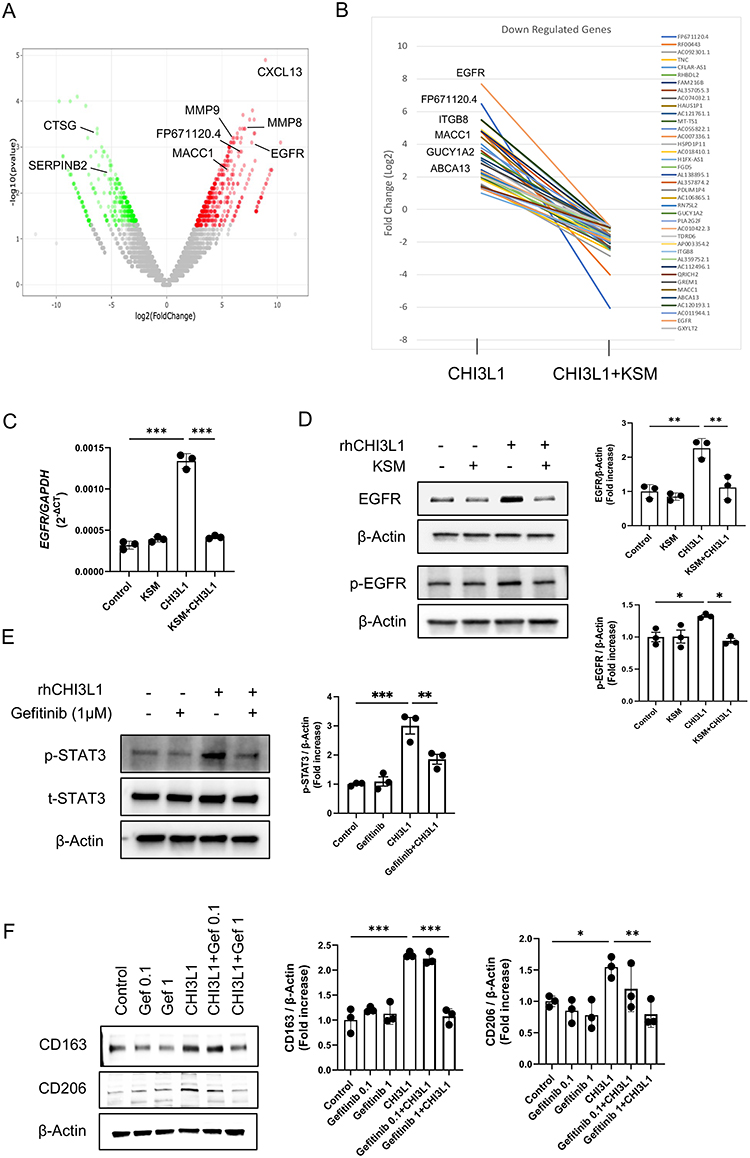 |
Figure 6 KSM inhibits CHI3L1-stimulated M2 macrophage activation via EGFR. Bulk RNA-seq sequencing analysis on the differentiated macrophages from THP-1 cells was used to evaluate mRNA expression in the macrophage differentiation regulated by CHI3L1 and KSM. (A). Volcano plots showing differentially expressed genes regulated by CHI3L1. (B) Representative plots of the top 20 genes that are upregulated (> 2-fold) by CHI3L1 stimulation but downregulated (< 2-fold) by KSM treatment. (C) Representative mRNA expression of EGFR in differentiated macrophages with CHI3L1 and KSM treatment was detected by real-time qRT-PCR. (D) Representative immunoblots showing p-EGFR (Tyr1068) and total EGFR expression in differentiated macrophages with CHI3L1 and KSM. Right panel, densitometric quantitation on the blots of EGFR and p-EGFR. (E) Effect of EGFR inhibition on the expression of p-STAT3 and total-STAT3 in CHI3L1-stimulated, differentiated macrophages by gefitinib (Tocris Bioscience, #3000) treatment (1 μM, 72 hours). Right panel, densitometric quantitation on the blots of p-STAT3 and total STAT3. (F) Effects of EGFR inhibition on the expression of CD163 and CD206 in CHI3L1-stimulated, differentiated macrophages by gefitinib treatment (0.1 and 1 μM, 72 hours). Right panel, densitometric quantitation on the blots of CD163 and CD206. The values in (C–F) are the mean ± SEM. *P<0.05, **P<0.01, ***P<0.001 (One-Way ANOVA, multiple comparisons). |
To further understand the mechanistic relationship between CHI3L1 and EGFR, we evaluated downstream signaling pathways in macrophages. These studies demonstrate that CHI3L1 induces EGFR phosphorylation at Tyr1068 (Figure 6D), and treatment with an EGFR inhibitor blocks CHI3L1-stimulated STAT3 activation (Figure 6E). Consistent with these signaling effects, treatment with the EGFR inhibitor gefitinib abrogated the CHI3L1-induced upregulation of the M2 macrophage markers CD163 and CD206 (Figure 6F), suggesting a functional role for EGFR-STAT3 signaling in CHI3L1-driven M2 macrophage differentiation. Similar inhibitory effects on CHI3L1-driven M2 marker expression were observed using a second, structurally distinct EGFR inhibitor, erlotinib (Figure S6C), further supporting the involvement of EGFR signaling in this process.
