")
Back to Journals » Drug Design, Development and Therapy » Volume 8
Ivabradine, coronary artery disease, and heart failure: beyond rhythm control
Authors Scicchitano P , Cortese F, Ricci G, Carbonara S, Moncelli M, Iacoviello M, Cecere A, Gesualdo M, Zito A, Caldarola P, Scrutinio D, Lagioia R, Riccioni G, Ciccone M
Received 13 January 2014
Accepted for publication 27 March 2014
Published 3 June 2014 Volume 2014:8 Pages 689—700
DOI https://doi.org/10.2147/DDDT.S60591
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Pietro Scicchitano,1 Francesca Cortese,1 Gabriella Ricci,1 Santa Carbonara,1 Michele Moncelli,1 Massimo Iacoviello,1 Annagrazia Cecere,1 Michele Gesualdo,1 Annapaola Zito,1 Pasquale Caldarola,2 Domenico Scrutinio,3 Rocco Lagioia,3 Graziano Riccioni,4 Marco Matteo Ciccone1
1Section of Cardiovascular Diseases, Department of Emergency and Organ Transplantation, University of Bari, School of Medicine, Policlinico, Bari, Italy; 2Section of Cardiovascular Diseases, Policlinic, San Paolo Hospital, Bari, Italy; 3Section of Cardiovascular Diseases, Fondazione Maugeri, Cassano Murge, Italy; 4Intensive Cardiology Care Unit, San Camillo de Lellis Hospital, Manfredonia, Foggia, Italy
Abstract: Elevated heart rate could negatively influence cardiovascular risk in the general population. It can induce and promote the atherosclerotic process by means of several mechanisms involving endothelial shear stress and biochemical activities. Furthermore, elevated heart rate can directly increase heart ischemic conditions because of its skill in unbalancing demand/supply of oxygen and decreasing the diastolic period. Thus, many pharmacological treatments have been proposed in order to reduce heart rate and ameliorate the cardiovascular risk profile of individuals, especially those suffering from coronary artery diseases (CAD) and chronic heart failure (CHF). Ivabradine is the first pure heart rate reductive drug approved and currently used in humans, created in order to selectively reduce sinus node function and to overcome the many side effects of similar pharmacological tools (ie, β-blockers or calcium channel antagonists). The aim of our review is to evaluate the role and the safety of this molecule on CAD and CHF therapeutic strategies.
Keywords: chronic heart failure, heart rate reduction, cardiac ischemic disease, heart-rate lowering drugs, funny current
Introduction
Elevated heart rate is a physiopathological condition that can exert a negative influence on the cardiovascular risk burden of the general population.1 Far from being an adaptation to many clinical conditions (anemia, sepsis, heart valve alterations, etc), heart rate elevation is even able to increase the therosclerotic process by means of several mechanisms involving endothelial shear stress and biochemical stress-related enzymatic cascades.1,2 Other mechanisms are involved in the genesis of cardiac impairment during elevated heart rate. In particular, the imbalance between oxygen supply and demand is the first, immediate cause of ischemic manifestations during elevated heart rate. Heart rate is one of the three determinants of oxygen cardiac demand, thus its increase will certainly increase oxygen demand, leading to ischemic consequences in the case of epicardial coronary stenosis.
Apart from the several regulatory systems involved in coronary blood flow controls, it is already known that the major coronary blood flow supply from epicardial coronary arteries happens during diastole: this phase of the cardiac cycle is characterized by a reduced compression of the coronary vessels by the surrounding muscular cardiac fibers as compared to the systolic period. Furthermore, the difference between the mean diastolic pressure in the aorta and the ventricle cavity pressure is responsible for the filling of the coronary vessels during the cardiac cycle, as well as the duration of diastole. Thus, the larger the increase in systolic time per minute due to high heart rate, the shorter the duration of the diastole: the consequence is a reduction in coronary artery blood supply. This situation can induce ischemia in patients suffering from coronary artery disease (CAD).
The scientific community has identified several drugs able to reduce heart rate: the main purpose was to reduce the left ventricular wall stress due to increased oxygen and to increase systolic time duration related to an augmented heart rate. Colin et al3 demonstrated that a reduction in heart rate improved cardiac oxygen consumption by reducing left ventricular wall stress and by increasing diastolic time. In particular, they analyzed eight dogs at rest and during a treadmill test. They were investigated during exercise at spontaneous and paced heart rate (250 bpm) after administration of saline, atenolol, or ivabradine. The results demonstrated the positive effects of both reducing heart rate drugs (atenolol or ivabradine) on oxygen cardiac demand and wall stress.
This is the pathophysiological rationale for adopting rate-control agents, such as β-adrenoceptor antagonists and calcium channel blockers in the management of some cardiovascular diseases. Nevertheless, these agents always showed side effects that sometimes could be considered unacceptable from the patients and in relation to their supposed clinical purpose.
Ivabradine seems to overcome the limitations of the previous drugs. It is an already extensively studied pure heart-rate reducing agent.4–8 Ivabradine acts on If current, ie, the inward Na+/K+ current, activated upon hyperpolarization and directly modulated by cyclic adenosine monophosphate (cAMP), and is responsible for sinus rhythm generation; thus, it is considered as the “pacemaker current”.1,7 Ivabradine decreases diastolic depolarization and inhibits If in a use-dependent manner. Reduction in heart rate both decreases myocardial oxygen consumption and increases its supply due to prolongation of diastolic perfusion time. These actions led physicians to adopt ivabradine in CAD treatment and in heart failure (HF) management.
Background: chemistry and pharmacodynamics of ivabradine
Ivabradine is a compound whose chemical structure is similar to that of verapamil. It is a 7,8-dimethoxy 3-{3-{[(1S)-(4,5-dimethoxybenzocyclobutan-1-yl)methyl]methylamino}propyl}1,3,4,5-tetrahydro-2H-3-benzazepin-2-one.9,10 The inhibition of the pace-maker current (If) mediated by such a molecule is able to inhibit the diastolic depolarization of the atrial myocardial cells in a use- and rate-dependent manner.2
The structure of ivabradine is similar to one of the first agents in this category of pharmacological compounds: zatebradine. Nevertheless, ivabradine is more specific and more reliable than zatebradine in selectively reducing heart rate.11 Zatebradine is able to exert pleiotropic effects on the currents involved in the generation of the action potential of the cardiac pacemaker cells: it could influence the potassium delayed-rectifier current (IK) pacemaker cells widespread in the heart, such as those present within Purkinje fibers. Thus, the effects of such a molecule can be observed in multiple sites in the heart.11,12 This effect is negative due to a prolongation in the duration of phase 2 and 3 of the action potential which could be the source for the further development of ventricular malignant arrhythmias. The influence on IK current was not demonstrated for ivabradine.11,12 Ivabradine, as well as zatebradine, are not able to influence the other currents involved in the genesis of action potentials in the sinus node cells: the low-threshold transient type calcium current (ICaT) and the high-threshold long-lasting type calcium current (ICaL).9,11,12
Thus, the specific action of ivabradine is the inhibition of the If current, which is a “mixed” conductance of Na+ and K+ ions. Its peculiarity is that this current starts to be effective when the cell membrane potential is around −40/−50 mV, while the conductance of the channels involved in the current generation stops at more positive voltages, ie, during the depolarization period.2
Ivabradine acts on a peculiar channel: the hyperpolarization activated-cyclic nucleotide (HCN)-gated channels which are tetramers, similar to voltage-gated potassium channels modulated by cyclic nucleotides. Among the four HCN known isoforms, HCN4 is the most represented on the membrane of the cardiac atrial sinus node cells and it is modulated by cAMP, whose binding to HCN channels induces a conformational change of the protein able to increase the probability that the channel is open during hyperpolarization.2
The action of ivabradine is a characteristic one: it enters the HCN channel when it is open (state-dependence property); on entry, it is driven to its binding domain within the HCN channel by electrostatic forces generated by the depolarization process: the outward ion current pulls ivabradine to the site of interaction, while the repolarization generates an inward current able to make ivabradine dissociate from the binding site. This characteristic of the drug is named current-dependence property and belongs only to ivabradine.13 Furthermore, it is known that the channel opens during repolarization and it is closed during depolarization: this means that the interaction between the drug and the binding site is dependent on the rate of closing/opening of the channels, ie, it is related to heart rate (use-dependency property).2,13
Selectivity and safety
Ivabradine is a well-tolerated drug. Clinical studies showed that <1% of patients withdraw because of side effects.8 This is due to the characteristic pharmacodynamics of this novel drug. It inhibits HCN-gated channels, ie, the main channels able to mediate If current. Its action is a typical use and rate-dependence type. This means that the drug was created in order to increase the modulation of the channel at higher heart rates:2,13 when heart rate is less than 50–60 bpm, the action of ivabradine on HCN channels is very poor. For this reason, patients do not experience extreme bradycardia as in those who are administered β-blockers.
