Back to Journals » International Journal of Nanomedicine » Volume 19
Ionizable Lipid Nanoparticle-Mediated TRAIL mRNA Delivery in the Tumor Microenvironment to Inhibit Colon Cancer Progression
Authors da Silva WN, Carvalho Costa PA, Scalzo Júnior SRA, Ferreira HAS ![]() , Prazeres PHDM
, Prazeres PHDM ![]() , Campos CLV, Rodrigues Alves MT, Alves da Silva NJ
, Campos CLV, Rodrigues Alves MT, Alves da Silva NJ ![]() , de Castro Santos AL, Guimarães LC, Ferris MEC, Thatte A, Hamilton A, Bicalho KA, Lobo AO
, de Castro Santos AL, Guimarães LC, Ferris MEC, Thatte A, Hamilton A, Bicalho KA, Lobo AO ![]() , Santiago HDC, da Silva Barcelos L, Figueiredo MM
, Santiago HDC, da Silva Barcelos L, Figueiredo MM ![]() , Teixeira MM, Vasconcelos Costa V, Mitchell MJ, Frézard F, Pires Goulart Guimaraes P
, Teixeira MM, Vasconcelos Costa V, Mitchell MJ, Frézard F, Pires Goulart Guimaraes P ![]()
Received 2 December 2023
Accepted for publication 5 March 2024
Published 14 March 2024 Volume 2024:19 Pages 2655—2673
DOI https://doi.org/10.2147/IJN.S452896
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Kamakhya Prakash Misra
Walison Nunes da Silva,1 Pedro Augusto Carvalho Costa,1 Sérgio Ricardo Aluotto Scalzo Júnior,1 Heloísa AS Ferreira,1 Pedro Henrique Dias Moura Prazeres,2 Caroline Leonel Vasconcelos Campos,3 Marco Túllio Rodrigues Alves,1 Natália Jordana Alves da Silva,1 Ana Luiza de Castro Santos,1 Lays Cordeiro Guimarães,1 Maria Eduarda Chen Ferris,1 Ajay Thatte,4 Alex Hamilton,4 Kelly Alves Bicalho,5 Anderson Oliveira Lobo,6 Helton da Costa Santiago,3 Lucíola da Silva Barcelos,1 Maria Marta Figueiredo,7 Mauro Martins Teixeira,3 Vivian Vasconcelos Costa,8 Michael J Mitchell,4 Frédéric Frézard,1 Pedro Pires Goulart Guimaraes1
1Department of Physiology and Biophysics, Federal University of Minas Gerais, Belo Horizonte, MG, Brazil; 2Department of Pathology, Federal University of Minas Gerais, Belo Horizonte, MG, 31270-901, Brazil; 3Department of Biochemistry and Immunology, Federal University of Minas Gerais, Belo Horizonte, MG, 31270-901, Brazil; 4Department of Bioengineering, University of Pennsylvania, Philadelphia, PA, USA; 5Instituto René Rachou, Fundação Oswaldo Cruz, Belo Horizonte, MG, Brazil; 6Department of Materials Engineering, Federal University of Piauí, Teresina, PI, Brazil; 7State University of Minas Gerais, Divinopolis, MG, Brazil; 8Department of Morphology, Federal University of Minas Gerais, Belo Horizonte, MG, Brazil
Correspondence: Pedro Pires Goulart Guimaraes, Department of Physiology and Biophysics, Federal University of Minas Gerais, Belo Horizonte, MG, Brazil, Email [email protected]
Introduction: Immunotherapy has revolutionized cancer treatment by harnessing the immune system to enhance antitumor responses while minimizing off-target effects. Among the promising cancer-specific therapies, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) has attracted significant attention.
Methods: Here, we developed an ionizable lipid nanoparticle (LNP) platform to deliver TRAIL mRNA (LNP-TRAIL) directly to the tumor microenvironment (TME) to induce tumor cell death. Our LNP-TRAIL was formulated via microfluidic mixing and the induction of tumor cell death was assessed in vitro. Next, we investigated the ability of LNP-TRAIL to inhibit colon cancer progression in vivo in combination with a TME normalization approach using Losartan (Los) or angiotensin 1– 7 (Ang(1– 7)) to reduce vascular compression and deposition of extracellular matrix in mice.
Results: Our results demonstrated that LNP-TRAIL induced tumor cell death in vitro and effectively inhibited colon cancer progression in vivo, particularly when combined with TME normalization induced by treatment Los or Ang(1– 7). In addition, potent tumor cell death as well as enhanced apoptosis and necrosis was found in the tumor tissue of a group treated with LNP-TRAIL combined with TME normalization.
Discussion: Together, our data demonstrate the potential of the LNP to deliver TRAIL mRNA to the TME and to induce tumor cell death, especially when combined with TME normalization. Therefore, these findings provide important insights for the development of novel therapeutic strategies for the immunotherapy of solid tumors.
Keywords: immunotherapy, TRAIL, mRNA, lipid nanoparticle, losartan, angiotensin (1– 7)
Introduction
Cancer therapy has undergone a remarkable transformation with the introduction of immunotherapy. This groundbreaking approach utilizes specialized therapies to enhance antitumor immune responses against cancerous cells with reduced effects on healthy tissues, ensuring more focused and safer treatments.1,2 Despite significant advances in treatments, cancer remains a significant public health issue globally, ranking as the second leading cause of death worldwide. According to projections for 2023, almost 2 million new cancer cases and over half a million cancer-related deaths are expected in the United States.3
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) has garnered significant interest as a potential targeted therapy for cancer, given its ability to induce apoptosis specifically in a variety of tumor cells by stimulating the immune system.1 TRAIL is an innate immune cytokine that is widely expressed in immune cells, including lymphocytes, natural killer cells, and neutrophils,1,4 and exhibits the ability to suppress tumor initiation and metastasis, playing an important role in the clearance of diseased cells.1,5–7 TRAIL binds to death receptors 4 and 5 (DR4 and DR5) overexpressed on the surface of a range of cancer cells to trigger apoptosis.1 While significant efforts have focused on administering TRAIL in protein form, clinical translation of such approaches is limited by the failure to deliver sufficient protein into the tumor to generate a therapeutic effect.1,7,8 As an alternative approach, gene therapy can potentially overcome these barriers by delivering plasmid DNA or messenger RNA (mRNA) coding for TRAIL in the tumor microenvironment (TME).
Numerous therapeutic approaches have been employed in the battle against cancer, aiming to improve patient outcomes and quality of life.9–11 Among these approaches, mRNA only requires delivery to the cytoplasm, bypassing the need to reach the nucleus for protein translation.12 This streamlined process improves efficiency and enhances safety, without risk of insertional mutagenesis, making mRNA an attractive option in cancer therapeutics.13 This makes mRNA therapeutics an attractive option in cancer immunotherapy as well as in vaccines, genome editing, and allergy tolerization.13,14
Although viral delivery systems have been extensively employed for gene delivery, their clinical application is hindered by immune responses, such as neutralizing antibodies and T-cell responses, which can compromise the efficacy and safety of the therapy.15,16 Therefore, non-viral delivery approaches, such as lipid nanoparticles (LNPs), are being actively explored to overcome these limitations and enhance mRNA delivery and immunogenicity control.17 Encapsulating nucleic acids, such as mRNA, in lipid nanoparticles is a promising strategy for safe and effective delivery, offering protection from degradation, longer circulation, and avoidance of renal clearance. Microfluidic devices have been employed to produce LNPs in a controlled and reproducible manner.18–20 These devices play a crucial role in advancing high-throughput in vivo screening methods, facilitating the development of optimized LNP formulations for various biomedical applications.12,18,21
Blood and lymphatic vasculature, fibroblasts, and immune cells are considered abnormal in the TME as well as the excessive extracellular matrix (ECM).22,23 This combination generates delivery barriers for therapeutics, even for delivery systems such as LNPs.24 Therefore, TME normalization is a promising strategy to improve delivery. The use of agents to induce normalization of TME can significantly enhance treatment outcomes.24,25 Various strategies have been explored to normalize the TME, including the vasculature, lymphatic system, and ECM, to improve cancer therapy delivery and slow tumor progression.17,25 Antiangiogenic therapies, such as anti-VEGF, have demonstrated vascular normalization effects and prolonged survival in clinical trials for diverse types of tumors,26 including breast cancer,27 non-small-cell lung cancer (NSCLC)28,29 and glioblastoma.30 Additionally, antagonists of angiotensin II, such as losartan, have been shown to reduce profibrotic stromal activity, decompress tumor blood vessels, enhance drug and oxygen delivery, and increase the effectiveness of chemotherapy.31–33 Furthermore, angiotensin 1–7 (Ang (1–7)), a vasoactive peptide, exhibits anticancer effects, including inhibiting angiogenesis, reducing tumor growth, and targeting cancer-associated fibroblasts and tumor fibrosis.34,35 Therefore, these approaches have been used in cancer therapy as promising tools to improve the delivery of bioactive molecules to the TME.
