Back to Journals » Drug Design, Development and Therapy » Volume 10
Intranasal administration of elastin-like polypeptide for therapeutic delivery to the central nervous system
Authors McGowan J, Shao Q, Vig P, Bidwell G
Received 11 February 2016
Accepted for publication 19 May 2016
Published 6 September 2016 Volume 2016:10 Pages 2803—2813
DOI https://doi.org/10.2147/DDDT.S106216
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Jeremy WD McGowan,1 Qingmei Shao,1 Parminder JS Vig,1,2 Gene L Bidwell III1,2
1Department of Neurology, 2Department of Biochemistry, University of Mississippi Medical Center, Jackson, MS, USA
Abstract: Bypassing the blood–brain barrier is one of the primary considerations when designing compounds intended to function in the central nervous system (CNS). Intranasal (IN) administration of otherwise blood–brain barrier impermeable molecules can result in high CNS concentrations and low systemic accumulation, indicating that IN administration may be a useful method of delivering therapeutics to the CNS. Elastin-like polypeptide (ELP) is a large, non-immunogenic, highly manipulable biopolymer with extensive evidence supporting its use as a carrier with the ability to improve drug pharmacokinetics and drug targeting. The ability of ELP to reach the CNS via IN administration has been shown previously. Previous studies have also identified the ability of cell penetrating peptides (CPPs) to increase the uptake of molecules in some instances, including via the IN route. Here, we compared and contrasted the biodistribution of ELPs with or without addition of the CPPs Tat or SynB1 via both the IN and intravenous routes. Administration of ELP via the IN route led to significant accumulation in the brain, especially in the olfactory bulbs. When injected intravenously, <3% of the ELP signal was present outside the vascular compartment. This contrasted with IN administration, which resulted in 79% of the fluorescence signal localized outside the vascular space. The fusion of Tat or SynB1 significantly altered the biodistribution of ELP, decreasing the total CNS accumulation following IN administration. The addition of CPPs to ELP increased their retention in the nasal epithelium. These results suggest ELP may represent an effective CNS delivery vector without further modification and that the addition of a CPP significantly influences biodistribution.
Keywords: central nervous system, cell penetrating peptide, elastin-like polypeptide, intranasal administration, drug delivery, blood–brain barrier
Introduction
Biopharmaceuticals represent potentially powerful therapeutics with high specificity and increased efficacy compared to many small molecule drugs. Unfortunately, the design and use of these macromolecules are complicated by restrictive biological barriers and limitations of parenteral administration, severely diminishing the applicability of biologics in many diseases. These complications quickly become more apparent when developing therapeutics intended to work within the central nervous system (CNS) due to the largely impermeable blood–brain barrier (BBB). To attempt to work around these limiting factors, we have investigated the use of an engineered biopolymer carrier, administered directly onto the nasal epithelium, in combination with peptides designed to increase uptake of fused molecules.
Methods to bypass the BBB via alternative administration routes, particularly intranasal (IN) delivery, have shown positive results with small molecule drugs in the past and with some larger biomolecules more recently.1,2 Direct contact with the nasal epithelium has been demonstrated to facilitate the movement of molecules down multiple pathways, resulting in significantly increased CNS levels compared to other non-invasive administration routes.3 The usefulness of transport directly into cerebrospinal fluid is debated, but transport along the olfactory and trigeminal nerves has been well documented and can result in therapeutically relevant CNS penetration with drastically decreased peripheral impact.2,4 While IN delivery can increase CNS uptake of non-BBB penetrating compounds when compared to other administration methods, in the case of macromolecules, concentrations are often still insufficient to be clinically relevant. Various methods have been employed to attempt to increase the CNS penetration of macromolecules via IN administration, including the use of gelling solutions that increase contact time with the nasal epithelium, encapsulation in liposomes, the use of different types of nanoparticles, and coadministration or conjugation with numerous molecules intended to increase absorption.5,6 Each of these methods have shown variable levels of success based on any number of factors, but the most influential is likely the administered therapeutic macromolecule itself. The molecular properties most likely to lead to efficient IN delivery are not fully defined, but similar to intravenous (IV), small lipophilic molecules have been shown to easily reach the CNS via IN administration.1 When larger molecules are considered, the properties leading to efficient CNS delivery are much more vague. Albumin, insulin, oxytocin, and nerve growth factor all vary in size and charge but have all successfully been delivered via IN administration.1,3 The distribution of these molecules varied a great deal, with the largest molecules tending to have the highest concentration in the olfactory bulbs compared to a slightly more uniform distribution with some of the smaller molecules. Even with all the differences in the properties of these molecules, all were able to show efficacy in vivo. Therefore, it becomes valuable to investigate the ability of a highly manipulable macromolecule with and without secondary modifications intended to increase CNS absorption via IN administration in order to understand and identify the physical properties most likely to lead to viable IN delivered therapeutics. Additionally, a comparison to the more common IV administration route will provide an understanding of the overall ability of the IN route to deliver large polypeptides to the CNS.