Discussion
The tumor microenvironment (TME), composed of diverse immune cells and mediators, plays a crucial role in tumor cell survival, metastasis, and patient prognosis.29–31 Among these, tumor-associated macrophages (TAMs) are the predominant innate immune cells in the TME and contribute significantly to immunosuppression, tumor cell evasion, cancer cell proliferation, survival, invasion, and metastasis.32,33 While exceptions exist, M2 macrophages or M2-like TAMs are generally regarded as pro-tumoral, whereas M1 macrophages or M1-like TAMs are associated with antitumor activity.34 Therefore, understanding the mechanisms driving pro-tumoral macrophage activation and identifying regulatory inhibitors is critical for developing interventions against tumor progression. Our findings demonstrated that CHI3L1 drives M2 or M2-like TAM differentiation both in vivo and in vitro, aligning with its well-established pro-tumoral activities. Moreover, we revealed that Kasugamycin (KSM), an aminoglycoside antibiotic, is an inhibitor of CHI3L1 signaling, bioactivity, and CHI3L1-induced M2 macrophage differentiation. Notably, KSM treatment significantly reduced melanoma lung metastases driven by CHI3L1 overexpression. Together, these results suggest a novel anti-tumor activity of KSM, which likely acts through the inhibition of CHI3L1-stimulated M2-like TAM differentiation. Our studies further identified that the CHI3L1-EGFR axis could be a mechanism underlying CHI3L1-induced M2 or M2-like TAM differentiation, as well as a target of the inhibitory effect of KSM.
Recent work from our laboratory has shown that CHI3L1 is a powerful immune modulator that increases tumor immune tolerance by regulating immune checkpoint molecules in lung macrophages.10,11 CHI3L1 also promotes alternative macrophage activation, contributing to conditions like pulmonary fibrosis and airway remodeling in asthma.8,35 However, its direct role in TAM differentiation and its contribution to tumor metastasis and progression had not been well studied. Our current results establish CHI3L1 as a key driver of pro-tumoral M2 macrophage differentiation in vitro and in a melanoma lung metastasis model in vivo. The role of CHI3L1 in pro-tumoral M2 macrophage activation has also been recognized in other malignancies, including gastric, breast, and lung cancers.36–38 For example, Cohen et al demonstrated that cancer-associated fibroblasts expressing high levels of CHI3L1 enhanced M2 macrophage recruitment and activation, thereby driving breast cancer progression.38 These findings emphasize that CHI3L1-induced pro-tumoral M2 macrophage differentiation represents a promising therapeutic target for disrupting tumor progression.21,39
Despite its translational potential, the development of CHI3L1-targeted therapies for lung and other cancers remains limited. Existing strategies, such as neutralizing anti-CHI3L1 antibodies or gene-specific siRNA approaches, have shown efficacy in preclinical studies.6,11 While natural compounds like resveratrol have demonstrated anti-CHI3L1 activity,40 their precise molecular mechanisms remain unclear. Targeting putative CHI3L1 receptors or coreceptors, such as IL-13Rα2, TMEM219, CD44, or galectin-3, has also been proposed to inhibit CHI3L1-mediated signaling.26,41–43 However, these receptors interact with multiple ligands essential for other biological processes, limiting their specificity and clinical use.
