Back to Journals » OncoTargets and Therapy » Volume 11
IRX-2 natural cytokine biologic for immunotherapy in patients with head and neck cancers
Authors Wolf GT, Moyer JS, Kaplan MJ ![]() , Newman JG, Egan JE, Berinstein NL, Whiteside TL
, Newman JG, Egan JE, Berinstein NL, Whiteside TL
Received 13 February 2018
Accepted for publication 8 April 2018
Published 28 June 2018 Volume 2018:11 Pages 3731—3746
DOI https://doi.org/10.2147/OTT.S165411
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Samir Farghaly
Gregory T Wolf,1 Jeffrey S Moyer,1 Michael J Kaplan,2 Jason G Newman,3 James E Egan,4 Neil L Berinstein,4 Theresa L Whiteside5
1Department of Otolaryngology-Head and Neck Surgery, University of Michigan, Ann Arbor, MI, 2Department of Otolaryngology-Head and Neck Surgery, Stanford University Medical Center, Stanford, CA, 3Department of Otorhinolaryngology, University of Pennsylvania, Philadelphia, PA, 4IRX Therapeutics, New York, NY, 5Department of Immunology, University of Pittsburgh Cancer Institute, Pittsburgh, PA, USA
Abstract: Head and neck squamous cell carcinoma (HNSCC) is an immunosuppressive malignancy characterized by tumor-driven immune-system abnormalities that contribute to disease progression. For patients with surgically resectable HNSCC, treatment is often curative surgery followed by irradiation or chemoradiation in high-risk settings to reduce the risk of recurrence. Poor survival and considerable morbidity of current treatments suggest the need for new therapeutic modalities that can improve outcomes. Defects in antitumor immunity of HNSCC patients include suppressed dendritic cell (DC) maturation, deficient antigen-presenting cell function, compromised natural killer (NK)-cell cytotoxicity, increased apoptosis of activated T lymphocytes, and impaired immune-cell migration to tumor sites. Strategies for relieving immunosuppression and restoring antitumor immune functions could benefit HNSCC patients. IRX-2 is a primary cell-derived biologic consisting of physiologic levels of T-helper type 1 cytokines produced by stimulating peripheral blood mononuclear cells of normal donors with phytohemagglutinin. The primary active components in IRX-2 are IL2, IL1β, IFNγ, and TNFα. In vitro, IRX-2 acts on multiple immune-system cell types, including DCs, T cells, and NK cells, to overcome tumor-mediated immunosuppression. In clinical settings, IRX-2 is administered as part of a 21-day neoadjuvant regimen, which includes additional pharmacologic agents (low-dose cyclophosphamide, indomethacin, and zinc) to promote anticancer immunoresponses. In a Phase IIA trial in 27 patients with surgically resectable, previously untreated HNSCC, neoadjuvant IRX-2 increased infiltration of T cells, B cells, and DCs into tumors and was associated with radiological reductions in tumor size. Event-free survival was 64% at 2 years, and overall 5-year survival was 65%. Follow-up and data analysis are under way in the multicenter, randomized, Phase IIB INSPIRE trial evaluating the IRX-2 regimen as a stand-alone therapy for activating the immune system to recognize and attack tumors.
Keywords: head and neck cancer, head and neck squamous cell carcinoma, immunotherapy, cytokines, lymphocytic infiltration, dendritic cells, T cells
Introduction
IRX-2 is a primary-cell-derived biologic consisting of physiologic levels of T-helper type 1 cytokines, produced by stimulating normal donor-derived peripheral blood mononuclear cells (PBMCs) with phytohemagglutinin. The primary active components in IRX-2 are IL2, IL1β, IFNγ, and TNFα. In vitro studies have demonstrated that IRX-2 acts on multiple immune-system cell types – including dendritic cells (DCs), T cells, and natural killer (NK) cells – to overcome tumor-mediated immunosuppression.1–6 In clinical settings, IRX-2 is administered as part of a 21-day neoadjuvant IRX-2 regimen, which includes additional pharmacologic agents (low-dose cyclophosphamide, indomethacin, and zinc) to promote an anticancer immunoresponse.7,8 In a Phase IIA trial in 27 patients with surgically resectable, previously untreated head and neck squamous cell carcinoma (HNSCC), neoadjuvant IRX-2 increased infiltration of T cells, B cells, and DCs into tumors and was associated with radiological reductions in tumor size. Event-free survival was 64% at 2 years. Overall survival was 65% at 5 years, and was longer for patients with lymphocytic infiltration greater versus lower than average.9
Currently under way, the multicenter, randomized, Phase IIB INSPIRE (IRX-2 Neoadjuvant Therapy in Head and Neck SCC to Provide Immune Response Enhancement) trial (NCT02609386) is being conducted to evaluate the IRX-2 regimen as a stand-alone agent for activating the immune system to recognize and attack tumors. Also in progress are trials assessing IRX-2 as neoadjuvant therapy in surgically operable breast cancer (NCT02950259) and cervical/vulvar intraepithelial neoplasia (NCT03267680).
Here, we briefly review the epidemiology of HN cancer (the clinical indication that has been the focus of initial research with IRX-2) and the background for the development of immunotherapy for treatment of HN cancer. We discuss the preclinical research on the complex mechanisms of IRX-2 activity and then summarize both the completed and the ongoing clinical research for this cancer immunotherapy regimen.
Overview of head and neck cancer
Epidemiological background
The category of HN carcinoma encompasses a group of related cancers of the upper aerodigestive tract primarily involving the oral cavity, pharynx (oropharynx, nasopharynx, hypopharynx), and larynx.10 More than 90% of tumors in this category are HNSCC or histologic variants of HNSCC.11 In 2012, there were an estimated 686,000 new cases of HN cancer worldwide: 529,000 were cancers of the lip, oral cavity, and pharynx; 157,000 were laryngeal cancers. The estimated number of deaths from cancers of the lip, oral cavity, and pharynx was 292,000. For laryngeal cancer, the estimated number of deaths was 83,000.11 Considered together, cancers of the lip, oral cavity, nasopharynx, and pharynx represent the seventh-most frequent type of cancer by incidence and the ninth-most common cause of cancer death.11 Although early detection of HN cancer has increased in the last 2 decades, 5-year survival has remained at 50%–59%, despite advances in surgical procedures and improved delivery of chemotherapy and radiation therapy (RT).8
Risk factors for head and neck cancer
Globally, alcohol consumption and tobacco use account for the development of approximately 65% of oral cavity cancers and 85% of larynx cancers. The combined use of tobacco products and alcohol has a multiplicative effect on the risk of developing HN cancer.11 For nasopharyngeal carcinomas, other risk factors include Epstein–Barr virus infection, occupational exposure to wood dust and formaldehyde, radiation exposure, consumption of specific preserved or salted foods, malaria infection, and genetic predisposition.11
A strong causal relationship has been established between human papillomavirus (HPV) type 16 and the development of oropharyngeal cancer.10 It is estimated that 60%–70% of newly diagnosed oropharyngeal cancers in the US and EU are HPV-positive.10 From 1988 to 2004, population-level incidence of HPV-positive oropharyngeal cancer increased by 225% (95% CI 208%–242%), from 0.8 per 100,000 to 2.6 per 100,000, whereas the incidence of HPV-negative cancers (primarily alcohol- and tobacco-related) declined by 50% (95% CI 47%–53%), from 2.0 per 100,000 to 1.0 per 100,000.12 Data from clinical trials suggest that patients with HPV-related cancer have improved response to treatment and live longer than patients with HPV-negative cancer, regardless of the treatment administered.10,13 The 3-year overall survival is approximately 75% for HPV-associated cancer, as opposed to <50% for HPV-negative cancer.13 Immunomodulation plays a significant role in the improvement of clinical outcomes in patients with HPV-associated HN cancer.14,15 Compared to HPV-negative tumors, HPV-positive tumors exhibit higher levels of immunoinfiltration, which correlate with a significantly better outcome.16 In a retrospective analysis of 270 consecutively treated patients with oropharyngeal cancer, 3-year survival for patients with HPV-positive/TILhigh tumors was 96% compared with 76% for HPV-positive/TILmoderate tumors and 59% for HPV-positive/TILlow tumors. For patients with HPV-positive/TILlow tumors, 3-year survival (56%) was similar to that for patients with HPV-negative tumors.17
Management and currently available treatment options for patients with head and neck cancer
Clinicians who treat patients with HN cancer use the specific site of disease, stage of disease, and pathological findings to help guide treatment selection (ie, the appropriate surgical procedure, radiation targets, dose and fractionation, indications for chemotherapy).10 For the approximately 30%–40% of patients who present with stage I or II disease, single-modality treatment with surgery or RT is generally advised.10 Multimodality therapy – most often surgery followed by adjuvant RT or chemoradiation – is recommended for the approximately 60% of patients diagnosed with locally or regionally advanced disease.10 For patients with advanced disease that is recurrent or persistent, the recommended approach is surgery plus concurrent systemic therapy with RT. All forms of treatment are associated with significant functional morbidity in voice, articulation, deglutition, swallowing, or esthetic appearance.
Therapeutic aims for patients diagnosed with unresectable disease or disease with distant metastases are palliation and prolongation of life. Clinicians treating such patients can use RT to target areas of symptomatic disease, administer single-agent or combination systemic chemotherapy, and employ measures to manage symptoms caused by disease spread.10 Such factors as comorbidities and availability of support services can have secondary impact on survival. Participation in clinical trials is recommended for all patients with advanced disease.10 HPV-related cancers have better prognoses, as recognized in the most recent American Joint Committee on Cancer (AJCC) staging system and clinical trials that have focused on the roles of immunotherapy, surgery, chemoradiation, and other treatment-related questions.10,18
Complications associated with currently available treatments for head and neck cancer
Current therapies for HN cancer have been associated with significant complications.13 Surgical resection of cancerous tissues can result in facial deformity, dysarthria, or dysphagia. The severity of these complications can be lessened by utilizing minimally invasive techniques, such as transoral laser microsurgery and transoral robotic surgery when feasible.13 While the proximity of adjacent critical anatomic structures can often limit the aggressiveness of surgical resections, modern techniques of free-tissue reconstruction and microvascular transplantation have extended the volume of tissue that can be removed with preservation of important voice and swallowing functions, as well as appearance.