Elastin-like polypeptide (ELP) represents this ideal macromolecule for these and future investigations. ELP is a biopolymer consisting of a repeated five amino acid motif (VPGXG, where X is any amino acid except proline) with several highly desirable characteristics.7 ELP is easily modified by basic molecular cloning techniques, allowing for changes in size, amino acid composition at the flexible X site, and the addition of either therapeutic peptides/proteins or reactive sites for small molecule attachment. ELP undergoes a temperature induced, reversible phase transition8 which facilitates purification of ELP and ELP-fusion proteins. The practical applications of this phase transition also include production of slow release aggregates not dissimilar to some of the IN gels previously investigated.9,10 Additionally, ELP has been demonstrated to be both non-immunogenic and nontoxic, offering improvement over many other proposed macromolecular carriers.11–13 Finally, ELP has been demonstrated to improve the half-life of conjugated small molecules and peptides, addressing one of the major hurdles of therapeutic peptide development and offering the ability to alter the kinetics of attached cargo.13,14 Modified ELPs have been used to successfully deliver peptides to the CNS in previous studies, further supporting its use as a platform for these investigations.15
Recent research suggests cell penetrating peptides (CPPs) may prove promising for IN administration as they have been demonstrated to significantly increase the penetration of some molecules through biological membranes.16,17 Many of the commonly used CPPs are short cationic amino acid sequences that mediate interaction with the plasma membrane and subsequent internalization, often via endocytosis, bringing with them their attached cargo.18 Some studies have used CPPs and macromolecules in tandem with IN delivery, but as with other methods intended to increase IN delivery, the results have been highly variable. One study using coadministration of the CPP penetratin and some of its analogs with several molecules including insulin and exendin-4 found significant differences between the penetratin analogs.17 The authors were also able to correlate the degree of binding between the CPPs and macromolecules with the degree of IN uptake, suggesting a higher degree of binding increased absorption.17 Another study using nano-micelles modified with the CPP Tat via IN administration found significantly increased CNS uptake and transport down the olfactory and trigeminal nerves with high concentrations in the brain stem.19 These data highlight the need for examination of multiple CPPs on the same macromolecular platform to better understand the effects of CPP-macromolecule combinations on IN administration. Here, we have fused Tat or SynB1, both common short cationic CPPs, to an ELP carrier and administered these by either IN or IV administration routes.20,21 We show in this article that both Tat and SynB1 alter the delivery of ELP to the CNS compared to ELP alone via the IN route, suggesting ELP by itself may be an effective delivery vector and that CPPs may adversely affect the delivery of some macromolecules.
Methods
Protein purification and labeling
ELP, SynB1-ELP, and Tat-ELP polypeptides were expressed in Escherichia coli using the pET expression system and were purified by inverse transition cycling as previously described.22 Polypeptides were labeled with tetramethylrhodamine-5-maleimide (Life Technologies, Thermo Fisher Scientific, Waltham, MA, USA) on cysteine residues as previously described.23
Animals
Male 5-week-old FVB/NJ mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA) and allowed to acclimate for a minimum of 7 days prior to experiments. Peptide administration was performed on 6–8-week-old mice. All animal use in this study was conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of the University of Mississippi Medical Center.