Here, we developed an LNP platform through microfluidic mixing to deliver TRAIL mRNA in the TME to induce tumor cell death (Figure 1). Rather than targeting a specific mutation in the tumor, our unique therapeutic strategy focuses on physically normalizing the TME via the administration of losartan (Los) or Ang (1–7) to specifically improve the delivery of TRAIL mRNA to the TME, inducing TRAIL secretion for the targeted induction of tumor cell apoptosis.
 |
Figure 1 Schematic workflow for the development of an ionizable lipid nanoparticle (LNP) platform to deliver TRAIL mRNA to the tumor microenvironment to induce tumor cell death. (A) Schematic representation of the components used for LNP synthesis. (B) Synthesis of LNP-TRAIL via microfluidic mixing. LNPs were characterized by dynamic light scattering (DLS), and their efficacy was assessed in vitro against Colo 205 cells. (C) Schematic representation of in vivo treatment of humanized NSG mice transplanted with human colon cancer cells. Mice were orally treated with Los or Ang (1–7) and intratumorally treated with LNP-TRAIL. Tumor volume was assessed during treatment, and tumor analysis of cell death and immune cell infiltration were carried out at the end of the experiment. |
Materials and Methods
Cell Culture and Cell Line
Colon cancer cells (Colo 205) expressing both luciferase and green fluorescent protein (GFP) were obtained from GeneCopoeia (Rockville, MD, USA) and cultured in RPMI-1640 medium (Gibco) supplemented with 10% (v/v) fetal bovine serum (Gibco) and 1% (v/v) Penicillin-Streptomycin solution (100 U/mL penicillin, and 100 μg/mL streptomycin; Gibco). Cells were grown at 37 °C with 5% CO2 in a humidified incubator.
Cell Viability and Apoptosis Assays
To investigate cell apoptosis mediated by delivery of TRAIL mRNA, Colo 205 cells were seeded into 96-well plates (ThermoFisher) at a density of 105 cells per well. After 24 h, cells were treated with 50 μM of the pan-caspase inhibitor Z-VAD-FMK (R&D Systems) for 4 h at 37 °C prior to treatment with LNP-TRAIL.36 Following the treatment, cells were incubated with 0.15 mg/well of D-luciferin (Invitrogen) for 10 min and luminescence was measured using the Cytation 5 Cell Imaging equipment from Biotek at the Center for Acquisition and Processing of Images (CAPI-UFMG).
In vitro Evaluation of TRAIL Production
To assess the TRAIL production induced by LNP-TRAIL, HS-5 stromal cells and Colo 205 tumor cells were seeded in 96-well plates (ThermoFisher) at a density of 105 cells per well. Following 24 h, cells were treated with 0.5 µg/well of LNP-TRAIL for 24 h. TRAIL monoclonal antibody (CD253) was utilized to assess TRAIL frequency via flow cytometry. Cell acquisition was conducted on an LSR Fortessa instrument, and data analysis was carried out using GraphPad Prism V7.0 and FlowJo V10.4.11. Combinations of fluorochromes and forward scatter area (FSC-A) versus forward scatter height (FSC-H) were employed to exclude debris and doublets, respectively (Table S1).
Preparation of 2-Hydroxypropyl-β-Cyclodextrin (HPβCD): Ang (1-7) Inclusion Complex
A freeze-drying technique was used to obtain inclusion complexes between HPβCD (Sigma-Aldrich) and Ang (1–7) (Bachem). A molar ratio of 1:1 was used as previously described.37 The required amounts of Ang (1–7) and HPβCD were accurately weighed and dispersed in Milli-Q water and the solution was magnetically stirred at room temperature for 48 h before freeze-drying.37
Ionizable Lipid Synthesis
The synthesis of ionizable lipid C12-200 was performed as previously described by reacting polyamine core 200 (Enamine Inc.) with 1,2-epoxydodecane (Sigma-Aldrich).12,38 This reaction was carried out in 100% ethanol at a temperature of 90 °C for 48 h. The final product was then purified using flash chromatography as previously described before use in LNP formulation.12
Preparation of LNPs
Ionizable lipid nanoparticles were prepared using a weight ratio of 10:1 of C12-200 to mRNA, following a previously described method.20 Briefly, an ethanol phase consisting of ionizable lipid, cholesterol (Avanti Polar Lipids), C14-PEG2000 (Avanti Polar Lipids), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE, Avanti Polar Lipids), was combined with an aqueous phase containing mRNA in 10 mM citrate buffer pH 5, using microfluidic mixing at a volume ratio of 3:1 to formulate LNPs. The LNPs were then dialyzed against 1x PBS for 2 h using dialysis cassettes with a molecular weight cut-off of 20 kDa (ThermoFisher), sterile filtered using a 0.22 μm filter, and stored at 4 °C.
LNP Characterization
LNP hydrodynamic diameter (HD), polydispersity index (PDI), and zeta potential were measured using a Zetasizer Nano ZS machine (DLS, Malvern Panalytical) at Laboratory of Biophysics of Nanostructured Systems at the UFMG. For the analysis of LNP structure, cryogenic-transmission electron microscopy (cryo-TEM) was employed. LNP samples were prepared at a temperature of 25 °C and approximately 100% humidity in a vitrification system. Vitrified samples were then examined using the Tecnai G2-20 - FEI Super Twin 200 kV microscope at the Microscopy Center of UFMG. The encapsulation efficiency and concentration of mRNA in LNPs for in vitro and in vivo applications were determined using a Ribogreen assay (ThermoFisher) and NanoDrop-OneC-UV-Vis.39 To determine the pKa of each LNP, the 2-(p-toluidine) naphthalene-6-sulfonic acid (TNS) assay was used. The experiment involved preparing a buffer solution by combining 150 mM sodium chloride, 20 mM sodium phosphate, 25 mM sodium citrate, and 20 mM sodium acetate, and then adjusting the pH from 2 to 12 in 0.5 increments. 140 µL of each pH-adjusted solution and 5 uL of each LNP were added in triplicate to a 96-well plate, followed by the addition of TNS to each well to achieve a final concentration of 6 µM. Fluorescence was measured on a Cytation 5 Cell Imaging (Biotek) with excitation and emission wavelengths of 325/435 nm. A sigmoidal curve fit was generated from the fluorescence emitted to determine the pH at which 50% protonation occurred, which was taken as the apparent pKa.
Human Samples
The human samples used in the study were approved by the Institutional Review Board of the Federal University of Minas Gerais (COEP: 51,183,421.3.0000.5149). Healthy adult donors provided blood samples and completed a comprehensive questionnaire. All donors provided their consent to participate in the study.
Ethics Statement
In vivo experiments were conducted following the regulations outlined in the National Institute of Health (NIH) Guide for the Care and Use of Laboratory Animals and authorized by the Ethical Committee for Animal Experimentation of the UFMG (CEUA 269/2022). The use of Human clinical samples complies with the Declaration of Helsinki and was performed with approval from the ethics committee.
Animal Models
For this study, we utilized 6–8-week-old transgenic mice NOD-SCID IL-2Rγnull (NSG) strain of both genders. These mice were bred at the Immunopharmacology Laboratory of the Institute of Biological Sciences (ICB) at the Federal University of Minas Gerais (UFMG). They were kept in ventilated cages in an animal biosafety level-2 facility (ICB, UFMG) and maintained under a 12 h light/12 h dark cycle at 24 °C ± 2 °C. They were provided with access to food and water ad libitum. We closely monitored the mice for any indications of illness, such as ruffled fur, arching of the back, loss of weight, and decreased activity.
Mouse Humanization Using Human Peripheral Blood Mononuclear Cells (PBMCs)
PBMCs were isolated from blood using Ficoll-Hypaque (GE-Healthcare; GE17-1440-02) gradient separation. PBMCs were rested overnight in RPMI medium (RPMI 1640 (Gibco; 31,870,074) with 10% human serum (Sigma Aldrich; H5667 or H4522), 1% penicillin/streptomycin (Sigma-Aldrich; P0781) at 37 °C and 5% CO2 before adoptive cell transfer. Cells were washed with PBS and 107 PBMCs were intravenously injected into each mouse. Peripheral blood from the humanized mice was examined using a plot of mCD45 versus human CD3. Different immune cell subpopulations, including CD4+ T cells (CD4+/CD3+) and CD8+ T cells (CD8+/CD3+), were assessed based on molecular markers specific to each cell subset. Antibodies listed in Table S1 were used for labeling, which was performed at room temperature for 20 min while protecting from light. Subsequently, the blood samples were lysed using an ammonium chloride solution, centrifuged at 400g for 5 min, and finally resuspended in 2% paraformaldehyde (PFA). Cell acquisition was performed using an LSR Fortessa (BD Biosciences), and data analysis was conducted using GraphPad Prism V7.0 and FlowJo V10.4.11.
In vivo Colo 205 Tumor Cell Study
Colo 205 tumor cells were injected subcutaneously (SC) on the flank (3 × 106 cells in 100 μL) of humanized and non-humanized NSG mice of both genders aged 16–20 weeks, weighing 25–30 g using a 30-gauge needle (Becton Dickson, Rutherford, NJ, USA). Tumor-bearing mice were weighed and randomly divided into six different groups when the tumor volume reached 100 mm3: (1) PBS; (2) Los; (3) Ang (1–7); (4) LNP-TRAIL; (5) Los + LNP-TRAIL; and (6) Ang (1–7) + LNP-TRAIL. The mice were treated with LNP-TRAIL intratumorally every 48 h at a dose of 5 µg of mRNA. Tumor growth was monitored over time using a caliper and IVIS bioluminescence image, as previously reported.40 Tumors were removed 18 and 24 days after transplantation and weighed. The length (L) and width (W) of the tumors were measured and used to calculate tumor volume (V) using the formula V = 0.5 × (L × W2).41
Oral Treatments for Normalization of TME
Non-humanized and humanized mice were orally treated with 121.2 mg/kg Losartan (Los) or 60 µg/kg CD-Ang (1–7), and the control mice received an equivalent volume of PBS. The treatment was initiated seven days (non-humanized mice) or fourteen days (humanized mice) after tumor induction and continued until the mice reached the endpoint of euthanasia.
Liver and Renal Function Assays
Alanine Transaminase (ALT) and Aspartate Aminotransferase (AST) were assessed in EDTA-treated plasma using the ALT Activity Assay Kit (Bioclin K049-6) and the AST Activity Assay Kit (Bioclin K048-6), respectively. Urea levels were determined using the Urea UV Kit (K056-1) from Bioclin. All assays were conducted in accordance with the manufacturer’s instructions. Duplicate wells per mouse sample were analyzed, and the mean value of these technical replicates was utilized for subsequent analysis.
