")
Back to Journals » Drug Design, Development and Therapy » Volume 11
Irinotecan and 5-fluorouracil-co-loaded, hyaluronic acid-modified layer-by-layer nanoparticles for targeted gastric carcinoma therapy
Authors Gao Z, Li Z, Yan J, Wang P
Received 1 May 2017
Accepted for publication 30 June 2017
Published 5 September 2017 Volume 2017:11 Pages 2595—2604
DOI https://doi.org/10.2147/DDDT.S140797
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jianbo Sun
Zhuanglei Gao,1 Zhaoxia Li,2 Jieke Yan,3 Peilin Wang1
1Department of General Surgery, 2Department of Pediatrics, 3Department of Renal Transplantation, The Second Hospital of Shandong University, Jinan, Shandong, People’s Republic of China
Abstract: For targeted gastric carcinoma therapy, hyaluronic acid (HA)-modified layer-by-layer nanoparticles (NPs) are applied for improving anticancer treatment efficacy and reducing toxicity and side effects. The aim of this study was to develop HA-modified NPs for the co-loading of irinotecan (IRN) and 5-fluorouracil (5-FU). A novel polymer–chitosan (CH)–HA hybrid formulation (HA–CH–IRN/5-FU NPs) consisting of poly(D,L-lactide-co-glycolide) (PLGA) and IRN as the core, CH and 5-FU as a shell on the core and HA as the outmost layer was prepared. Its morphology, average size, zeta potential and drug encapsulation ability were evaluated. Human gastric carcinoma cells (MGC803 cells) and cancer-bearing mice were used for the testing of in vitro cytotoxicity and in vivo antitumor efficiency of NPs. HA–CH–IRN/5-FU NPs displayed enhanced antitumor activity in vitro and in vivo than non-modified NPs, single drug-loaded NPs and drugs solutions. The results demonstrate that HA–CH–IRN/5-FU NPs can achieve impressive antitumor activity and the novel targeted drug delivery system offers a promising strategy for the treatment of gastric cancer.
Keywords: gastric carcinoma, irinotecan, 5-fluorouracil, hyaluronic acid, layer-by-layer nanoparticles
Introduction
Gastric cancer (GC) is the fourth most common cancer worldwide and the third leading cause of global cancer mortality, with an estimated 951,600 new cases and 723,100 deaths in 2012.1,2 Depending on GC classification and stage, clinical therapeutic regimens include surgery, systemic therapy, radiation therapy and multimodality treatments.3–5 However, GC is often diagnosed at an advanced stage, at which surgical techniques are not suitable for these patients. It has been demonstrated that chemotherapy can provide both palliation and improved survival in patients with advanced and metastatic GC.6
Older agents such as fluoropyrimidines, platinum compounds and, recently, taxanes and irinotecan (IRN) have shown the most activity as a single active ingredient and in combination regimens in patients with advanced GC.7–10 Combination chemotherapy regimens have been widely applied in clinics and bring superior time-to-treatment failure (TTF), progression-free survival (PFS) and overall survival (OS) than single ones.11,12 These first-line combination regimens contain cisplatin and fluorouracil (CF); epirubicin, cisplatin and fluorouracil (ECF); epirubicin, oxaliplatin and capecitabine (ECX); fluorouracil, leucovorin and irinotecan (FOLFIRI), etc. The results of a randomized Phase III study comparing FOLFIRI to ECX in patients with advanced gastric or esophagogastric junction (EGJ) adenocarcinoma showed longer TTF and better toleration with FOLFIRI than with ECX.12 To optimize both or multi-drugs’ synergistic therapeutic efficacies and reduce side effects, recent efforts have been devoted to developing novel combination nanomedicines. Therefore, we designed multifunctional nanoparticles (NPs) to co-deliver the hydrophilic drug (5-fluorouracil [5-FU]) and the hydrophobic drug (IRN) for gastric carcinoma therapy.
Layer-by-layer (LBL) assembly is a versatile technique to develop multilayer films by the electrostatic attraction of oppositely charged polyelectrolytes.13–15 LBL techniques possess the ability to incorporate various agents with different physicochemical properties.16,17 In addition, LBL-based NPs could be engineered as the active targeting drug delivery system by the layering materials, which are targeting moieties.18,19 Moreover, the assembled polymer layers could carry a large amount of agents and control the release of them, thus having long blood circulation time.13
The main purpose of this study was to specifically target and kill GC cells through co-delivery of 5-FU and IRN. As a result, we designed a novel polymer–chitosan (CH)–hyaluronic acid (HA) hybrid formulation (HA–CH–IRN/5-FU NPs) consisting of poly(D,L-lactide-co-glycolide) (PLGA) and IRN as the core, CH and 5-FU as a shell on the core and HA as the outmost layer to target GC cells. CH, the natural linear polysaccharide, is a biocompatible, biodegradable and mucoadhesive cationic polymer that exhibits increased and rapid uptake by cancer cells.20 Recently, CH NPs have been exploited extensively in the effective delivery of anticancer agents to the tumor area.21,22 HA is a nontoxic, biocompatible, biodegradable and negative polymer that could actively target the variant CD44 receptor commonly overexpressed on various cancers, including breast cancer and GC.23,24 HA-decorated NPs have been developed for CD44-targeted GC therapy.25–27 Results showed that HA-coated NPs can be effectively used as a targeted antitumor drug delivery system to enhance therapeutic effects and overcome multi-drug resistance. In order to evaluate HA–CH–IRN/5-FU NPs, we further investigated the physical–chemical and biological characteristics, especially the in vitro and in vivo antitumor efficacy.
Materials and methods
Materials
PLGA (50:50, molecular weight 5,000–15,000) was purchased from the Jinan Daigang Biomaterial Co., Ltd. (Jinan, China). HA (molecular weight 66–90 kDa) was obtained from Shandong Freda Biochem Co., Ltd. (Jinan, China). CH (degree of deacetylation: 80%; molecular weight 400 kDa), 5-FU, IRN, Pluronic F-68 and 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Roswell Park Memorial Institute-1640 (RPMI-1640) and fetal bovine serum (FBS) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). All chemicals and reagents used were of analytical grade or better.
Cells and culture
Human gastric carcinoma cells (MGC803 cells) were purchased from American Type Culture Collection (Manassas, VA, USA) and were grown in an RPMI-1640 medium containing 10% FBS and 1% antibiotic antimycotic at 37°C and a humidified 5% CO2 atmosphere.
Animals and guidelines
Balb/c nude mice (8 weeks old, 20 g weight) were purchased from Shanghai Slack Laboratory Animal Co., Ltd. (Shanghai, China) and housed in ventilated cages at a temperature of 20°C±2°C and a relative humidity of 60%±5%. All animal experiments comply with the National Institutes of Health Guide for the care and use of laboratory animals (NIH Publication No 8023, revised 1978), and the animal experiments was approved by the medical ethics committee of Shandong University (No 100120160189).