IN and IV administration
Animals were anesthetized using 3% isoflurane (Henry Schein, Melville, NY, USA) for 5 minutes, and depth of anesthesia was verified by the absence of toe-pinch reflex. Both IV and IN groups received a total dose of 50 mg/kg for all assays. Mice in the IN group were placed in the supine position, and polypeptides were administered in 0.5–2 μL drops into the right nostril every 1 minute (range: 8.5–13.6 μL) (n=4). Mice continued to lie in the supine position under anesthesia for a total of 10 minutes from the start of administration before being returned to their home cage. One hour after the initiation of administration, mice were sacrificed, with or without perfusion, and blood, urine, brain, spinal cord, heart, lungs, stomach, spleen, liver, and kidneys were collected. Mice in the IV groups were anesthetized as before and placed in the supine position (n=4). Polypeptides were suspended in 100 μL phosphate-buffered saline (PBS) and injected into the femoral vein. Mice remained in the supine position for a total of 10 minutes including administration before being returned to their home cage for 1 hour. Mice were then sacrificed and tissues collected as in the IN group.
Polypeptide biodistribution
Whole organ ex vivo fluorescence was determined using an IVIS® Spectrum Imager (PerkinElmer Inc., Waltham, MA, USA), and fluorescence measurements in average radiant efficiency were determined using Living Image software (PerkinElmer Inc.). Fluorescence intensity of saline-treated mice was subtracted to correct for background fluorescence, and standard curves were generated by placing a serial dilution of each polypeptide (from the same labeling batch as administered to animals) into a 96 well plate and measuring fluorescence using identical imaging parameters. After imaging, tissues were frozen in liquid nitrogen and stored at −80°C until slicing for histological analysis.
For quantitative fluorescence analysis, brains were sliced axially in 20 μm thick sections using a Thermo Cryostar NX50 (Thermo Scientific). Brains were sliced in entirety and slices taken every ~250 μm were placed onto slides for scanning using a ScanArray® Express Microarray Scanner (Packard Bioscience, Meriden, CT, USA) at 541 nm excitation. A standard curve was created by mixing polypeptide solutions directly in optimal cutting temperature freezing medium (Fisher Healthcare, Pittsburgh, PA, USA) at various concentrations, drawing the solution into 1 mL syringes, and freezing the syringes in liquid nitrogen. The resulting frozen cylinders were sliced 20 μm thick and scanned under the same settings as the brain sections. Finally, regions of interest including the olfactory bulbs, both cerebral hemispheres, and the cerebellum were selected using ImageJ software (version 1.47, National Institutes of Health, USA) and analyzed for mean gray value. In order to mark blood vessels for images, one additional animal for each group was treated as described earlier before being injected by IV with 1 mg of 500,000 MW fluorescein isothiocyanate–dextran (Sigma-Aldrich Co., St Louis, MO, USA) suspended in 100 μL PBS 1 minute prior to sacrifice. The resulting brains were treated and sliced, as described earlier, before being scanned with both 541 and 488 nm excitation.
Direct fluorescence analysis of plasma was determined using a Nanoquant® plate in a fluorescent plate reader (Tecan, Männedorf, Switzerland) as previously described.10 Plasma samples of 2 μL were measured at 543 nm excitation and 575 nm emission wavelengths at a gain of 90. Fluorescence values were then plotted onto a standard curve created from the same protein used in the experiments, measured at the same settings as the plasma samples in the Nanoquant plate. All fluorescence measurements were corrected for background fluorescence as described earlier.
Statistics
Differences in organ levels among polypeptide treatments and administration routes were assessed using two-way analysis of variance (ANOVA) with a post-hoc Tukey’s multiple comparison. Polypeptide levels in the nasal cavity after IN administration were assessed using a one-way ANOVA with a post-hoc Tukey’s multiple comparison. Alpha <0.05 was taken as an overall statistically significant difference among groups, and 95% confidence intervals were used to assess differences among mean values in multiple comparisons. Statistics were performed using Graphpad Prism (Version 6, GraphPad Software, La Jolla, CA, USA).
Results
CNS delivery of ELP and CPP-ELPs by IN and IV administration
Past studies using small peptides with CPPs have suggested CPPs may be able to increase CNS uptake via IN administration.2,24 Figure 1A shows representative IVIS images of brains after treatment with each construct via IN or IV administration. The heat map clearly shows regional differences in peptide concentrations in IN-treated animals, with the highest concentrations around the olfactory bulbs. In contrast, the IV-treated animals displayed a much more even distribution as would be expected. Figure 1B and C represents the measurement of polypeptide levels in the whole brain and spinal cord, respectively. Analysis of the mean fluorescence of the entire brain showed that for all polypeptides, IN administration resulted in less total polypeptide in the brain than did IV administration. The addition of a CPP to ELP resulted in a significant decrease in brain levels following both IN and IV administration. SynB1-ELP levels were nearly six-fold lower than ELP levels after IN administration and 2.5-fold lower after IV administration. Tat-ELP levels were undetectable over autofluorescence in both IN and IV-administered animals. Very similar trends were found in the spinal cord (Figure 1C). IV injection resulted in higher total polypeptide levels than did IN administration, and the addition of CPPs to ELP resulted in lower overall spinal cord levels. These data suggest the addition of Tat or SynB1 can significantly modify the biodistribution of ELP resulting in reduced transport to the CNS. Also, although IV injection resulted in higher overall polypeptide levels in the brain and spinal cord, this assay does not differentiate between intravascular polypeptide and polypeptide localized across the BBB.