Blood Flow Evaluation
To assess the blood flow measures in tumors, Laser-Doppler perfusion imaging (LDPI) was used (Moor Instruments, Devon, UK). LDPI was conducted in anesthetized mice at a minimal level of ambient light to avoid any influence on the laser light and recorded signals. The animals were maintained at a constant temperature of 37 °C for 5 min before and during the imaging procedure. Hot colors indicate higher blood flow.42,43 The MoorLDI V5.3 software (Moor Instruments, Axminster, UK) was used to calculate the mean pixel value of each scanned image, and the mean flux was expressed as relative units, representing the average blood flow of the tumor area.
Bioluminescence Imaging of Tumor Cell Burden in Mice
To assess tumor burden, mice were treated with 100mg/kg D-Luciferin via intraperitoneal administration (Perkin Elmer), and tumor burden was quantified by luminescence using whole-animal imaging with an In Vivo Imaging System (IVIS, Perkin Elmer) 25 days following transplantation at the René Rachou Institute, Fiocruz Minas. The acquired images were analyzed with the Living Image Acquisition and analysis software (Xenogen).
Immunofluorescence and Confocal Microscopy
To investigate cell death, vessel morphology, and collagen content, tumors were fixed in 4% buffered paraformaldehyde overnight at 4 °C after dissection. Samples were then incubated overnight at 4 °C in a solution of 30% sucrose diluted in PBS, embedded in optimal cutting temperature compound (OCT, Tissue-Tek), and frozen at −80 °C. Cryosections of 20 μm were prepared, blocked in PBS with 4% BSA and 0.5% Triton x-100 for 2 h, and then immunostained with CD31-PE (1:200; BioLegend), Collagen I (1:100; ThermoFisher), Caspase-3 (1:500; Cell Signaling Technology), and anti-Guinea pig-AlexaFluor-647 as a secondary antibody (1:1000; Life Technologies). The sections were subsequently washed with PBS containing 5 μg/mL DAPI (Invitrogen) and mounted using Dako fluorescence mounting medium (Dako, Santa Clara, CA). Confocal microscopy was used to image and analyze the stained tumor sections with an inverted Zeiss LSM 880 confocal microscope (Oberkochen, Germany), carried out at the Center for Acquisition and Processing of Images (CAPI-UFMG). Fiji software®, version 1.53 (National Institute of Health),44 was used to quantify the CD31, Caspase-3, and Collagen I area, with multiple random fields from each section used for quantification.
Histopathological and Immunohistochemical Analysis
To investigate the histopathological features associated with treatments, collected tumors were fixed using a 4% formaldehyde solution and then dehydrated and embedded in paraffin. Tissue sections, 5 μm in thick, were prepared and stained with hematoxylin and eosin (H&E) for microphotograph analysis. To determine tissue morphological alterations in the tumors, an inflammatory and necrosis score system was used. Chronic inflammatory reactions in all samples were based on the presence or absence of plasma cells, epithelioid and giant macrophages, and lymphocytes. Some parameters as vessels number, cellular atypia, hemorrhage, fibrosis, necrosis, mitosis figures, pyknosis, caryorrhexis and inflammatory infiltrate, were evaluated in stained slides using a 40X microscope objective. H&E staining was used to characterize the semi-quantitative analyses. The scoring system was performed using 10 images as follow: 0 = no injury (based on histological control tissue); 1 = discrete and slight, when the lesion occupies less than 25% of the tissue; 2 = moderate, when the lesion occupies 25 to 50% of the tissue; 3 = intense, when the lesion occupies 50 to 75%; 4 = intense, when the lesion occupies more than 75% of the tissue.
Tumor-Infiltrating Leukocyte Immunophenotyping
Mouse tumors and spleens were harvested, minced, and filtered through cell strainers of 40 μm (BD FALCON) to isolate the cells used for immunophenotyping. The isolated cells were washed in PBS and treated with Live/Dead solution to exclude dead cells. Monoclonal antibodies were used for staining, and the cells were fixed and permeabilized according to the manufacturer’s instructions (ThermoFisher). LSR Fortessa instrument was used for cell acquisition, and data analysis was performed using GraphPad Prism V7.0 and FlowJo V10.4.11. To ensure the accuracy of our analyses, we utilized a combination of fluorochromes to exclude debris from the samples. A gate based on forward scatter area (FSC-A) versus forward scatter height (FSC-H) was implemented to eliminate doublets. Only live cells were considered in our study, identified using a Live/Dead Kit (Invitrogen) designed for 405 nm excitation. In particular, nonviable tumor cells were identified through a Live/Dead Fixable Acqua gate against FSC-A. Subpopulations of tumor cells were then gated within mCD45-hCD45+Colo205GFP+ cells. Various T-cell subpopulations were distinguished based on molecular markers such as CD3, CD4, CD8 (Table S1). Within each T-cell subset, the frequencies of cells expressing checkpoint inhibitors CTLA-4 and PD1 were assessed. Tumor-infiltrating leukocytes were stimulated with autologous tumor cells in the presence of Brefeldin A (ThermoFisher) and Monensin (ThermoFisher) for 4 h. Subsequently, the tumor-infiltrating leukocytes were treated with monoclonal antibodies (mAbs) targeting specific surface proteins, followed by fixation and permeabilization.
Statistical Analysis
Data are presented as the mean value with error bars representing standard deviation (SD). Statistical analyses such as the student’s t test and one-way or two-way analysis of variance (ANOVA) were performed, followed by post hoc tests using Tukey’s or Dunnett’s method to compare two groups or among multiple groups. These analyses were conducted using GraphPad Prism 7.0. p < 0.05 was considered statistically significant. Each experiment was repeated at least three times independently with similar results and a representative dataset is presented.
Data Availability
The data that support the findings of this study are available within the paper and its Supplementary Information. All data generated in this study are available from the corresponding author on request.
Results
Characterization and in vitro Testing of mRNA-LNPs
Ionizable lipid nanoparticles encapsulating mRNA coding for TRAIL protein (LNP-TRAIL) were synthesized using the ionizable lipid C12-200, a PEG-lipid conjugate (C14-PEG2000), a phospholipid (DOPE), and cholesterol via controlled microfluidic mixing (Figure 2A). The LNP-TRAILs were characterized based on their size, polydispersity, zeta potential and apparent pKa. The LNP-TRAILs exhibited an average hydrodynamic diameter of 121.1 ± 0.56 nm and a polydispersity index of 0.146 ± 0.04 (Figure 2B). The cryo-TEM data revealed that LNPs exhibited a homogeneous distribution and well-defined structure, as indicated by the presence of an electron-dense nucleus (Figure 2C). The LNP-TRAILs exhibited a slightly negative zeta potential of −2.82 ± 0.60 mV (Figure 2D). Encapsulation efficiency measurements, determined through a RiboGreen assay, revealed that LNPs produced using microfluidic mixing exhibited an encapsulation efficiency of TRAIL at around 84%. We also measured the apparent pKa of LNP-TRAILs via TNS assay, which demonstrated a pKa value of 7.96, suggesting strong ionizability at endosomal pH (Figure 2E). For the in vitro assessment of TRAIL production, HS-5 stromal cells and Colo 205 tumor cells were treated with LNP-TRAIL. Our results indicate that LNP-TRAIL effectively induced TRAIL production in both cellular lineages. (Figure 2F–H). To investigate the ability of LNPs to induce tumor cell death, we performed an in vitro viability assay. Colo 205 cells were treated with LNP-TRAIL with or without the apoptosis inhibitor Z-VAD-FMK. Our results revealed a decrease in the cell viability of Colo 205 cells treated with LNP-TRAIL by approximately 38% compared to control, indicating an increase in cell death (Figure 2I–J). Additionally, we found that caspase inhibitor Z-VAD-FMK reduced TRAIL-mediated tumor cell death in the presence of LNP-TRAIL. These findings suggested that LNP-TRAIL has the potential to trigger TRAIL-mediated tumor cell death.
