Back to Journals » OncoTargets and Therapy » Volume 13
Irinotecan Induces Autophagy-Dependent Apoptosis and Positively Regulates ROS-Related JNK- and P38-MAPK Pathways in Gastric Cancer Cells
Authors Zhu Q, Guo Y, Chen S, Fu D, Li Y, Li Z, Ni C
Received 2 December 2019
Accepted for publication 5 March 2020
Published 2 April 2020 Volume 2020:13 Pages 2807—2817
DOI https://doi.org/10.2147/OTT.S240803
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Yong Teng
Qingyun Zhu,1,2 Yuehui Guo,2 Shiwei Chen,2 Daiquan Fu,2 Yanxiang Li,2 Zhi Li,1 Caifang Ni1
1Department of Interventional Radiology, The First Affiliated Hospital of Soochow University, Suzhou 215006, People’s Republic of China; 2Department of Intervention, Gongli Hospital of Shanghai Pudong New Area, Shanghai 200135, People’s Republic of China
Correspondence: Caifang Ni; Zhi Li Email [email protected]; [email protected]
Background: Irinotecan (IRI) is considered an option for second-line treatment of advanced gastric cancer; however, acquired drug resistance currently limits its clinical application. Recently, many researchers have shown that autophagy plays a crucial role in the resistance of tumor cells to chemotherapy and radiotherapy. In this study, we investigated the relationship between autophagy and antitumor activity of IRI in gastric cancer cells.
Methods: We used MTT assay, flow cytometry and immunofluorescence staining to detect viability, apoptosis and autophagy in gastric cancer. Western blotting assay was used to determine the expression of LC3, Beclin-1, P62, cleaved PARP and Caspase 3. In vivo animal study was performed finally.
Results: We found that IRI treatment dose- and time-dependently inhibited growth and induced apoptosis in gastric cancer cells. Moreover, IRI treatment caused autophagy in these cells, whereas autophagy inhibitors— 3-methyladenine (3-MA), chloroquine (CQ), and Beclin-1 small interfering RNA (siRNA)—suppressed cytotoxicity of IRI. A mechanistic analysis showed that IRI-induced autophagy and apoptosis were related to increased reactive oxygen species (ROS) accumulation and activation of the JNK- and p38-MAPK pathways. Further in vivo experiments revealed that IRI suppressed tumor growth, induced autophagy, and stimulated the JNK- and p38-MAPK pathways, whereas 3-MA attenuated these effects.
Conclusion: Taken together, these results indicate that IRI stimulates the ROS-related JNK- and p38-MAPK pathways to promote autophagy-dependent apoptosis. Thus, a combination of IRI with a pharmacological autophagy enhancer may be a promising therapeutic strategy against gastric cancer.
Keywords: gastric cancer, irinotecan, autophagy, ROS, JNK, p38
Introduction
Gastric cancer is the fourth most common malignant tumor and the third most common cause of cancer-related deaths worldwide.1 Over 50% of newly diagnosed gastric cancer cases occur in developing countries, and approximately 70% of deaths occur in less economically developed areas despite declining incidence and mortality rates.2 Despite the advances in diagnosis and treatment, the median 5-year overall survival (OS) rate among patients with advanced gastric cancer after surgical resection is still low.3 Hence, there is an urgent need to find promising therapeutic techniques for treatment of gastric cancer.
Irinotecan (IRI) is a semisynthetic derivative of camptothecin, which was originally isolated from Camptotheca acuminata.4 As a camptothecin derivative, IRI directly inhibits the activity of topoisomerase I (TOP I), a nuclear enzyme that is essential for replication and transcription. TOP I unwinds the supercoiled DNA, and subsequently, IRI leads to lethal replication-mediated double-strand breaks and eventually causes cell death.5 As a chemotherapeutic agent, IRI has been used to treat various cancers, such as colorectal cancer, hepatocellular carcinoma, and pancreatic cancer.6–8 It has also been applied to gastric cancer treatment.9,10 Nonetheless, the therapeutic effect of IRI is still hampered by acquired chemotherapy resistance.11 Therefore, it is necessary to devise a promising and safe treatment regimen to overcome drug resistance and reduce drug side effects in gastric cancer.
Autophagy is a highly conserved physiological mechanism that eliminates needless proteins or damaged organelles as well as degrades and recycles cellular components under conditions of low nutrient supply (or other analogous conditions) to ensure homeostasis.12 Autophagy is regulated by autophagy-related genes (ATG) and initiated by a double-membrane organelle formation, which is triggered by autophagy-related proteins, such as LC3, Beclin-1 and P62.13 The role of autophagy in cancer remains in debate because it can either exhibit a cytoprotective or cytotoxic role depending on cancer type. Some studies show that autophagy facilitates cell survival and serves a cytoprotective function in cancer treatment.14,15 Furthermore, autophagy has been identified as an important mechanism of resistance to IRI treatment in cancers. It has been reported that autophagy and cellular metastasis are enhanced by the resistance to IRI in LoVo colon cancer cells.16 Survival-promoting autophagy is activated to protect tumor cells against the cytotoxic effects of IRI in colon cancer cells.17 Nevertheless, the relationship between autophagy and resistance to IRI treatment in gastric cancer has not been clearly demonstrated.