Some studies pointed out different percentages of bradycardia events in case of ivabradine administration.14,15 When compared to β-blockers, ivabradine demonstrated reduced incidence of sinus bradycardia (considered as heart rate lower than 40–50 bpm), while the drug reveals its non-inferiority to β-blockers according to hypotension and/or dizziness incidence and no incidences of syncope.15 Nevertheless, the data about such a specific side effect of the drug are very poor. For this reason, the recommendation is to reduce the dose of ivabradine in the case of heart rate <50 bpm or when bradycardia symptoms occur.
In contrast to β-blockers and calcium channel antagonists, ivabradine selectively reduces heart rate without affecting myocardial contractility or the conduction system.16 This characteristic is important due several conditions that could be negatively affected when administering other drugs other than ivabradine: for example, nondihydropyridine calcium channel antagonists can worsen both acute and CHF conditions or can slow atrioventricular nodal conduction, leading to severe side effects.
On the contrary, there is no significant prolongation of the PR, QRS, or corrected QT intervals while using ivabradine.17,18 Interesting data have emerged from studies evaluating the influence of ivabradine administration on ventricular repolarization, and thus on QT interval.14,19,20 The main action of ivabradine is to reduce the heart rate. The reduction in heart rate is usually associated with a prolongation in QT interval: ivabradine is effectively able to prolong the mean uncorrected QT interval for up to 37.5 milliseconds14 or to 18–30 milliseconds.19 A prolongation to values higher than 500 milliseconds or 60 milliseconds higher than baseline was found in 0.2%–1.6% of cases.19 Nevertheless, when corrected for heart rate, independently of the kind of formula adopted, there was a normalization of the QT intervals.19,20 This prompts us to think about the safety of the drug on the duration of the ventricular repolarization. Despite such data and results, there is still little evidence on the absolute absence of the pro-arrhythmic risk of ivabradine in terms of torsade de pointes incidence. For this reason, ivabradine is not recommended in patients suffering from congenital QT prolongation or in those using drugs able to prolong QT interval.
Visual disturbances were detected as a main side effect of the drug and are explained by the possible action of ivabradine on retinal channels mediating the Ih current; these channels show a structure similar to that of cardiac If channels. The blockage of the Ih current can result in a transient, enhanced brightness in a limited visual field area, an effect called phosphenes.8,21 These disturbances were detected in 3% of the patients enrolled in the Systolic Heart Failure Treatment with the I(f) Inhibitor Ivabradine Trial (SHIFT).22 In particular, the authors found an increased percentage of visual disturbances in patients treated with ivabradine as compared to those that underwent placebo administration (phosphenes: 3% in ivabradine group versus (vs) 1% in placebo group, P<0.0001; blurred vision: 1% in ivabradine group vs <1% in placebo group, P=0.042).22 Nevertheless, these events did not force patients to withdraw from taking the drug (phosphines inducing drug withdrawal: <1% in ivabradine group vs <1% in placebo group, P=0.224; blurred vision inducing drug withdrawal: <1% in ivabradine group vs <1% in placebo group, P=1.000).22
Additional beneficial pharmacological properties
Ivabradine can reduce the evolution of the atherosclerotic process. Animal models involving apolipoprotein E knockout mice fed a cholesterol-rich diet showed that ivabradine demonstrated such a protective action.23 This reduction may go beyond heart rate reduction (HRR) as demonstrated by Custodis et al23 in their animal model: ivabradine administration reduced oxidative stress by reducing nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity, superoxide production, and lipid peroxidation, although the mechanisms are still not well understood. These data were confirmed in several human studies.18,24–34
Ivabradine is effectively able to negatively influence the evolution of the atherosclerotic process in human coronary arteries. An increased heart rate is associated with increased progression of coronary atherosclerosis, as demonstrated in animal models and human research.35 This is due to the combination of pulsatile flow and parallel increased curvature of the these arteries. As Prosi et al36 demonstrated in their “bifurcation model”, coronary arteries undergo geometrical three-dimensional changes during the cardiac cycle: the coronary vessels show their higher curvature during systole when they reach lower wall tension and shear stress; the opposite happens during diastole. The low and oscillatory shear stress stimulates specific endothelial mechanoreceptors (integrins, ion channels, caveolae, etc) able to activate nuclear factors that result in the upregulation of pro-atherosclerotic genes and downregulation of athero-protective ones.1 Thus, if the patient’s heart spends much time in systole, as happens during elevated heart rates, the risk of atherosclerosis development dramatically increases. Furthermore, an increased heart rate causes endothelial dysfunction,23,37 maybe by means of an induction of inflammation that is able to enhance pathogenesis and the progression of atherosclerosis.38
Furthermore, interesting data come from literature studies about the role of ivabradine and HRR in stunned myocardium.39–41 Monnet et al39 demonstrated that ivabradine ameliorated subendocardial perfusion, a condition associated with an improvement of the stunning of the myocardium. Left ventricular wall thickening was enhanced by HRR after ivabradine administration, a condition which was associated with reduction in the severity of myocardial stunning.39 This particular effect of ivabradine is maintained during recovery from exercise.40 While atenolol has poor effects on left ventricular wall thickening during recovery, ivabradine is more effective in ameliorating myocardial stunning during all phases of exercise.40 Lucats et al41 evaluated the influence of HRR (obtained by means of ivabradine and atenolol) on post-systolic wall-thickening (PSWT). PSWT occurs after the closure of the aortic valve and is a waste of energy for the heart because it does not contribute to the ejection fraction. During myocardial stunning, an increasing in PSWT occurs. Ivabradine is both able to reduce PSWT and to convert PSWT to ejectional thickening, more so than atenolol, resulting in an improvement in myocardial stunning.
CAD
CAD represent the most common cause of death in middle-aged and older adults in the world.42 CAD due to atherosclerosis are one of the main determinants of cardiovascular disease development.