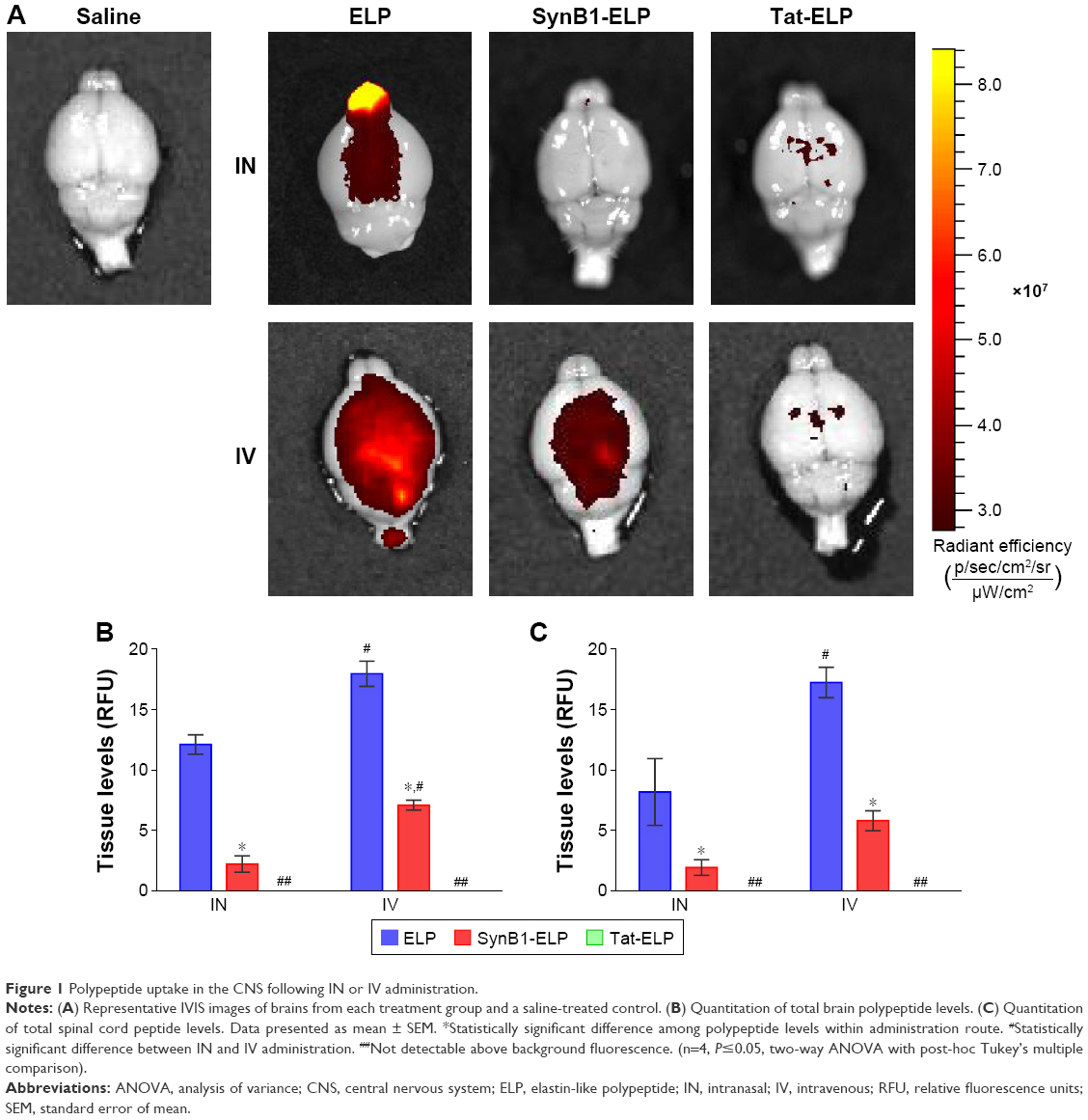 | Figure 1 Polypeptide uptake in the CNS following IN or IV administration. |
Peripheral organ biodistribution of ELP and CPP-ELPs following IN and IV administration
In addition to determining CNS levels, we also assessed the peripheral biodistribution of each polypeptide after IN and IV administration. IN administration resulted in very little polypeptide reaching systemic circulation. The plasma concentration of all constructs was high in the IV groups as would be expected (Figure 2A), and differences among polypeptides likely reflects differences in the pharmacokinetics.25 Following IN administration and consistent with the low plasma levels, systemic organ levels of all constructs were quite low with one notable exception, the lungs (Figure 2B). Lung levels of ELP were highly variable but higher than levels in any other organ, and SynB1-ELP lung levels in particular were increased more than seven times compared to ELP or Tat-ELP. This very likely represents aspiration of the polypeptide by the mice, and these data suggest that the addition of SynB1 to ELP increases its propensity for aspiration. In the IV administration group, the kidneys and stomach accumulated polypeptides at the highest levels (Figure 2C). Both SynB1-ELP and Tat-ELP accumulated to significantly higher levels in the kidneys compared to ELP, consistent with previous studies by our group.26
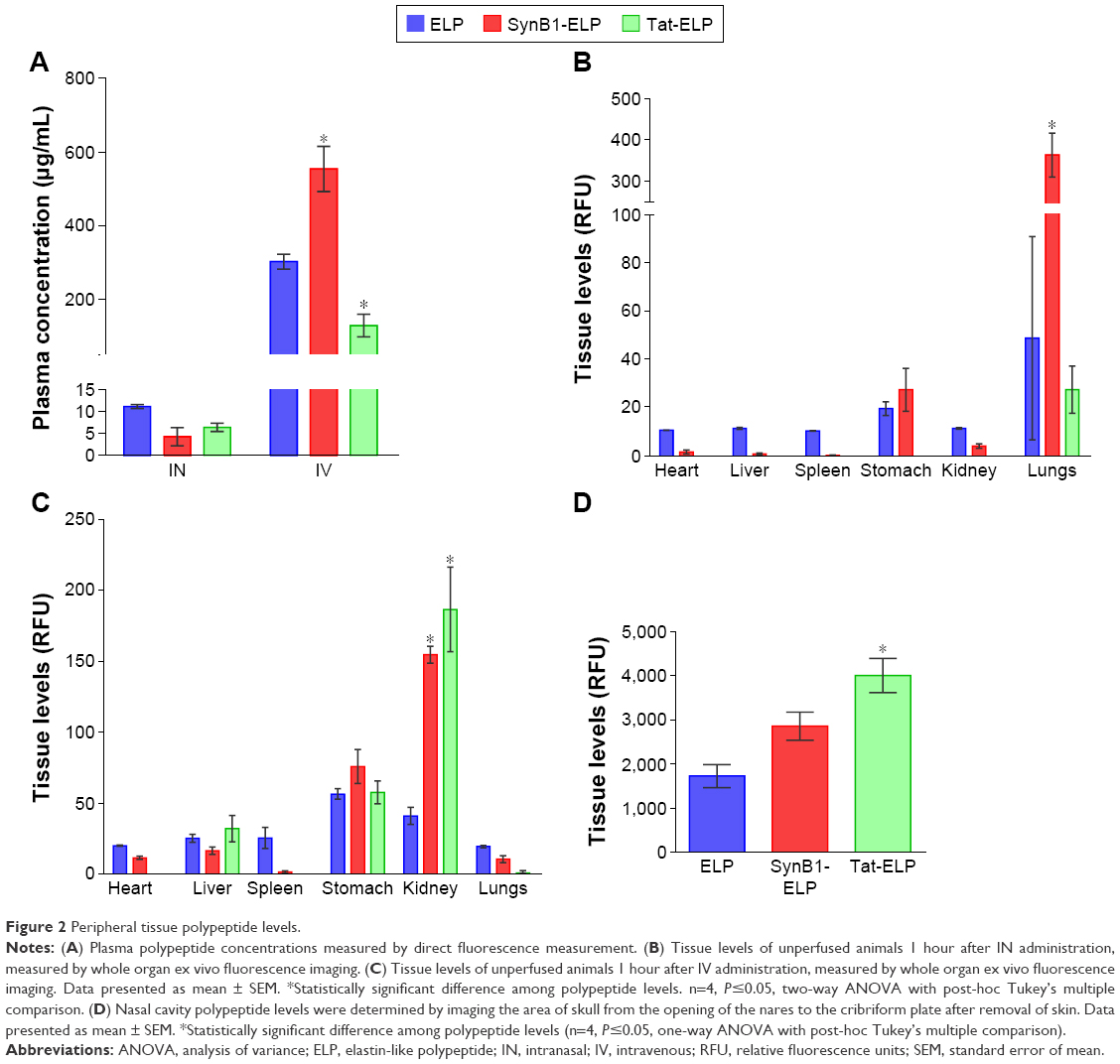 | Figure 2 Peripheral tissue polypeptide levels. |