Acute radiation-induced toxicities include xerostomia, dysphagia, mucositis, dermatitis, and pain.19 Chronic comorbidities associated with RT include xerostomia (60%–90% incidence), grade 3 dysphagia (15%–30%), mandibular osteoradionecrosis (5%–15%), skin fibrosis, chondronecrosis, and lymphedema.13,19 HNSCC patients treated with RT also have a higher likelihood of experiencing occlusive carotid artery disease.13 Radiation-induced sarcomas occur in survivors of HNSCC, with a risk of up to 0.3%.20 Modern intensity-modulated RT can (to some extent) lessen treatment toxicity to normal adjacent tissues by more precisely targeting the radiation to the tumor and reducing the delivery of radiation to healthy tissues. However, since intensity-modulated RT may increase the volume of tissue receiving low-dose radiation,21 the risk of radiation-induced malignancy may be increased. On the other hand, underdosing in an effort to spare the organ can increase the risk of disease recurrence.22
Usually administered concurrently with RT, the two currently preferred chemotherapeutic agents for HN cancers are high-dose cisplatin10,23 and cetuximab, an IgG1 monoclonal antibody that inhibits EGFR and signal transduction.24 Cisplatin-associated complications include xerostomia, dysphagia, neurotoxicity, and renal failure.13 Cetuximab-associated side effects include acneiform rash, mucositis/stomatitis, impaired nutrition, and hypomagnesemia. One of the most serious reported side effects of cetuximab is an anaphylactic reaction that occurs primarily during the first infusion of the drug.24 Cumulative toxicities along with patient fatigue, poor performance status, and the development of drug resistance limit the prolonged use and effectiveness of cisplatin and other agents.
Addressing immunosuppression in head and neck cancer
Impairments of cellular immunity in HNSCC
Profound defects in cellular immunity are frequently identified in patients with HNSCC. These impairments are associated with poor prognosis.25 An analysis of lymph nodes from patients with advanced HNSCC and noncancer controls revealed that uninvolved regional lymph nodes from the HNSCC patients were significantly smaller than those from the healthy controls. While the percentage cross-sectional area of T cells was reduced to a nonsignificant degree for HNSCC nodes compared to control nodes, the density of T cells was significantly reduced. The lymph nodes from HNSCC patients showed pronounced sinus histiocytosis, a pathology characterized by accumulation of myeloid-related cells displaying markers associated with immature DCs and macrophages.26 The accumulation of DCs in lymph-node sinuses suggests a forestalled antitumor response. Having migrated to the lymph-node sinuses from the area draining the tumor, DCs are unable to enter the parenchyma and mature into cells that can present tumor antigens to T cells and empower a targeted immunoresponse.26
As in other cancer types, including lung, renal cell, breast, and colorectal, DC impairment in HNSCC appears to be tumor-mediated.27–31 The number of DCs in the peripheral blood (PB) of HNSCC patients is reduced compared to cancer-free controls, and the degree of reduction is associated with the stage of disease. In one study, a twofold decrease in the total number of DCs was observed for patients with stage 1–2 HNSCC; patients with advanced disease (stages 3–4) showed a fourfold decrease in the number of DCs. In both patient groups, reductions in DC levels were accompanied by accumulation of immature DCs and cells that lacked mature lymphoid- and myeloid-lineage markers.31,32 Based on both in vitro and in vivo evidence, several soluble tumor-derived factors – including IL10, IL6, MCSF, VEGF, and gangliosides and/or prostanoids – are capable of inhibiting DC differentiation, maturation, and/or antigen-presentation functions.33 That DCs isolated from cancer patients are reduced in their capacity to present antigens effectively to T cells is due in large part to reduced expression of antigen-processing components and costimulatory molecules.1,2,30,34 Also downregulated in DCs isolated from HNSCC patients is CCR7, a chemokine receptor required for directing DCs to secondary lymphoid tissues, where they can initiate an adaptive immunoresponse by presenting antigens to T cells.1,2 Epigenetic hypermethylation of CD1A, a gene that encodes an immunoprotein responsible for presenting antigens by dendritic Langerhans cells to T lymphocytes, has been associated with patient survival in HNSCC.35
A key mechanism used by tumors to thwart attack by the immune system is targeted apoptosis of activated T lymphocytes, both at the tumor site and in peripheral circulation.36,37 The proportion of circulating CD8+ T cells that undergo spontaneous apoptosis is elevated in patients with HNSCC compared to sex- or age-matched healthy controls.36 It is likely that antigen-specific effector T cells are eliminated indirectly through the release of tumor-derived microvesicles (MVs; currently referred to as extracellular vesicles) bearing the death-receptor ligand FasL.3,38,39 More recent data indicate that extracellular vesicles isolated from plasma of patients with HNSCC carry a broad range of immunosuppressive proteins – such as FasL, TGFβ1, TRAIL, COX2, CD39/CD73, PDL1, CTLA4, and others – which are biologically active and induce apoptosis in CD8+ T cells and immunosuppression of other immunoeffector cells.40 While CD8+ T cells are significantly more sensitive to apoptosis than CD4+ T cells,36 absolute numbers of both CD8+ and CD4+ T cells are significantly reduced in the PB of HNSCC patients compared to healthy controls. Patients with HN cancer also have fewer invariant NK T cells than healthy individuals,41 and low levels of these cells in the peripheral circulation are significantly associated with inferior clinical outcomes in terms of locoregional recurrence and disease-specific survival.42 These various deficits in cell-mediated immunity occur early in disease and are long-lasting after treatment.
In HNSCC, as in other cancer types, the numbers of tumor-infiltrating lymphocytes, their functions, and their locations in the tumor microenvironment are associated with response to treatment and overall prognosis, and may reflect underlying host immunocompetence.43,44 Although immunosuppressive mechanisms can limit their effectiveness, tumor infiltration by CD8+ T cells is generally associated with positive patient prognosis. In a study of 278 patients with previously untreated HNSCC, higher levels of CD8+ T cells were associated with improved overall survival (HR 0.77, 95% CI 0.64–0.94; P=0.008) and relapse-free survival (HR 0.85, 95% CI 0.72–1.00; P=0.05).44 When age, clinical stage, disease site, comorbidities, HPV status, treatment, and smoking were controlled for in a multivariable Cox model, higher levels of CD8+ T cells were still associated with improved overall survival (HR 0.81, 95% CI 0.67–0.99; P=0.003).44 Likewise, in 83 patients with tonsillar SCC, a significant positive correlation was observed between CD8+ T-cell infiltration and good clinical outcome, defined as 3-year relapse-free survival (P<0.001).45 Higher CD8+ levels have also been associated with favorable outcomes for metastatic disease. In a study of 33 patients with oropharyngeal and hypopharyngeal carcinoma, 5-year disease-free survival was 91% when there was increased intraepithelial infiltration of CD8+ T cells (>1.29 CD8+ cells per 100 tumor cells) in metastatic lymph nodes versus 53% when there was not.46
Evaluation of infiltration by CD4+ T cells is complicated by the fact that in tumors, CD4+ cells can be activated, anergic, or regulatory.43 Secondary markers like CD69 (a cell-surface glycoprotein that is rapidly upregulated on T cells upon activation)47 and FOXP3 (a transcription factor that serves as a regulatory T-cell [Treg] lineage marker)48 can be helpful in differentiating among these cellular states. In 84 newly diagnosed, previously untreated HNSCC patients, univariate analysis uncovered positive associations between high levels of CD4+ CD69+ T cells and both better locoregional tumor control and overall survival.49 For patients lacking CD4+ CD69+ T cells at diagnosis, survival at 2 years was 42% (95% CI 25%–68%) versus 89% (95% CI 77%–100%) for patients with the highest levels of CD4+ CD69+ T cells (P=0.01). In multivariate analysis, the level of CD4+ CD69+ T cells influenced overall survival probability (relative risk 0.79, P=0.031).49 In this same study, infiltration by CD4+ FOXP3+ T cells was also positively correlated with better locoregional control, but exhibited no correlation with overall survival.49 In a study of 278 patients with previously untreated HNSCC, higher levels of FOXP3+ tumor-infiltrating lymphocytes were associated with improved overall survival (HR 0.83, 95% CI 0.69–1.00; P=0.05).44 However, in a study in patients with local recurrence of resected HN cancer, recurrence-free survival was significantly worse for patients with tumors that contained a median of six or more FOXP3+ cells per ten high-power fields compared to those that contained fewer than or equal to six FOXP3+ cells.50 In an analysis of 83 patients with tonsillar SCC, no differences in levels of FOXP3+ T cells were observed in tumors from patients who achieved 3-year relapse-free survival versus those who did not (mean 27.9 vs 24.6 tumor-infiltrating cells). At the same time in this study, a high CD8+:FOXP3+ ratio was linked to good clinical outcomes when adjusted for prognosis (P=0.04).45 These somewhat discrepant results can be explained by the fact that to date, no specific marker for Treg exists and by the finding that FOXP3 is not expressed in all induced Treg (although FOXP3 is expressed, albeit transiently, in activated effector CD4+ T cells).51
Abnormalities in B cells, mediators of humoral immunity, have also been reported in patients with HNSCC. Decreased frequency of CD19+ B lymphocytes in the peripheral circulation of HNSCC patients is associated with an increase in percentage of activated CD19+ HLA-DR+ B cells.52 Because activated B cells are CD39+/CD73+, produce immunoinhibitory adenosine, and inhibit functions of activated T cells,53 they are considered “regulatory B cells.” Their accumulation in the periphery and lymph nodes of patients with HNSCC downregulates immunoresponses. On the other hand, accumulations of other B-cell subsets (eg, CD20+ B cells) in lymph nodes of HNSCC patients have been associated with favorable outcomes.46 Therefore, changes in the ratio of B-cell subsets (the B cell:Breg ratio) represent an important component of antitumor immunity.