Ischemic episodes can be enhanced by an increased heart rate because this condition induces an imbalance between myocardial oxygen supply and consumption. Heusch pointed out such a point in his review on this topic.43 It is already known that coronary blood flow is influenced by several mechanisms: extravascular compression, neurohumoral regulation, metabolic regulation, endothelial function, and blood vessel geometry during cardiac cycle, etc. Heart rate is able to influence all these aspects of coronary blood flow regulation.43,44 Low heart rate reduces extravascular compression, reduces the time the heart stays in systole (which increases diastole time and consequentially, coronary blood flow), and meliorates the curvature angles of the coronary arteries during the cardiac cycle. All these changes improve shear stress and ameliorates endothelial function, etc.43,44
An interesting study on the pleiotropic effects of ivabradine comes from Skalidis et al.45 These authors demonstrated that a reduction in heart rate by means of ivabradine was able to improve coronary blood flow at rest. Although this corroborated the classical pathophysiology of coronary blood vessel filling, the authors went further by infusing adenosine in order to evaluate coronary flow reserve (CFR) in their CAD patients. After ivabradine treatment, the patients showed an improvement in CFR as compared to baseline. Skalidis et al45 concluded that ivabradine was able to ameliorate the endothelial function of coronary vessels, thus improving the microvascular function of such vessels.
In patients suffering from CAD, these events are fundamental in order to guarantee a good perfusion despite stenosis.43,44 Thus, HRR influences the outcome of patients suffering from CAD. A meta-analysis involving the GISSI (Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico)-2 trial showed an increase in the in-hospital mortality (3.3%–10.1%) rate after myocardial infarction in patients with heart rate >100 bpm on admission as compared to those with heart rates <60 bpm, independently of HF condition.46
Ivabradine demonstrated its efficacy in the treatment of chronic stable angina pectoris and myocardial ischemia,7,17,47,48 which was similar to the action of β-blockers.49 Ivabradine (5, 7.5, and 10 mg twice daily) is non-inferior to atenolol (50 or 100 mg per day) if used for reducing coronary ischemic symptoms, as demonstrated in 939 patients suffering from chronic stable angina pectoris.18 In ST-segment elevation myocardial infarction (STEMI) patients (left ventricular ejection fraction [LVEF] <50%) ivabradine improved LVEF rather than metoprolol.50,51
Another aspect to be considered is the potentiating of α-adrenergic receptors within the cellular membranes of vascular walls of cells when β-blocker administration is undertaken.52 The administration of β-blockers could inhibit the activity of the corresponding β-receptors, but on the opposing side, α-adrenergic receptor activity could be increased.52 The consequence of this double action of beta-blockers on beta and alpha receptors is an increase in the vasoconstriction properties of α-adrenergic receptors. Such an action cannot be achieved by using ivabradine due to its selective action on the If current in sinus node cells.
Experimental studies demonstrated a reduction in post-ischemic remodeling after ivabradine use.49,53 In a dog model, coronary artery perfusion is 30%–40% greater with ivabradine than with β-blockers during exercise.54 Linka et al55 and Van Camp et al56 demonstrated through microbubbles that the endocardium is more affected by reduced driving blood pressure than the epicardium; thus, it is more susceptible to ischemia than the epicardium. Thus, ischemic patients exhibit more damage at the endocardium level than the epicardium level. It is well-known that the endocardium is the major determinant of ventricular systolic wall thickness. During ischemia, one of the first characteristics detectable are losses in cardiac wall thickness and in ejection fraction.57 Ivabradine is able to improve regional blood flow40 and to convert post-systolic wall thickening in ejectional thickening.41 Furthermore, it is already known that, in normal healthy conditions, myocardial oxygen consumption undergoes a transmural gradient that is responsible for a major increase in oxygen consumption in the endocardial layer but not in the epicardial one.58 The combination of reduced driving pressure blood flow55,56,59 and increased oxygen consumption58 in the endocardium makes this layer at particularly high risk of ischemia. Thus, the positive effects of ivabradine on regional flow and cardiac chamber performance34,43,44 can enhance endocardial perfusion and ameliorate the ischemic condition of CAD patients.
Ivabradine is a fundamental molecule for the treatment of CAD patients with or without left ventricular dysfunction, as indicated in the European Society of Cardiology guidelines.60 Ivabradine effectively controls heart rate both at rest and during exercise and improves the symptoms of angina and the exercise capacity of patients with chronic stable angina, while preserving other cardiovascular functions and electrophysiological parameters.61 Studies have tried to outline the importance of ivabradine in the clinical management of patients suffering from cardiovascular diseases.
The Morbidity–Mortality Evaluation of the If Inhibitor Ivabradine in Patients with Coronary Disease and Left Ventricular Dysfunction (BEAUTIFUL) study tested ivabradine influence on cardiovascular death and morbidity in CAD and left ventricular systolic dysfunction (LVSD; LVEF <40%) patients (Table 1).25 Ten thousand, nine hundred and seventeen patients were enrolled: 5,479 receiving 5 mg of ivabradine (progressively increased) and 5,438 receiving placebo. The trial failed to demonstrate ivabradine as being able to improve cardiac outcomes in the population enrolled; nevertheless, the drug decreased admissions to hospital secondary to fatal and non-fatal myocardial infarction by about 36% and coronary revascularization of about 30% in the subgroup of patients showing heart rates ≥70 bpm.26 These results pointed out the need of recording baseline heart rate in addition to other risk factors in CAD patients.27 In a post-hoc analysis, Fox et al28 considered BEAUTIFUL patients showing limiting angina symptoms at baseline: ivabradine was able to reduce major cardiovascular events in patients with stable CAD and LVSD if presenting with limiting angina symptoms.28 The BEAUTIFUL Holter substudy showed that treatment with ivabradine was associated with a significant decrease in 24-hour heart rate than placebo (6.3±9.4 bpm vs 0.4±7.2 bpm, P<0.001 between-group difference). Nevertheless, there was a significant decrease in waking heart rate with ivabradine than during sleep.29 This means that ivabradine is almost safe in CAD patients suffering from left ventricular dysfunction because there is no risk of excessive heart rate drop during sleep; furthermore, no statistically significantly changes were detected in the atrioventricular node, His–Purkinje system, and atrial and ventricular refractoriness, the latter demonstrated by a normal corrected QT interval.29