Previously, we identified KSM as a chitinase inhibitor through high-throughput screening of chemical and drug libraries.24 The broad-spectrum anti-chitinase activity of KSM has since been validated against various human and microbial chitinases.25 Although CHI3L1 lacks enzymatic activity, its unique chitin-binding domain (CBD), shared with true chitinases, led us to hypothesize that KSM could similarly modulate CHI3L1 bioactivity. It has been shown that chitin-binding domain (CBD) determines the biological function of CHIT1.44 Evidence of direct physical interactions between KSM and CHIT1 supports this notion, as does the structural similarity of the CBD in CHI3L1 and CHIT1. Recent studies further highlight the critical role of CBD in the biological activity of CHI3L1, particularly in lung and colon cancers.17,39,45 However, future studies are needed to elucidate the precise KSM binding site on CHI3L1 and how this interaction inhibits its signaling and biological activities.
KSM is a naturally occurring aminoglycoside antibiotic isolated from Streptomyces kasugaensis.46 Unlike other aminoglycosides, KSM has a unique carbohydrate structure comprising two sugars, a D-chiro-inositol moiety and a kasugamine moiety (2,4-diamino-2,3,4,6-tetradeoxy-D-arabino-hexose) with a carboxy-imino-methyl group.47 These distinctive features raise the possibility of a direct interaction between KSM and the carbohydrate-binding domains of CHI3L1. While originally developed as an antibiotic effective against a broad range of microorganisms, KSM is currently used as a fungicide in organic agriculture due to its low toxicity in plants, humans, fish, and animals.48,49 Its past use in treating respiratory and urinary tract infections50,51 highlights its potential clinical utility. Developing KSM as a repurposed anticancer drug offers significant advantages, including drug accessibility for patients with lung cancer. However, as an aminoglycoside, KSM’s impact on the microbiome warrants careful consideration. Future efforts should focus on developing KSM derivatives with minimized antibiotic activity while retaining their anti-chitinase and antitumor properties.
Transcriptomic analysis of CHI3L1-stimulated genes in M2 macrophage differentiation provided further insight into the molecular basis of KSM’s anti-tumoral activity. CHI3L1 upregulated several pro-tumoral genes while downregulating genes with antitumor activity. Among these, EGFR was particularly noteworthy, as its expression was significantly stimulated by CHI3L1 and suppressed by KSM. EGFR is a well-established driver of tumor progression in lung and other cancers,52–54 and its signaling in macrophage activation has been implicated in infection and tumor development.52,55 Recently, our lab reported a feed-forward relationship between CHI3L1 and EGFR expression in normal or EGFR mutant lung epithelial cells.56 The current studies further provide a mechanistic understanding of CHI3L1-EGFR interaction by demonstrating that the EGFR inhibitor treatment blocks the well-characterized CHI3L1 signaling of STAT3 activation, one of the downstream signaling molecules of the EGFR pathway associated with M2 macrophage polarization.57–60 Collectively, these studies establish a novel signaling cascade underlying the interaction of CHI3L1 and EGFR: CHI3L-EGFR-STAT3, in addition to the already established feed-forward relationship in protumor activity of CHI3L1.56
Interestingly, KSM has also demonstrated antiviral immune activity by enhancing type I interferon responses,16 suggesting that it may indirectly augment macrophage-mediated antitumor immunity. Taken together, these studies suggest that the antitumor effect of KSM was potentially mediated through interruption of the CHI3L1-EGFR axis that plays an essential role in macrophage polarization and or differentiation. The effects of KSM on macrophage functions, such as phagocytosis and interactions with cytotoxic T cells, remain to be determined. As a limitation of this study, it should be noted that the effects of KSM may not be entirely specific to CHI3L1, as KSM has additional biological targets, including interferon-stimulated genes (ISGs),16 which exert strong immunomodulatory effects in antiviral responses and the tumor microenvironment.