Key mediators of innate immunosurveillance of cancer, NK cells are also functionally compromised in HNSCC patients. NK cells isolated from the PB of cancer patients undergo spontaneous apoptosis and exhibit reduced necrotic and apoptotic tumoricidal activity.54–57 These impairments have demonstrated clinical repercussions. In a study of 263 patients with previously untreated HNSCC, quantitated NK-cell cytotoxicity was correlated inversely with subsequent death from the disease (P=0.05), regional metastases (P=0.008), and distant metastases (P=0.03).58
Immunotherapeutic approaches for HNSCC
Given that numerous immunosuppressive mechanisms operating in patients with HNSCC interfere with cellular and humoral antitumor immunity and have a negative impact on responses to therapy and outcome, novel strategies designed to rejuvenate, restore, and activate antitumor immunoresponses are a rational therapeutic approach to HNSCC. Immunotherapeutic approaches currently under investigation for the treatment of HNSCC include anticancer vaccines that sensitize the immune system to tumor-associated antigens, oncolytic viruses that target and selectively kill tumor cells, monoclonal antibodies against immunoregulatory or tumor-growth-promoting signaling molecules, cell-based therapies, such as adoptive transfer of ex vivo–activated T cells and NK cells, and immunomodulators that boost host antitumor immunoresponse through generalized immune-system stimulation.25,59
The expression of inhibitory receptors, such as PD1 and CTLA4, marks exhausted T cells that have lost normal function, exhibiting reduced proliferative capacity or cytolytic activity. Blocking these inhibitory receptors (or targeting their ligands), and thereby enhancing antitumor immunity, has thus emerged as a promising immunotherapeutic approach for the treatment of HNSCC and other cancers. Pembrolizumab and nivolumab, monoclonal antibodies against PD1, have both been approved by the US Food and Drug Administration for treatment of platinum-refractory recurrent or metastatic HNSCC. However, since the majority of patients do not benefit from anti-PD1 therapy alone, combination therapy employing these agents along with vaccines or other immunomodulators is under active study.60,61
Cytokines, a diverse group of small proteins secreted by cells for signaling and communication, represent one class of immunomodulators. The many functions of cytokines include control of cell proliferation and differentiation as well as regulation of angiogenesis and immune and inflammatory responses. The cytokine IL2 – produced by antigen-stimulated CD4+ T-helper cells and (to a lesser extent) CD8+ and NK T cells and activated DCs – was initially viewed as the primary factor driving augmentation of an effector lymphocyte immunoresponse;62 more recently, it has been determined that IL2 also plays a crucial role in thymic Treg differentiation.48 IL2 was thus evaluated as therapy for various cancers in several clinical trials. Based on data from clinical and experimental studies demonstrating that locally injected IL2 could modulate host immunoreactivity and mediate antitumor effects, locoregional administration of IL2 was considered a promising therapy for HNSCC, a malignancy that tends to grow and recur locally surrounded by an extensive lymph-node network.63 Ultimately, however, clinical outcomes were not significantly improved, despite evidence in some studies of upregulated antitumor responses in HNSCC patients following peritumoral, intranodal injection of high-dose recombinant IL2.63 Nevertheless, when nonrecombinant IL2 was administered in low doses, temporary regression of recurrent HNSCC was observed.64–66 These results suggested that more positive clinical outcomes might be achieved if IL2 were administered perilymphatically to tumor-involved lymph nodes as a component of a low-dose mixture with other human cytokines.
IRX-2 is a primary-cell-derived biologic consisting of physiologic levels of T-helper type 1 cytokines, produced by stimulating normal donor PBMCs with phytohemagglutinin. The primary active components in IRX-2 are IL2, IL1β, IFNγ, and TNFα. Other IRX-2 components include IL6, IL8, GCSF, and GMCSF (Table 1).1 In vitro, IRX-2 acts on multiple immune system cell types – including DCs, T cells, and NK cells – to overcome tumor-mediated immunosuppression.1–6 Insights into the mechanism of action of IRX-2 gleaned from these in vitro studies are discussed in more detail in the next section.
 | Table 1 Components of the IRX-2 regimen |
In clinical trials, the IRX-2 biologic is administered as part of the neoadjuvant IRX-2 regimen, which includes additional pharmacologic agents to promote an anticancer immunoresponse (Table 1):7 a noncytotoxic dose of cyclophosphamide for depleting and inhibiting Treg cells,67 indomethacin for inhibiting the synthesis of immunosuppressing prostaglandins by suppressor monocytes/macrophages,68 zinc gluconate for mitigating impairments of cell-mediated immunity observed in zinc-deficient cancer patients,69,70 and omeprazole, a proton-pump inhibitor, for protecting the stomach against the erosive effects of indomethacin8 and at the same time perhaps contributing to improved survival.71 The standard IRX-2-immunotherapy regimen is administered for 21 days (Figure 1). On treatment day 1, patients receive the low-dose intravenous infusion of cyclophosphamide, followed by 21 days of oral indomethacin, oral zinc, and oral omeprazole. For 10 days, starting on day 4, the IRX-2 biologic is administered via subcutaneous injections bilaterally into the upper neck near the sternocleidomastoid muscle-insertion regions. Injection of the IRX-2 biologic into these mastoid regions ensures drainage into the regional lymphatic vessels that are primary or secondary basins for the primary cancer and where physiologic quantities of the IRX-2 biologic can restore the lymph-node microenvironment and enable DC activation of T cells against endogenous tumor peptides.8
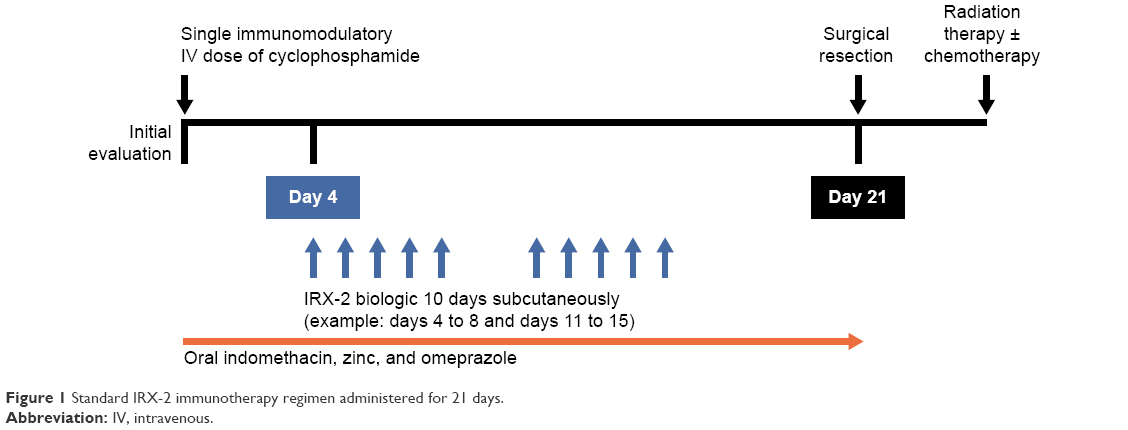 | Figure 1 Standard IRX-2 immunotherapy regimen administered for 21 days. |
Mechanisms responsible for IRX-2 immunorestorative activity
Data from preclinical studies demonstrate that IRX-2 acts on many cell types through multiple mechanisms to augment the immunoresponse and counteract tumor-induced immunosuppression (Figure 2). In patients treated with IRX-2, sinus histiocytosis regresses, lymph nodes increase in size, nodal T cells expand, and T-cell infiltration of tumors is increased.8,9,26 In vitro, treatment with IRX-2 promotes maturation of immature DCs into antigen-presenting DCs.1,2 These findings suggest that one way in which IRX-2 overcomes immunomediated suppression is by directly activating and maturing the nodal DC population, thereby promoting their productive interactions with T cells.26 This model is supported by in vitro data.1 Monocyte-derived DCs isolated from PBMCs of healthy human donors and then incubated with IRX-2 exhibit morphology characteristic of mature DCs: they become nonadherent and develop extensive cellular projections. Consistent with these morphological changes, monocyte-derived DCs treated with IRX-2 downregulate CD1a (a marker expressed at high levels by immature DCs) and upregulate CD83 (a defining marker of DC maturation). Hypermethylation of the CD1A promoter, presumably leading to downregulation of CD1A expression, has been associated with improved survival in HNSCC.35 Also increased is the expression of factors required for effective T-cell activation: major histocompatibility complex (MHC) class II molecules, costimulatory molecules CD86 and CD40, and ICAM1.1
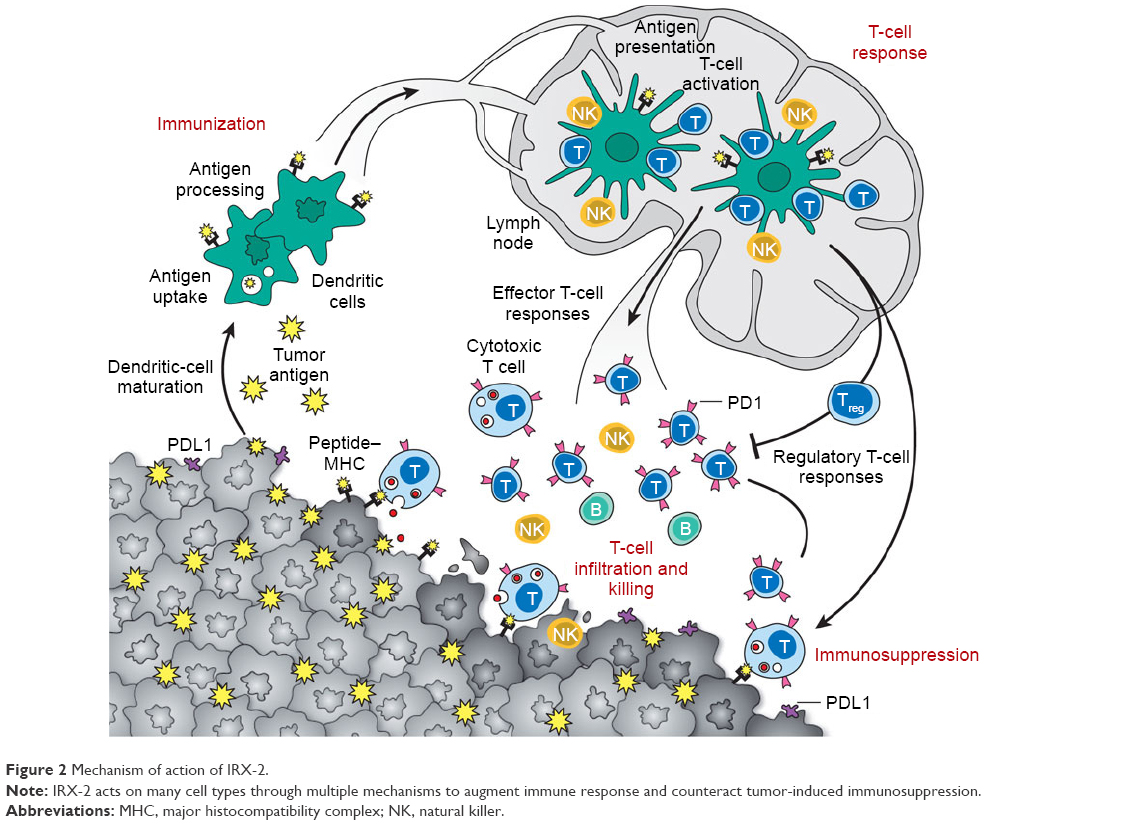 | Figure 2 Mechanism of action of IRX-2. |
Incubation with IRX-2 induces critical functional changes in monocyte-derived DCs that are indicative of maturation, including dose-dependent reductions in endocytic capability1 and upregulation of the components of DC antigen-presenting machinery (LMP2, TAP1, TAP2, tapasin, and calreticulin).2 Monocyte-derived DCs treated with IRX-2 can stimulate the proliferation of T cells, increase production of IL121 (a cytokine integral to the promotion of a T-helper 1 response), and induce high-potency cytotoxic T lymphocytes.2 When these IRX-2-treated cells are pulsed with tumor-cell lysates, they carry a high density of tumor antigen-derived peptides on their surfaces; when primed with these monocyte-derived DCs, CD8+ T lymphocytes isolated from patients with HNSCC demonstrate high cytotoxicity, efficiently killing target tumor cells.2