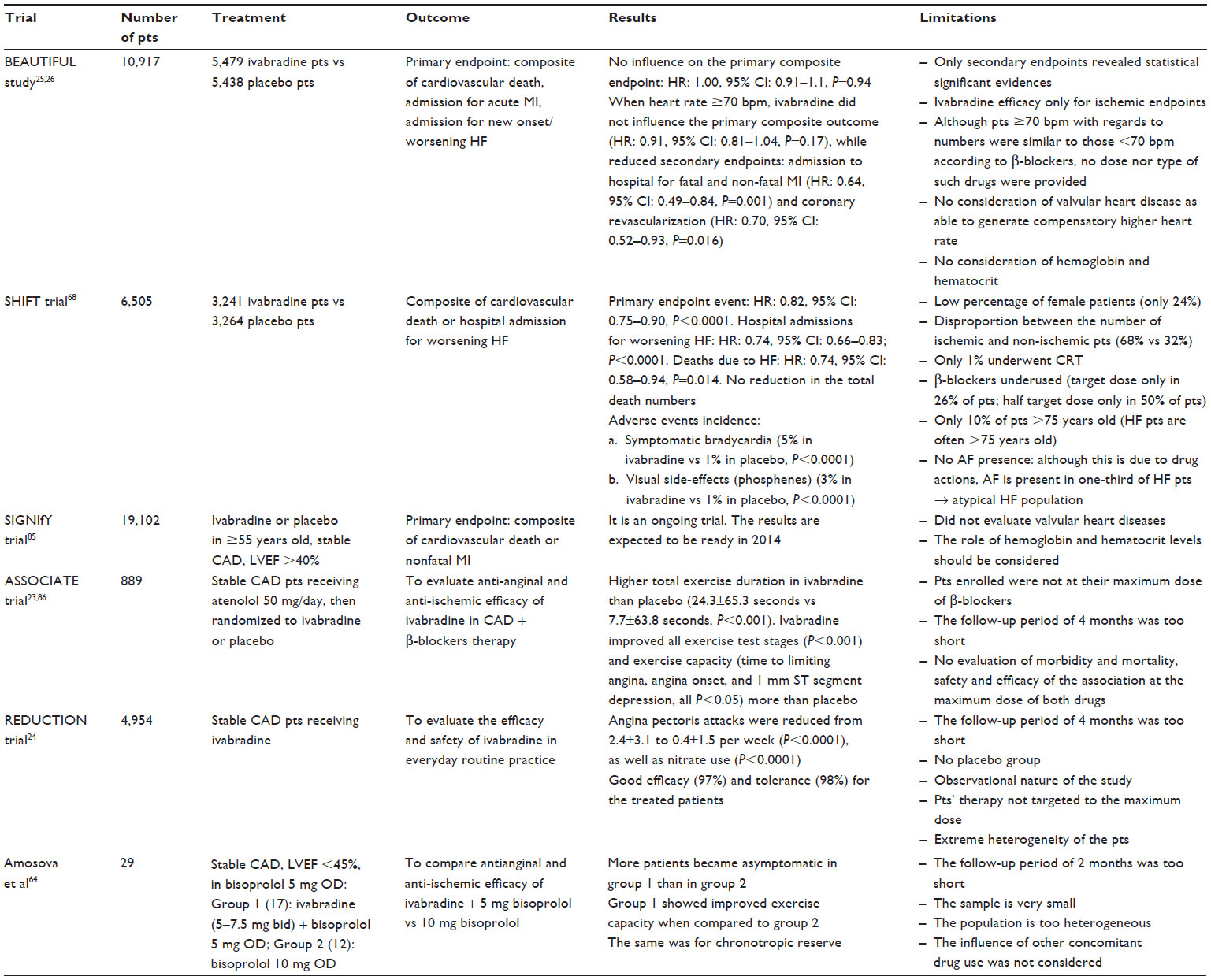 | Table 1 Relevant clinical trials on ivabradine use: an overview about their characteristics and limitations |
Nevertheless, despite the data coming from the BEAUTIFUL trial, the scientific community would like to better select the population of patients treated with ivabradine and their outcomes. The Study Assessing the Morbidity–Mortality Benefits of the If Inhibitor Ivabradine in Patients with Coronary Artery Disease (SIGNIfY) trial was managed in order to achieve such an aim, ie, the influence of this drug on cardiovascular mortality and hospitalization for myocardial infarction.30 This is an ongoing trial and we are awaiting its final results (Table 1).62
Nevertheless, many other scientific observations validate the positive influence of ivabradine in CAD patients. Tardif et al18 compared the antianginal and anti-ischemic effects of ivabradine and atenolol. Nine hundred and thirty-nine patients with stable angina were randomized to receive ivabradine 5 mg twice daily for 12 weeks or atenolol 50 mg once daily for 4 weeks and 100 mg once daily for 12 weeks. Patients underwent a treadmill exercise test. An increase in total exercise duration was observed after ivabradine 7.5 and 10 mg administration. Angina attacks were two-thirds less in the ivabradine group when compared to the β-blockers group.18 The Efficacy and Safety of Ivabradine on Top of Atenolol in Stable Angina Pectoris (ASSOCIATE) trial randomized CAD patients to ivabradine or placebo for 4 months, both in addition to atenolol 50 mg once daily.63 Ivabradine significantly improved exercise capacity as compared to placebo when administered in patients with CAD on β-blocker therapy (Table 1).32 Köster et al33 evaluated the efficacy and safety of ivabradine in everyday routine practice in the REDUCTION study. Four thousand, nine hundred and fifty-four patients with stable angina pectoris in ivabradine therapy underwent a 4-month follow-up. Ivabradine reduced heart rate and was well-tolerated by patients suffering from symptomatic CAD during every day routine use (Table 1).33
Furthermore, ivabradine combined with a low dose of bisoprolol, in stable angina patients confirmed amelioration of anginal and ischemic benefits, as well as improvement in chronotropic reserve.64 Another study compared ivabradine and the calcium channel antagonist amlodipine.65 Ivabradine improved exercise tolerance as well as amlodipine, although it showed a superior effect on the reduction of rate pressure product.65
Thus, the treatment of stable angina pectoris by means of ivabradine is as effective in reducing angina and ischemic symptoms as β-blockers and calcium channel antagonists, although did not show the typical adverse reactions associated with β-blockers or other anti-angina drugs.66
It is interesting to note that ivabradine is able to decrease myocardial damage, which is a condition that protects against primary ischemic ventricular fibrillation.67 It may exert a sort of cardioprotection from ischemia-induced ventricular fibrillation by increasing regional myocardial blood flow and preserving cardiomyocytes and mitochondrial ultrastructure.68 Moreover, ivabradine, unlike propranolol, delays the time to onset of ischemia-induced ventricular fibrillation by preserving myocardial energy status.69
A study from Heusch et al34 pointed out that ivabradine is able to reduce myocardial damage by improving regional blood flow, regional function, and infarct size in pigs subjected to 90 minutes of controlled left anterior descending coronary artery hypoperfusion and 120 minutes of reperfusion. This skill can explain the great impact of ivabradine when administered in patients with CAD and left ventricular dysfunction with a resting heart rate >70 bpm.70 This hypothesis contributes to the comprehension of the results of the BEAUTIFUL trial,25,26 which were not explained by the authors in their manuscripts.70
CHF
CHF is a common, disabling disease, roughly affecting 2%–3% of the population in many industrialized countries, resulting in high mortality and morbidity rates.71 Heart rate is tightly related to HF development, as many studies have already proven.72 In an elegant review from Heusch,72 the relationship between heart rate and HF was clearly explained. This author pointed out that rapid atrial pacing can lead to heart dysfunction. Furthermore, elevated heart rate is able to induce tachycardiomyopathy, ie, a transient cardiac dysfunction due to an increased heart rate, which could be reverted if the heart rate returns to normal values.72
Thus, heart rate reduction is particularly important in CHF because it attenuates the effect of energy starvation of the myocardium.73 A great number of agents, such as β-blockers and angiotensin-converting enzyme inhibitors, have proven benefits in patients with LVSD, which may be due to positive heart remodeling.74–76 The heart rate is also directly related to the risk of all causes of death, cardiovascular death, or admission to hospital in patients with HF: heart rate reduction is associated with improved outcomes.77
The BEAUTIFUL study showed that ivabradine did not affect the primary endpoint, with similar results between the ivabradine and placebo groups. Nevertheless, according to a sub-analysis of this trial, patients suffering from CAD and left ventricular dysfunction, showing a heart rate of 70 bpm or higher, had a 34% increased risk of cardiovascular death and a 53% increase in hospital admission for HF as compared to patients with a heart rate lower than 70 bpm.26 Unlike β-blockers, ivabradine does not modify myocardial contractility and intracardiac conduction, even in patients with impaired systolic function (Table 1).14
The SHIFT study was designed in order to evaluate ivabradine action only in CHF patients suffering from LVSD, regardless of etiology.22,78 Six thousand, five hundred and fifty-eight symptomatic CHF patients with left ventricular dysfunction (LVEF <35%) in optimal medical therapy were randomly assigned to ivabradine or placebo. They had to be in sinus rhythm, with a heart rate 70 bpm or higher. Ivabradine reduced heart rate by 15 bpm from baseline. Moreover, most cardiovascular endpoints (death from HF, hospital admission for worsening of HF, any cardiovascular admission) were significantly reduced by ivabradine treatment. After a median follow-up of nearly 23 months, there was a significant reduction in the primary endpoint of cardiovascular death and hospital admission for worsening HF. The drug was well-tolerated, with relatively few mechanism-related adverse events, such as bradycardia, atrial fibrillation, and visual disturbances like phosphenes (Table 1).79
The SHIFT trial presents some critical points. The SHIFT investigators attempted to enroll a population on optimum and stable therapy with the maximally tolerated therapy. Nevertheless, about 50% patients assuming target β-blocker doses at baseline did not reach any significant benefit from ivabradine in relation to the primary endpoint. Furthermore, the same use of β-blockers was underused: patients at target dose were only 26%, while only 50% of patients were at half target dose. Once more, only 1% of enrolled patients underwent cardiac resynchronization therapy.