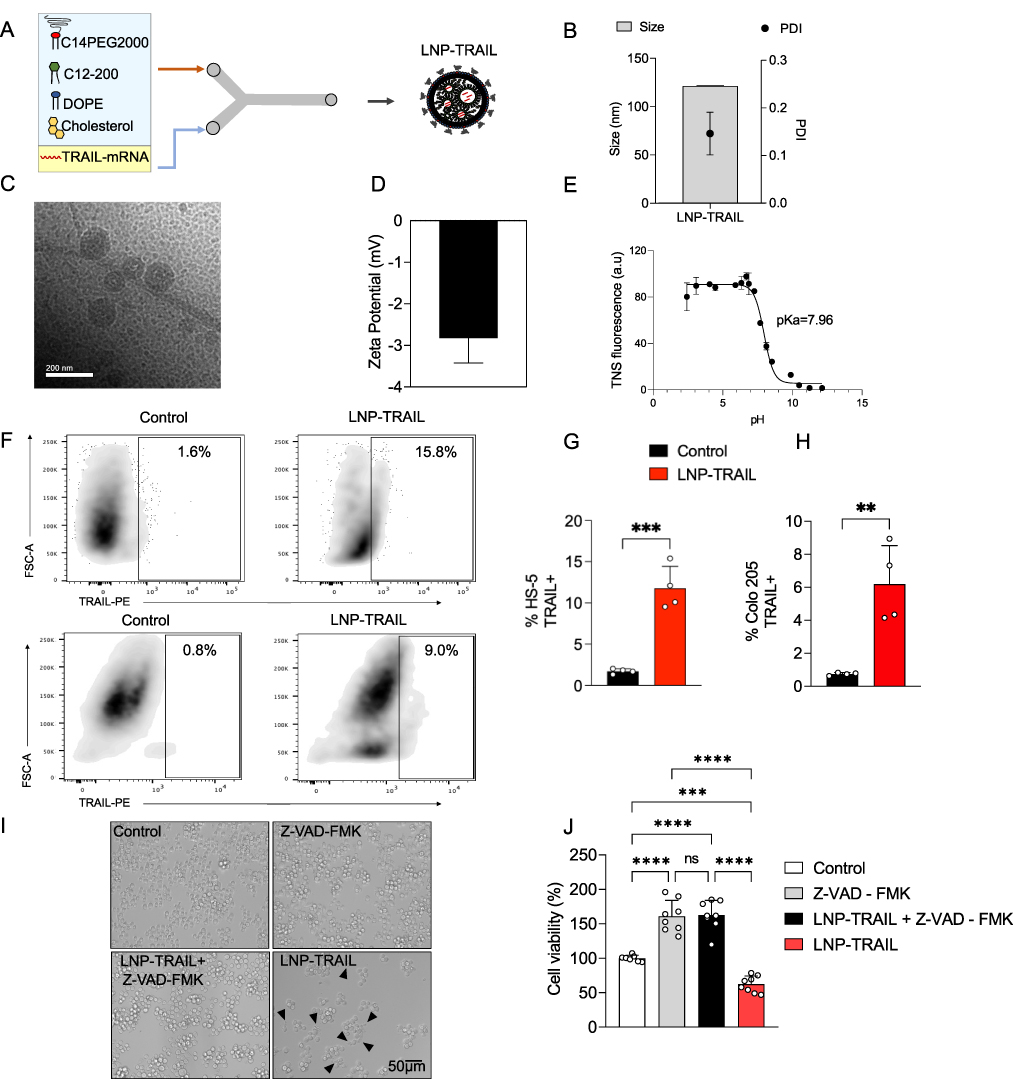 |
Figure 2 Synthesis, characterization, and in vitro testing of LNP-TRAIL. (A) Schematic illustrating the synthesis process for LNPs encapsulating mRNA-TRAIL. LNPs were synthesized via microfluidic mixing, which involved the combination of an aqueous phase containing mRNA coding for TRAIL protein and an ethanol phase comprising lipids. (B) Representative cryo-TEM image of LNPs. (C) Hydrodynamic diameter measurements of LNPs with the corresponding polydispersity index (PDI), measured by dynamic light scattering (DLS). (D) Zeta potential of LNP-TRAIL. (E) Apparent pKa measurement of LNP-TRAIL. (F) Gating strategy to determine the frequency of HS-5 stromal cells and live Luc+ Colo 205 tumor releasing TRAIL. (G) Quantification of live HS-5 stromal cells expressing TRAIL after 24 h of in vitro treatment with LNP-TRAIL. (H) Percentage of live Luc+ Colo 205 tumor cells expressing TRAIL after 24 h of in vitro treatment with LNP-TRAIL. (I) Brightfield micrographs of Colo 205 tumor cells treated with 0.5 μg/well of LNP-TRAIL in the presence or absence of 50 μM pan-caspase inhibitor Z-VAD-FMK. Black arrows represent apoptotic bodies. (J) Cell viability of Colo 205 tumor cells following treatment with 0.5 μg mRNA/well of LNP-TRAIL in the presence or absence of 50 μM pan-caspase inhibitor Z-VAD-FMK. Data are presented as mean ± SD. One-way ANOVA followed by Tukey’s multiple comparison test (ns not significant; **p < 0.01; ***p < 0.001 and ****p < 0.0001). |
LNP-TRAIL Combined with TME Normalization Inhibited Tumor Progression
To investigate the normalization of TME, NSG mice were treated with Los or Ang (1–7) (Figure 3A). Our study revealed significantly increased vessel perfusion within the tumors after administration of Los or Ang (1–7) (Figure 3B, and C). Furthermore, we found decreased area occupied by collagen I (Figure 3D, and E) and increased intratumoral vasculature (Figure 3F, and G) after treatment with Los or Ang (1–7). We then questioned whether the reduction of solid stress and increased vasculature in TME would favor the delivery of intratumoral LNP-TRAIL. LNP-TRAIL was administered every two days starting from day 11 following tumor cell injection (Figure 4A). Our results indicated that the normalization of TME improved the intratumoral delivery of LNP-TRAIL, resulting in reduced volume, weight, and tumor area in NSG mice treated with Los or Ang (1–7) in combination with LNP-TRAIL (Figure 4B–E) compared to the other groups. Specifically, at the 18-day time point, there was a substantial decrease of approximately 75% and 80% in tumor volume in the groups treated with LNP-TRAIL combined with Los or Ang (1–7), respectively. These results were in comparison to the control group treated with LNP-TRAIL alone (Figure 4C). Together, these findings suggested that TEM normalization may have facilitated the intratumoral delivery of LNP-TRAIL, resulting in asubstantial inhibition of tumor growth. Additionally, to assess the potential hepatic or renal toxicity induced by the treatments, we measured the levels of Alanine ALT, AST, and urea in the serum of mice from each group. Our findings reveal no toxicity in groups treated with LNP-TRAIL, Los, Ang(1–7), whether administered individually or in combination, in comparison to the control group (Figure S5).
 |
Figure 3 LNP-TRAIL combined with TME normalization enhanced blood perfusion and reduced collagen I deposition in tumor tissue. (A) Schematic representation of subcutaneous transplantation of Colo 205 cells and treatments with Los, Ang (1–7), and LNP-TRAIL in NSG mice. (B) Representative tumor blood flow images in grayscale and visual intensity scale 18 days post-treatment. Warmer colors represent higher perfusion levels, while the dashed area denotes the tumor region. (C) Quantification of blood flow perfusion within tumor tissue 18 days post-treatment (n = 7–8/group). (D) Representative fluorescence images of the tumor marked with anti-collagen I antibody (magenta) and DAPI (blue), acquired using a 20X objective. (E) Quantification of collagen I area within tumor tissue (n = 3 samples/group). (F) Representative immunofluorescence images of tumors labeled for endothelial cells (CD31; red) to identify blood vessels and nuclei (DAPI; blue), acquired using a 20X objective. (G) Quantification of the vasculature area in tumor tissue (n = 3–4 samples/group). Data are presented as mean ± SD. One-way ANOVA followed by Tukey’s multiple comparison test (*p < 0.05; **p < 0.01; ***p < 0.001 and ****p < 0.0001). |
 |
Figure 4 LNP-TRAIL combined with TME normalization inhibits tumor progression. (A) Schematic representation of subcutaneous transplantation of Colo 205 cells and treatments with Los, Ang (1–7), and LNP-TRAIL in NSG mice. (B) Macroscopic image of tumor growth 18 days after Colo 205 injection. (C) Quantitative analysis of tumor volume over time (n = 7–8/group). Treatment with LNP-TRAIL combined with Los or Ang (1–7) reduced tumor volume compared to Control and LNP-TRAIL groups. (D) Tumor weight and (E) tumor area after treatment with LNP-TRAIL combined with Los or Ang (1–7) (n = 7–8/group). Data are presented as mean ± SD. One-way ANOVA followed by Tukey’s multiple comparison test (*p < 0.05; **p < 0.01; ***p < 0.001 and ****p < 0.0001). |
Establishment of Humanized Mice Using Human Peripheral Blood Mononuclear Cells
To assess whether the treatment with LNP-TRAIL combined with TME normalization would enhance immune cell infiltration, we developed a humanized mouse model by transplanting PBMCs from healthy donors in NSG mice (Figure S1A). The frequency of CD3+ cells exhibited around 90% increase on the fourteenth day following humanization compared to the seventh day (Figure S1B and C). Our findings indicated the presence of human CD8+ and CD4+ T cells in the peripheral blood of transplanted NSG mice. Furthermore, there was a surge in the frequency of circulating lymphocytes after 14 days, which suggests the proliferation of these cells (Figure S1 D– F). These results serve as confirmation of the successful establishment of a humanized model.
LNP-TRAIL Combined with TME Normalization Induced Potent Tumor Cell Death in Humanized Mouse Model
To validate the inhibition of tumor progression in humanized mice, we injected Colo 205 tumor cells into the flank region of humanized NSG mice. After 14 days of humanization, we began daily treatment with Los or Ang (1–7). LNP-TRAIL treatment was administered every two days, starting on the day 18 after tumor cell injection (Figure 5A). Our results demonstrated that the administration of Los or Ang (1–7) effectively increased the perfusion of blood vessels within Colo 205 tumors (Figure S2A–C). Again, we observed reduced tumor progression in NSG mice treated with Los or Ang (1–7) in combination with LNP-TRAIL (Figure 5B–G). Specifically, in the humanized mice, we observed a remarkable decrease in tumor volume, achieving reductions of 83% and 85% (Figure 5E) in the groups treated with LNP-TRAIL combined with Los or Ang (1–7), respectively. These outcomes were notably different compared to the control group treated with LNP-TRAIL alone. In addition, we evaluated the frequency of dead cells within the tumor after treatment using flow cytometry (Figure 6A). Our findings demonstrated that the combination of LNP-TRAIL treatment with Los or Ang (1–7), significantly increased the frequency of dead tumor cells (mCD45-, hCD45-, Colo 205 GFP+ cells) in humanized mice by approximately 26% and 39%, respectively (Figure 6A–B). These results were in comparison to the control group treated with LNP-TRAIL alone. Given the importance of caspase-3 in the apoptotic signaling pathways triggered by TRAIL,45,46 we evaluate the marked area of caspase-3 intratumorally (Figure 6C). Our findings revealed a substantial increase in caspase-3 staining, with approximately 15- and 12-fold increments in the groups treated with LNP-TRAIL combined with Los or Ang, respectively, compared to the control group treated solely with LNP-TRAIL (Figure 6D). Furthermore, an increased area of necrosis was observed in the group treated with the combination of LNP-TRAIL and Los or Ang (1–7) (Figure S3A–C). Collectively, our results demonstrated that the combination of LNP-TRAIL treatment and normalization of the TME in humanized mice significantly contributed to the inhibition of tumor progression.