In immune-impaired patients with HNSCC, NK-cell function is restored by IRX-2 treatment. The frequency of NK cells in PBMCs isolated from HNSCC patients is comparable to the frequency of NK cells in PBMCs isolated from healthy age- and sex-matched controls. However, the frequency of NK cells from HNSCC patients that express the activating receptors NKG2D, NKp30, and NKp46 is significantly lower than the frequency of NK cells from matched controls. In NK cells that do express these receptors, expression levels are lower in HNSCC patients.5 Conversely, the frequency of NK cells from HNSCC patients that express the inhibitory receptor NKG2A is greater than that in matched controls. Extending these findings, in flow-based cytotoxicity assays, culturing PBMCs from HNSCC patients with IRX-2 for 16 hours restores expression levels of NKp30 and Nkp46 and increases cytotoxicity against K562 cells (cells of human leukemic origin). Further, NK cells isolated by magnetic bead separation and treated with IRX-2 demonstrate enhanced cytotoxicity against cells from PCI-13 (an HN cancer cell line).5 IRX-2 has also been shown to insulate against TGFβ1-mediated suppression and downregulation of NKp30 and NKG2D surface proteins.5
In an in vitro model simulating the human tumor microenvironment, IRX-2 influenced T-cell polarization, promoting the proliferation of T-effector cells without inducing the expansion of Treg. In this model, coculturing conventional CD4+CD25− T cells with autologous immature DCs and irradiated tumor cells in the presence of rhIL2, IL10, and IL15 promotes their proliferation and differentiation into Treg. The addition of IRX-2 to the coculture medium decreases the proportion of the cells in the system exhibiting a Treg phenotype without changing the rate of T-cell proliferation or affecting cell viability. When conventional T cells were cultured for 10 days in the absence or presence, respectively, of IRX-2, the mean percentages of expressing cells were 53% versus 24% for CD25, 55% versus 20% for CD122, 57% versus 25% for CD132, 57% versus 29% for CD152, and 49% versus 28% for FOXP3. In the absence of IRX-2, the mean percentages of T cells expressing the immunoregulatory cytokines IL10 and TGFβ1 were 57% and 62%, respectively, while the mean percentage of IFNγ-expressing cells was only 10%. When cultured in the presence of IRX-2, the percentages of IL10- and TGFβ1-expressing cells were significantly lower (22% [P<0.01] and 34% [P<0.01], respectively), whereas the percentage of IFNγ-expressing cells increased significantly to 34% (P<0.01). These findings were supported by measurements of cytokine levels in cell supernatants.6
It has been reported that inhibition of the Akt–mTOR pathway with rapamycin promotes Treg expansion,72,73 and conversely that activation of the Akt–mTOR pathway antagonizes the induction of FOXP3+ cells in mice.74 Through relief of inhibition by the P13K–Akt pathway and via other mechanisms, IRX-2 protects CD8+ effector T cells from apoptosis mediated by tumor-derived FasL+ MV.3,4 Normally, when primary-blood-derived CD8+ T cells are cocultured with MVs isolated from culture supernatants of the FasL-transfected PCI-13 cell line, they undergo apoptosis, as evidenced by decreases in caspase activation and annexin binding. CD4+ T cells also exhibit sensitivity to MV-mediated apoptosis, although to a lesser extent than CD8+ T cells. For both CD8+ and CD4+ T cells, pretreatment with IRX-2 provides protection against MV-induced apoptosis by blocking the signaling cascade at multiple levels.3,4 1) IRX-2 downregulates the expression of Fas on the surface of T cells, making these cells more resistant to targeted apoptosis mediated by Fas-loaded MVs. When activated primary CD4+ and CD8+ T cells are incubated with MVs, both the percentage of cells expressing Fas and the expression levels of Fas increase. When these cells are instead treated with IRX-2, Fas expression is significantly decreased compared to the outcome in untreated controls (P<0.02). Pretreating cells with IRX-2 prior to incubation with MVs blocks upregulation of Fas (P<0.02).4 2) IRX-2 increases expression of cFLIP4 (a modulator of the extrinsic apoptotic pathway), inhibiting interaction of the CASP8 prodomain with the RIP1 death domain and thereby regulating CASP8-dependent NFκB activation.75 Consistent with this result, IRX-2-mediated protection from MV-induced apoptosis is enhanced in Jurkat cells overexpressing cFLIP. Cells overexpressing cFLIP have been found to be almost entirely resistant to MV-induced cytochrome C release and caspase activation. 3) IRX-2-induced modulation of survival proteins is mediated by Akt. Pretreatment of Jurkat cells with IRX-2 abolishes time-dependent Akt dephosphorylation induced by MVs,3 whereas blocking Akt signaling through use of an antibody with specificity to Akt1/2 cripples the ability of IRX-2 to protect against apoptosis. In the presence of the Akt inhibitor, IRX-2-pretreatment-induced increases in the expression of the antiapoptotic factors cFLIP and Bcl2 and decreases in the expression of proapoptotic factors, such as Bax, were abrogated.4
IRX-2 has also been shown to increase antigen-specific T-cell responses to several tumor antigens using a number of different vaccination platforms in murine models.76,77 In one study, coadministration of IRX-2 enhanced the T-cell response to a WT1 synthetic long-peptide vaccine containing multiple MHC class I and class II binding epitopes.77 In another study, when administered as an adjuvant, IRX-2 amplified the T-cell-specific response (measured in spleen or lymph-node cells by IFNγ ELISpot assay) to a dominant mouse peptide (NFT) derived from human PSMA.76 Immunoresponses increased, regardless of whether the antigen was delivered via irradiated cells expressing human PSMA, via NFT peptide conjugated to KLH, or via NFT peptide emulsified in incomplete Freund’s adjuvant. Whereas one adjuvant administration of IRX-2 with antigen did not enhance T-cell response, the response was amplified with a minimum of four additional daily administrations of IRX-2 (without the antigen), and increasing the number of additional IRX-2 administrations to nine had no further impact. The amplified immunoresponse also mediated tumor protection in vivo: mice immunized with IRX-2 and antigen exhibited delayed tumor progression following challenge with PSMA-expressing tumor cells.76
IRX-2 clinical trials already completed
Phase I and Phase IIA trials and substudies in HNSCC
The safety and efficacy of the IRX-2 regimen have been evaluated in several clinical studies. An initial Phase I trial of the IRX-2 regimen was performed by the Instituto Nacional de Cancerología in Mexico City. A total of 42 patients were treated with either a 10-day course of 1 mL IRX-2 biologic (200 units/mL IL2 equivalence) or a 20-day course of 2 mL IRX-2 biologic administered perilymphatically.78 Of those patients, 39 subsequently underwent surgical resection and 22 stage IV patients received additional RT. Prior to surgical resection, a partial clinical response (defined as >50% tumor reduction) or a complete clinical response was observed in 42% of patients. Comparison of post-treatment biopsies or surgical specimens with pretreatment samples showed that 90% of patients had a reduction in tumor volume and an increase in leukocyte infiltration. An improvement in tumor-related symptoms characterized by reduced pain, reduced bleeding from the tumor surface, improved breathing and phonation, and reduced dysphagia was observed in 24 of the 42 patients. After 36 months, IRX-2 patients showed reduced rates of recurrence, increased mean survival times, and increased overall survival compared to institutional controls. There were no differences in outcome parameters between the 10-day and 20-day treatment regimens. No significant toxicities were observed.78
In a second Phase I trial, the 21-day IRX-2 regimen was administered to 13 patients with advanced HNSCC who had failed surgery and/or RT. At the conclusion of treatment, patients were followed for up to 4 weeks. Over the course of the study, all 13 study participants experienced at least one treatment-emergent adverse event (AE), but the majority of these were categorized as mild or moderate in terms of severity. Lymphopenia was reported in four patients, and anemia, abdominal pain, dysphagia, anorexia, headache, and dyspnea were each reported in three patients. No deaths or study discontinuations were attributed to treatment with the IRX-2 biologic. On treatment day 21, clinical response to the IRX-2 regimen was evaluated by computed tomography (CT). Of the eight patients who had interpretable scans, one had a complete response to treatment, five showed stable disease, and two exhibited progressive disease. In this study, the IRX-2 regimen was well tolerated in patients with HN cancer and capable of effecting tumor responses and stabilizing disease, even in advanced cases.7