Thus, despite the possible beneficial role that ivabradine could exert over β-blockers, one cannot conclude that the former could easily substitute the latter drug. To the best of our knowledge, no prospective clinical trial has been managed in order to clearly evaluate the efficacy and long-term outcomes of ivabradine administration versus β-blockers. Thus, further studies are needed in order to evaluate the reciprocal relationship between ivabradine and β-blockers not only in HF patients, but even in those suffering from CAD.
It should be noted that enhanced heart rate is a compensatory mechanism in CHF, by preserving cardiac output. Nevertheless, it increases left ventricular oxygen consumption. The HRR induced by ivabradine prevents LVSD in CHF. Long-term HRR induced by ivabradine improves diastolic left ventricle function, likely by attenuating hypoxia, reducing remodeling, and/or preserving nitric oxide bioavailability. HRR initiation effectively seems to trigger processes as angiogenesis and/or preservation of endothelial nitric oxide synthase expression, as demonstrated in a rat model by Fang et al.80
Furthermore, ivabradine reduces heart rate and simultaneously improves stroke volume and cardiac output without any adverse effects on myocardial contractility, left ventricular relaxation, and coronary tone in contrast to β-blockers and other similar drugs’ effects.50,54 Left ventricular diastolic diameter is influenced by ivabradine, potentially due to the drug’s skill in decreasing left ventricular collagen density. Several experimental studies found that chronic treatment with ivabradine reduced cardiac collagen accumulation in rats with congestive HF.16,81,82
Couvreur et al83 demonstrated a characteristic action of ivabradine on calcium metabolism in cardiac cells from HF rabbits. In particular, they observed that ivabradine was able to induce a two-fold increase in the expression of FKBP12/12.6. Such a condition was associated with the amelioration of cardiac function. FKBP12/12.6 is a protein able to modulate ryanodine receptor (RyR)2 activity; thus, it regulates calcium metabolism in cardiac cells. The positive action of ivabradine in HF could be due to not only a simple reduction in heart rate, but rather several mechanisms, which are not at all understood and probably involve calcium metabolism.
It is interesting to evaluate the relationship between occlusive CAD and left ventricle remodeling, systolic dysfunction, and HF. The BEAUTIFUL Echo substudy aimed at evaluating the effects of HRR with ivabradine on left ventricular size (primary endpoint: change in left ventricular end-systolic volume index [LVESVI] and function and the cardiac biomarker N-terminal pro-brain natriuretic peptide [NT-proBNP]).84 Echocardiography evaluation was performed at baseline, and after 3 and 12 months. Ivabradine showed a significant impact on systolic volume but not on diastolic volume.84 Ivabradine decreased LVESVI much more than placebo (−1.48±13.00 mL/m2 vs 1.85±10.54 mL/m2, P=0.018). LVEF increased more with ivabradine than placebo (2.00%±7.02% vs 0.01%±6.20%, P=0.009).84 Ivabradine had no impact on NT-proBNP levels because of the great variability in NT-proBNP values or because left ventricle diastolic volumes were unchanged, which induced a prolonged diastolic filling able to impair diastolic wall stress and the consequent production of NT-proBNP.85 Nevertheless, further studies are needed in order to better highlight all the positive and partial results obtained by international trials that adopted ivabradine in CHF patients.
Future development
The advent of CAD and/or CHF conditions is unfortunately related to the genesis of cardiac rhythm disorders that impair the natural function of the heart. Despite new advances in pharmacological and interventional therapies, poor results still persist due to the poor knowledge about the molecular mechanisms underlining the genesis of cardiac arrhythmias. An elegant review from Difrancesco86 pointed out that the dysfunctional activity of HCN channels is able to contribute to rhythm disorders. In particular, the author pointed out the possible role of HCN dysfunction in the genesis of symptomatic sinus bradycardia, tachycardia–bradycardia syndrome, atrioventricular node block, and atrial fibrillation, etc. The use of ivabradine in counteracting such rhythm disorders can be taken into account due to the promising results that it showed in human studies.87 Some studies outlined the overexpression of HCN2 channel in the ventricles of failing hearts and the predisposition of these structures to ventricular arrhythmias.88 Research from Suenari et al89 revealed that ivabradine is able to exert its pharmacological effects on the pacemaker cells cardiomyocytes present in the pulmonary veins. Whole-cell patch-clamp techniques and the indo-1 fluorimetric ratio technique demonstrated that ivabradine was able to decrease the If and ICa2+ transient currents in single isolated rabbit pulmonary vein cardiomyocytes endowed with pacemaker activity.89 This research will open the way to new developments in the comprehension of atrial fibrillation genesis and in the complex mechanism underlining the functions of pacemaker pulmonary vein cardiomyocytes.
In mice suffering from HF due to dilated cardiomyopathy, ivabradine administration is able to reduce the occurrence of lethal arrhythmias.88 Plotnikov et al90 demonstrated that ventricular tachyarrhythmias originating from HCN212 injection were suppressed by ivabradine faster than its action on sinus node: thus, ectopic electric disorders could be influenced by HCN4 drugs more easily than the natural pacemaker cells. Further studies are needed in order to confirm such results.
Furthermore, studies91,92 have observed that amiodarone and other antiarrhythmic drugs were able to exert their pharmacological action even by interacting with HCN channels of the heart: in particular, amiodarone and bepridil were able to inhibit HCN4 channels of HEK 293 cells at their therapeutic concentrations.91
The future development of scientific pharmacological research will be the evaluation of the role of ivabradine and other drugs able to act on HCN channels on the development and/or on counteracting cardiac arrhythmia expression and maintenance by means of well-structured clinical trials.