In this study, we investigated the connection between autophagy and antitumor activity of IRI in gastric cancer cells. We demonstrated that IRI causes growth inhibition, apoptosis, and autophagy in gastric cancer cells. Autophagy was found to play a cytotoxic role in IRI-treated cells. Additionally, the JNK- and p38-MAPK signaling pathways were involved in IRI-induced autophagy and were mediated by reactive oxygen species (ROS).
Materials and Methods
Reagents and Antibodies
IRI, 3-MA, CQ, N-acetyl-
Cell Lines and Cell Culture
Gastric cancer cell lines MGC803 and SGC7901 were purchased from the Cell Bank of the Chinese Academy of Medical Sciences (Shanghai, China). All the cells were cultured in RPMI 1640 containing 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in a 5% CO2 incubator.
Cell Viability Assay
Cell viability was determined by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. In brief, cells (104 per well) were seeded in 96-well microplates and then exposed to various concentrations of IRI for various periods. The MTT solution was added to each well for an additional 4 h incubation. The culture medium was removed, and 100 μL of dimethyl sulfoxide was added. Finally, absorbance at 490 nm was measured on an ELISA microplate reader.
Western Blotting
After treatment, the cells were lysed in ice-cold lysis buffer.18 The samples were clarified by centrifugation for 15 min at 4°C. The protein concentrations were measured with the BCA protein assay (Pierce, Waltham, MA, USA), and 20 μg of each protein sample was resolved on a 15% gel by denaturing sodium dodecyl sulfate polyacrylamide gel electrophoresis and then electrophoretically transferred onto polyvinylidene fluoride membranes (Millipore, Bedford, MA, USA). The blotted membranes were blocked in 5% nonfat milk, incubated with primary antibodies, and then probed with the secondary antibodies. β-actin served as a loading control. The signals were visualized with the ECL detection solution (Thermo Scientific, Rockford, IL, USA). The relative densities of bands were quantified using Image J software.
Transmission Electron Microscopy (TEM)
Cells were collected and fixed with 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) at 4°C for 1 h. After the cells were washed with phosphate buffer, they were post-fixed in 1% osmium tetroxide (OsO4) for 1 h. The cells were then washed with water, dehydrated in a graded series of ethanol solutions and propylene oxide, and embedded in araldite (Merck, Rahway, NJ, USA). Ultrathin sections (40–60 nm) were prepared on an ultramicrotome. The sections were mounted on 200-mesh nickel grids and double-stained with uranyl acetate and lead citrate. Ultrastructure of the cells was examined under a transmission electron microscope (JEM-1200EX, JEOL, Tokyo, Japan).
Immunofluorescence Assay
The cells were treated with 20 μM IRI, seeded in 24-well plates containing glass coverslips, and incubated for 24 h. Next, the cells were washed with ice-cold phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde in PBS for 30 min at room temperature (RT), allowed to permeate in 1% Triton X-100/PBS for 30 min at RT, and blocked with 10% FBS/PBS overnight at 4°C. The cells were incubated with the anti–LC3-II antibody for 4 h at RT, and then with a secondary antibody for 1 h in the dark at RT. The nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI, Sigma) and observed under a Zeiss LSM710 confocal microscope (Zeiss, Oberkochen, Germany).19
Measurement of Intracellular ROS Levels
The ROS levels in gastric cancer cells were measured by means of 2,7-dichlorofluorescein diacetate (DCFH-DA), an oxidant-sensitive fluorescent reagent. Briefly, cells cultured in the RPMI 1640 medium supplemented with 10% of FBS were treated with 20 μM IRI for 24 h. The cells were then harvested, washed twice with PBS, and incubated in serum-free RPMI 1640 containing 10 μM DCFH-DA at 37°C for 30 min in the dark. The cells were washed three times with ice-cold PBS, and DCF fluorescence intensity was analyzed immediately on a FACS Canto flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA), at an excitation wavelength of 488 nm and emission at 525 nm.