In a Phase IIA trial, neoadjuvant IRX-2 was well tolerated and prolonged overall survival in patients with surgically resectable, therapy-naïve HNSCC (stages II–IV). All 27 patients enrolled in this study completed the 21-day IRX-2 regimen, and 26 completed surgery. Nineteen patients received postsurgical RT, and ten patients received both postsurgical RT and chemotherapy. During treatment and the 30-day postoperative period, seven patients experienced eight serious AEs. With the exception of a single postoperative wound infection, no serious AEs were deemed treatment-related. Patients were followed for more than 3 years. Disease-free survival was 72% at 1 year, 64% at 2 years, and 63% at 3 years. Overall survival was 92% at 1 year, 73% at 2 years, 69% at 3 years, and 65% at 5 years.8
Since tumor infiltration by certain lymphocyte subsets can predict improved clinical outcomes in HN cancer,43,44 pretreatment tumor biopsies from 25 Phase IIA study participants were compared to matched primary-tumor surgical specimens for lymphocytic infiltration using hematoxylin and eosin staining and immunohistochemistry (IHC) with antibodies against CD3, CD4, CD8, CD20, and CD68. Increases in lymphocytic infiltration following IRX-2 treatment occurred in the majority of patients (20 of 25) with CD3+CD4+ T cells and CD20+ B cells primarily localized to peristromal tumors and CD3+CD8+ T cells and CD68+ macrophages localized within the tumor. High average values of lymphocytic infiltration at surgery were associated with increases in tumor necrosis and reductions in primary-tumor size, both indicators of improved clinical outcomes. The strongest correlation was observed for CD20+ B-cell infiltration. A positive correlation was also observed between survival at 5 years and high overall lymphocytic infiltration. Overall survival was longer for patients with lymphocytic infiltration greater than the median (Figure 3). The level of CD3+ T lymphocytes was the strongest predictor of survival.9
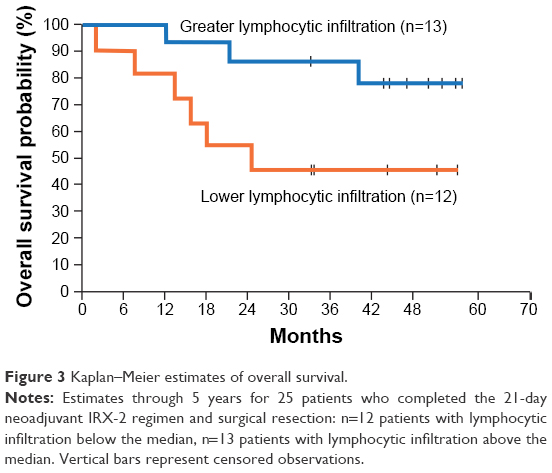 | Figure 3 Kaplan–Meier estimates of overall survival. |
In a substudy undertaken to define the mechanisms responsible for the increase in lymphocytic infiltration with neoadjuvant IRX-2, matched pre- and post-treatment tumor specimens from seven Phase IIA trial patients were interrogated with two immunoprofiling technologies, multiplex IHC (PerkinElmer, Waltham, MA, USA) and transcriptome analysis (NanoString Technologies, Seattle, WA, USA). Multiplex IHC provided detailed visualization and quantification of various immune cells in the tumor microenvironment. Supporting the Phase IIA pathology findings, five of the seven patients exhibited substantial increases in CD68+ T cells and four had substantial increases in the density of CD4+ T cells. Extending Phase IIA study results, expression of the immunoregulatory protein PDL1 was increased in four patients. Transcriptome analysis provided a global snapshot of the tumor microenvironment and quantitative information on immune-cell subsets. The expression levels of 770 genes, including 109 that define 24 different immune-cell types, 30 encoding known cancer/testis antigens, >500 encoding critical proteins in immunoresponse pathways, and 40 pan-cancer reference genes, were assayed using the NanoString Pancancer Immune Profiling panel. Consistent with the preclinical findings that IRX-2 activates multiple immune cells, there were two- to threefold increases in gene expression for most immune-cell types following IRX-2 treatment. The most marked increases were for B cells and DCs. Induction of multiple gene-family members within the chemokine profile (such as CCLs, CXCLs, and CCRs) after IRX-2 treatment suggest that lymphocytic infiltration may be driven in part by IRX-2-mediated production of chemoattractants. In keeping with the multiplex IHC finding of an increase in PDL1 protein expression, transcriptome analysis revealed upregulation of PDL1 gene expression after treatment with IRX-2.79 Upregulation of CTLA4 was also noted. Increased expression of these checkpoint inhibitors is indicative of an adaptive immunoresponse triggered by successful IRX-2 activation of antitumor immunofunctions.
In a second substudy, multiparameter flow cytometry was used to investigate IRX-2-induced changes in PB from 25 Phase IIA patients. Following treatment with IRX-2, there was a significant decrease in the number of circulating B cells (P<0.005). This result, along with increases in tumor infiltration by B cells and upregulation of chemokines, supports the premise that IRX-2 promotes migration of B cells from the blood to tumor sites and tumor-draining lymph nodes. A role for IRX-2 in promoting the differentiation of naïve (CD3+CD45RA+CCR7+) T cells into memory T cells was also elucidated. Whereas the total number of T lymphocytes did not change following treatment with IRX-2, the mean number of peripherally circulating naïve T cells and central memory T cells (CD3+CD45RA−CCR7+CD27+) decreased, and the mean number of effector memory T cells (CD3+CD45RA+CCR7−CD27−) increased moderately.80
Pilot study of IRX-2 treatment for cervical cancer
While the majority of IRX-2 studies have been conducted in patients with HN cancer, IRX-2 has also been evaluated for the treatment of cervical cancer. In a pilot study, the 21-day IRX-2 regimen (minus omeprazole) was administered to ten consecutive patients with clinically staged IB1, IB2, and IIA therapy-naïve cervical cancer. Patients were scheduled for radical hysterectomy on treatment day 21. All ten patients completed the IRX-2 regimen, and none experienced toxic side effects attributable to treatment with the biologic. However, all ten experienced indomethacin-related gastric symptoms and discontinued the nonsteroidal anti-inflammatory drug prior to treatment day 21. Clinical response to the IRX-2 regimen was assessed on treatment day 21, prior to scheduled surgery. A partial response to treatment, defined as >50% reduction in the product of the two longest perpendicular diameters of the tumor, was identified in three patients. A minor response to treatment, defined as >25% reduction, occurred in two patients. Seven patients underwent scheduled hysterectomies, and tumor fragmentation was detected in the resected tumors from all five clinical responders. At a median follow-up of 30 months (6–31 months), all but two patients were disease-free. The results of this pilot study provisionally support the ability of IRX-2 to induce tumor responses in cervical cancer.81
Ongoing IRX-2 clinical trials
The Phase IIB INSPIRE trial
Follow-up and data analysis are under way in an international, multicenter, randomized trial – INSPIRE (NCT02609386), conducted with enrollment of 105 patients. The primary aim of INSPIRE was to determine the event-free survival of subjects treated with IRX-2 versus controls. Secondary aims were to evaluate further the safety of the IRX-2 regimen, elaborate the mechanisms by which IRX-2 restores and activates the immune system to recognize and attack tumors, and determine overall survival.
The design of the INSPIRE trial is summarized in Figure 4. To be eligible for enrollment, patients had to be at least 18 years old, have previously untreated stage II, III, or IVa cancer surgically resectable with curative intent, and have a Karnofsky Performance Status ≥70%. Enrolled patients were randomized 2:1 to either the IRX-2 biologic regimen arm or the standard IRX-2-regimen control arm (the IRX-2 regimen minus the biologic). During the neoadjuvant stage of INSPIRE, at least 21 days before resection, patients in both arms were to receive the standard IRX-2 regimen daily (Figure 2). On 10 days between days 4 and 21 of the neoadjuvant treatment period, patients in the IRX-2 biologic regimen arm were to receive the IRX-2 biologic administered bilaterally by subcutaneous injection at or near the mastoid-insertion sites of the left and right sternocleidomastoid muscles. By delivering IRX-2 in the area of tumor-draining lymph nodes rather than systemically, the biologic is positioned to mobilize antigen-presenting cells and enhance DC function, as well as directly to promote the proliferation of T cells and the maturation of T cells into cytotoxic lymphocytes.