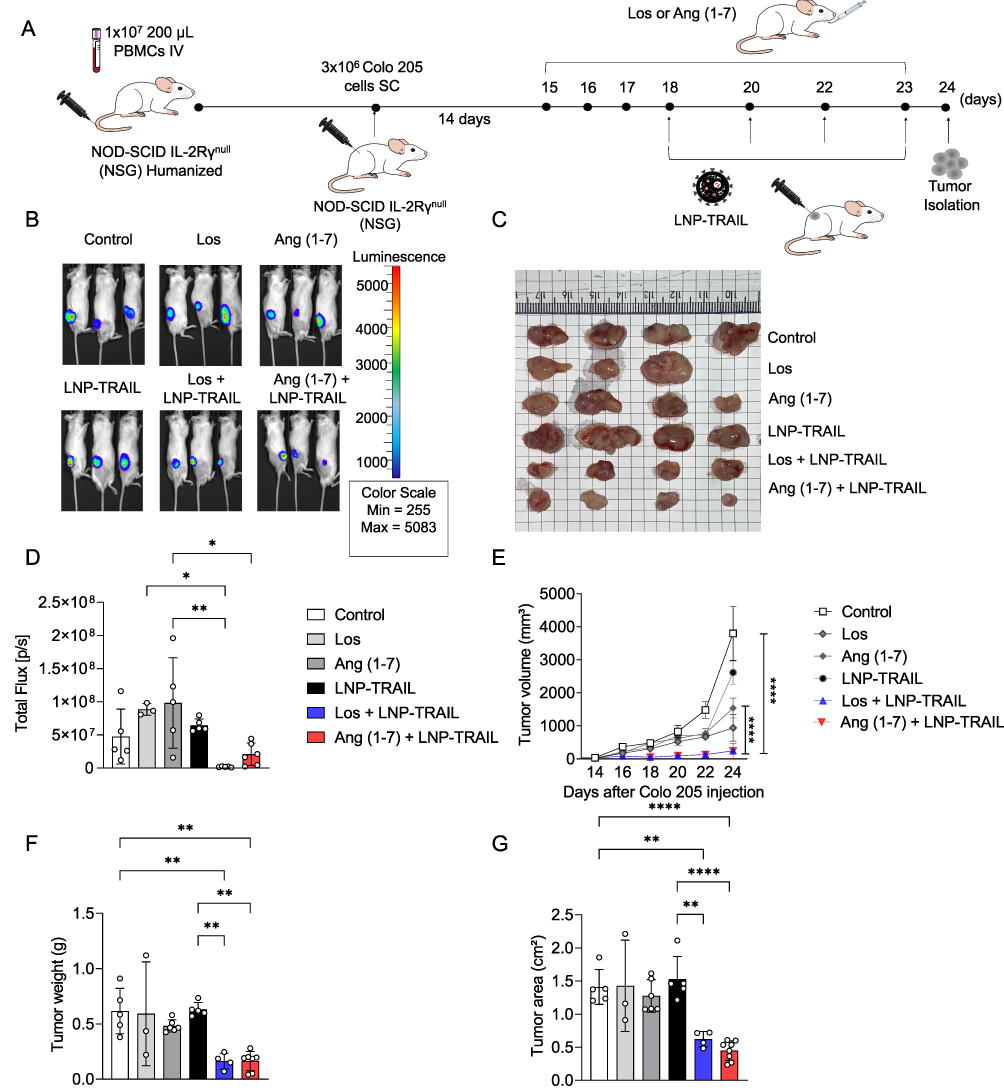 |
Figure 5 LNP-TRAIL therapy in a humanized mouse model results in a potent reduction of tumor growth. (A) Schematic representation of NSG mice humanization and subcutaneous Colo 205 tumor cells transplantation following treatments with Los, Ang (1–7), and LNP-TRAIL. (B) Representative images of bioluminescence detection of tumor growth in NSG mice 24 days after treatment with Los, Ang (1–7), and LNP-TRAIL. Color scale represents the luminescence signal emitted by Luc+ Colo 205 cells. (C) Macroscopic image of tumor growth 24 days after Colo 205 injection. (D) Tumor burden quantification by total flux from bioluminescence imaging (n = 3–6/group). (E) Quantitative analysis of the tumor volume over time. Treatment with LNP-TRAIL combined with Los or Ang (1–7) reduced tumor volume compared to Control and LNP-TRAIL groups (F) Tumor weight and (G) area after LNP-TRAIL treatment combined with Los or Ang (1–7) (n = 3–7/group). Data are presented as mean ± SD. One-way ANOVA followed by Tukey’s multiple comparison test (*p < 0.05; **p < 0.01; and ****p < 0.0001). |
 |
Figure 6 LNP-TRAIL combined with TME normalization induced potent tumor cell death. (A) Gating strategy to determine the frequency of dead tumor cells identified through a Live/Dead Fixable Acqua gate against FSC-A (mCD45-, hCD45-, Colo 205 GFP+ cells) (B) Frequency quantification of dead tumor cells after LNP-TRAIL treatment combined with TME normalization (n = 3–7 samples/group). (C) Representative fluorescence images of the tumor marked with anti-caspase-3 antibody (yellow) and DAPI (blue) were acquired using a 40x objective. (D) Quantification of normalized caspase-3 area in tumor tissues (n = 3 samples/group). Data are presented as mean ± SD. One-way ANOVA followed by Tukey’s multiple comparison test (*p < 0.05; ***p < 0.001 and ****p < 0.0001). |
LNP-TRAIL Combined with TME Normalization Increases Cytotoxic T Lymphocyte (CTL) Response and Inhibited Tumor Progression
The process of tumor rejection initiates with the infiltration of immune cells, particularly T cells, into the tumor site following the initial CTL response.47,48 Therefore, we used humanized mice to assess the infiltration of immune cells into the tumor following treatment with LNP-TRAIL combined with Los or Ang (1–7). Our data indicate that normalization of the TME not only enhanced the efficiency and delivery of LNP-TRAIL in the TME but also stimulated the infiltration of cytotoxic CD8+ T cells (Figure 7A–C) and inflammatory infiltrate (Figure S4A–C), which could be associated with enhanced death of tumor cells. We then assessed the co-expression of immune checkpoint molecules CTLA-4 and PD-1 (Figure 7D–G) as elevated levels of these molecules in cytotoxic T cells can lead to dysfunction and hinder their effector function.49–51 We observed increased CTLA-4 expression in infiltrating CD8+ and CD4+ T cells (Figure 7E and H) in mice treated with LNP-TRAIL combined with Los or Ang (1–7). There were no significant differences detected in the co-expression of CTLA-4 and PD-1 molecules on infiltrating CD8+ and CD4+ T cells within the tumor (Figure 7D, F, G, I, and J).
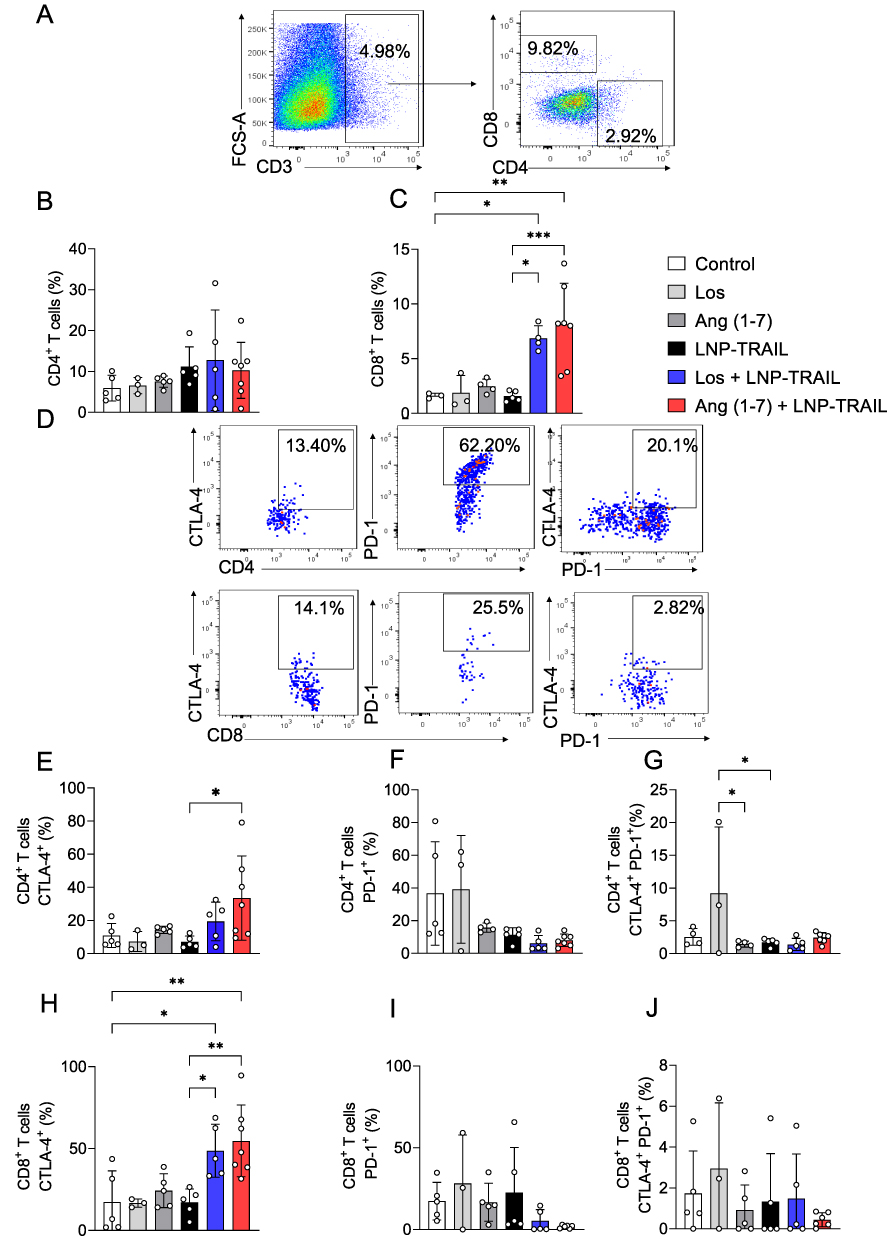 |
Figure 7 LNP-TRAIL combined with TME normalization enhances cytotoxic T lymphocyte response and reduces tumor progression. (A) Gating strategy for identification of CD8+ T cells within CD3+ live cells. Frequency quantification of live tumor-infiltrating (B) CD4+ T and (C) CD8+ T cells (n = 3–7 samples/group). (D) Representative dot plots of the relative levels of CTLA-4 and PD-1 in tumor-infiltrating CD4+ and CD8+ T cells of the Los + LNP-TRAIL group. Frequency quantification of (E) CTLA-4 and (F) PD-1 in tumor-infiltrating CD4+ human T cells (n = 3–7 samples/group). (G) Co-expression of CTLA-4 and PD-1 in tumor-infiltrating CD4+ T cells (n = 3–7 samples/group). Frequency quantification of (H) CTLA-4 and (I) PD-1 in tumor-infiltrating CD8+ human T cells (J) Co-expression of CTLA-4 and PD-1 in tumor-infiltrating CD8+ T cells (n = 3–7 samples/group). Data are presented as mean ± SD. One-way ANOVA followed by Tukey’s multiple comparison test (*p < 0.05; **p < 0.01; ***p < 0.001). |
In addition, we evaluated Ki67 expression, a marker used to assess the rate of cell proliferation.52,53 Previously reported results demonstrated the antitumor activity of CD8+ T cells Ki67+.54 We found increased proliferation rate of infiltrating CD8+ and CD4+ T cells within the tumor after treatment with LNP-TRAIL combined with Los or Ang (1–7) Figure 8A–D). Collectively, our findings suggest that TME normalization is a promising strategy to improve the delivery of LNP-encapsulated mRNA to the TME. Additionally, TME normalization enhanced not only the delivery efficacy of LNP-TRAIL in the TME but also the infiltration of cytotoxic CD8+ T cells. From a clinical perspective, this approach can be utilized to deliver mRNA to the TME to inhibit tumor growth.