Xenograft Experiments and Immunohistochemical Staining
BALB/c nude mice (6 weeks old) were purchased from Shanghai Slac Laboratory Animal Co., Ltd. (Shanghai, China). The animal experiments were performed with the approval of the Animal Ethics Committee of the Second Military Medical University. Animal experiments were carried out according to the National Institutes of Health guidelines to the Care and Use of Laboratory Animals. In total, 2 × 106 viable SGC7901 cells in 100 μL of PBS were injected subcutaneously into the nude mice. When the resultant tumors reached approximately 100 mm3, the animals were injected intraperitoneally with either 1) physiological saline (100 μL) or 2) IRI (20 mg/kg), 3-MA (30 mg/kg), or their combination once weekly for 15 days. Tumor volume (V) was calculated using the following formula: V = longest diameter × shortest diameter2 × 0.5. The excised tumors were weighed and prepared for subsequent Western blotting and immunohistochemical staining.
Statistical Analysis
All statistical analyses were performed in the SPSS 17.0 statistical software (SPSS, Inc., Chicago, IL, USA). The data are presented as means ± standard deviation. The significance of differences between two groups was evaluated with two-tailed Student’s t test. Probabilities of P < 0.05 were considered statistically significant.
Results
IRI Inhibits Growth and Induces Apoptosis in Gastric Cancer Cells
IRI has been reported to cause growth inhibition and apoptosis in tumor cells.16,20 To confirm that IRI has the same effects on gastric cancer cell lines, MGC803 and SGC7901 cells were treated with IRI at various concentrations and for different periods. Two important apoptosis-related signaling molecules, cleaved caspase 3 and cleaved PARP, were analyzed next. As shown in Figure 1A and B, the expression of cleaved caspase 3 and cleaved PARP increased in a dose-dependent and time-dependent manner, suggesting that apoptosis in MGC803 and SGC7901 cells was induced by IRI. Additionally, the MTT assay was conducted using cells treated with various concentrations of IRI for various periods. Cell viability significantly decreased as the IRI dose or action duration increased (Figure 1C). These results indicated that IRI inhibits growth and induces apoptosis in gastric cancer cells.
 |
Figure 1 IRI induces cytotoxicity and apoptosis in gastric cancer cells. (A) MGC803 and SGC7901 cells were treated with IRI (0, 20, or 40 μM) for 24 h or (B) with 20 μM IRI for 0, 12, or 24 h, and cleaved PARP and cleaved caspase 3 protein expression levels were examined by Western blotting. β-actin served as the internal control. (C) MGC803 and SGC7901 cells were incubated with various concentrations of IRI for the indicated periods, and cell viability was determined by MTT assay. *P < 0.05. |
IRI Induces Autophagy in Gastric Cancer Cells
Autophagy has been demonstrated to take part in the drug resistance of gastric cancer,21 but whether IRI induces autophagy to mediate this drug resistance in gastric cancer cells remains unknown. The conversion of LC3 from LC3-I to LC3-II is a specific indicator of the autophagy process. We performed Western blotting to evaluate the expression of autophagy marker protein LC3-I/II in MGC803 and SGC7901 cells treated with different concentrations of IRI. We found that IRI treatment upregulated the proteins LC3-II in a concentration- and time-dependent manner (Figure 2A). To confirm the induction of autophagy by IRI, TEM analysis was performed. As shown in Figure 2B, cells treated with IRI showed accelerated autophagosome formation, a major phenomenon of autophagy. Consistent with these results, IRI treatment dramatically promoted the formation and aggregation of LC3-positive vesicles (Figure 2C). Moreover, IRI treatment increased levels of Beclin-1 and decreased protein abundance of P62, both of which are markers of autophagy (Figure 2D). These results provided evidence that autophagy can be induced by IRI in gastric cancer cells.