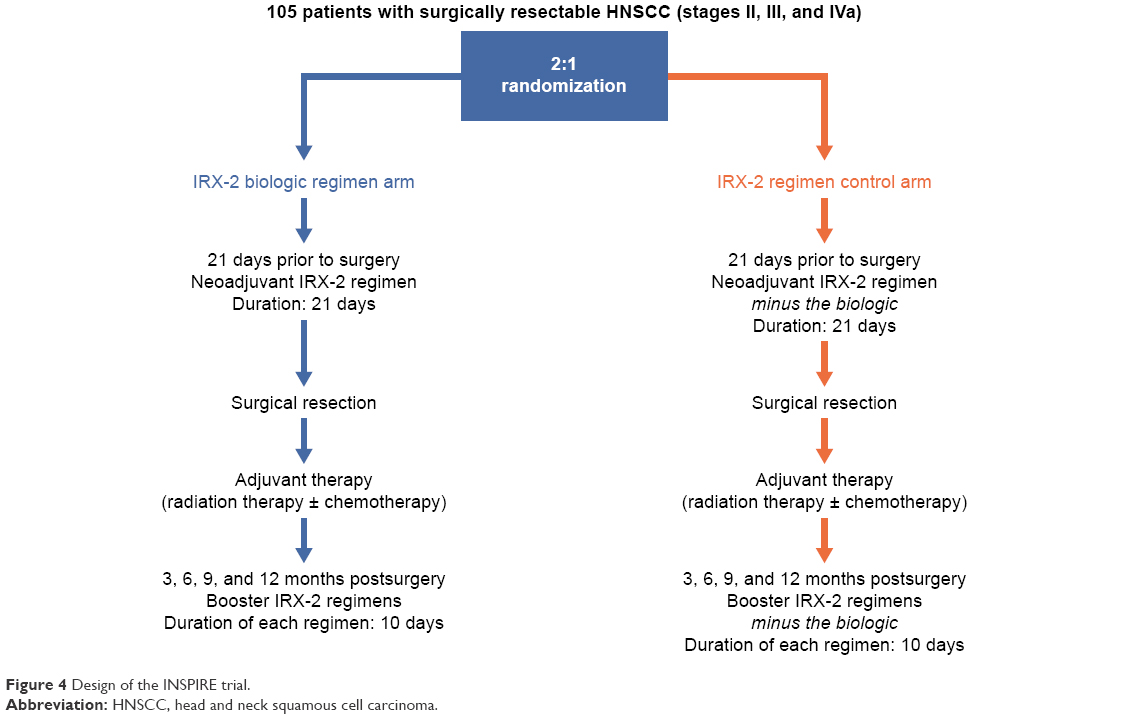 | Figure 4 Design of the INSPIRE trial. |
After tumor resection, patients in both arms of INSPIRE were eligible to receive standard adjuvant RT or chemotherapy according to National Comprehensive Cancer Network guidelines, followed by an adjuvant standard IRX-2 booster regimen at 3, 6, 9, and 12 months. During each booster period, patients in the IRX-2 biologic regimen arm were to receive the IRX-2 biologic administered by subcutaneous injection for 5 consecutive days into bilateral deltoid regions (Figure 4).
The end points of INSPIRE are summarized in Table 2. The primary end point for INSPIRE is event-free survival assessed by chest CT scans performed annually for 4 years and by physical examinations administered at routine follow-up visits every 3 months after surgery for the first 2 years and every 6 months during years 3 and 4. Secondary end points include overall survival and the safety of neoadjuvant and booster regimens.
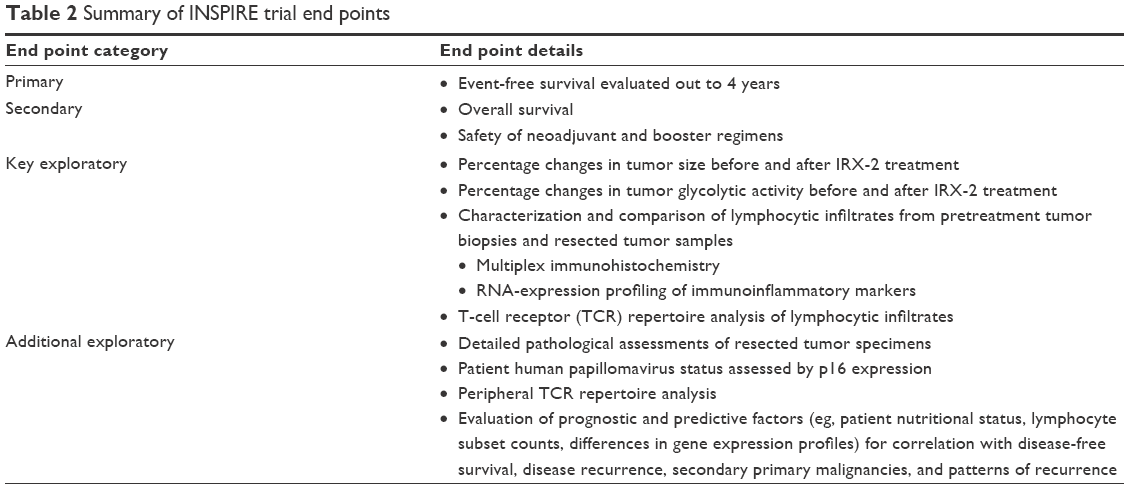 | Table 2 Summary of INSPIRE trial end points |
Several exploratory end points have been defined for the INSPIRE trial to further understanding of the mechanism of action of the biologic, to quantify changes in the tumor microenvironment induced by IRX-2, and to identify a pretreatment prognostic phenotype predictive of responsiveness to the IRX-2 immunotherapy regimen. The exploratory end points include percentage changes in tumor size assessed via comparison of CT or magnetic resonance imaging obtained before treatment with the IRX-2 regimen and just prior to surgery, percentage changes in tumor glycolytic activity assessed via comparison of fludeoxyglucose positron-emission tomography obtained pretreatment and just prior to surgery, and characterization and comparison of lymphocytic infiltrates from pretreatment tumor biopsies and resected tumor samples. Multiplex IHC will be employed to quantify cellular infiltrates and determine their degree of activation. Markers of interest for lymphocytes include but are not limited to CD3, CD4, CD8, CD68, CD45RO, and FOXP3. Tumor-cell markers include PDL1 and MHC class I and II molecules. Transcriptome analysis will provide quantitative information on immune-cell subsets and insights regarding mechanisms possibly responsible for any changes in lymphocytic infiltration. To determine the effects of IRX-2 on antigen-receptor diversity of the infiltrating T-cell population, T-cell receptor repertoire analysis will be performed on extracted RNA.
Detailed pathological assessments of resected tumor specimens will be performed in the INSPIRE trial, with attention to the size and extent of the tumor, degree of tumor differentiation, number and location of involved lymph nodes, extracapsular extension of the tumor, proximity of the tumor to resection margins, perineural involvement, and vascular invasion. Further, since data from clinical trials suggest that patients with HPV-related cancer have a better prognosis regardless of treatment administered than patients with HPV-negative cancer, the HPV status of each patient will be determined on the basis of p16 expression, and the relationship between HPV status and the clinical benefit conferred by IRX-2 treatment will be explored.
T-cell receptor-repertoire analysis will be performed on T cells isolated from PB prior to and following treatment with IRX-2 during the first year of the trial. In addition, such factors as nutritional status (body-mass index, albumin levels), absolute lymphocyte counts, lymphocyte-subset counts, and differences in histology of surgical specimens and gene expression profiles will be evaluated for correlation with disease-free survival, disease recurrence, secondary primary malignancies, and patterns of recurrence.
Phase I trial of neoadjuvant IRX-2 in surgically operable breast cancer
A Phase IB trial of the neoadjuvant IRX-2 regimen in women with early-stage breast cancer is under way at the Providence Portland Medical Center in Oregon (NCT02950259). Up to 40 patients with surgically operable invasive breast cancer of any receptor subtype (including triple-negative) are being treated in this trial. The primary objective is to establish the safety and tolerability of the treatment. Secondary objectives include evaluation of changes in lymphocyte infiltration by IHC, transcriptome analysis, and deep sequencing of the T-cell repertoire.
Phase II randomized trial of IRX-2 in cervical or vulvar intraepithelial neoplasia
A Phase II randomized controlled trial of the IRX-2 regimen delivered into the cervix or vulva of patients with HPV-positive cervical intraepithelial neoplasia grade 3 (CIN3) or vulvar intraepithelial neoplasia grade 3 (VIN3) is under way at the University of Southern California (NCT03267680). Thirty patients with CIN3 will be randomized to the IRX-2 regimen or placebo, and 30 patients with VIN3 will be randomized to the IRX-2 regimen or placebo. The primary end point is pathological response through 25 weeks. Secondary end points include incidence and severity of AEs, measures of cervical or vulvar lymphocytic infiltration, changes in HPV viral load, and induction of cellular and humoral responses to HPV.
Conclusion
Patients with HNSCC have multiple profound defects in cellular immunity, likely mediated by tumor-induced immunosuppression.25 Therefore, a sensible treatment strategy for HNSCC is multifaceted immunotherapy with the goal of relieving immunosuppression and restoring antitumor immune functions. The IRX-2 biologic is an injectable cancer immunotherapy composed of multiple cytokines purified from the stimulated PBMCs of normal donors. IRX-2 acts on multiple immune-cell types through several molecular mechanisms to counteract tumor-induced immunosuppression. IRX-2 induces activation and maturation of DCs,1,2 enhances NK-cell cytotoxicity,5 promotes the expansion and antitumor activity of effector T cells,6 and protects those same T cells from targeted tumor-induced apoptosis.3,4 In clinical settings, the IRX-2 biologic is administered as part of a multi-agent immunotherapy regimen that includes low-dose cyclophosphamide, indomethacin, and zinc to increase immunoactivation further. In a Phase IIA trial, neoadjuvant treatment of HNSCC patients with the IRX-2 immunotherapy regimen was associated with enhanced infiltration of lymphocytes into tumors and a favorable safety profile.8 Lymphocytic infiltration was associated with radiological reductions in tumor size and improvement in overall survival.9 Data from a Phase IIA substudy employing two immunoprofiling technologies – sequential fluorescent multiplex IHC and transcriptome analysis – provide confirmatory evidence that neoadjuvant treatment with IRX-2 increases the infiltration of T cells, B cells, and DCs into tumors, that this infiltration may be mediated by increased expression of multiple chemokine pathways, and that these cells are functionally active. Successful reestablishment of antitumor functions by IRX-2 stimulates an adaptive immune response characterized by upregulation of checkpoint molecules, such as CTLA4 and PDL1.79
Follow-up and data analysis are under way in INSPIRE (NCT02609386), a Phase IIB multicenter randomized controlled trial in previously untreated oral-cavity HNSCC patients evaluating the clinical activity of the IRX-2 regimen as stand-alone therapy for restoring and activating the immune system to recognize and attack tumors. Clinical outcome data will be available beginning in 2019. Preliminary data from exploratory studies – eg, histochemical, immune-cell signature, and T-cell-receptor profiling analyses – may be available in 2018. Other indications for which the IRX-2 regimen is being assessed include neoadjuvant therapy in surgically operable breast cancer (NCT02950259) and therapy for CIN and VIN (NCT03267680). Clinical studies evaluating combination therapy with IRX-2 and immune checkpoint inhibitors are being designed to counteract the adaptive upregulation of molecules like CTLA4 and PDL1 triggered by IRX-2-mediated restoration of a coordinated, multicellular, antitumor immunoresponse.