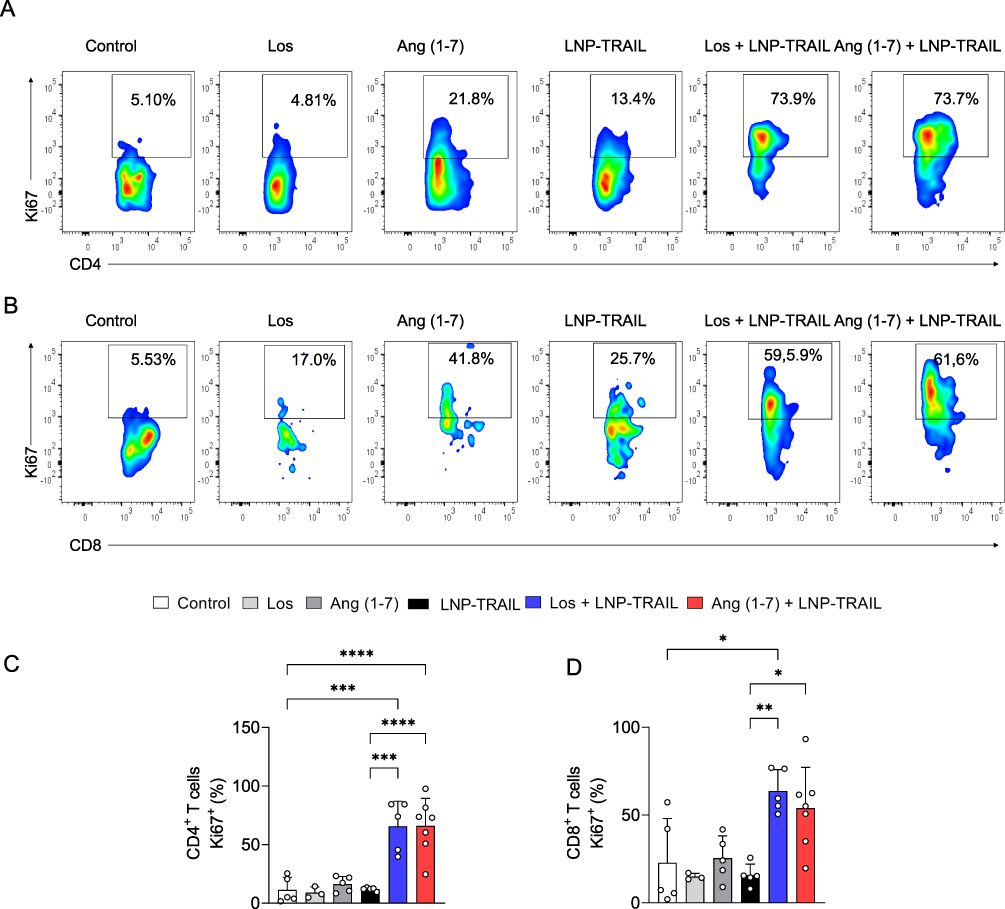 |
Figure 8 LNP-TRAIL combined with TME normalization increases the proliferation rate of tumor-infiltrating CD4+ and CD8+ T cells. Gating strategy for identifying (A) Ki67+/CD4+ and (B) Ki67+/CD8+ T cells. Frequency of tumor-infiltrating (C) CD4+ T and (D) CD8+ T cells expressing Ki67 cell proliferation marker (n = 3–7 samples/group). Data are presented as mean ± SD. One-way ANOVA followed by Tukey’s multiple comparison test (*p < 0.05; **p < 0.01; and ***p < 0.001 and ****p < 0.0001). |
Discussion
The development of targeted therapies for cancer remains a significant challenge due to the complexity of cancer biology, heterogeneity among tumor types, and the acquisition of resistance mechanisms.55 TRAIL therapy has gained substantial interest as a selective cancer therapeutic, owing to its ability to induce programmed cell death selectively in cancer cells.1 Nevertheless, the use of soluble protein TRAIL in most studies is limited by its short half-life, off-target effects, inability to provide enough protein to the tumor, and acquired resistance.1 Consequently, there is a rising interest in exploring mRNA-based strategies to generate TRAIL in situ for cancer therapy.1,2 However, mRNA is unstable in vivo and requires a carrier to cross cell membranes.56,57 The encapsulation of mRNA-TRAIL in lipid nanoparticles offers the potential for protection and efficient delivery to induce tumor cell death through TRAIL-mediated mechanisms, highlighting the therapeutic capabilities of this approach.2 Here, we have developed LNPs comprising ionizable lipids commonly employed for mRNA delivery2 in combination with helper lipids, cholesterol, and a lipid-PEG (Figure 2A). LNPs maintain a neutral charge at physiological pH, minimizing toxicity in comparison to cationic lipids.38 The presence of ionizable lipid is essential for LNPs to effectively escape the acidic endosomal environment after endocytosis.38 Within the endosome, protonation occurs, leading to the fusion of LNP membrane lipids with the anionic lipids of the endosome.38 This process facilitates the release of the mRNA cargo into the cytosol. The LNP formulated with mRNA-TRAIL was characterized by monodisperse size, exhibiting a dense core and a spherical morphology, with an average hydrodynamic diameter of approximately 120.5 nm and similar pKa to our previous studies.12,20 Our in vitro findings illustrate the effective induction of TRAIL production in both stromal and tumor cells by the formulated LNP-TRAIL, highlighting the therapeutic potential of these LNPs (Figure 2F–H). Furthermore, our in vitro findings indicated that LNP-TRAIL has the potential to induce tumor cell death through TRAIL-mediated mechanisms (Figure 2I and J). These data have shown the promising therapeutic capability of LNPs as a delivery system for mRNA-based treatment of cancer. Likewise, utilizing mRNA makes it possible to achieve sustained expression of TRAIL within tumors, enhancing efficacy while minimizing systemic side effects. mRNA-based approaches allow for customization of expression levels and timing, as well as co-delivery of therapeutics and modulation of target gene expression to overcome resistance mechanisms2.
Emerging immunotherapies face a significant challenge known as extracellular matrix confinement (EMC), which impedes their delivery efficacy.56,57 The presence of aberrant blood and lymphatic vessels contributes to the development of a hostile tumor microenvironment, marked by conditions such as hypoxia, low pH, and elevated interstitial fluid pressure. Furthermore, the presence of collagen within the matrix can hinder the delivery of macromolecules like nucleic acids, peptides, and nanomedicines, primarily due to steric interactions. In addition, the matrix indirectly delays therapeutic delivery by compressing the vasculature25. These abnormalities play a crucial role in driving tumor progression, fostering immunosuppression, and promoting resistance to treatment. One proposed strategy to address these challenges involves the transient normalization of the TME, creating a favorable window during which the administration of immunotherapy can yield improved outcomes. To address these issues, here we administered Los or Ang (1–7) to mice aiming to reduce solid stress and increase vascular perfusion as previously described.26,58,59 We observed that Los and Ang (1–7) rapidly reduced established tumor matrix levels (Figure 3D and E). Nonetheless, the mechanisms by which these drugs degrade/destabilize the matrix and inhibit collagen I deposition are poorly understood.25,26,59 Our findings reveal that treatment with Los and Ang (1–7) reduced collagen I deposition and increased blood perfusion within the tumor, improving the functional delivery and antitumor effect of LNP-TRAIL.