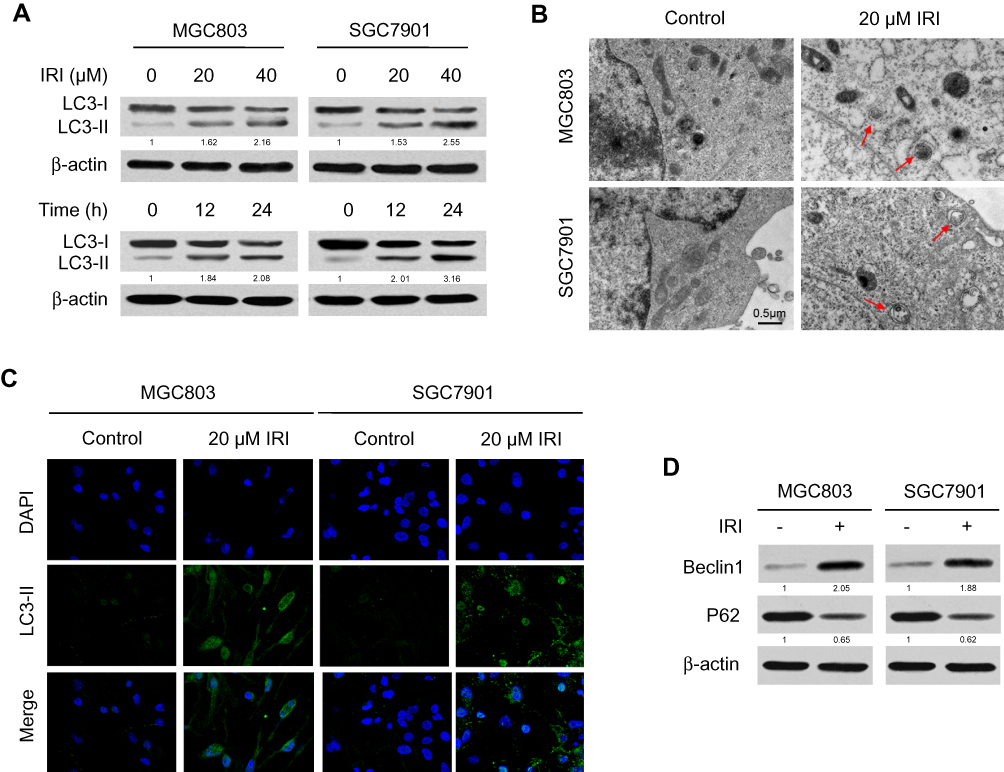 |
Figure 2 IRI induces autophagy in gastric cancer cells. (A) MGC803 and SGC7901 cells were treated with IRI (0, 20, or 40 μM) for 24 h, or with 20 μM IRI for 0, 12, or 24 h, and LC3 protein expression was examined by Western blotting. β-actin served as the internal control. (B) TEM detection of autophagosome formation in MGC803 and SGC7901 cells treated with 20 μM IRI for 24 h (red arrows indicate autophagosomes). Scale bar: 0.5 μm. (C) Representative images of LC3-II immunostaining in MGC803 and SGC7901 cells incubated with 20 μM IRI for 24 h. (D) Western blotting analysis of the protein levels of Beclin-1 and P62 in MGC803 and SGC7901 cells incubated with 20 μM IRI for 24 h. |
Blocking Autophagy Suppresses the Antitumor Activity of IRI in Gastric Cancer Cells
To evaluate the role of autophagy in the therapeutic effects of IRI on gastric cancer cell growth, two autophagy inhibitors, 3-methyladenine (3-MA) and chloroquine (CQ), were applied for subsequent experiments. The former was able to suppress formation of the autophagosome during the initial stages of autophagy, and the latter blocked the transition of autophagosomes to autolysosomes. As depicted in Figure 3A, the influence of IRI on the LC3-II level in MGC803 and SGC7901 cells was attenuated by 3-MA treatment but augmented by the presence of CQ. Additionally, the expression of Beclin-1 and P62—after treatment with 3-MA or CQ following IRI exposure—dramatically decreased and increased, respectively, as compared with IRI treatment alone (Figure 3A). The MTT assay results revealed that inhibition of autophagy using 3-MA or CQ significantly increased the proliferation of IRI-treated cells (Figure 3B). Furthermore, knockdown of Beclin-1 suppressed LC3-II and Beclin-1 expression, and enhanced P62 expression in the presence of IRI and reversed the suppressive growth effect of IRI in gastric cancer cells (Figure 3C and D). Moreover, the levels of cleaved PARP and cleaved caspase 3 decreased in cells treated with IRI+3-MA, IRI+CQ, or IRI+siBeclin1 in comparison with IRI-treated cells (Figure 3E and F). Taken together, these results indicated that autophagy exerted cytotoxic action, whereas inhibition of autophagy weakened the antitumor effects of IRI on human gastric cancer cells.
 |
Figure 3 Autophagy inhibition suppresses the antitumor activity of IRI in gastric cancer cells. (A) MGC803 and SGC7901 cells were pretreated with 2 mM 3-MA for 24 h or 10 μM CQ for 4 h, and then exposed to 20 μM IRI for 24 h. Western blotting was carried out to analyze LC3, Beclin-1 and P62 protein expression. β-actin was used as the internal control. (B) MTT assay was conducted to analyze cell viability. (C) MGC803 and SGC7901 cells were transfected with Beclin-1 siRNA or siNC, and then exposed to 20 μM IRI for 24 h. Western blotting was carried out to analyze LC3, Beclin-1 and P62 protein expression. β-actin was used as the internal control. (D) Cell viability was determined by the MTT assay. (E, F) Cleaved-PARP and cleaved-caspase-3 protein expression was examined by Western blotting in the indicated cells. β-actin was employed as the internal control. *P < 0.05 compared to Control group; #P < 0.05 compared to IRI group. |
ROS Mediate IRI-Induced Autophagy and Growth Inhibition in Gastric Cancer Cells
Some studies have shown that ROS stimulate autophagy.22 To determine whether ROS are involved in the IRI-induced autophagy, we measured ROS levels via the DCF-DA assay kit with quantitation of DCF fluorescence. As expected, ROS levels increased after treatment with IRI alone, but the accumulation of ROS was attenuated when the cells were cotreated with NAC, an antioxidant (Figure 4A). Subsequently, Western blotting results showed that the expression level of LC3-II markedly diminished in the cells cotreated with IRI and NAC as compared with the cells treated with IRI alone (Figure 4B). Furthermore, cell viability was enhanced while the protein amounts of cleaved caspase 3 and cleaved PARP were reduced by this cotreatment (Figure 4C and D). Altogether, these results indicated that IRI-induced autophagy and growth inhibition in gastric cancer cells were mediated by ROS.