Preparation of IRN-loaded PLGA NPs
IRN-loaded PLGA NPs (IRN NPs) was prepared by solvent displacement technique.28 Briefly, IRN (20 mg) and PLGA (100 mg) were dissolved in acetone (5 mL) to get the organic phase. In all, 1% F68 (w/v) was dissolved in Milli-Q water (50 mL) to obtain the aqueous solution. The organic phase was added drop by drop into the aqueous solution (50 mL) and stirred by a laboratory magnetic stirrer (400 rpm at room temperature) for 8 h to completely evaporate the organic solvent. The obtained NP suspensions were collected by centrifugation at 10,000 rpm for 15 min, resuspended in Milli-Q water, washed three times and filtered through a 0.45 μm membrane. The obtained NPs were stored at 2°C–8°C.
Preparation of 5-FU-loaded CH NPs
5-FU-loaded CH NPs (5-FU NPs) were prepared by the self-assembly technique.29 Briefly, 5-FU (20 mg) was dissolved in Milli-Q water (5 mL) added drop by drop into the CH (100 mg) in acetic acid (1% v/v) solution. The obtained NP suspensions were collected by centrifugation at 10,000 rpm for 15 min, resuspended in Milli-Q water, washed three times and filtered through a 0.45 μm membrane. The obtained NPs were stored at 2°C–8°C.
Preparation of HA–CH–IRN/5-FU NPs
PLGA–CH hybrid NPs (CH–IRN/5-FU NPs) were prepared as follows: 5-FU (20 mg) was mixed with CH (100 mg) in acetic acid (1% v/v) solution and then added drop by drop into the IRN NP suspensions.
HA–CH–IRN/5-FU NPs (Figure 1A) was fabricated by adding HA solution drop by drop into the CH–IRN/5-FU NP suspensions.30 The obtained CH–IRN/5-FU NP and HA–CH–IRN/5-FU NP suspensions were collected by centrifugation at 10,000 rpm for 15 min, resuspended in Milli-Q water, washed three times and filtered through a 0.45 μm membrane. The obtained NPs were stored at 2°C–8°C.
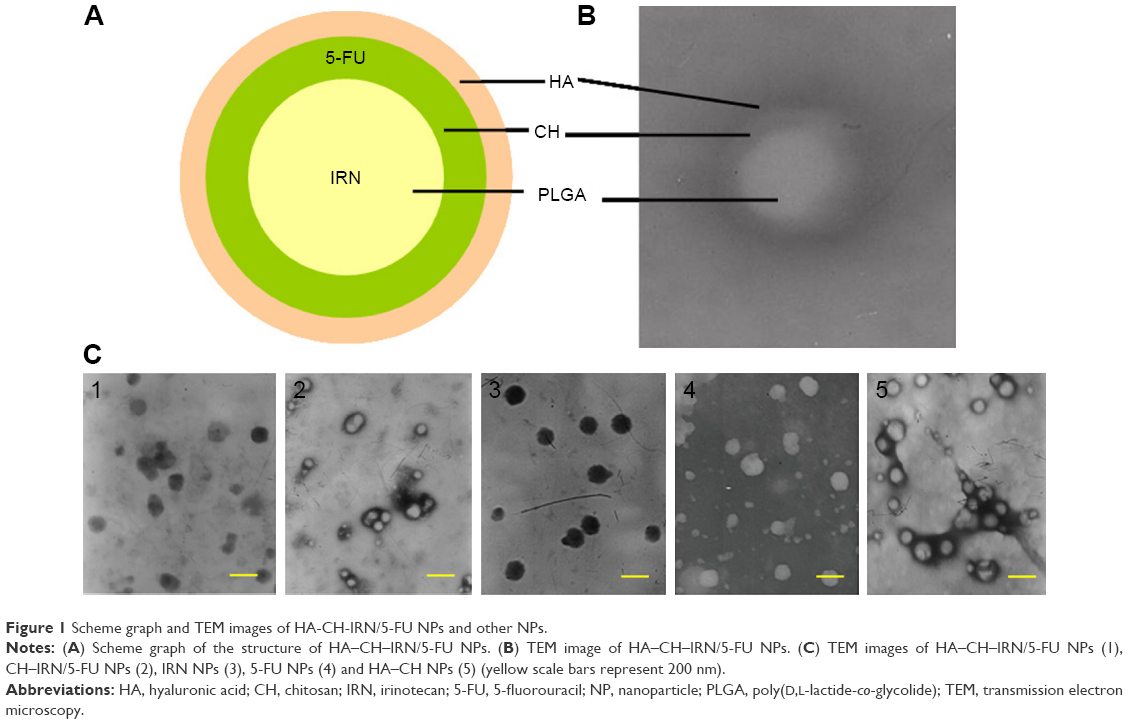 | Figure 1 Scheme graph and TEM images of HA-CH-IRN/5-FU NPs and other NPs. |
Blank HA- and CH-containing NPs not including IRN and 5-FU (HA–CH NPs) were prepared by the same procedure without adding the two drugs.
Characterization of NPs
Transmission electron microscopy (TEM) was used to investigate the morphology of HA–CH–IRN/5-FU NPs, using JEM-1200EX transmission electron microscope (JEOL, Tokyo, Japan).31 One drop of HA–CH–IRN/5-FU NPs was placed onto a copper grid and air-dried. A drop of 2% aqueous solution of sodium phosphotungstate was applied as a negative stain, and the NPs were then examined under the microscope.
Mean particle size (volume mean diameter), polydispersity index (PDI) and zeta potential of NPs were characterized using photon correlation spectroscopy with a Zetasizer 3000 (Malvern Instruments, Malvern, UK) as described in our previous study.32
The IRN and 5-FU entrapment efficiency (EE) in NPs was determined by high-performance liquid chromatography (HPLC; LC-20A; Shimadzu, Tokyo, Japan). 5-FU content was quantified using 250×4.6 mm C18 column.29 The mobile phase (flow rate 0.6 mL/min) was KH2PO4 0.05 M triethylamine 0.1%, and ultraviolet detection was set at 266 nm. Quantitative analysis of IRN was carried out by spectrophotometric assay (UV-1800, UV–VIS spectrophotometer; Shimadzu) at 370 nm.33 The EE of the NPs was calculated according to the following equation: EE (%) = drug amount in NPs/drug feeding ×100.