In order to determine the amount of peptide still in the nasal cavity after IN administration, the skull was stripped of soft tissues and the entire section of skull encompassing the nasal cavity from the cribriform plate to the opening of the nares was measured intact in the IVIS spectrum imager. The data show Tat-ELP levels were significantly higher in the nasal cavity, and SynB1-ELP levels tended to be higher but did not reach statistical significance (Figure 2D). These results suggest that the decreased CNS levels seen with the CPP-ELPs results from the CPP mediating cellular uptake in the nasal cavity and/or increasing the propensity for aspiration (in the case of SynB1) (Figure 2C and D).
Determination of extravascular CNS ELP levels
The ex vivo whole organ imaging assay cannot distinguish between intravascular and extravascular polypeptide. To determine the amount of extravascular polypeptide, the experiment was repeated, but the animals were perfused with heparinized PBS (10 units/mL) to clear all blood and intravascular polypeptide prior to ex vivo brain imaging. The total fluorescence of non-perfused brains was compared to the total fluorescence of perfused brains to calculate the percent extravascular. Following IN administration, total brain levels remained stable even after perfusion. In contrast, the vast majority of the polypeptide in the CNS of IV injected animals was sequestered within the vasculature (Figure 3A). While over 79% of IN-administered peptide was extravascular, <3% IV-administered peptide remained post perfusion (Figure 3B). The data clearly demonstrated that while IV injection may result in higher overall CNS polypeptide levels, nearly all of the signal is from within the vasculature and not past the BBB.
 | Figure 3 Total extravascular ELP levels in the brain. |
Quantitative fluorescence histology to determine polypeptide levels within the brain
The ex vivo whole organ analysis provides a good estimate of total tissue polypeptide levels but does not provide the resolution necessary for assessing regional polypeptide levels within a tissue. In order to determine the amount of each ELP or CPP-ELP in the olfactory bulb, cerebral hemispheres, and cerebellum, brains of the perfused mice from the previous experiment were assessed by quantitative fluorescence histology using axial sections. Perfused brains were used to eliminate contribution from intravascular polypeptide, and levels in each brain region were quantified relative to standards of the injected protein. As shown in Figure 4A, IN administration resulted in very high ELP levels in the olfactory bulb, with the cerebellum accumulating the second highest ELP levels and very low levels in the cerebral hemispheres. The addition CPPs reduced the amount of polypeptide that reached the brain in both the olfactory bulbs and the cerebellum. Following IV injection, polypeptide levels were distributed much more evenly throughout the brain (Figure 4B), and olfactory bulb levels were much lower after IV injection than after IN administration. The addition of CPPs also caused a reduction in most brain regions after IV injection.