To investigate immune response in the TME, we treated humanized mice with LNP-TRAIL via intratumoral injection. This approach provides the advantage of attaining in situ therapeutic effects through localized delivery of high concentrations of mRNA, minimizing systemic exposure.60 The intratumoral delivery of mRNA, as demonstrated by OX40L in current clinical trials (NCT03739931), has resulted in complete responses in tumors that exhibit only partial responsiveness to systemic checkpoint inhibitor.61
The successful implementation of a humanized model was confirmed by the levels of CD8+ and CD4+ T cells in the peripheral blood of NSG mice transplanted with PBMCs (Figure S1D–F). Our study revealed that normalization of the TME resulted in enhanced frequency of CD4+ and CD8+ T cell infiltration in human colon cancer (Figure 7A–C), and this T cell infiltration is a significant prognostic factor in this type of cancer.47,62,63 Combining LNP-TRAIL treatment with normalization of the TME reduced tumor volume (Figure 5C–E) and increased frequency of tumor cell death in humanized mice (Figure 6A and B). Furthermore, our results demonstrated that treatment with LNP-TRAIL combined with TME normalization increased caspase-3 (Figure 6C and D). Previous studies1,64,65 have demonstrated that caspase-3 activation cleaves many targets intracellular proteins, resulting in the morphological and biochemical characteristics of apoptosis, which was observed in our results (Figure 6A and B).
The co-expression of immune checkpoint molecules CTLA-4 and PD-1 is critical in regulating the immune response by preventing the immune system from attacking healthy cells. These molecules act as a “brake” on the immune system, and their role is to prevent an excessive or uncontrolled immune response that could damage healthy tissues.50 However, cancer cells can exploit this mechanism to evade immune detection by expressing ligands for checkpoint molecules, which leads to T-cell exhaustion and dysfunction.50 Studies have shown that the combination of anti-CTLA-4 plus anti-PD-1 therapy led to a more effective tumor immune response, generating proliferation and activation of CD4+ and CD8+ CD45RO+/EOMES+/CD69+ T cells within the tumor.49–51 Here we found that combining LNP-TRAIL treatment with TME normalization prevented the co-expression of immune checkpoint markers by tumor-infiltrating CD4+ and CD8+ T cells. Finally, our results revealed that treatment with LNP-TRAIL combined with TME normalization also stimulated the expression of Ki67, a marker of activation and proliferation of tumor-infiltrating cytotoxic CD8+ and CD4+ T cells (Figure 8A–D). This intensification of the effect of LNP-TRAIL immunotherapy resulted in the notable death of tumor cells.
Conclusion
In this work, we developed an LNP platform to deliver TRAIL mRNA directly to the tumor microenvironment, aiming to induce tumor cell death. Additionally, this study highlights the importance of normalizing the TME to optimize the delivery and efficacy of LNP-TRAIL, promoting increased infiltration of immune cells. Specifically, the combination of LNP-TRAIL immunotherapy with TME normalization resulted in notable reduction in tumor volume and the induction of tumor cell death. While our focus revolves around the intratumoral injection of LNP-TRAIL, further investigations are crucial to explore its delivery and efficacy in systemic treatment. This includes assessing long-term effects, identifying potential resistance mechanisms, and optimizing the dosage and schedule of combination therapy. Collectively, our study demonstrates the potential of integrated therapeutic approaches to enhance immune response against tumors, offering crucial insights for the development of innovative strategies in cancer therapy.
Acknowledgment
This work was funded by National Council for Scientific and Technological Development-CNPq (401390/2020-9; 442731/2020-5; 305932/2022-5; 422002/2023- 830 2; 408482/2022-2), PRPq-UFMG, CAPES (88887.506690/2020-00; 38/2022 Programa de desenvolvimento da pós-graduação parcerias estratégicas nos estados III), and FAPEMIG (APQ-00826-21; APQ-02402-23; RED-00202-22 Rede de Pesquisa em Imunobiológicos e Biofármacos para terapias avançadas e inovadoras). P.P.G.G. is supported by CNPq (442731/2020-5; 305932/2022-5; 422002/2023-2; 408482/2022-2),FAPEMIG (APQ-00826-21; APQ-02402-23), and MCTI/FINEP—MS/SCTIE/DGITIS/CGITS (6205283B-BB28-4F9C-AA65-808FE4450542). The authors thank the Network Technological Platforms from FIOCRUZ, for the support and financing of the services provided by the Microscopy and Image Microanalysis Platform - René Rachou Institute/Fiocruz Minas.
Disclosure
The authors declare that they have no conflicts of interest in this work.
References
1. Guimarães PPG, Gaglione S, Sewastianik T, Carrasco RD, Langer R, Mitchell MJ. Nanoparticles for immune cytokine TRAIL-based cancer therapy. ACS Nano. 2018;12(2):912–931. doi:10.1021/acsnano.7b05876
2. Riley RS, June CH, Langer R, Mitchell MJ. Delivery technologies for cancer immunotherapy. Nat Rev Drug Discov. 2019;18(3):175–196. doi:10.1038/s41573-018-0006-z
3. Bethesda M. SEER Cancer Statistics Factsheets: Common Cancer Sites. National Cancer Institute; 2023.
4. Noh JY, Yoon SR, Kim TD, Choi I, Jung H. Toll-like receptors in natural killer cells and their application for immunotherapy. J Immunol Res. 2020;2020:1–9. doi:10.1155/2020/2045860
5. Atsavapranee ES, Billingsley MM, Mitchell MJ. Delivery technologies for T cell gene editing: applications in cancer immunotherapy. EBioMedicine. 2021;67:103354. doi:10.1016/j.ebiom.2021.103354
6. El-Mayta R, Zhang Z, Hamilton AG, Mitchell MJ. Delivery technologies to engineer natural killer cells for cancer immunotherapy. Cancer Gene Ther. 2021;28(9):947–959. doi:10.1038/s41417-021-00336-2
7. Perlstein B, Finniss SA, Miller C, et al. TRAIL conjugated to nanoparticles exhibits increased anti-tumor activities in glioma cells and glioma stem cells in vitro and in vivo. Neuro Oncol. 2013;15(1):29–40. doi:10.1093/neuonc/nos248
8. Kim TH, Jo YG, Jiang HH, et al. PEG-transferrin conjugated TRAIL (TNF-related apoptosis-inducing ligand) for therapeutic tumor targeting. J Control Release. 2012;162(2):422–428. doi:10.1016/j.jconrel.2012.07.021
9. Chen L, Han X. Anti–PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Investig. 2015;125(9):3384–3391. doi:10.1172/JCI80011
10. Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front Oncol. 2018;8. doi:10.3389/fonc.2018.00086
11. Al-Haideri M, Tondok SB, Safa SH, et al. CAR-T cell combination therapy: the next revolution in cancer treatment. Cancer Cell Int. 2022;22(1):365. doi:10.1186/s12935-022-02778-6
12. Guimaraes PPG, Zhang R, Spektor R, et al. Ionizable lipid nanoparticles encapsulating barcoded mRNA for accelerated in vivo delivery screening. J Control Release. 2019;316:404–417. doi:10.1016/j.jconrel.2019.10.028
13. Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG. Non-viral vectors for gene-based therapy. Nat Rev Genet. 2014;15(8):541–555. doi:10.1038/nrg3763
14. Nayak S, Herzog RW. Progress and prospects: immune responses to viral vectors. Gene Ther. 2010;17(3):295–304. doi:10.1038/gt.2009.148
15. Paunovska K, Loughrey D, Dahlman JE. Drug delivery systems for RNA therapeutics. Nat Rev Genet. 2022;23(5):265–280. doi:10.1038/s41576-021-00439-4
16. Roberts TC, Langer R, Wood MJA. Advances in oligonucleotide drug delivery. Nat Rev Drug Discov. 2020;19(10):673–694. doi:10.1038/s41573-020-0075-7
17. Buck J, Grossen P, Cullis PR, Huwyler J, Witzigmann D. lipid-based DNA therapeutics: hallmarks of non-viral gene delivery. ACS Nano. 2019;13(4):3754–3782. doi:10.1021/acsnano.8b07858
18. Guimarães PPG, Figueroa-Espada CG, Riley RS, et al. In vivo bone marrow microenvironment siRNA delivery using lipid–polymer nanoparticles for multiple myeloma therapy. Proc Natl Acad Sci. 2023;120(25). doi:10.1073/pnas.2215711120
19. Prazeres PHDM, Ferreira H, Costa PAC, et al. Delivery of plasmid DNA by ionizable lipid nanoparticles to induce CAR expression in t cells. Int J Nanomed. 2023;18:5891–5904. doi:10.2147/IJN.S424723
20. Scalzo S, Santos AK, Ferreira HA, et al. ionizable lipid nanoparticle-mediated delivery of plasmid DNA in cardiomyocytes. Int J Nanomed. 2022;17:2865–2881. doi:10.2147/IJN.S366962
21. Guimaraes LC, Costa PAC, Scalzo Júnior SRA, et al. Nanoparticle-based DNA vaccine protects against SARS-CoV-2 variants in female preclinical models. Nat Commun. 2024;15(1):590. doi:10.1038/s41467-024-44830-1
22. Padera TP, Stoll BR, Tooredman JB, Capen D, di TE, Jain RK. Cancer cells compress intratumour vessels. Nature. 2004;427(6976):695. doi:doi:10.1038/427695a
23. Binnewies M, Roberts EW, Kersten K, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541–550. doi:10.1038/s41591-018-0014-x
24. Tiwari A, Trivedi R, Lin SY. Tumor microenvironment: barrier or opportunity towards effective cancer therapy. J Biomed Sci. 2022;29(1):83. doi:10.1186/s12929-022-00866-3
25. Jain RK. Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. J Clin Oncol. 2013;31(17):2205–2218. doi:10.1200/JCO.2012.46.3653
26. Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell. 2014;26(5):605–622. doi:10.1016/j.ccell.2014.10.006
27. Tolaney SM, Boucher Y, Duda DG, et al. Role of vascular density and normalization in response to neoadjuvant bevacizumab and chemotherapy in breast cancer patients. Proc Natl Acad Sci. 2015;112(46):14325–14330. doi:10.1073/pnas.1518808112
28. US National Library of Medicine. Carboplatin a and B in PUPWANSCLCancerAA 1. US National Library of Medicine; 2023. Available from: https://www.clinicaltrials.gov/ct2/show/NCT00642759.