Serum stability of HA–CH–IRN/5-FU NPs and single drug-loaded NPs was evaluated in phosphate-buffered saline (PBS) containing 10% FBS (v/v) at 37°C for 72 h. At times 0, 2, 4, 8, 24, 48 and 72 h, 1 mL of each sample was diluted with 2 mL of triethylamine and the mixture was bath sonicated for 5 min, followed by centrifugation at 10,000 rpm for 5 min. The variation trends of the particle size were evaluated.
In vitro drug release studies
In vitro drug release studies were performed on IRN and/or 5-FU-co-loaded NPs.34 Briefly, 50 mg of NPs were resuspended in 10 mL of PBS and placed at 37°C in a shaker incubator shaking at the speed of 100 rpm. At predetemined times (1, 2, 4, 8, 12, 24, 48 and 72 h), the suspension was centrifuged at 12,000 rpm for 10 min, the supernatant was harvested and the amount of IRN and 5-FU was quantified by the method mentioned in the “characterization of NPs” section. The pellets of NPs were resuspended in fresh PBS in the original tube for further incubation.
Cellular uptake studies
The uptake rates of IRN and/or 5-FU-co-loaded NPs by MGC803 cells were tested using coumarin 6 as a model fluorescent molecule, which can be encapsulated into various NPs for quantitative investigation.31 Kinds of coumarin 6-loaded NPs were prepared by the same method as described in the “Preparation of IRN-loaded PLGA NPs”, “Preparation of 5-FU-loaded CH NPs” and “Preparation of HA–CH–IRN/5-FU NPs” sections, with the adding of 20 mg coumarin 6 along with the drugs into each formula. Coumarin 6-loaded NPs were added at concentrations of 200 mg/mL into MGC803 cells equilibrated with Hank’s buffered salt solution (HBSS) at 37°C. After incubation for 24 and 72 h, the medium was removed and the wells were washed three times with cold PBS solution. Then, cells were washed once with 1 mL of PBS; detached with trypsin/EDTA, centrifuged at 1,500 rpm, 4°C for 5 min; resuspended in 300 μL of PBS and directly introduced to a flow cytometer.
In vitro cytotoxicity assays
In vitro cytotoxicity of NPs was estimated in MGC803 cells using MTT assay.35 Briefly, MGC803 cells were seeded in 96-well plates at a density of 2,000 cells/well and incubated for 24 h. IRN- and/or 5-FU-contained solutions or NPs were added at various concentrations for 48 h. MTT was then added to the media at a concentration of 5 mg/mL and incubated for 4 h. The formazan crystals were dissolved in 100 μL dimethyl sulfoxide (DMSO), and optical absorbance was recorded at 570 nm with a Model 680 Microplate Reader (Bio-Rad Laboratories Inc., Hercules, CA, USA). Cell viability was calculated as percentage of the untreated control.
Synergistic effect evaluation and selection of the ratio of drugs
To evaluate the synergistic effects and select the suitable ratio of drug co-loaded in the NPs, the combination index (CI) was measured according to the Chou and Talalay’s method.36,37 Based on the cell viability results calculated in the “Cellular uptake studies” section, the median inhibitory concentration (IC50) values were calculated using Origin 8.0 (OriginLab, Northampton, MA, USA). CI was calculated by the equation: CI = [(C)IRN/(C50)IRN] + [(C)5-FU/(C50)5-FU]. (C)IRN and (C)5-FU represents the concentration of IRN and 5-FU in the combination system at the IC50 value. (C50)IRN and (C50)5-FU represent the IC50 value of IRN alone and 5-FU alone, respectively. CI <1, CI =1 and CI >1 indicate synergism, additive effect and antagonism, respectively.
Tumor induction
Tumors were induced in mice by subcutaneous injection of MGC803 cells (107 cells suspended in 100 μL normal saline) into the right and left flanks on the dorsal side of the Balb/c nude mice.38 The measurements were taken in two perpendicular dimensions, and tumor volumes were calculated by the following equation: tumor volume (mm3) = (longest diameter × shortest diameter2)/2. When the volume of the tumor reached ~50 mm3 (~1 day), the GC-bearing mice were divided into eight groups (with eight mice in each group) that were ready for the treatment.
In vivo tissue distribution study
The GC-bearing mice model was used to investigate the in vivo tissue distribution of IRN- and/or 5-FU-contained solutions or NPs.39 The mice were divided into several groups and administered 1 mL of solutions or NPs through the tail vein separately. At predetermined time intervals, mice were sacrificed and the heart, liver, spleen, lung, kidney, stomach, colon and tumor of mice were collected. The tissues were cut into small pieces and homogenized with physiological saline. After appropriate dilution of supernatants, the content of IRN and 5-FU was quantified by the method mentioned in the “Characterization of NPs” section.
In vivo antitumor effect and system toxicity evaluation
In vivo antitumor effect and system toxicity of the NPs were evaluated in terms of tumor volume and body weight changes.40 The mice were divided into several groups and administered IRN- and/or 5-FU-contained solutions or NPs every other day for 3 weeks. Tumor sizes and mouse body weights were measured. Tumor volumes were calculated using the equation presented in the “Synergistic effect evaluation and selection of the ratio of drugs” section.
Statistical analysis
The level of significance in all statistical analyses was set at a probability of P<0.05. Experiments were performed at least three times (n=3) and expressed as mean ± SD. Statistical differences were determined using Student’s t-test for comparison of two groups and one-way ANOVA for multiple groups.
Results
Characterization of HA–CH–IRN/5-FU NPs
Morphology of HA–CH–IRN/5-FU NPs exhibited an LBL type of spherical particle (Figure 1B). It appeared as a white core, a gray first layer over the core and a darker outside second layer. Morphology of other NPs is presented in Figure 1C. Mean particle size, PDI and zeta potential were also tested (Table 1). The size of NPs was increased from 91 to 153 along with the LBL formation procedure, indicated that the coating of CH and HA layer enlarged the particles. Zeta potential of the NPs varied due to the positive or negative charges of the materials used. The EE of IRN- and/or 5-FU-loaded NPs was >90%. Changes in size in the presence of serum are described in Figure 2. The NPs tested were stable up to 72 h without any significant size changes.