Conclusion
Ivabradine is a reliable pharmacological tool for physicians to reduce the morbidity and mortality related to CAD and CHF. Its positive effects, which go beyond simple HRR are related to its limited side effects: this characteristic makes ivabradine more acceptable to patients and with less risk due to its administration. Nevertheless, more research is needed in order to obtain more solid evidence about ivabradine use in clinical medical therapeutic practice.
Disclosure
The authors report no conflicts of interest in this work.
References
Giannoglou GD, Chatzizisis YS, Zamlis C, Parcharidis GE, Mikhailidis DP, Louridas GE. Elevated heart rate and atherosclerosis. An overview of the pathogenetic mechanisms. Int J Cardiol. 2008; 126(3):302–312. | |
Scicchitano P, Carbonara S, Ricci G, et al. HCN channels and heart rate. Molecules. 2012;17(4):4225–4235. | |
Colin P, Ghaleh B, Monnet X, et al. Contributions of heart rate and contractility to myocardial oxygen balance during exercise. Am J Physiol Heart Circ Physiol. 2003;284(2):H676–H682. | |
Parakh N, Bhargara B. Rate control with Ivabradine. Am J Cardiovasc Drugs. 2011;11(1):1–12. | |
Thollon C, Villaine JP. If inhibition in cardiovascular diseases. Adv Pharmacol. 2010;59:53–92. | |
Tardif JC. Clinical results of If current inhibitor by ivabradine. Drugs. 2007;67 Suppl 2:S35–S41. | |
Riccioni G, Vitulano N, D’Orazio N. Ivabradine: beyond heart rate control. Adv Ther. 2009;26(1):12–24. | |
Menown IB. Ivabradine: a new strategy for management of stable angina. Br J Hosp Med. 2007;68(6):21–25. | |
Thollon C, Bidouard JP, Cambarrat C, et al. Stereospecific in vitro and in vivo effects of the new sinus node inhibitor (+) -S 16257. Eur J Pharmacol. 1997;339(1):43–51. | |
Ragueneau I, Laveille C, Jochemsen R, Resplandy G, Funck-Brentano C, Jaillon P. Pharmacokinetic-pharmacodynamic modeling of the effects of ivabradine, a direct sinus node inhibitor, on heart rate in healthy volunteers. Clin Pharmacol Ther. 1998;64(2):192–203. | |
Thollon C, Cambarrat C, Vian J, Prost JF, Peglion JL, Vilaine JP. Electrophysiological effects of S 16257, a novel sino-atrial node modulator, on rabbit and guinea-pig cardiac preparations: comparison with UL-FS 49. Br J Pharmacol. 1994;112(1):37–42. | |
Bois P, Bescond J, Renaudon B, Lenfant J. Mode of action of bradycardic agent, S 16257, on ionic currents of rabbit sinoatrial node cells. Br J Pharmacol. 1996;118(4):1051–1057. | |
Bucchi A, Baruscotti M, DiFrancesco D. Current-dependent block of rabbit sino-atrial node If channels by ivabradine. J Gen Physiol. 2002;120(1):1–13. | |
Savelieva I, Camm AJ. If inhibition with ivabradine: electrophysiological effects and safety. Drug Saf. 2008;31(2):95–107. | |
Rosa GM, Ferrero S, Ghione P, Valbusa A, Brunelli C. An evaluation of the pharmacokinetics and pharmacodynamics of ivabradine for the treatment of heart failure. Expert Opin Drug Metab Toxicol. 2014;10(2): 279–291. | |
DiFrancesco D, Camm JA. Heart rate lowering by specific and selective I(f) current inhibition with ivabradine: a new therapeutic perspective in cardiovascular disease. Drugs. 2004;64(16):1757–1765. | |
Borer JS, Fox K, Jaillon P, Lerebours G; Ivabradine Investigators Group. Antianginal and antiischemic effects of ivabradine, an If inhibitor, in stable angina: a randomized, double-blind, multicentered, placebo-controlled trial. Circulation. 2003;107(6):817–823. | |
Tardif JC, Ford I, Tendera M, Bourassa MG, Fox K; INITIATIVE Investigators. Efficacy of ivabradine, a new selective I(f) inhibitor, compared with atenolol in patients with chronic stable angina. Eur Heart J. 2005;26(23):2529–2536. | |
Camm AJ. How does pure heart rate lowering impact on cardiac tolerability? Eur Heart J Suppl. 2006;8 (Suppl D):D9–D15. | |
Murat SN, Orcan S, Akdemir R, Dogan M, Kara E, Balci M. Arrhythmic effects of ivabradine in patients with coronary artery disease. Clin Invest Med. 2009;32(5):E322–E326. | |
Scott AE, Kruszewski K, Leslie SJ. Sinus node If channel inhibition – a new therapeutic approach to heart rate lowering. Curr Drug Therapy. 2009;4(1):12–18. | |
Swedberg K, Komajda M, Bohm M, et al; SHIFT Investigators. Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo-controlled study. Lancet. 2010;376(9744):875–885. | |
Custodis F, Baumhäkel M, Schlimmer N, et al. Heart rate reduction by ivabradine reduces oxidative stress, improves endothelial function, and prevents atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2008;117(18):2377–2387. | |
Dominguez-Rodriguez A, Blanco-Palacios G, Abreu-Gonzales P. Increased heart rate and atherosclerosis: potential implications of ivabradine therapy. World J Cardiol. 2011;3(4):101–104. | |
Fox K, Ford I, Steg PG, Tendera M, Ferrari R; BEAUTIFUL Investigators. Ivabradine for patients with stable coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): a randomised, double-blind, placebo-controlled trial. Lancet. 2008;372(9641):807–816. | |
Fox K, Ford I, Steg PG, Tendera M, Robertson M, Ferrari R; BEAUTIFUL investigators. Heart rate as a prognostic risk factor in patients with coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): a subgroup analysis of a randomised controlled trial. Lancet. 2008;372(9641):817–821. | |
Bruguera Cortada J, Varela A. Role of heart rate in cardiovascular diseases: how the results of the BEAUTIFUL study change clinical practice. Am J Cardiovasc Drugs. 2009;9 Suppl:9–12. | |
Fox K, Ford I, Steg PG, Tendera M, Robertson M, Ferrari R; BEAUTIFUL Investigators. Relationship between ivabradine treatment and cardiovascular outcomes in patients with stable coronary artery disease and left ventricular systolic dysfunction with limiting angina: a subgroup analysis of the randomized, controlled BEAUTIFUL trial. Eur Heart J. 2009;30(19):2337–2345. | |
Tendera M, Talajic M, Robertson M, et al; BEAUTIFUL Investigators. Safety of ivabradine in patients with coronary artery disease and left ventricular systolic dysfunction (from the BEAUTIFUL Holter Substudy). Am J Cardiol. 2011;107(6):805–811. | |
Ferrari R. A step further with ivabradine: SIGNIfY (Study assessInG the morbidity–mortality beNefits of the If inhibitor ivabradine in patients with coronarY artery disease). Eur Heart J. 2009;11(Suppl D): 19–27. | |
Lucats L, Ghaleh B, Colin P, Monnet X, Bizé A, Berdeaux A. Heart rate reduction by inhibition of If or by beta-blockade has different effects on postsystolic wall thickening. Br J Pharmacol. 2007;150(3): 335–341. | |