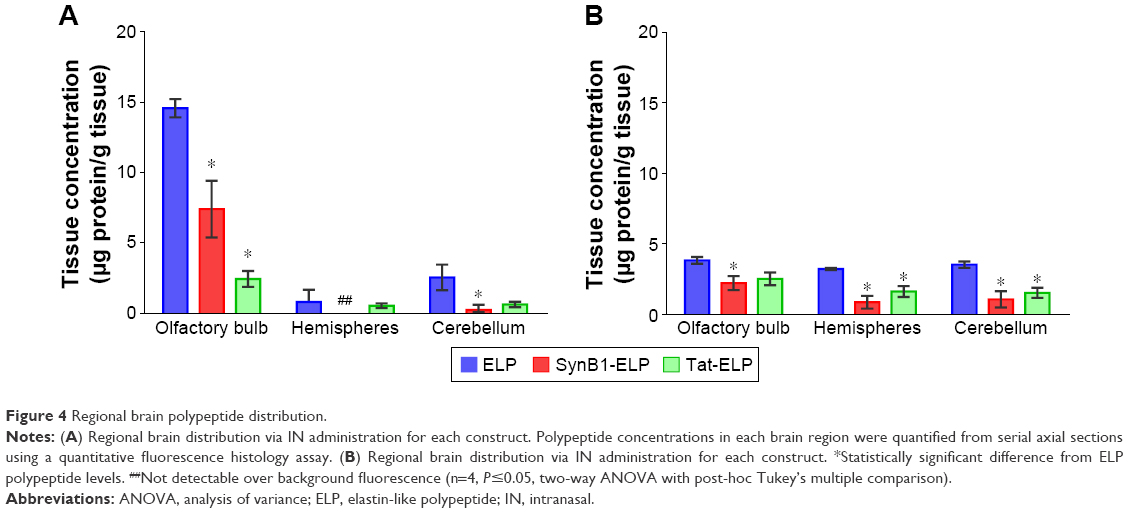 | Figure 4 Regional brain polypeptide distribution. |
Polypeptide biodistribution within the brain
To investigate the regional polypeptide distributions within the CNS, animals were treated by IN or IV administration of ELP as earlier, then were given a bolus IV injection of high molecular weight fluorescein labeled dextran to mark the perfused vasculature immediately prior to sacrifice. Brains were removed without perfusion, sectioned both axially and sagittally, and scanned using a fluorescence slide scanner. Figure 5 clearly shows the increased signal in the olfactory bulbs of the ELP-administered IN, as well as the vascular localization of the IV-administered ELP. Sagittal sections elucidated distribution of ELP in the ventral brain, with very high levels in the olfactory bulb and diffuse signal along the ventral border (Figure 5B). When administered IV, ELP was present throughout the brain, but the signal was only present overlaying the dextran signal from the perfused vasculature.
 | Figure 5 Brain distribution of ELP following IV and IN administration. |
Discussion
Options for the targeted delivery of therapeutic compounds to the CNS are limited. While there is a host of recent research into methods to accomplish this goal, which show a great deal of promise in terms of BBB penetration, in many cases CNS specificity remains as a hurdle. Delivery systems based on some nanoparticles or lipidization often result in high delivery to less tightly regulated organs.27,28 Additional concerns about toxicity or particle build up are entirely relevant given the complexity of some of the materials and modifications used.29,30 The ELP IN delivery route manages to successfully side step many of these potential complications, displaying a high CNS/peripheral organ concentration ratio using a non-immunogenic, low toxicity material.
The beneficial properties of ELP as a drug carrier have been well demonstrated and strongly support its use as both a molecule to facilitate the delivery of attached cargo and to address the half-life barrier to the development of many peptide or small molecule drugs. These data further support the use of ELP as a potential therapeutic delivery platform, as the results show unaltered ELP is able to reach the CNS via IN administration in μg/mL quantities while keeping plasma levels 20 times lower than IV-administered levels. These lower plasma levels result in minimal impact to peripheral systems, decreasing the likelihood of off target drug effects and protecting clearance organs from potential toxicity due to rapid accumulation of macromolecules as seen in the kidneys of the IV-treated animals. The addition of molecules to ELP is likely to alter the kinetics and biodistribution depending on the properties of the chosen molecule itself, but previous data does support CNS delivery of ELP with fused peptides.15
One limitation of the use of preclinical rodent models to study IN drug delivery results from anatomical species differences, raising questions about translatability.31 The relative surface area of the nasal cavity and olfactory epithelium is significantly higher in most model organisms than it is in humans, suggesting data gathered from experiments involving mice or rats may significantly overestimate the ability of the IN route to facilitate delivery into the CNS. However, in spite of the limitations of anatomical differences, there is an increasing evidence that these preclinical models are still of value. Recent human clinical trials have demonstrated effective IN administration of proteins with measurable therapeutic benefit, most notably in the area of IN insulin delivery for Alzheimer’s disease or mild cognitive impairment.32
Another potential limitation of IN drug delivery is that the amount of drug that reaches the CNS versus the amount absorbed into systemic circulation is highly dependent on the physical properties of the therapeutic molecule. In the case of macromolecules such as the proteins described here, the consensus in the literature is that significant amounts of protein are absorbed centrally with relatively little absorbed into the blood.1 Of primary importance for these non-BBB permeable macromolecules, the amount that passes the BBB via the IN route is significantly more than would pass by via even high dose systemic injection, resulting in lower levels throughout most peripheral tissues as shown by our data. Finally, even given optimization of the delivery parameters, low overall CNS levels of IN-delivered therapeutics can limit the practicality of this route. Therefore, IN delivery may be especially useful for delivery of highly potent therapeutics such as modern biologics, hormones, or growth factors.32 Here, we found central ELP levels of 5–15 μg/mL, which corresponds to 80–250 nM, well within the concentration range in which we have shown activity of ELP-fused growth factors.8,33