In view of the evidence presented in the current review from mechanistic as well as clinical studies, IRX-2 emerges as a promising immunotherapeutic for treatment of HNSCC and other malignancies. At a time when oncoimmunology is capturing the attention of the medical community, the range of options available for immediate consideration can be expanded to include IRX-2 monotherapy or a combination of IRX-2 with immune checkpoint inhibitors.
Acknowledgments
The authors wish to thank Galen Press Inc (Austerlitz, NY) for editorial support services, which were paid for by IRX Therapeutics, New York, NY.
Disclosure
GTW is a consultant for and has received research support from IRX Therapeutics, New York, NY. MJK, JSM, JGN, and TLW have received research support from IRX Therapeutics. JEE is a member of the management team for IRX Therapeutics. NLB is a member of the management team for and has received research support from IRX Therapeutics. The authors report no other conflicts of interest in this work.
References
Egan JE, Quadrini KJ, Santiago-Schwarz F, Hadden JW, Brandwein HJ, Signorelli KL. IRX-2, a novel in vivo immunotherapeutic, induces maturation and activation of human dendritic cells in vitro. J Immunother. 2007;30(6):624–633. | ||
Schilling B, Harasymczuk M, Schuler P, Egan J, Ferrone S, Whiteside TL. IRX-2, a novel immunotherapeutic, enhances functions of human dendritic cells. PLoS One. 2013;8(2):e47234. | ||
Czystowska M, Han J, Szczepanski MJ, et al. IRX-2, a novel immunotherapeutic, protects human T cells from tumor-induced cell death. Cell Death Differ. 2009;16(5):708–718. | ||
Czystowska M, Szczepanski MJ, Szajnik M, et al. Mechanisms of T-cell protection from death by IRX-2: a new immunotherapeutic. Cancer Immunol Immunother. 2011;60(4):495–506. | ||
Schilling B, Halstead ES, Schuler P, Harasymczuk M, Egan JE, Whiteside TL. IRX-2, a novel immunotherapeutic, enhances and protects NK-cell functions in cancer patients. Cancer Immunol Immunother. 2012;61(9):1395–1405. | ||
Schilling B, Harasymczuk M, Schuler P, Egan JE, Whiteside TL. IRX-2, a novel biologic, favors the expansion of T effector over T regulatory cells in a human tumor microenvironment model. J Mol Med (Berl). 2012;90(2):139–147. | ||
Freeman SM, Franco JL, Kenady DE, et al. A phase 1 safety study of an IRX-2 regimen in patients with squamous cell carcinoma of the head and neck. Am J Clin Oncol. 2011;34(2):173–178. | ||
Wolf GT, Fee WE Jr, Dolan RW, et al. Novel neoadjuvant immunotherapy regimen safety and survival in head and neck squamous cell cancer. Head Neck. 2011;33(12):1666–1674. | ||
Berinstein NL, Wolf GT, Naylor PH, et al. Increased lymphocyte infiltration in patients with head and neck cancer treated with the IRX-2 immunotherapy regimen. Cancer Immunol Immunother. 2012;61(6):771–782. | ||
National Comprehensive Cancer Network. NCCN guidelines version 1.2017: head and neck cancers. Available from: https://www.nccn.org/professionals/physician_gls/PDF/head-and-neck.pdf. Accessed May 5, 2017. | ||
Stewart BW, Wild CP. World Cancer Report 2014. Lyon: IARC Press; 2014. | ||
Chaturvedi AK, Engels EA, Pfeiffer RM, et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol. 2011;29(32):4294–4301. | ||
Whang SN, Filippova M, Duerksen-Hughes P. Recent progress in therapeutic treatments and screening strategies for the prevention and treatment of HPV-associated head and neck cancer. Viruses. 2015;7(9):5040–5065. | ||
Spence T, Bruce J, Yip KW, Liu FF. HPV associated head and neck cancer. Cancers (Basel). 2016;8(8):E75. | ||
Andersen AS, Sølling AS, Ovesen T, Rusan M. The interplay between HPV and host immunity in head and neck squamous cell carcinoma. Int J Cancer. 2014;134(12):2755–2763. | ||
Heusinkveld M, Goedemans R, Briet RJ, et al. Systemic and local human papillomavirus 16-specific T-cell immunity in patients with head and neck cancer. Int J Cancer. 2012;131(2):E74–E85. | ||
Ward MJ, Thirdborough SM, Mellows T, et al. Tumour-infiltrating lymphocytes predict for outcome in HPV-positive oropharyngeal cancer. Br J Cancer. 2014;110(2):489–500. | ||
American Joint Committee on Cancer. Implementation of AJCC 8th edition cancer staging system. 2017. Available from: https://cancerstaging.org/About/news/Pages/Implementation-of-AJCC-8th-Edition-Cancer-Staging-System.aspx. Accessed February 5, 2018. | ||
Bhide SA, Newbold KL, Harrington KJ, Nutting CM. Clinical evaluation of intensity-modulated radiotherapy for head and neck cancers. Br J Radiol. 2012;85(1013):487–494. | ||
Thiagarajan A, Iyer NG. Radiation-induced sarcomas of the head and neck. World J Clin Oncol. 2014;5(5):973–981. | ||
Gutiontov SI, Shin EJ, Lok B, Lee NY, Cabanillas R. Intensity-modulated radiotherapy for head and neck surgeons. Head Neck. 2016;38 Suppl 1:E2368–E2373. | ||
Daly-Schveitzer N, Julieron M, Tao YG, Moussier A, Bourhis J. Intensity-modulated radiation therapy (IMRT): toward a new standard for radiation therapy of head and neck cancer? Eur Ann Otorhinolaryngol Head Neck Dis. 2011;128(5):241–247. | ||
Busch CJ, Tribius S, Schafhausen P, Knecht R. The current role of systemic chemotherapy in the primary treatment of head and neck cancer. Cancer Treat Rev. 2015;41(3):217–221. | ||
Saba NF, Mody MD, Tan ES, et al. Toxicities of systemic agents in squamous cell carcinoma of the head and neck (SCCHN): a new perspective in the era of immunotherapy. Crit Rev Oncol Hematol. 2017;115:50–58. | ||
Ferris RL. Immunology and Immunotherapy of head and neck cancer. J Clin Oncol. 2015;33(29):3293–3304. | ||
Meneses A, Verastegui E, Barrera JL, de la Garza J, Hadden JW. Lymph node histology in head and neck cancer: impact of immunotherapy with IRX-2. Int Immunopharmacol. 2003;3(8):1083–1091. | ||
Thurnher M, Radmayr C, Ramoner R, et al. Human renal-cell carcinoma tissue contains dendritic cells. Int J Cancer. 1996;68(1):1–7. | ||
Hoffmann TK, Müller-Berghaus J, Ferris RL, Johnson JT, Storkus WJ, Whiteside TL. Alterations in the frequency of dendritic cell subsets in the peripheral circulation of patients with squamous cell carcinomas of the head and neck. Clin Cancer Res. 2002;8(6):1787–1793. | ||
Chaux P, Moutet M, Faivre J, Martin F, Martin M. Inflammatory cells infiltrating human colorectal carcinomas express HLA class II but not B7-1 and B7-2 costimulatory molecules of the T-cell activation. Lab Invest. 1996;74(5):975–983. | ||
Gabrilovich DI, Corak J, Ciernik IF, Kavanaugh D, Carbone DP. Decreased antigen presentation by dendritic cells in patients with breast cancer. Clin Cancer Res. 1997;3(3):483–490. | ||
Almand B, Resser JR, Lindman B, et al. Clinical significance of defective dendritic cell differentiation in cancer. Clin Cancer Res. 2000;6(5):1755–1766. | ||
Almand B, Clark JI, Nikitina E, et al. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001;166(1):678–689. | ||
Pinzon-Charry A, Maxwell T, Lopez JA. Dendritic cell dysfunction in cancer: a mechanism for immunosuppression. Immunol Cell Biol. 2005;83(5):451–461. | ||
Whiteside TL, Stanson J, Shurin MR, Ferrone S. Antigen-processing machinery in human dendritic cells: up-regulation by maturation and down-regulation by tumor cells. J Immunol. 2004;173(3):1526–1534. | ||
Virani S, Bellile E, Bradford CR, et al. NDN and CD1A are novel prognostic methylation markers in patients with head and neck squamous carcinomas. BMC Cancer. 2015;15:825. | ||
Hoffmann TK, Dworacki G, Tsukihiro T, et al. Spontaneous apoptosis of circulating T lymphocytes in patients with head and neck cancer and its clinical importance. Clin Cancer Res. 2002;8(8):2553–2562. | ||
Reichert TE, Strauss L, Wagner EM, Gooding W, Whiteside TL. Signaling abnormalities, apoptosis, and reduced proliferation of circulating and tumor-infiltrating lymphocytes in patients with oral carcinoma. Clin Cancer Res. 2002;8(10):3137–3145. | ||
Kim JW, Wieckowski E, Taylor DD, Reichert TE, Watkins S, Whiteside TL. Fas ligand-positive membranous vesicles isolated from sera of patients with oral cancer induce apoptosis of activated T lymphocytes. Clin Cancer Res. 2005;11(3):1010–1020. | ||
Bergmann C, Strauss L, Wieckowski E, et al. Tumor-derived microvesicles in sera of patients with head and neck cancer and their role in tumor progression. Head Neck. 2009;31(3):371–380. | ||