Tardif JC, Ponikowski P, Kahan T; ASSOCIATE Investigators. Effects of ivabradine in patients with stable angina receiving beta-blockers according to baseline heart rate: an analysis of the ASSOCIATE study. Int J Cardiol. 2013;168(2):789–794. | |
Köster R, Kaehler J, Meinertz T; REDUCTION Study Group. Treatment of stable angina pectoris by ivabradine in every day practice: the REDUCTION study. Am Heart J. 2009;158(4):51–57. | |
Heusch G, Skyschally A, Gres P, van Caster P, Schilawa D, Schulz R. Improvement of regional myocardial blood flow and function and reduction of infarct size with ivabradine: protection beyond heart rate reduction. Eur Heart J. 2008;29(18):2265–2275. | |
Beere PA, Glagov S, Zarins CK. Retarding effect of lowered heart rate on coronary atherosclerosis. Science. 1984;226(4671):180–182. | |
Prosi M, Perktold K, Ding Z, Friedman MH. Influence of curvature dynamics on pulsatile coronary artery flow in a realistic bifurcation model. J Biomech. 2004;37(11):1767–1775. | |
Drouin A, Gendron ME, Thorin E, Gillis MA, Mahlberg-Gaudin F, Tardif JC. Chronic heart rate reduction by ivabradine prevents endothelial dysfunction in dyslipidaemic mice. Br J Pharmacol. 2008;154(4):749–757. | |
Rogowski O, Shapira I, Shirom A, Melamed S, Toker S, Berliner S. Heart rate and microinflammation in men: a relevant atherothrombotic link. Heart. 2007;93(8):940–944. | |
Monnet X, Ghaleh B, Colin P, de Curzon OP, Giudicelli JF, Berdeaux A. Effects of heart rate reduction with ivabradine on exercise-induced myocardial ischemia and stunning. J Pharmacol Exp Ther. 2001;299(3): 1133–1139. | |
Monnet X, Colin P, Ghaleh B, Hittinger L, Giudicelli JF, Berdeaux A. Heart rate reduction during exercise-induced myocardial ischaemia and stunning. Eur Heart J. 2004;25(7):579–586. | |
Lucats L, Ghaleh B, Monnet X, Colin P, Bizé A, Berdeaux A. Conversion of post-systolic wall thickening into ejectional thickening by selective heart rate reduction during myocardial stunning. Eur Heart J. 2007;28(7):872–879. | |
Pagidipati NJ, Gaziano TA. Estimating deaths from cardiovascular disease: a review of global methodologies of mortality measurement. Circulation. 2013;127(6):749–756. | |
Heusch G. Heart rate in the pathophysiology of coronary blood flow and myocardial ischaemia: benefit from selective bradycardic agents. Br J Pharmacol. 2008;153(8):1589–1601. | |
Custodis F, Schirmer SH, Baumhäkel M, Heusch G, Böhm M, Laufs U. Vascular pathophysiology in response to increased heart rate. J Am Coll Cardiol. 2010;56(24):1973–1983. | |
Skalidis EI, Hamilos MI, Chlouverakis G, Zacharis EA, Vardas PE. Ivabradine improves coronary flow reserve in patients with stable coronary artery disease. Atherosclerosis. 2011;215(1):160–165. | |
Zuanetti G, Mantini L, Hernández-Bernal F, et al. Relevance of heart rate as a prognostic factor in patients with acute myocardial infarction: insights from the GISSI-2 study. Eur Heart J. 1998;19 Suppl F:F19–F26. | |
Riccioni G. Ivabradine: an intelligent drug for the treatment of ischemic heart disease. Molecules. 2012;17(11):13592–13604. | |
Riccioni G. Ivabradine: the hope for a good treatment of ischemic heart disease. Curr Med Chem. 2013;20(4):1817–1823. | |
Mulder P, Barbier S, Chagraoui A, et al. Long-term heart rate reduction induced by the selective I(f) current inhibitor ivabradine improves left ventricular function and intrinsic myocardial structure in congestive heart failure. Circulation. 2004;109(13):1674–1679. | |
Fasullo S, Cannizzaro S, Maringhini G, et al. Comparison of ivabradine versus metoprolol in early phases of reperfused anterior myocardial infarction with impaired left ventricular function: preliminary findings. J Card Fail. 2009;15(10):856–863. | |
Riccioni G. Ivabradine: from molecular basis to clinical effectiveness. Adv Ther. 2010;27(3):160–167. | |
Rozanski A, Qureshi E, Bauman M, Reed G, Pillar G, Diamond GA. Peripheral Arterial Responses to treadmill exercise among healthy subjects and atherosclerotic patients. Circulation. 2001;103:2084–2089. | |
Böhm M, Reil JC. Perspectives of I(f) inhibition by ivabradine in cardiology. Drugs. 2007;67 Suppl 2:43–49. | |
Simon L, Ghaleh B, Puybasset L, Giudicelli JF, Berdeaux A. Coronary and hemodynamic effects of S16257, a new bradycardic agent, in resting and exercising conscious dogs. J Pharmacol Exp Ther. 1995;275(2): 659–666. | |
Linka AZ, Sklenar J, Wei K, Jayaweera AR, Skyba DM, Kaul S. Assessment of transmural distribution of myocardial perfusion with contrast echocardiography. Circulation. 1998;98(18):1912–1920. | |
Van Camp G, Ay T, Pasquet A, et al. Quantification of myocardial blood flow and assessment of its transmural distribution with real-time power modulation myocardial contrast echocardiography. J Am Soc Echocardiogr. 2003;16(3):263–270. | |
Schulz R, Heusch G. The relationship between regional blood flow and contractile function in normal, ischemic, and reperfused myocardium. Basic Res Cardiol. 1998;93(6):455–462. | |
Decking UK, Schrader J. Spatial heterogeneity of myocardial perfusion and metabolism. Basic Res Cardiol. 1998;93(6):439–445. | |
Hsu LY, Groves DW, Aletras AH, Kellman P, Arai AE. A fully quantitative pixel-wise measurement of myocardial blood flow by contrast-enhanced first-pass CMR perfusion imaging: microsphere validation in dogs and feasibility study in humans. JACC Cardiovasc Imaging. 2012;5(2):154–166. | |
Fox K, Garcia MA, Ardissino D, et al; Task Force on the Management of Stable Angina Pectoris of the European Society of Cardiology; ESC Committee for Practice Guidelines (CPG). Guidelines on the management of stable angina pectoris: executive summary: The Task Force on the Management of Stable Angina Pectoris of the European Society of Cardiology. Eur Heart J. 2006;27(11):1341–1381. | |
Canet E, Lerebours G, Vilaine JP. Innovation in coronary artery disease and heart failure: clinical benefits of pure heart rate reduction with ivabradine. Ann N Y Acad Sci. 2011;1222:90–99. | |
Fox K, Ford I, Steg PG, Tardif JC, Tendera M, Ferrari R. Rationale, design, and baseline characteristics of the Study assessInG the morbidity-mortality beNefits of the If inhibitor ivabradine in patients with coronarY artery disease (SIGNIFY trial): a randomized, double-blind, placebo-controlled trial of ivabradine in patients with stable coronary artery disease without clinical heart failure. Am Heart J. 2013;166(4):654–661. e6. | |
Tardif JC, Ponikowski P, Kahan T; ASSOCIATE Study Investigators. Efficacy of the I(f) current inhibitor ivabradine in patients with chronic stable angina receiving beta-blocker therapy: a 4-month, randomized, placebo-controlled trial. Eur Heart J. 2009;30(5):540–548. | |