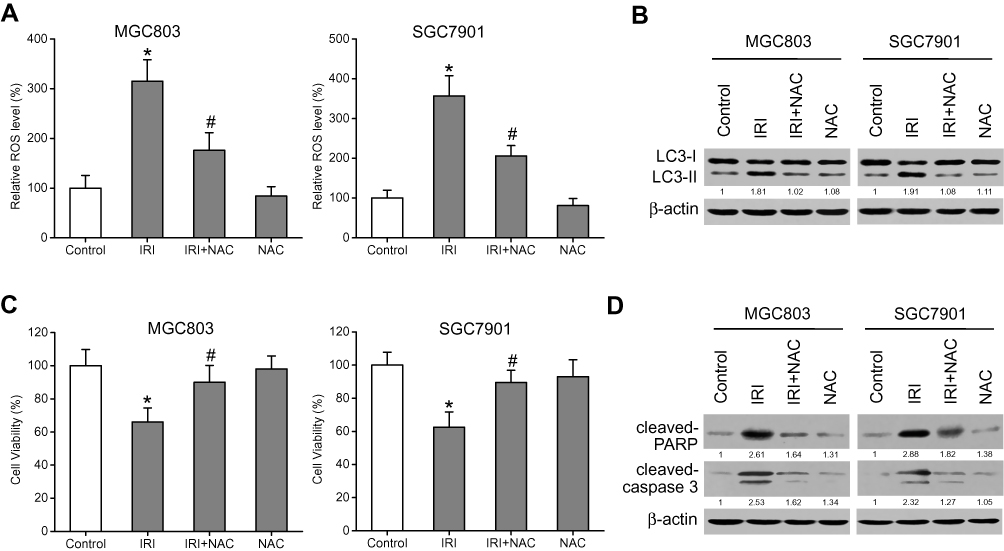 |
Figure 4 ROS mediate the IRI-induced autophagy and growth inhibition in gastric cancer cells. (A) MGC803 and SGC7901 cells were pretreated with 5 mM NAC for 1 h and then exposed to 20 μM IRI for 24 h. ROS levels were evaluated with a DCF-DA assay kit, and DCF fluorescence was measured. (B) LC3-I and LC3-II protein amounts were evaluated by Western blotting. β-actin was used as the internal control. (C) The MTT assay was carried out to analyze cell viability. (D) Cleaved-PARP and cleaved-caspase-3 protein expression was examined by Western blotting. β-actin served as the internal control. *P < 0.05 compared to Control group; #P < 0.05 compared to IRI group. |
IRI-Induced Autophagy Is Related to the ROS-Mediated JNK- and P38-MAPK Pathways
Given that the MAPK pathway is crucial for the initiation and induction of autophagy,23 we tested whether this cascade is involved in IRI-induced autophagy. As illustrated in Figure 5A, IRI treatment increased the amounts of phospho- (p-)JNK and p-p38 in MGC803 and SGC7901 cells. By contrast, cotreatment with NAC blocked IRI-induced p-JNK and p-p38 formation. In addition, both inhibition of p-p38 by SB203580 and inhibition of p-JNK by SP600125 decreased the expression of LC3-II, which was enhanced by IRI treatment alone (Figure 5B). Overall, these results mean that the JNK- and p38-MAPK signaling pathways are involved in IRI-induced autophagy and are mediated by ROS.