Ludwig S, Floros T, Theodoraki MN, et al. Suppression of lymphocyte functions by plasma exosomes correlates with disease activity in patients with head and neck cancer. Clin Cancer Res. 2017;23(16):4843–4854. | ||
Molling JW, Kölgen W, van der Vliet HJ, et al. Peripheral blood IFN-γ-secreting Vα24+Vβ11+ NKT cell numbers are decreased in cancer patients independent of tumor type or tumor load. Int J Cancer. 2005;116(1):87–93. | ||
Molling JW, Langius JA, Langendijk JA, et al. Low levels of circulating invariant natural killer T cells predict poor clinical outcome in patients with head and neck squamous cell carcinoma. J Clin Oncol. 2007;25(7):862–868. | ||
Jochems C, Schlom J. Tumor-infiltrating immune cells and prognosis: the potential link between conventional cancer therapy and immunity. Exp Biol Med (Maywood). 2011;236(5):567–579. | ||
Nguyen N, Bellile E, Thomas D, et al. Tumor infiltrating lymphocytes and survival in patients with head and neck squamous cell carcinoma. Head Neck. 2016;38(7):1074–1084. | ||
Nasman A, Romanitan M, Nordfors C, et al. Tumor infiltrating CD8+ and Foxp3+ lymphocytes correlate to clinical outcome and human papillomavirus (HPV) status in tonsillar cancer. PLoS One. 2012;7(6):e38711. | ||
Pretscher D, Distel LV, Grabenbauer GG, Wittlinger M, Buettner M, Niedobitek G. Distribution of immune cells in head and neck cancer: CD8+ T-cells and CD20+ B-cells in metastatic lymph nodes are associated with favourable outcome in patients with oro- and hypopharyngeal carcinoma. BMC Cancer. 2009;9:292. | ||
Ziegler SF, Ramsdell F, Alderson MR. The activation antigen CD69. Stem Cells. 1994;12(5):456–465. | ||
Li X, Zheng Y. Regulatory T cell identity: formation and maintenance. Trends Immunol. 2015;36(6):344–353. | ||
Badoual C, Hans S, Rodriguez J, et al. Prognostic value of tumor-infiltrating CD4+ T-cell subpopulations in head and neck cancers. Clin Cancer Res. 2006;12(2):465–472. | ||
Sun DS, Zhao MQ, Xia M, Li L, Jiang YH. The correlation between tumor-infiltrating Foxp3+ regulatory T cells and cyclooxygenase-2 expression and their association with recurrence in resected head and neck cancers. Med Oncol. 2012;29(2):707–713. | ||
Whiteside TL. The role of regulatory T cells in cancer immunology. Immunotargets Ther. 2015;54:159–171. | ||
Andrade M, Ferreira S, Gonçalves L, et al. Cell surface markers for T and B lymphocytes activation and adhesion as putative prognostic biomarkers for head and neck squamous cell carcinoma. Hum Immunol. 2013;74(12):1563–1574. | ||
Saze Z, Schuler PJ, Hong CS, Cheng D, Jackson EK, Whiteside TL. Adenosine production by human B cells and B cell-mediated suppression of activated T cells. Blood. 2013;122(1):9–18. | ||
Bauernhofer T, Kuss I, Henderson B, Baum AS, Whiteside TL. Preferential apoptosis of CD56dim natural killer cell subset in patients with cancer. Eur J Immunol. 2003;33(1):119–124. | ||
Melioli G, Semino C, Margarino G, et al. Expansion of natural killer cells in patients with head and neck cancer: detection of “noninhibitory” (activating) killer Ig-like receptors on circulating natural killer cells. Head Neck. 2003;25(4):297–305. | ||
Mickel RA, Kessler DJ, Taylor JM, Lichtenstein A. Natural killer cell cytotoxicity in the peripheral blood, cervical lymph nodes, and tumor of head and neck cancer patients. Cancer Res. 1988;48(17):5017–5022. | ||
Baskic D, Vujanovic L, Arsenijevic N, Whiteside TL, Myers EN, Vujanovic NL. Suppression of natural killer-cell and dendritic-cell apoptotic tumoricidal activity in patients with head and neck cancer. Head Neck. 2013;35(3):388–398. | ||
Schantz SP, Ordonez NG. Quantitation of natural killer cell function and risk of metastatic poorly differentiated head and neck cancer. Nat Immun Cell Growth Regul. 1991;10(5):278–288. | ||
Bann DV, Deschler DG, Goyal N. Novel immunotherapeutic approaches for head and neck squamous cell carcinoma. Cancers (Basel). 2016;8(10):E87. | ||
Moskovitz J, Moy J, Ferris R. Immunotherapy for head and neck squamous cell carcinoma. Curr Oncol Rep. 2018;20(2):22. | ||
Economopoulou P, Kotsantis I, Psyrri A. The promise of immunotherapy in head and neck squamous cell carcinoma: combinatorial immunotherapy approaches. ESMO Open. 2017;1(6):e000122. | ||
Floros T, Tarhini AA. Anticancer cytokines: biology and clinical effects of interferon-α2, interleukin (IL)-2, IL-15, IL-21, and IL-12. Semin Oncol. 2015;42(4):539–548. | ||
Whiteside TL, Letessier E, Hirabayashi H, et al. Evidence for local and systemic activation of immune cells by peritumoral injections of interleukin 2 in patients with advanced squamous cell carcinoma of the head and neck. Cancer Res. 1993;53(23):5654–5662. | ||
Cortesina G, De Stefani A, Giovarelli M, et al. Treatment of recurrent squamous cell carcinoma of the head and neck with low doses of interleukin-2 injected perilymphatically. Cancer. 1988;62(12):2482–2485. | ||
Cortesina G, De Stefani A, Galeazzi E, et al. Interleukin-2 injected around tumor-draining lymph nodes in head and neck cancer. Head Neck. 1991;13(2):125–131. | ||
Cortesina G, De Stefani A, Galeazzi E, et al. Temporary regression of recurrent squamous cell carcinoma of the head and neck is achieved with a low but not with a high dose of recombinant interleukin 2 injected perilymphatically. Br J Cancer. 1994;69(3):572–576. | ||
Berd D, Mastrangelo MJ, Engstrom PF, Paul A, Maguire H. Augmentation of the human immune response by cyclophosphamide. Cancer Res. 1982;42(11):4862–4866. | ||
Berlinger NT. Deficient immunity in head and neck cancer due to excessive monocyte production of prostaglandins. Laryngoscope. 1984;94(11 Pt 1):1407–1410. | ||
Hadden JW. The treatment of zinc deficiency is an immunotherapy. Int J Immunopharmacol. 1995;17(9):697–701. | ||
Prasad AS, Beck FW, Snell DC, Kucuk O. Zinc in cancer prevention. Nutr Cancer. 2009;61(6):879–887. | ||
Papagerakis S, Bellile E, Peterson LA, et al. Proton pump inhibitors and histamine 2 blockers are associated with improved overall survival in patients with head and neck squamous carcinoma. Cancer Prev Res (Phila). 2014;7(12):1258–1269. | ||
Strauss L, Whiteside TL, Knights A, Bergmann C, Knuth A, Zippelius A. Selective survival of naturally occurring human CD4+CD25+Foxp3+ regulatory T cells cultured with rapamycin. J Immunol. 2007;178(1):320–329. | ||
Zeiser R, Leveson-Gower DB, Zambricki EA, et al. Differential impact of mammalian target of rapamycin inhibition on CD4+CD25+Foxp3+ regulatory T cells compared with conventional CD4+ T cells. Blood. 2008;111(1):453–462. | ||
Haxhinasto S, Mathis D, Benoist C. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J Exp Med. 2008;205(3):565–574. | ||
Matsuda I, Matsuo K, Matsushita Y, Haruna Y, Niwa M, Kataoka T. The C-terminal domain of the long form of cellular FLICE-inhibitory protein (c-FLIPL) inhibits the interaction of the caspase 8 prodomain with the receptor-interacting protein 1 (RIP1) death domain and regulates caspase 8-dependent nuclear factor κB (NF-κB) activation. J Biol Chem. 2014;289(7):3876–3887. | ||
Naylor PH, Hernandez KE, Nixon AE, et al. IRX-2 increases the T cell-specific immune response to protein/peptide vaccines. Vaccine. 2010;28(43):7054–7062. | ||
Naylor PH, Egan JE, Berinstein NL. Peptide based vaccine approaches for cancer: a novel approach using a WT-1 synthetic long peptide and the IRX-2 immunomodulatory regimen. Cancers (Basel). 2011;3(4):3991–4009. | ||
Hadden J, Verastegui E, Barrera JL, et al. A trial of IRX-2 in patients with squamous cell carcinomas of the head and neck. Int Immunopharmacol. 2003;3(8):1073–1081. | ||
Berinstein N, McNamara M, Nguyen A, Egan J, Wolf G. Increased immune infiltration and chemokine receptor expression in head and neck epithelial tumors after neoadjuvant immunotherapy with the IRX-2 regimen. Oncoimmunology. 2018;7(5):e1423173. | ||
Whiteside TL, Butterfield LH, Naylor PH, et al. A short course of neoadjuvant IRX-2 induces changes in peripheral blood lymphocyte subsets of patients with head and neck squamous cell carcinoma. Cancer Immunol Immunother. 2012;61(6):783–788. | ||
Duenas-Gonzalez A, Verastegui E, Lopez-Graniel C, et al. A pilot study of perilymphatic leukocyte cytokine mixture (IRX-2) as neoadjuvant treatment for early stage cervical carcinoma. Int Immunopharmacol. 2002;2(7):1007–1016. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.