Amosova E, Andrejev E, Zaderey I, Rudenko U, Ceconi C, Ferrari R. Efficacy of ivabradine in combination with Beta-blocker versus uptitration of Beta-blocker in patients with stable angina. Cardiovasc Drugs Ther. 2011;25(6):531–537. | |
Ruzyllo W, Tendera M, Ford I, Fox KM. Antianginal efficacy and safety of ivabradine compared with amlodipine in patients with stable effort angina pectoris: a 3-month randomised, double-blind, multicentre, noninferiority trial. Drugs. 2007;67(3):393–405. | |
Marquis-Gravel G, Tardif JC. Ivabradine: the evidence of its therapeutic impact in angina. Core Evid. 2008;3(1):1–12. | |
Vaillant F, Timour Q, Descotes J, et al. Ivabradine induces an increase in ventricular fibrillation threshold during acute myocardial ischemia: an experimental study. J Cardiovasc Pharmacol. 2008;52(6):548–554. | |
Vaillant F, Dehina L, Mazzadi A, et al. Heart rate reduction with ivabradine increases ischaemia-induced ventricular fibrillation threshold: role of myocyte structure and myocardial perfusion. Resuscitation. 2011;82(8):1092–1019. | |
Vaillant F, Dehina L, Dizerens N, et al. Ivabradine but not propranolol delays the time to onset of ischaemia-induced ventricular fibrillation by preserving myocardial metabolic energy status. Resuscitation. 2013;84(3):384–390. | |
Heusch G. A BEAUTIFUL lesson – ivabradine protects from ischaemia, but not from heart failure: through heart rate reduction or more? Eur Heart J. 2009;30(19):2300–2301. | |
Dickstein K, Cohen-Solal A, Filippatos G, et al; ESC Committee for Practice Guidelines (CPG). ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2008: the Task Force for the diagnosis and treatment of acute and chronic heart failure 2008 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association of the ESC (HFA) and endorsed by the European Society of Intensive Care Medicine (ESICM). Eur J Heart Fail. 2008;10(10):933–989. | |
Heusch G. Heart rate and heart failure. Not a simple relationship. Circ J. 2011;75(2):229–236. | |
McAlister FA, Wiebe N, Ezekowitz JA, Leung AA, Armstrong PW. Meta-analysis: beta-blockers dose, heart rate reduction, and death in patients with heart failure. Ann Intern Med. 2009;150:784–794. | |
Doughty RN, Whalley GA, Walsh HA, Gamble GD, López-Sendón J, Sharpe N; CAPRICORN Echo Substudy Investigators. Effects of carvedilol on left ventricular remodeling after acute myocardial infarction: the CAPRICORN Echo Substudy. Circulation. 2004;109(2):201–206. | |
Ferrari R; Perindopril and Remodeling in Elderly with Acute Myocardial Infarction Investigators. Effects of angiotensin-converting enzyme inhibition with perindopril on left ventricular remodeling and clinical outcome: results of the randomized Perindopril and Remodeling in Elderly with Acute Myocardial Infarction (PREAMI) Study. Arch Intern Med. 2006;166(6):59–66. | |
Remme WJ, Riegger G, Hildebrandt P, et al. The benefits of early combination treatment of carvedilol and an ACE-inhibitor in mild heart failure and left ventricular systolic dysfunction. The carvedilol and ACE-inhibitor remodelling mild heart failure evaluation trial (CARMEN). 2004;18(1):57–66. | |
Flannery G, Gehrig-Mills R, Billah B, Krum H. Analysis of randomized controlled trials on the effect of magnitude of heart rate reduction on clinical outcomes in patients with systolic chronic heart failure receiving beta-blockers. Am J Cardiol. 2008;101(6):865–869. | |
Ekman I, Chassany O, Komajda M. Heart rate reduction with ivabradine and health related quality of life in patients with chronic heart failure: results from the SHIFT study. Eur Heart J. 2011;32(19):2395–2404. | |
Teerlink JR. Ivabradine in heart failure – no paradigm SHIFT … yet. Lancet. 2010;376(9744):847–849. | |
Fang Y, Debunne M, Vercauteren M, et al. Heart rate reduction induced by the if current inhibitor ivabradine improves diastolic function and attenuates cardiac tissue hypoxia. J Cardiovasc Pharmacol. 2012;59(3): 260–267. | |
Reil JC, Reil GH, Böhm M. Heart rate reduction by I(f)-channel inhibition and its potential role in heart failure with reduced and preserved ejection fraction. Trends Cardiovasc Med. 2009;19(5):152–157. | |
Setakis E, Kassianos G, Cockle S. Adverse events and subsequent changes in beta-blocker treatment in coronary heart disease patients in UK primary care. Eur Heart J. 2009;30:974. Suppl 1. Abstract P5476. | |
Couvreur N, Tissier R, Pons S, et al. Chronic heart rate reduction with ivabradine improves systolic function of the reperfused heart through a dual mechanism involving a direct mechanical effect and a long-term increase in FKBP12/12.6 expression. Eur Heart J. 2010;31(12):1529–1537. | |
Ceconi C, Freedman SB, Tardif JC, et al; BEAUTIFUL Echo-BNP Investigators. Effect of heart rate reduction by ivabradine on left ventricular remodeling in the echocardiographic substudy of BEAUTIFUL. Int J Cardiol. 2011;14:408–414. | |
Troughton RW, Richards AM, Yandle TG, Frampton CM, Nicholls MG. The effects of medications on circulating levels of cardiac natriuretic peptides. Ann Med. 2007;39(4):242–260. | |
Difrancesco D. Funny channel gene mutations associated with arrhythmias. J Physiol. 2013;591(17):4117–4124. | |
Benezet-Mazuecos J, Rubio JM, Farré J, Quiñones MÁ, Sanchez-Borque P, Macía E. Long-term outcomes of ivabradine in inappropriate sinus tachycardia patients: appropriate efficacy or inappropriate patients. Pacing Clin Electrophysiol. 2013;36(7):830–836. | |
Kuwabara Y, Kuwahara K, Takano M, et al. Increased expression of HCN channels in the ventricular myocardium contributes to enhanced arrhythmicity in mouse failing hearts. J Am Heart Assoc. 2013;2(3): e000150. | |
Suenari K, Cheng CC, Chen YC, et al. Effects of ivabradine on the pulmonary vein electrical activity and modulation of pacemaker currents and calcium homeostasis. J Cardiovasc Electrophysiol. 2012;23(2): 200–206. | |
Plotnikov AN, Bucchi A, Shlapakova I, et al. HCN212-channel biological pacemakers manifesting ventricular tachyarrhythmias are responsive to treatment with I(f) blockade. Heart Rhythm. 2008;5(2):282–288. | |
Tamura A, Ogura T, Uemura H, et al. Effects of antiarrhythmic drugs on the hyperpolarization-activated cyclic nucleotide-gated channel current. J Pharmacol Sci. 2009;110(2):150–159. | |
Fan X, Chen Y, Wu P, et al. Novel electropharmacological activity of amiodarone on human HCN channels heterologously expressed in the Xenopus oocytes. Eur J Pharmacol. 2011;669(1–3):15–23. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.