 |
Figure 5 IRI-induced autophagy is related to a ROS-mediated MAPK pathway. (A) MGC803 and SGC7901 cells were pretreated with 5 mM NAC for 1 h and then exposed to 20 μM IRI for 24 h. The expression levels of p-JNK and p-p38 were analyzed by Western blotting. β-actin was used as the internal control. (B) MGC803 and SGC7901 cells were pretreated with 10 µM SP600125 or 10 µM SB203580 for 1 h and then exposed to 20 μM IRI for 24 h. The expression of LC3-I and LC3-II proteins was determined by Western blotting. β-actin served as the internal control. |
IRI Induces Autophagy and Inhibits Tumor Growth in a Murine Xenograft Model of Gastric Cancer
Finally, a xenograft model was set up to verify the influence of IRI-mediated autophagy on tumor growth in vivo. As shown in Figure 6A and B, IRI treatment significantly reduced the tumor volume relative to the control group, whereas treatment with IRI and 3-MA yielded inferior tumor suppression when compared with IRI alone. Furthermore, IRI plus 3-MA treatment decreased the percentage of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive tumor cells and increased the proportion of Ki67-positive cells in tumor tissues compared with IRI-treated tissues (Figure 6C). Moreover, Western blotting results revealed that IRI enhanced the expression of LC3-II, cleaved caspase 3, and cleaved PARP as well as the amounts of p-JNK and p-p38 in tumor tissues, whereas 3-MA dramatically attenuated these effects (Figure 6D). These findings indicated that IRI inhibited tumor growth and induced autophagy, whereas suppression of autophagy attenuated the inhibitory action of IRI.
 |
Figure 6 Autophagy participates in IRI-induced antitumor effects in a SGC7901 xenograft. A total of 2 × 106 viable SGC7901 cells in 100 μL of PBS were injected subcutaneously into nude mice. When the resultant tumors reached approximately 100 mm3, the animals were injected intraperitoneally with either 1) physiological saline (100 μL) or 2) IRI (20 mg/kg), 3-MA (30 mg/kg), or their combination once weekly for 15 days (n=4). (A) Representative xenografts from mice after subcutaneous injection of SGC7901 cells and subsequent systemic (intraperitoneal) treatment of the mice with IRI, 3-MA, or the combination. (B) The tumor growth was measured every 5 days. (C) TUNEL staining and Ki67 immunohistochemical staining of the tumors. (D) Tumor tissues in each treatment group were examined by Western blotting with the indicated antibodies. *P < 0.05 compared to Control group; #P < 0.05 compared to IRI group. |
Discussion
It has been reported that IRI is an effective therapeutic agent as a second-line treatment of advanced gastric and gastroesophageal adenocarcinomas.10 Besides, IRI therapy alone is considered a salvage-line treatment of advanced gastric cancer in clinical practice.24 Even though cytotoxic chemotherapy of gastric cancer patients with IRI is capable of enhancing the antitumor effects of other treatments, acquired chemoresistance remains a serious clinical problem in gastric cancer therapy. In addition, autophagy is reported to be activated to protect tumor cells from the cytotoxic action of IRI and is associated with drug resistance,16 but the underlying molecular mechanisms in gastric cancer still need deeper exploration. In this study, we tested two gastric cancer cell lines, MGC803 and SGC7901, to investigate IRI-induced autophagy and the response to antitumor activity of IRI. We observed upregulation of cleaved PARP and cleaved caspase 3 and a decrease of viability in IRI-treated MGC803 and SGC7901 cells, indicating that IRI exerts its cytotoxic action by inducing apoptosis and growth inhibition.
Autophagy has been regarded as a cellular metabolic response to many metabolic stressors, including chemotherapeutic agents. It is known as an important mechanism of resistance to IRI treatment in cancer cells.16,17 In the present study, we aimed to find out whether IRI could induce autophagy and whether autophagy participates in the antitumor activity of IRI in MGC803 and SGC7901 cells. TEM, immunostaining, and a Western blot assay showed that autophagy could be induced by IRI treatment in gastric cancer cell lines. Other studies indicate that the blocking of autophagy using 3-MA or CQ enhances the sensitivity of tumor cells to chemotherapeutics, e.g., in lung adenocarcinoma, head and neck cancer, and breast cancer.25–27 By contrast, in the present study, we found that suppression of autophagy by the specific inhibitor 3-MA remarkably attenuated IRI-induced cytotoxicity and apoptosis in MGC803 and SGC7901 cells. These data confirm the cytotoxic role of autophagy in IRI-treated gastric cancer cells.
ROS are small highly reactive compounds that are produced naturally during the normal metabolism of oxygen.28 It is now widely accepted that ROS play an important role in the regulation of autophagy.29 Some anticancer drugs cause accumulation of intracellular ROS, which in turn induce autophagy and apoptosis.30,31 In the present study, we observed an increased level of ROS in IRI-treated cells. After obliteration of ROS with NAC, the accumulation of ROS was attenuated, along with the decline of LC3-II and cleaved-PARP levels, and the cell viability recovered. These results suggest that ROS generated after IRI treatment promote autophagosome formation and exert a cytotoxic action on MGC803 and SGC7901 cells. Notably, MAPKs such as JNK participate in the induction of autophagy by phosphorylating BCL-2 and disrupting the BCL-2/Beclin 1 complex.32 p38 also mediates cell autophagy by reducing the phosphorylation of mTOR.33 In the present study, we also observed upregulation of MAPK signaling proteins p-JNK and p-p38 when the cells were treated with IRI, and this upregulation was impaired by cotreatment with NAC. This finding indicates that the MAPK signaling pathway is associated with ROS. Furthermore, inhibition of p-p38 by SB203580 or suppression of p-JNK activity by SP600125 attenuated the upregulation of LC3-II induced by IRI, suggesting that the JNK- and p38-MAPK pathways participate in IRI-induced autophagy.