In summary, our findings demonstrate that KSM inhibits CHI3L1-mediated signaling and oncogenic M2 macrophage differentiation, highlighting its potential to counteract CHI3L1’s protumor activities. These results suggest that re-purposing of KSM as a promising small molecule for development as a first-in-class chitinase-inhibitor-based anticancer agent that can be used alone or in combination with already developed EGFR inhibitors, particularly for cancers where CHI3L1 plays a critical role.
Data Sharing Statement
The data generated in this study and the supplementary materials are contained within the article.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Institute of Health (NIH) grants PO1 HL114501 (JAE), R01HL155558 (CGL), T32 HL134625 (TS), and Department of Defense grant W81XWH-22-1-0101 (CGL). This work was partially supported by the National Research Foundation (NRF) grant funded by the Korean government (MSIT) (#RS-2024-00405542) (SJS and CGL).
Disclosure
JAE is a cofounder of Sakonnet Biomedical, which develops inhibitors of 18 glycosyl hydrolases as therapeutics. JAE, CGL, and SK have composition-of-matter and use patents relating to antibodies against CHI3L1. CGL serves as a consultant for siRNAgen Inc., which develops RNA therapeutics. The other authors have declared that there is no conflict of interest in this work.
This manuscript has been made available as a preprint on bioRxiv (https://doi.org/10.1101/2025.05.26.654629).
References
1. Society AC. Key statistics for lung cancer; 2023. Available from: https://www.cancer.org/cancer/types/lung-cancer/about/key-statistics.html.
2. Organization WH. Lung cancer: fact sheet; 2023. Available from: https://www.who.int/news-room/fact-sheets/detail/lung-cancer.
3. Putzu C, Canova S, Paliogiannis P, et al. Duration of immunotherapy in non-small cell lung cancer survivors: a lifelong commitment? Cancers. 2023;15(3):689. doi:10.3390/cancers15030689
4. Wang J, Sheng Z, Yang W, Cai Y. Elevated serum concentration of chitinase 3-like 1 is an independent prognostic biomarker for poor survival in lung cancer patients. Cell Physiol Biochem. 2016;38(2):461–16. doi:10.1159/000438643
5. Zhao T, Su Z, Li Y, Zhang X, You Q. Chitinase-3 like-protein-1 function and its role in diseases. Signal Transduct Targe Ther. 2020;5(1):201.
6. Kim D-H, Park H-J, Lim S, et al. Regulation of chitinase-3-like-1 in T cell elicits Th1 and cytotoxic responses to inhibit lung metastasis. Nat Commun. 2018;9(1):503. doi:10.1038/s41467-017-02731-6
7. Ma B, Herzog EL, Moore M, et al. RIG-like helicase regulation of chitinase 3-like 1 axis and pulmonary metastasis. Sci Rep. 2016;6:26299. doi:10.1038/srep26299
8. Lee CG, Hartl D, Lee GR, et al. Role of breast regression protein 39 (BRP-39)/chitinase 3-like-1 in Th2 and IL-13-induced tissue responses and apoptosis. J Exp Med. 2009;206(5):1149–1166. doi:10.1084/jem.20081271
9. Ma B, Herzog EL, Lee CG, et al. Role of chitinase 3-like-1 and semaphorin 7a in pulmonary melanoma metastasis. Cancer Res. 2015;75(3):487–496. doi:10.1158/0008-5472.CAN-13-3339
10. Ma B, Kamle S, Akosman B, et al. CHI3L1 enhances melanoma lung metastasis via regulation of T cell co-stimulators and CTLA-4/B7 axis. Front Immunol. 2022;13:1056397. doi:10.3389/fimmu.2022.1056397
11. Ma B, Akosman B, Kamle S, et al. CHI3L1 regulates PD-L1 and anti–CHI3L1–PD-1 antibody elicits synergistic antitumor responses. J Clin Invest. 2021;131(21). doi:10.1172/JCI137750
12. Xu N, Bo Q, Shao R, et al. Chitinase-3-like-1 promotes M2 macrophage differentiation and induces choroidal neovascularization in neovascular age-related macular degeneration. Invest Ophthalmol Vis Sci. 2019;60(14):4596–4605. doi:10.1167/iovs.19-27493
13. USEPA. Human health risk assessment for proposed food uses of the fungicide kasugamycin on importing fruiting vegetables. Health Effects Division; 2005. Available from: https://www.federalregister.gov/documents/2014/08/29/2014-20502/kasugamycin-pesticide-tolerances.