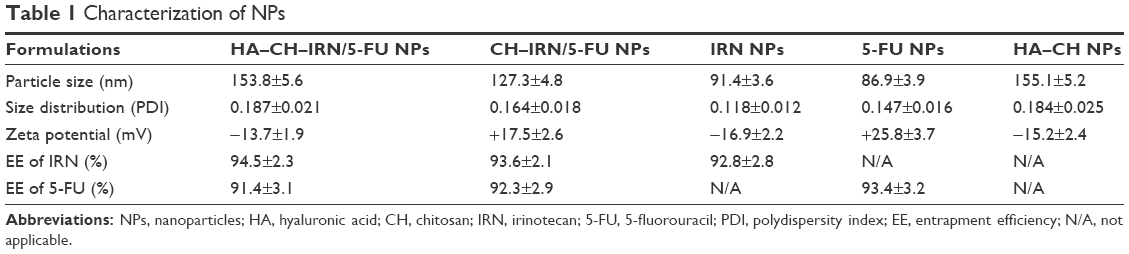 | Table 1 Characterization of NPs |
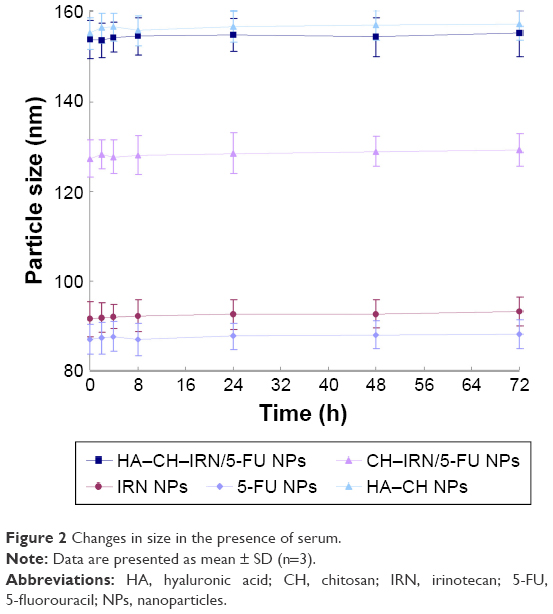 | Figure 2 Changes in size in the presence of serum. |
In vitro drug release
In vitro release of IRN and 5-FU from NPs was in a sustained behavior (Figure 3). The release behavior of 5-FU (Figure 3B) from the NPs was faster than that of IRN (Figure 3A). The release rate of HA–CH–IRN/5-FU NPs was the slowest. The release rate of drugs from CH–IRN/5-FU NPs was slower than that of IRN NPs. 5-FU released from 5-FU NPs was more slower than from the CH–IRN/5-FU NPs.
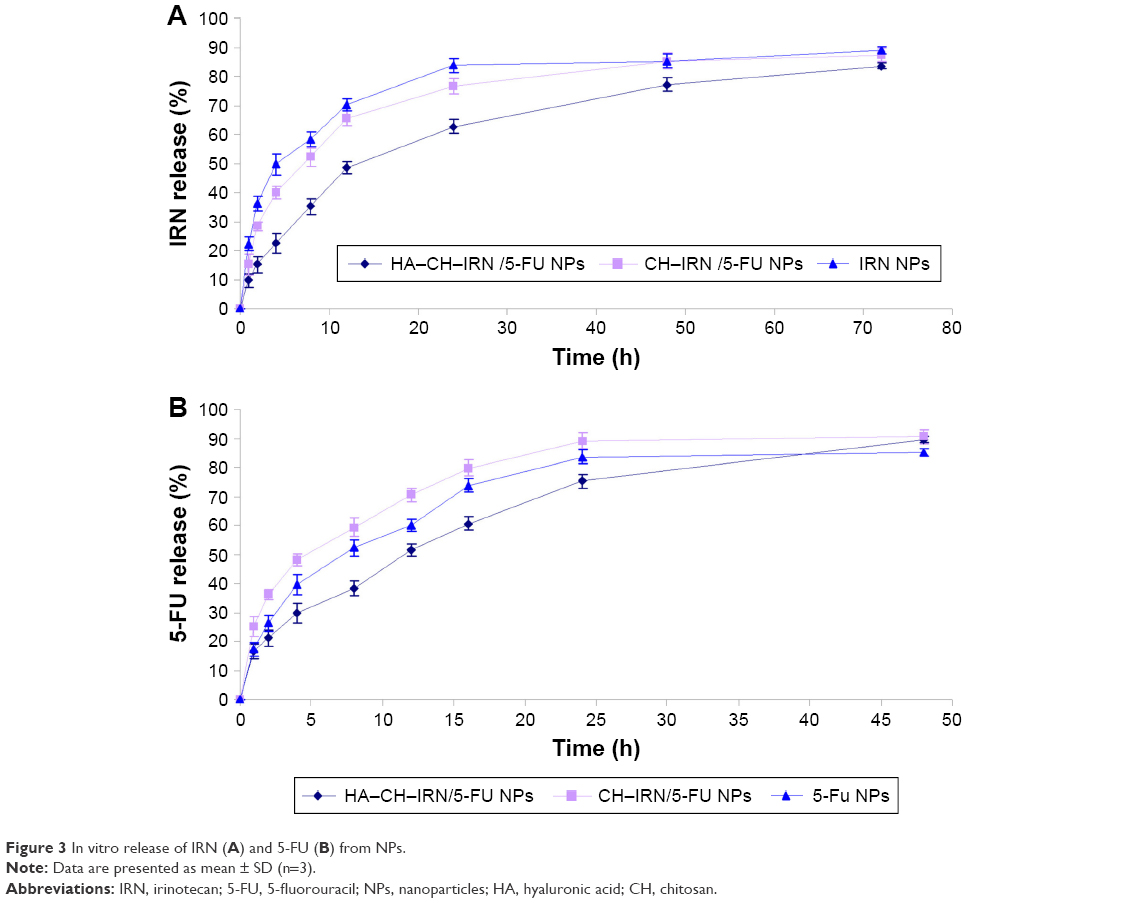 | Figure 3 In vitro release of IRN (A) and 5-FU (B) from NPs. |
Cellular uptake
Cellular uptake efficiency of the NPs was tested. As shown in Figure 4, cellular uptake efficiency of HA–CH–IRN/5-FU NPs and HA–CH NPs was significantly higher than other NPs, reached >80% at 72 h post treatments (P<0.05). Cellular uptake efficiency of CH–IRN/5-FU NPs, IRN NPs and 5-FU NPs showed no obvious difference compared with each other.