In conclusion, our study indicates that IRI stimulates the ROS-related JNK- and p38-MAPK pathways to promote autophagy-dependent apoptosis. A combination of IRI with a pharmacological autophagy enhancer may be a promising therapeutic strategy against gastric cancer. Further work on direct target(s) of IRI as well as more detailed studies of its molecular mechanism of action are in progress.
Acknowledgments
This research was supported by the “Six One Projects” for high-level health personnel in Jiangsu Province (LGY2018077) and Talent Project of Shanghai Pudong New Area Gongli Hospital (Grant No: GLRq2-17-04).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359. doi:10.1002/ijc.29210
2. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. doi:10.3322/caac.21262
3. Jimenez Fonseca P, Carmona-Bayonas A, Hernandez R, et al. Lauren subtypes of advanced gastric cancer influence survival and response to chemotherapy: real-world data from the AGAMENON national cancer registry. Br J Cancer. 2017;117(6):775–782. doi:10.1038/bjc.2017.245
4. Wen F, Zheng H, Wu Y, et al. Cost-effectiveness Analysis of Fluorouracil, Leucovorin, and Irinotecan versus Epirubicin, Cisplatin, and Capecitabine in Patients with Advanced Gastric Adenocarcinoma. Sci Rep. 2016;6:.36060. doi:10.1038/srep36060.
5. Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6(10):789. doi:10.1038/nrc1977
6. Del Rio M, Mollevi C, Bibeau F, et al. Molecular subtypes of metastatic colorectal cancer are associated with patient response to irinotecan-based therapies. Eur J Cancer. 2017;76:68–75. doi:10.1016/j.ejca.2017.02.003
7. Xu L, Zhu Y, Shao J, et al. Dasatinib synergises with irinotecan to suppress hepatocellular carcinoma via inhibiting the protein synthesis of PLK1. Br J Cancer. 2017;116(8):1027–1036. doi:10.1038/bjc.2017.55
8. Huang HC, Mallidi S, Liu J, et al. Photodynamic therapy synergizes with irinotecan to overcome compensatory mechanisms and improve treatment outcomes in pancreatic cancer. Cancer Res. 2016;76(5):1066–1077. doi:10.1158/0008-5472.CAN-15-0391
9. Makiyama A, Arimizu K, Hirano G, et al. Irinotecan monotherapy as third-line or later treatment in advanced gastric cancer. Gastric Cancer. 2017.
10. Ochenduszko S, Puskulluoglu M, Konopka K, et al. Clinical effectiveness and toxicity of second-line irinotecan in advanced gastric and gastroesophageal junction adenocarcinoma: a single-center observational study. Ther Adv Med Oncol. 2017;9(4):223–233. doi:10.1177/1758834016689029
11. Jensen NF, Agama K, Roy A, et al. Characterization of DNA topoisomerase I in three SN-38 resistant human colon cancer cell lines reveals a new pair of resistance-associated mutations. J Exp Clin Cancer Res. 2016;35(1):56. doi:10.1186/s13046-016-0335-x
12. Codogno P, Meijer AJ. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ. 2005;12(2):1509–1518. doi:10.1038/sj.cdd.4401751
13. Booth LA, Roberts JL, Dent P. The role of cell signaling in the crosstalk between autophagy and apoptosis in the regulation of tumor cell survival in response to sorafenib and neratinib. Semin Cancer Biol. 2019. doi:10.1016/j.semcancer.2019.10.013
14. Shen W, Zhang X, Fu X, et al. A novel and promising therapeutic approach for NSCLC: recombinant human arginase alone or combined with autophagy inhibitor. Cell Death Dis. 2017;8(3):e2720. doi:10.1038/cddis.2017.137
15. Chen Y, Zhou X, Qiao J, Bao A. MiR-142-3p overexpression increases chemo-sensitivity of NSCLC by inhibiting HMGB1-mediated autophagy. Cell Physiol Biochem. 2017;41(4):1370–1382. doi:10.1159/000467896
16. Chen MC, Lee NH, Ho TJ, et al. Resistance to irinotecan (CPT-11) activates epidermal growth factor receptor/nuclear factor kappa B and increases cellular metastasis and autophagy in LoVo colon cancer cells. Cancer Lett. 2014;349(1):51–60. doi:10.1016/j.canlet.2014.03.023