14. Lee JH, Lee CM, Lee JH, et al. Kasugamycin is a novel chitinase 1 inhibitor with strong antifibrotic effects on pulmonary fibrosis. Am J Respir Cell Mol Biol. 2022;67(3):309–319. doi:10.1165/rcmb.2021-0156OC
15. Lee CG, Herzog EL, Ahangari F, et al. Chitinase 1 is a biomarker for and therapeutic target in scleroderma-associated interstitial lung disease that augments TGF-β1 signaling. J Immunol. 2012;189(5):2635–2644. doi:10.4049/jimmunol.1201115
16. Gopinath S, Kim MV, Rakib T, et al. Topical application of aminoglycoside antibiotics enhances host resistance to viral infections in a microbiota-independent manner. Nat Microbiol. 2018;3(5):611–621. doi:10.1038/s41564-018-0138-2
17. Park KR, Yun HM, Yoo K, Ham YW, Han SB, Hong JT. Chitinase 3 like 1 suppresses the stability and activity of p53 to promote lung tumorigenesis. Cell Commun Signal. 2020;18(1):5. doi:10.1186/s12964-019-0503-7
18. Chaudhuri S, Li L, Zimmerman M, et al. Kasugamycin potentiates rifampicin and limits emergence of resistance in Mycobacterium tuberculosis by specifically decreasing mycobacterial mistranslation. Elife. 2018;7. doi:10.7554/eLife.36782
19. Benner B, Scarberry L, Suarez-Kelly LP, et al. Generation of monocyte-derived tumor-associated macrophages using tumor-conditioned media provides a novel method to study tumor-associated macrophages in vitro. J Immunother Cancer. 2019;7(1):140. doi:10.1186/s40425-019-0622-0
20. Sohn MH, Kang MJ, Matsuura H, et al. The chitinase-like proteins breast regression protein-39 and YKL-40 regulate hyperoxia-induced acute lung injury. Am J Respir Crit Care Med. 2010;182(7):918–928. doi:10.1164/rccm.200912-1793OC
21. Zhu W, Chen Q, Li Y, Wan J, Li J, Tang S. HIF-1α-overexpressing mesenchymal stem cells attenuate colitis by regulating M1-like macrophages polarization toward M2-like macrophages. Biomedicines. 2023;11(3):825. doi:10.3390/biomedicines11030825
22. Wen D, Xiao H, Gao Y, Zeng H, Deng J. N6-methyladenosine-modified SENP1, identified by IGF2BP3, is a novel molecular marker in acute myeloid leukemia and aggravates progression by activating AKT signal via de-SUMOylating HDAC2. Mol Cancer. 2024;23(1):116. doi:10.1186/s12943-024-02013-y
23. Lee CG, Cho SJ, Kang MJ, et al. Early growth response gene 1-mediated apoptosis is essential for transforming growth factor beta1-induced pulmonary fibrosis. J Exp Med. 2004;200(3):377–389. doi:10.1084/jem.20040104
24. Lee J-H, Lee C-M, Kim M-O, et al. Kasugamycin is a novel chitinase 1 inhibitor with strong antifibrotic effects on pulmonary fibrosis. bioRxiv. 2021;
25. Qi H, Jiang X, Ding Y, Liu T, Yang Q. Discovery of kasugamycin as a potent inhibitor of glycoside hydrolase family 18 chitinases. Front Mol Biosci. 2021;8:640356. doi:10.3389/fmolb.2021.640356
26. He CH, Lee CG, Dela Cruz CS, et al. Chitinase 3-like 1 regulates cellular and tissue responses via IL-13 receptor α2. Cell Rep. 2013;4(4):830–841. doi:10.1016/j.celrep.2013.07.032
27. Li W, Wu F, Zhao S, Shi P, Wang S, Cui D. Correlation between PD-1/PD-L1 expression and polarization in tumor-associated macrophages: a key player in tumor immunotherapy. Cytokine Growth Factor Rev. 2022;67:49–57. doi:10.1016/j.cytogfr.2022.07.004
28. Wang L, Guo W, Guo Z, et al. PD-L1-expressing tumor-associated macrophages are immunostimulatory and associate with good clinical outcome in human breast cancer. Cell Rep Med. 2024;5(2):101420. doi:10.1016/j.xcrm.2024.101420