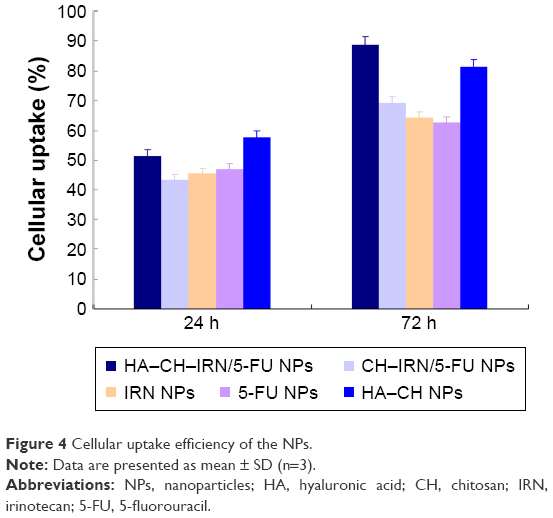 | Figure 4 Cellular uptake efficiency of the NPs. |
In vitro cytotoxicity
In vitro cytotoxicity of IRN- and/or 5-FU-contained solutions or NPs was investigated using MGC803 cells. Following incubation with samples for 48 h, cell viability was summarized (Figure 5). HA–CH–IRN/5-FU NPs exhibited the most effective tumor cell inhibition ability among all samples tested. The results illustrated that the drug-loaded NPs were more efficient than free drug solutions (P<0.05). Double drugs contained NPs, and the solution also had a higher cytotoxicity than single drug-contained NPs and solution (P<0.05).
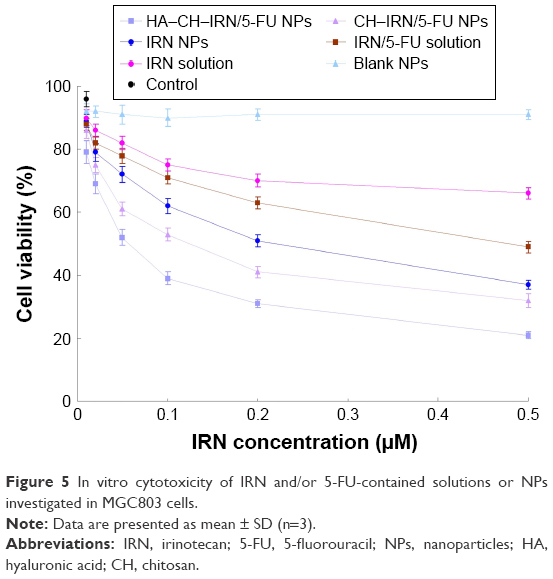 | Figure 5 In vitro cytotoxicity of IRN and/or 5-FU-contained solutions or NPs investigated in MGC803 cells. |
Synergistic effect and selection of the ratio of the two drugs
Dual drug-co-loaded NPs with various IRN to 5-FU weight ratios were applied to MGC803 cells using MTT assay to determine the synergistic effect and selection of the ratio of the two drugs (Table 2). The combination therapy showed synergistic effect when IRN to 5-FU ratio was 2:1. The CI50 values for HA–CH–IRN/5-FU NPs, CH–IRN/5-FU NPs and IRN/5-FU solutions were 0.521, 0.884 and 0.787, respectively. The IRN to 5-FU weight ratio of 2:1 exhibited stronger synergism in both NPs and solution formulations; thus, this ratio was determined in this study. So the ration of 2:1 (IRN to 5-FU, w/w) was used for all the in vitro and in vivo studies.
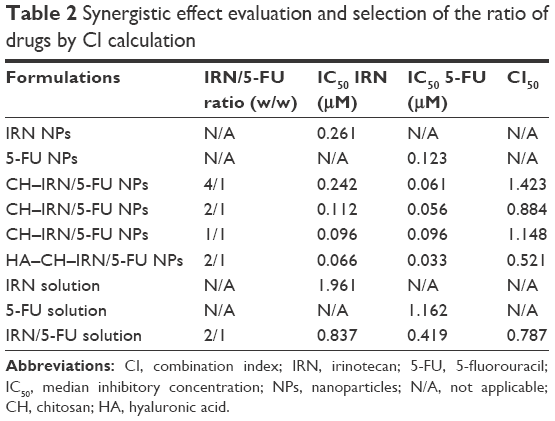 | Table 2 Synergistic effect evaluation and selection of the ratio of drugs by CI calculation |
In vivo tissue distribution
In vivo tissue distribution of HA–CH–IRN/5-FU NPs, CH–IRN/5-FU NPs and IRN/5-FU solution was investigated in GC-bearing mice model (Figure 6). IRN and 5-FU distributions of HA–CH–IRN/5-FU NPs were higher in the tumor tissue than those of CH–IRN/5-FU NPs and IRN/5-FU solution (P<0.05). Drugs loaded in NPs had relatively lower accumulation in heart and kidney than drugs’ solution (P<0.05).
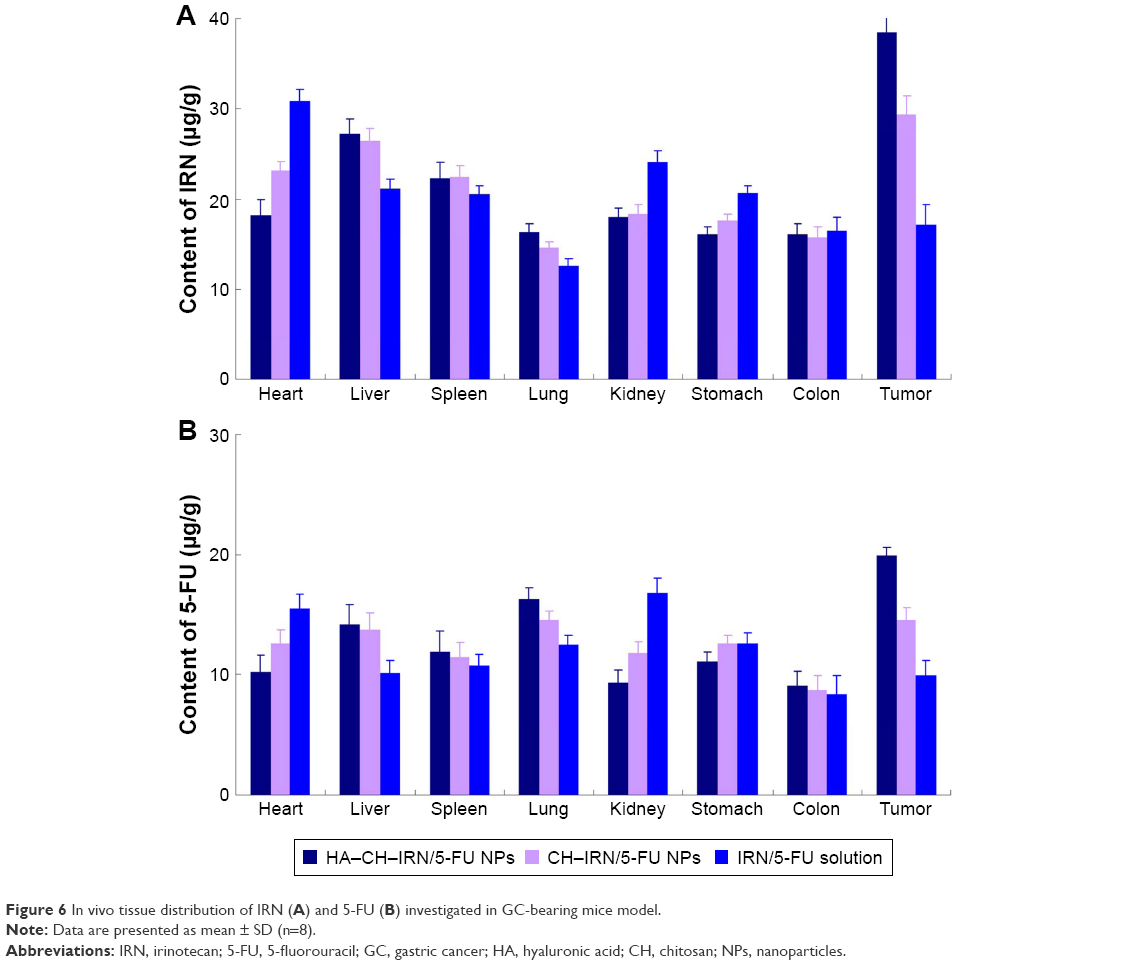 | Figure 6 In vivo tissue distribution of IRN (A) and 5-FU (B) investigated in GC-bearing mice model. |
In vivo antitumor effect and system toxicity
In vivo antitumor efficiency was evaluated in GC-bearing mice model (Figure 7A). The most obvious tumor inhibition was clearly observed in the HA–CH–IRN/5-FU NPs group; the tumor growth was prominently delayed, which attained ~217 mm3 on 21-day posttreatment. The 0.9% saline control group had a tumor volume of 1,530 mm3 on day 21. Tumor growth was more significantly inhibited by drug-loaded NPs than free drug solutions (P<0.05). Dual drug-co-loaded NPs and