17. Paillas S, Causse A, Marzi L, et al. MAPK14/p38alpha confers irinotecan resistance to TP53-defective cells by inducing survival autophagy. Autophagy. 2012;8(7):1098–1112. doi:10.4161/auto.20268
18. Lu KV, Zhu S, Cvrljevic A, et al. Fyn and SRC are effectors of oncogenic epidermal growth factor receptor signaling in glioblastoma patients. Cancer Res. 2009;69(17):6889–6898. doi:10.1158/0008-5472.CAN-09-0347
19. Fernandez S, Risolino M, Mandia N, et al. miR-340 inhibits tumor cell proliferation and induces apoptosis by targeting multiple negative regulators of p27 in non-small cell lung cancer. Oncogene. 2015;34(25):3240–3250. doi:10.1038/onc.2014.267
20. Keyvani-Ghamsari S, Rabbani-Chadegani A, Sargolzaei J, Shahhoseini M. Effect of irinotecan on HMGB1, MMP9 expression, cell cycle, and cell growth in breast cancer (MCF-7) cells. Tumour Biol. 2017;39(4):1010428317698354. doi:10.1177/1010428317698354
21. Zhao Z, Han F, Yang S, Wu J, Zhan W. Oxamate-mediated inhibition of lactate dehydrogenase induces protective autophagy in gastric cancer cells: involvement of the Akt-mTOR signaling pathway. Cancer Lett. 2015;358(1):17–26. doi:10.1016/j.canlet.2014.11.046
22. Hu W, Chen SS, Zhang JL, Lou XE, Zhou HJ. Dihydroartemisinin induces autophagy by suppressing NF-kappaB activation. Cancer Lett. 2014;343(2):239–248. doi:10.1016/j.canlet.2013.09.035
23. Duan P, Hu C, Quan C, et al. 4-Nonylphenol induces apoptosis, autophagy and necrosis in Sertoli cells: involvement of ROS-mediated AMPK/AKT-mTOR and JNK pathways. Toxicology. 2016;341–343:28–40. doi:10.1016/j.tox.2016.01.004
24. Subhash VV, Tan SH, Yeo MS, et al. ATM expression predicts veliparib and irinotecan sensitivity in gastric cancer by mediating P53-independent regulation of cell cycle and apoptosis. Mol Cancer Ther. 2016;15(12):3087–3096. doi:10.1158/1535-7163.MCT-15-1002
25. Hu X, Shi S, Wang H, et al. Blocking autophagy improves the anti-tumor activity of afatinib in lung adenocarcinoma with activating EGFR mutations in vitro and in vivo. Sci Rep. 2017;7(1):4559. doi:10.1038/s41598-017-04258-8
26. Shin D, Kim EH, Lee J, Roh JL. RITA plus 3-MA overcomes chemoresistance of head and neck cancer cells via dual inhibition of autophagy and antioxidant systems. Redox Biol. 2017;13:219–227. doi:10.1016/j.redox.2017.05.025
27. Wang Z, Shi X, Li Y, et al. Blocking autophagy enhanced cytotoxicity induced by recombinant human arginase in triple-negative breast cancer cells. Cell Death Dis. 2014;5(12):e1563. doi:10.1038/cddis.2014.503
28. Fan D, Liu SY, van Hasselt CA, et al. Estrogen receptor alpha induces prosurvival autophagy in papillary thyroid cancer via stimulating reactive oxygen species and extracellular signal regulated kinases. J Clin Endocrinol Metab. 2015;100(4):E561–571. doi:10.1210/jc.2014-3257
29. Cheng P, Ni Z, Dai X, et al. The novel BH-3 mimetic apogossypolone induces Beclin-1- and ROS-mediated autophagy in human hepatocellular carcinoma [corrected] cells. Cell Death Dis. 2013;4(2):e489. doi:10.1038/cddis.2013.17
30. Mi Y, Xiao C, Du Q, Wu W, Qi G, Liu X. Momordin Ic couples apoptosis with autophagy in human hepatoblastoma cancer cells by reactive oxygen species (ROS)-mediated PI3K/Akt and MAPK signaling pathways. Free Radic Biol Med. 2016;90:230–242. doi:10.1016/j.freeradbiomed.2015.11.022
31. Hung AC, Tsai CH, Hou MF, et al. The synthetic beta-nitrostyrene derivative CYT-Rx20 induces breast cancer cell death and autophagy via ROS-mediated MEK/ERK pathway. Cancer Lett. 2016;371(2):251–261. doi:10.1016/j.canlet.2015.11.035
32. Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30(6):678–688. doi:10.1016/j.molcel.2008.06.001
33. Tang G, Yue Z, Talloczy Z, et al. Autophagy induced by Alexander disease-mutant GFAP accumulation is regulated by p38/MAPK and mTOR signaling pathways. Hum Mol Genet. 2008;17(11):1540–1555. doi:10.1093/hmg/ddn042
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.