29. Ben-Baruch A. Inflammation-associated immune suppression in cancer: the roles played by cytokines, chemokines and additional mediators. Semin Cancer Biol. 2006;16(1):38–52. doi:10.1016/j.semcancer.2005.07.006
30. de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer. 2006;6(1):24–37. doi:10.1038/nrc1782
31. Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res. 2014;2014:149185. doi:10.1155/2014/149185
32. Sedighzadeh SS, Khoshbin AP, Razi S, Keshavarz-Fathi M, Rezaei N. A narrative review of tumor-associated macrophages in lung cancer: regulation of macrophage polarization and therapeutic implications. Transl Lung Cancer Res. 2021;10(4):1889–1916. doi:10.21037/tlcr-20-1241
33. Mei J, Xiao Z, Guo C, et al. Prognostic impact of tumor-associated macrophage infiltration in non-small cell lung cancer: a systemic review and meta-analysis. Oncotarget. 2016;7(23):34217–34228. doi:10.18632/oncotarget.9079
34. Jackute J, Zemaitis M, Pranys D, et al. Distribution of M1 and M2 macrophages in tumor islets and stroma in relation to prognosis of non-small cell lung cancer. BMC Immunol. 2018;19(1):3. doi:10.1186/s12865-018-0241-4
35. Zhou Y, Peng H, Sun H, et al. Chitinase 3-like 1 suppresses injury and promotes fibroproliferative responses in Mammalian lung fibrosis. Sci Transl Med. 2014;6(240):240ra76. doi:10.1126/scitranslmed.3007096
36. Huang J, Gu Z, Xu Y, Jiang L, Zhu W, Wang W. CHI3L1 (Chitinase 3 Like 1) upregulation is associated with macrophage signatures in esophageal cancer. Bioengineered. 2021;12(1):7882–7892. doi:10.1080/21655979.2021.1974654
37. Chen Y, Zhang S, Wang Q, Zhang X. Tumor-recruited M2 macrophages promote gastric and breast cancer metastasis via M2 macrophage-secreted CHI3L1 protein. J Hematol Oncol. 2017;10(1):36. doi:10.1186/s13045-017-0408-0
38. Cohen N, Shani O, Raz Y, et al. Fibroblasts drive an immunosuppressive and growth-promoting microenvironment in breast cancer via secretion of Chitinase 3-like 1. Oncogene. 2017;36(31):4457–4468. doi:10.1038/onc.2017.65
39. Chen A, Jiang Y, Li Z, et al. Chitinase-3-like 1 protein complexes modulate macrophage-mediated immune suppression in glioblastoma. J Clin Invest. 2021;131(16). doi:10.1172/JCI147552
40. Zhang W, Murao K, Zhang X, et al. Resveratrol represses YKL-40 expression in human glioma U87 cells. BMC Cancer. 2010;10:593. doi:10.1186/1471-2407-10-593
41. Lee CM, He CH, Nour AM, et al. IL-13Ralpha2 uses TMEM219 in chitinase 3-like-1-induced signalling and effector responses. Nat Commun. 2016;7:12752. doi:10.1038/ncomms12752
42. Zhou Y, He CH, Yang DS, et al. Galectin-3 interacts with the CHI3L1 axis and contributes to Hermansky-Pudlak syndrome lung disease. J Immunol. 2018;200(6):2140–2153. doi:10.4049/jimmunol.1701442
43. Geng B, Pan J, Zhao T, et al. Chitinase 3-like 1-CD44 interaction promotes metastasis and epithelial-to-mesenchymal transition through β-catenin/Erk/Akt signaling in gastric cancer. J Exp Clin Cancer Res. 2018;37(1):208. doi:10.1186/s13046-018-0876-2
44. Crasson O, Courtade G, Léonard RR, et al. Human chitotriosidase: catalytic domain or carbohydrate binding module, who’s leading HCHT’s biological function. Sci Rep. 2017;7(1):2768. doi:10.1038/s41598-017-02382-z
45. Chen CC, Llado V, Eurich K, Tran HT, Mizoguchi E. Carbohydrate-binding motif in chitinase 3-like 1 (CHI3L1/YKL-40) specifically activates Akt signaling pathway in colonic epithelial cells. Clin Immunol. 2011;140(3):268–275. doi:10.1016/j.clim.2011.04.007
46. Umezawa H, Hamada M, Suhara Y, Hashimoto T, Ikekawa T. Kasugamycin, a new antibiotic. Antimicrob Agents Chemother. 1965;5:753–757.