solutions showed better tumor suppression efficacy than the single drug containing NPs and solutions (P<0.05). HA–CH–IRN/5-FU NPs exhibited better tumor inhibition ability than non-modified CH–IRN/5-FU NPs (P<0.05). The obvious emaciation could be observed in the free drug solution groups, with an obvious decrease in the body weight (Figure 7B). The NP groups did not cause a significant difference in body weight changes. A significant body weight increase was found in the 0.9% saline control group.
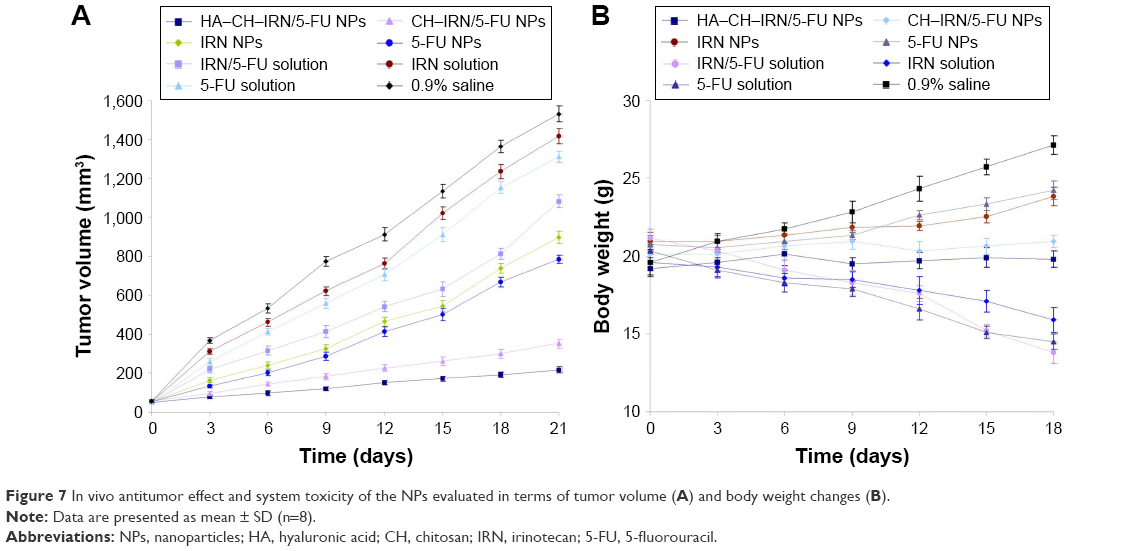 | Figure 7 In vivo antitumor effect and system toxicity of the NPs evaluated in terms of tumor volume (A) and body weight changes (B). |
Discussion
LBL assembly techniques are promising strategies for incorporating various agents with different physicochemical properties, and LBL-based NPs could be engineered as the targeted drug delivery system to provide enhanced stability, cellular uptake, regulation of drug release and targeting capabilities.41 In this study, we used the LBL technique to develop multilayer NPs by the electrostatic interaction of oppositely charged HA (negative) and CH (positive). Moreover, because of the specific binding between HA and CD44, our NPs can be used for targeted delivery of anticancer drugs into GC that are commonly overexpressed with CD44.
Electrostatic interactions are widely reported to be the main driving force for the multilayer formation process between two oppositely charged polyelectrolytes for the LBL technique.42,43 Morphology, size and zeta potential of NPs are often characterized to determine whether an additional layer could be successfully formulated onto the previous carriers. Figure 1B illustrates the layered core–shell structure of HA–CH–IRN/5-FU NPs; this could be the evidence that the system was successfully assembled. Moreover, the increased particle size indicated that the coating of CH and HA layers enlarged the particles. Zeta potential of the NPs was reversed due to the positive or negative charged materials used. The NPs tested were stable up to 72 h without any significant size changes in serum and suggest in a period of time that this formulation may be stable when intravenously administrated.
Sustained release behavior of IRN and 5-FU from NPs could significantly improve the therapeutic efficacy of the drugs loaded.27 Faster release behavior of 5-FU from the NPs than that from IRN could be explained by stating that the 5-FU was loaded in the outer layer of the NPs and the IRN was loaded in the inner core. More slowly release of 5-FU from 5-FU NPs than from the CH–IRN/5-FU NPs may be due to the preparation of 5-FU NPs using a large amount of CH that may stuck the 5-FU release.