29. Heist RS, Duda DG, Sahani DV, et al. Improved tumor vascularization after anti-VEGF therapy with carboplatin and nab-paclitaxel associates with survival in lung cancer. Proc Natl Acad Sci. 2015;112(5):1547–1552. doi:10.1073/pnas.1424024112
30. Batchelor TT, Gerstner ER, Emblem KE, et al. Improved tumor oxygenation and survival in glioblastoma patients who show increased blood perfusion after cediranib and chemoradiation. Proc Natl Acad Sci. 2013;110(47):19059–19064. doi:10.1073/pnas.1318022110
31. Nia HT, Datta M, Seano G, Huang P, Munn LL, Jain RK. Quantifying solid stress and elastic energy from excised or in situ tumors. Nat Protoc. 2018;13(5):1091–1105. doi:10.1038/nprot.2018.020
32. Murphy JE, Jyl W, Ferrone C, et al. TGF-B1 inhibition with losartan in combination with FOLFIRINOX (F-NOX) in locally advanced pancreatic cancer (LAPC): preliminary feasibility and R0 resection rates from a prospective phase II study. J Clin Oncol. 2017;35(4_suppl):386. doi:10.1200/JCO.2017.35.4_suppl.386
33. Diop-Frimpong B, Chauhan VP, Krane S, Boucher Y, Jain RK. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc Natl Acad Sci. 2011;108(7):2909–2914. doi:10.1073/pnas.1018892108
34. Chappell MC, Al Zayadneh EM. Angiotensin-(1-7) and the regulation of anti-fibrotic signaling pathways. J Cell Signal. 2017;02(01). doi:10.4172/2576-1471.1000134
35. Soto-Pantoja DR, Menon J, Gallagher PE, Tallant EA. Angiotensin-(1-7) inhibits tumor angiogenesis in human lung cancer xenografts with a reduction in vascular endothelial growth factor. Mol Cancer Ther. 2009;8(6):1676–1683. doi:10.1158/1535-7163.MCT-09-0161
36. Mitchell MJ, Webster J, Chung A, Guimarães PPG, Khan OF, Langer R. Polymeric mechanical amplifiers of immune cytokine-mediated apoptosis. Nat Commun. 2017;8(1):14179. doi:10.1038/ncomms14179
37. Lula I, Denadai ÂL, Resende JM, et al. Study of angiotensin-(1–7) vasoactive peptide and its β-cyclodextrin inclusion complexes: complete sequence-specific NMR assignments and structural studies. Peptides. 2007;28(11):2199–2210. doi:10.1016/j.peptides.2007.08.011
38. Kauffman KJ, Dorkin JR, Yang JH, et al. Optimization of lipid nanoparticle formulations for mRNA delivery in vivo with fractional factorial and definitive screening designs. Nano Lett. 2015;15(11):7300–7306. doi:10.1021/acs.nanolett.5b02497
39. Han X, Alameh MG, Butowska K, et al. Adjuvant lipidoid-substituted lipid nanoparticles augment the immunogenicity of SARS-CoV-2 mRNA vaccines. Nat Nanotechnol. 2023;18(9):1105–1114. doi:10.1038/s41565-023-01404-4
40. Krishnan B, Smith TL, Dubey P, et al. Angiotensin‐(1‐7) attenuates metastatic prostate cancer and reduces osteoclastogenesis. Prostate. 2013;73(1):71–82. doi:10.1002/pros.22542
41. Gast CE, Silk AD, Zarour L, et al. Cell fusion potentiates tumor heterogeneity and reveals circulating hybrid cells that correlate with stage and survival. Sci Adv. 2018;4(9). doi:10.1126/sciadv.aat7828
42. Kloppenberg FWH, Beerthuizen GIJM, ten Duis HJ. Perfusion of burn wounds assessed by Laser Doppler Imaging is related to burn depth and healing time. Burns. 2001;27(4):359–363. doi:10.1016/S0305-4179(00)00138-8
43. Sousa GF, Afewerki S, Dittz D, et al. Catalyst-free click chemistry for engineering chondroitin sulfate-multiarmed peg hydrogels for skin tissue engineering. J Funct Biomater. 2022;13(2):45. doi:10.3390/jfb13020045
44. Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–682. doi:10.1038/nmeth.2019
45. Raman D, Tay P, Hirpara JL, Liu D, Pervaiz S. TRAIL sensitivity of nasopharyngeal cancer cells involves redox dependent upregulation of TMTC2 and its interaction with membrane caspase-3. Redox Biol. 2021;48:102193. doi:10.1016/j.redox.2021.102193
46. Jana S, Hsieh AC, Gupta R. Reciprocal amplification of caspase-3 activity by nuclear export of a putative human RNA-modifying protein, PUS10 during TRAIL-induced apoptosis. Cell Death Dis. 2017;8(10):e3093–e3093. doi:10.1038/cddis.2017.476
47. Talhouni S, Fadhil W, Mongan NP, et al.. Activated tissue resident memory T-cells (CD8+CD103+CD39+) uniquely predict survival in left sided “immune-hot” colorectal cancers. Front Immunol. 2023:14. doi:10.3389/fimmu.2023.1057292.
48. Kather JN, Halama N. Harnessing the innate immune system and local immunological microenvironment to treat colorectal cancer. Br J Cancer. 2019;120(9):871–882. doi:10.1038/s41416-019-0441-6
49. Gupta PK, Godec J, Wolski D, et al. CD39 expression identifies terminally exhausted cd8+ t cells. PLoS Pathog. 2015;11(10):e1005177. doi:10.1371/journal.ppat.1005177
50. Wei SC, Anang NAAS, Sharma R, et al. Combination anti–CTLA-4 plus anti–PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies. Proc Natl Acad Sci. 2019;116(45):22699–22709. doi:10.1073/pnas.1821218116
51. Li H, van der Merwe PA, Sivakumar S. Biomarkers of response to PD-1 pathway blockade. Br J Cancer. 2022;126(12):1663–1675. doi:10.1038/s41416-022-01743-4
52. Uxa S, Castillo-Binder P, Kohler R, Stangner K, Müller GA, Engeland K. Ki-67 gene expression. Cell Death Differ. 2021;28(12):3357–3370. doi:10.1038/s41418-021-00823-x
53. Warth A, Cortis J, Soltermann A, et al. Tumour cell proliferation (Ki-67) in non-small cell lung cancer: a critical reappraisal of its prognostic role. Br J Cancer. 2014;111(6):1222–1229. doi:10.1038/bjc.2014.402
54. Simoni Y, Becht E, Fehlings M, et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018;557:7706):575–579. doi:10.1038/s41586-018-0130-2
55. Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12(1):31–46. doi:10.1158/2159-8290.CD-21-1059
56. Swingle KL, Hamilton AG, Mitchell MJ. Lipid nanoparticle-mediated delivery of mRNA therapeutics and vaccines. Trends Mol Med. 2021;27(6):616–617. doi:10.1016/j.molmed.2021.03.003
57. Hamilton AG, Swingle KL, Mitchell MJ. Biotechnology: overcoming biological barriers to nucleic acid delivery using lipid nanoparticles. PLoS Biol. 2023;21(4):e3002105. doi:10.1371/journal.pbio.3002105
58. Goel S, Duda DG, Xu L, et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91(3):1071–1121. doi:10.1152/physrev.00038.2010
59. Chauhan VP, Martin JD, Liu H, et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun. 2013;4(1):2516. doi:10.1038/ncomms3516
60. Di Trani CA, Cirella A, Arrizabalaga L, et al. Intratumoral injection of IL-12-encoding mRNA targeted to CSF1R and PD-L1 exerts potent anti-tumor effects without substantial systemic exposure. Mol Ther Nucleic Acids. 2023;33:599–616. doi:10.1016/j.omtn.2023.07.020
61. Hewitt SL, Bai A, Bailey D, et al. Durable anticancer immunity from intratumoral administration of IL-23, IL-36γ, and OX40L mRNAs. Sci Transl Med. 2019;11(477). doi:10.1126/scitranslmed.aat9143
62. Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–1964. doi:10.1126/science.1129139
63. Simpson JAD, Al-Attar A, Watson NFS, Scholefield JH, Ilyas M, Durrant LG. Intratumoral T cell infiltration, MHC class I and STAT1 as biomarkers of good prognosis in colorectal cancer. Gut. 2010;59(7):926–933. doi:10.1136/gut.2009.194472
64. De Miguel D, Gallego-Lleyda A, Anel A, Martinez-Lostao L. Liposome-bound TRAIL induces superior DR5 clustering and enhanced DISC recruitment in histiocytic lymphoma U937 cells. Leuk Res. 2015;39(6):657–666. doi:10.1016/j.leukres.2015.03.019
65. Wajant H, Moosmayer D, Wüest T, et al. Differential activation of TRAIL-R1 and −2 by soluble and membrane TRAIL allows selective surface antigen-directed activation of TRAIL-R2 by a soluble TRAIL derivative. Oncogene. 2001;20(30):4101–4106. doi:10.1038/sj.onc.1204558
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.