47. Flatt PM, Mahmud T. Biosynthesis of aminocyclitol-aminoglycoside antibiotics and related compounds. Nat Prod Rep. 2007;24(2):358–392. doi:10.1039/B603816F
48. Hamada M, Hashimoto T, Takahashi T, et al. Antimicrobial activity of kasugamycin. J Antibiot. 1965;18:104–106.
49. Takeuchi T, Ishizuka M, Takayama H, Kureha K, Hamada M, Umezawa H. Pharmacology of kasugamycin and the effect on Pseudomonas infection. J Antibiot. 1965;18:107–110.
50. Hiro Y, Okuda M, Sakamoto H, Tanaka M, Koga H. Effect of kasugamycin (KSM) on respiratory tract infection due to Pseudomonas aeruginosa. Iryo. 1967;21(8):906–911.
51. Tada S, Hakamada T, Mori O, Yamazaki Y. Use of kasugamycin in urinary tract infection, especially against Pseudomonas aeruginosa. Ann Urol. 1968;14(1):50–56.
52. Zhang W, Chen L, Ma K, et al. Polarization of macrophages in the tumor microenvironment is influenced by EGFR signaling within colon cancer cells. Oncotarget. 2016;7(46):75366–75378. doi:10.18632/oncotarget.12207
53. Wang Z, Li Z, Wu C, et al. MACC1 overexpression predicts a poor prognosis for non-small cell lung cancer. Med Oncol. 2014;31(1):790. doi:10.1007/s12032-013-0790-6
54. O’Leary C, Gasper H, Sahin KB, et al. Epidermal Growth Factor Receptor (EGFR)-mutated non-small-cell lung cancer (NSCLC). Pharmaceuticals. 2020;13(10):273. doi:10.3390/ph13100273
55. Hardbower DM, Singh K, Asim M, et al. EGFR regulates macrophage activation and function in bacterial infection. J Clin Invest. 2016;126(9):3296–3312. doi:10.1172/JCI83585
56. Kamle S, Ma B, Schor G, et al. Chitinase 3-like-1 (CHI3L1) in the pathogenesis of epidermal growth factor receptor mutant non-small cell lung cancer. Transl Oncol. 2024;49:102108. doi:10.1016/j.tranon.2024.102108
57. Xia T, Zhang M, Lei W, et al. Advances in the role of STAT3 in macrophage polarization. Front Immunol. 2023;14:1160719. doi:10.3389/fimmu.2023.1160719
58. Xu M, Li X, Song L. Baicalin regulates macrophages polarization and alleviates myocardial ischaemia/reperfusion injury via inhibiting JAK/STAT pathway. Pharm Biol. 2020;58(1):655–663. doi:10.1080/13880209.2020.1779318
59. Li X, Jiang M, Chen X, Sun W. Etanercept alleviates psoriasis by reducing the Th17/Treg ratio and promoting M2 polarization of macrophages. Immun Inflamm Dis. 2022;10(12):e734. doi:10.1002/iid3.734
60. He W, Zhu Y, Mu R, et al. A Jak2-selective inhibitor potently reverses the immune suppression by modulating the tumor microenvironment for cancer immunotherapy. Biochem Pharmacol. 2017;145:132–146. doi:10.1016/j.bcp.2017.08.019
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.