The therapeutic effects of the NPs would depend on internalization of the NPs to the cancer cells. Coumarin 6, a fluorescent probe, was used to represent the drug in the NP formulation to analyze cellular uptake of the NPs. Cellular uptake efficiency of HA–CH–IRN/5-FU NPs was significantly higher than other NPs. This could be attributed to enhanced cancer cell-specific adherence of the HA ligands. This behavior may improve the activity and overcome drug resistance.31 In order to investigate the cytotoxicity and synergistic effect of dual drug-co-loaded NPs, we have performed a cell viability study using MGC803 cells in an in vitro MTT assay. Evaluation of drug–drug interaction is important in all areas of medicine and particularly in combination cancer chemotherapy.44 Chou and Talalay45 in 1983 introduced the term CI for quantification of synergism or antagonism for two drugs. If the CI value is <1, synergism is indicated, and if the CI value is 1 to infinity, antagonism is indicated.46 When IRN to 5-FU weight ratio was 2:1, the CI50 values for HA–CH–IRN/5-FU NPs, CH–IRN/5-FU NPs and IRN/5-FU solution were <1. This section confirmed the synergistic effect and the best IRN to 5-FU ratio of the two drug-containing systems.
In vivo drug distribution of NPs was higher in the tumor tissue and lower in heart and kidney, which could decrease the side effects during the tumor therapy.47 On the contrary, the drug solution samples mainly distributed in heart and kidney. This may lead to systemic toxicity. Higher drug distribution of NPs formulations in tumor might be due to the sustained release behavior and targeted ability of the NPs thus prolonged the blood circulation time and could better target the tumor site.
It was observed that in vivo antitumor efficiency of the dual drugs could be better after loading in the NPs because of the higher tumor volume suppression. The higher antitumor efficiency of drugs after co-loaded in HA–CH–IRN/5-FU NPs than non-modified CH–IRN/5-FU NPs is related to the targeted ability of HA.48 Based on the healthy body weight of NP-treated mice, the NPs constructed were proposed as safe carriers for the delivery of anticancer drugs. Minimizing the toxicity and side effects of drugs could prove the targeting efficiency of NPs. The in vivo antitumor results suggested the best anti-tumor effect of folate decorated double drugs contained NLCs due to the synergetic effect of the two drugs, and the least systemic toxic side effect of the NLC formulations for the head and neck cancer treatment.
Conclusion
HA–CH–IRN/5-FU NPs consisting of PLGA and IRN as the core, CH and 5-FU as a shell on the core and HA as the outmost layer were prepared. HA–CH–IRN/5-FU NPs displayed enhanced antitumor activity in vitro and in vivo than non-modified NPs, single drug-loaded NPs and drug solutions. The results demonstrate that HA–CH–IRN/5-FU NPs can achieve impressive antitumor activity and the novel targeted drug delivery system offers a promising strategy for the treatment of GC.
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Apicella M, Corso S, Giordano S. Targeted therapies for gastric cancer: failures and hopes from clinical trials. Oncotarget. Epub 2017 Jan 26. | ||
Dikken JL, van Sandick JW, Maurits Swellengrebel HA, et al. Neo-adjuvant chemotherapy followed by surgery and chemotherapy or by surgery and chemoradiotherapy for patients with resectable gastric cancer (CRITICS). BMC Cancer. 2011;11:329. | ||
Hulshof MC, van Laarhoven HW. Chemoradiotherapy in tumours of the oesophagus and gastro-oesophageal junction. Best Pract Res Clin Gastroenterol. 2016;30(4):551–563. | ||
Merrett ND. Multimodality treatment of potentially curative gastric cancer: geographical variations and future prospects. World J Gastroenterol. 2014;20(36):12892–12899. | ||
Galdy S, Cella CA, Spada F, et al. Systemic therapy beyond first-line in advanced gastric cancer: an overview of the main randomized clinical trials. Crit Rev Oncol Hematol. 2016;99:1–12. | ||
Bugat R. Irinotecan in the treatment of gastric cancer. Ann Oncol. 2003;14(suppl 2):ii37–ii40. | ||
Ajani JA. Evolving chemotherapy for advanced gastric cancer. Oncologist. 2005;10(suppl 3):49–58. | ||
Gadgeel SM, Shields AF, Heilbrun LK, et al. Phase II study of paclitaxel and carboplatin in patients with advanced gastric cancer. Am J Clin Oncol. 2003;26(1):37–41. | ||
Gravalos C, Jimeno A. HER2 in gastric cancer: a new prognostic factor and a novel therapeutic target. Ann Oncol. 2008;19(9):1523–1529. | ||
Dank M, Zaluski J, Barone C, et al. Randomized phase III study comparing irinotecan combined with 5-fluorouracil and folinic acid to cisplatin combined with 5-fluorouracil in chemotherapy naive patients with advanced adenocarcinoma of the stomach or esophagogastric junction. Ann Oncol. 2008;19(8):1450–1457. | ||
Guimbaud R, Louvet C, Ries P, et al. Prospective, randomized, multicenter, phase III study of fluorouracil, leucovorin, and irinotecan versus epirubicin, cisplatin, and capecitabine in advanced gastric adenocarcinoma: a French intergroup (Fédération Francophone de Cancérologie Digestive, Fédération Nationale des Centres de Lutte Contre le Cancer, and Groupe Coopérateur Multidisciplinaire en Oncologie) study. J Clin Oncol. 2014;32(31):3520–3526. | ||
Ramasamy T, Tran TH, Choi JY, et al. Layer-by-layer coated lipid-polymer hybrid nanoparticles designed for use in anticancer drug delivery. Carbohydr Polym. 2014;102:653–661. | ||
Suh MS, Shen J, Kuhn LT, Burgess DJ. Layer-by-layer nanoparticle platform for cancer active targeting. Int J Pharm. 2017;517(1–2):58–66. | ||
Go DP, Hung A, Gras SL, O’Connor AJ. Use of a short peptide as a building block in the layer-by-layer assembly of biomolecules on polymeric surfaces. J Phys Chem B. 2012;116(3):1120–1133. | ||
Pang ST, Lin FW, Chuang CK, Yang HW. Co-delivery of docetaxel and p44/42 MAPK siRNA using PSMA antibody-conjugated BSA-PEI layer-by-layer nanoparticles for prostate cancer target therapy. Macromol Biosci. Epub 2017 Jan 27;17(5). | ||
Ramasamy T, Haidar ZS, Tran TH, et al. Layer-by-layer assembly of liposomal nanoparticles with PEGylated polyelectrolytes enhances systemic delivery of multiple anticancer drugs. Acta Biomater. 2014;10(12):5116–5127. | ||
Maiolino S, Moret F, Conte C, et al. Hyaluronan-decorated polymer nanoparticles targeting the CD44 receptor for the combined photo/chemo-therapy of cancer. Nanoscale. 2015;7(13):5643–5653. | ||
Poon Z, Chang D, Zhao X, Hammond PT. Layer-by-layer nanoparticles with a pH-sheddable layer for in vivo targeting of tumor hypoxia. ACS Nano. 2011;5(6):4284–4292. | ||
Campos J, Varas-Godoy M, Haidar ZS. Physicochemical characterization of chitosan-hyaluronan-coated solid lipid nanoparticles for the targeted delivery of paclitaxel: a proof-of-concept study in breast cancer cells. Nanomedicine (Lond). 2017;12(5):473–490. | ||
Kamath PR, Sunil D. Nano-chitosan particles in anticancer drug delivery: an up-to-date review. Mini Rev Med Chem. Epub 2017 Feb 27. | ||
Wang T, Hou J, Su C, Zhao L, Shi Y. Hyaluronic acid-coated chitosan nanoparticles induce ROS-mediated tumor cell apoptosis and enhance antitumor efficiency by targeted drug delivery via CD44. J Nanobiotechnology. 2017;15(1):7. | ||
Nascimento TL, Hillaireau H, Vergnaud J, Fattal E. Lipid-based nanosystems for CD44 targeting in cancer treatment: recent significant advances, ongoing challenges and unmet needs. Nanomedicine (Lond). 2016;11(14):1865–1887. | ||
Mattheolabakis G, Milane L, Singh A, Amiji MM. Hyaluronic acid targeting of CD44 for cancer therapy: from receptor biology to nanomedicine. J Drug Target. 2015;23(7–8):605–618. | ||
Kim E, Yang J, Kim HO, et al. Hyaluronic acid receptor-targetable imidazolized nanovectors for induction of gastric cancer cell death by RNA interference. Biomaterials. 2013;34(17):4327–4338. | ||
Lee H, Yang SH, Heo D, et al. Molecular Imaging of CD44-Overexpressing Gastric Cancer in Mice Using T2 MR Imaging. J Nanosci Nanotechnol. 2016;16(1):196–202. | ||
Yang Z, Luo H, Cao Z, et al. Dual-targeting hybrid nanoparticles for the delivery of SN38 to Her2 and CD44 overexpressed human gastric cancer. Nanoscale. 2016;8(22):11543–11558. | ||
Zou W, Liu C, Chen Z, Zhang N. Studies on bioadhesive PLGA nanoparticles: a promising gene delivery system for efficient gene therapy to lung cancer. Int J Pharm. 2009;370(1–2):187–195. | ||
Fabiano A, Bizzarri R, Zambito Y. Thermosensitive hydrogel based on chitosan and its derivatives containing medicated nanoparticles for transcorneal administration of 5-fluorouracil. Int J Nanomedicine. 2017;12:633–643. | ||
Zhang L, Wang J, Chi H, Wang S. Local anesthetic lidocaine delivery system: chitosan and hyaluronic acid-modified layer-by-layer lipid nanoparticles. Drug Deliv. 2016;23(9):3529–3537. | ||
Liu B, Han L, Liu J, Han S, Chen Z, Jiang L. Co-delivery of paclitaxel and TOS-cisplatin via TAT-targeted solid lipid nanoparticles with synergistic antitumor activity against cervical cancer. Int J Nanomedicine. 2017;12:955–968. | ||
Yan J, Wang Y, Jia Y, et al. Co-delivery of docetaxel and curcumin prodrug via dual-targeted nanoparticles with synergistic antitumor activity against prostate cancer. Biomed Pharmacother. 2017;88:374–383. | ||
Wang H, Agarwal P, Zhao S, et al. Hyaluronic acid-decorated dual responsive nanoparticles of Pluronic F127, PLGA, and chitosan for targeted co-delivery of doxorubicin and irinotecan to eliminate cancer stem-like cells. Biomaterials. 2015;72:74–89. | ||
Rafiei P, Haddadi A. Docetaxel-loaded PLGA and PLGA-PEG nanoparticles for intravenous application: pharmacokinetics and biodistribution profile. Int J Nanomedicine. 2017;12:935–947. | ||
Wang F, Li L, Liu B, Chen Z, Li C. Hyaluronic acid decorated pluronic P85 solid lipid nanoparticles as a potential carrier to overcome multidrug resistance in cervical and breast cancer. Biomed Pharmacother. 2017;86:595–604. | ||
Zhang X, Li J, Yan M. Targeted hepatocellular carcinoma therapy: transferrin modified, self-assembled polymeric nanomedicine for co-delivery of cisplatin and doxorubicin. Drug Dev Ind Pharm. 2016;42(10):1590–1599. | ||
Li C, Ge X, Wang L. Construction and comparison of different nanocarriers for co-delivery of cisplatin and curcumin: a synergistic combination nanotherapy for cervical cancer. Biomed Pharmacother. 2017;86:628–636. | ||
Abou-ElNaga A, Mutawa G, El-Sherbiny IM, et al. Novel nano-therapeutic approach actively targets human ovarian cancer stem cells after xenograft into nude mice. Int J Mol Sci. 2017;18(4):E813. | ||
Zhang N, Miao J, Sun P, et al. Pharmacokinetics, tissue distribution and anti-tumor effect of low density lipoprotein peptide conjugated submicron emulsions. Biomed Pharmacother. 2016;82:614–619. | ||
Kim KM, Lim HK, Shim SH, Jung J. Improved chemotherapeutic efficacy of injectable chrysin encapsulated by copolymer nanoparticles. Int J Nanomedicine. 2017;12:1917–1925. | ||
Dubas ST, Schlenoff JB. Factors controlling the growth of polyelectrolyte multilayers. Macromolecules. 1999;32(24):8153–8160. | ||
Li P, Liu D, Miao L, et al. A pH-sensitive multifunctional gene carrier assembled via layer-by-layer technique for efficient gene delivery. Int J Nanomedicine. 2012;7:925–939. | ||
Chai F, Sun L, He X, et al. Doxorubicin-loaded poly (lactic-co-glycolic acid) nanoparticles coated with chitosan/alginate by layer by layer technology for antitumor applications. Int J Nanomedicine. 2017;12:1791–1802. | ||
Qu CY, Zhou M, Chen YW, Chen MM, Shen F, Xu LM. Engineering of lipid prodrug-based, hyaluronic acid-decorated nanostructured lipid carriers platform for 5-fluorouracil and cisplatin combination gastric cancer therapy. Int J Nanomedicine. 2015;10:3911–3920. | ||
Chou TC, Talalay P. Analysis of combined drug effects: a new look at a very old problem. Trends Pharmacol Sci. 1983;4:450–454. | ||
Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58(3):621–681. | ||
Nazari-Vanani R, Moezi L, Heli H. In vivo evaluation of a self-nanoemulsifying drug delivery system for curcumin. Biomed Pharmacother. 2017;88:715–720. | ||
Yu D, Li W, Zhang Y, Zhang B. Anti-tumor efficiency of paclitaxel and DNA when co-delivered by pH responsive ligand modified nanocarriers for breast cancer treatment. Biomed Pharmacother. 2016;83:1428–1435. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.