While we have shown IN administration to deliver ELP to the CNS at potentially therapeutic levels, there are important considerations with regard to the use of the IN route. First, the concentrations of administered molecules were not evenly distributed throughout the CNS, with high signal localized to the olfactory bulbs, moderate signal in the cerebellum, and almost no signal in the hemispheres. This distribution mirrors what some others have seen via IN administration of large molecules, supporting the olfactory and trigeminal nerve pathways as the major conduits down which molecules are transported.19 In the case of work done previously on cerebellar neuronal degeneration, this distribution could be beneficial, resulting in less effect of the therapeutics on areas outside of the cerebellum.34,35 Conversely, if the target brain areas are within the cerebrum, changes to therapeutic design may be needed to facilitate a more even distribution via IN or a different delivery method may be required. Additionally, because very high concentrations are delivered to the olfactory bulb, special considerations need to be payed to the potential for toxicity in olfactory bulb neurons as well as excessive or sustained effects of the delivered therapeutics. Given ELP’s low toxicity, any neurotoxic effects are likely to be modulated by the conjugated therapeutics. While no behavioral effects, such as anosmia, in mice were reported in previous experiments using repeated doses of ELP-conjugated molecules via IN, no studies have also been reported on the direct effects of IN-delivered ELP on sense of smell.
These data also show a significant decrease in CNS delivery via IN administration with the addition of the Tat and SynB1 CPPs. This suggests there are additional considerations when using CPPs with larger molecules. One possibility is that the addition of a short cationic peptide binds the ELP to the outer membrane of the nasal epithelial cells, but either a lack of internalization or intracellular sequestration prevent it from going any farther. It’s also possible that IN administration of these CPP compounds alters mucous properties or production, resulting in decreased CNS delivery by retention of compounds in the mucous layer or the promotion of aspiration. While increased aspiration was seen with the addition of CPPs, all IN treatments resulted in high signal localized to the lungs which could lead to adverse effects. One way to minimize the potential for aspiration may be the use of higher concentrations and smaller volumes or by changing ELP’s characteristic transition temperature, both of which could either increase the viscosity of the solution reducing the propensity for aspiration or cause the formation of an ELP gel which could both reduce aspiration and increase contact time within the nasal cavity. Future studies are needed to investigate these possibilities.
These results like all other are not without caveats. It is worth considering the data from Xu et al, which showed the CNS levels of TAT-HaFGF increased very rapidly after IN administration before rapidly decreasing by 1 hour.2 We chose our time point based on our previous data suggesting CNS levels peak at approximately 1 hour, and we would conjecture the difference in time course is likely due to the large differences in size between the administered molecules. Also, while we did keep the polypeptide concentrations in the administered solution at 100 mg/mL ±10 mg/mL, it is possible that differences in viscosity of the polypeptide solutions could have played a role. We are currently considering the addition of viscosity measures to future experiments to rule out this potential effect.
Future experiments will seek to identify the peptide characteristics ideally suited for optimal IN delivery. Studies including additional time points and ELP constructs of smaller or larger size will help determine the effect of molecular size on CNS levels and clearance kinetics. Incorporation of the thermal properties of ELP are also likely to prove useful in investigating the effects of increased contact times with the nasal epithelium and the effects of thermal targeting on IN administration. In summary, these data create effective baseline information which may be used to evaluate the kinetics of large polypeptides administered intranasally.
Acknowledgments
Partial salary support for GLB was provided by National Institutes of Health (NIH) grant R01HL121527. Access to ex vivo specimen imaging was supported by the Animal Imaging Core Facility of the University of Mississippi Medical Center. The authors would like to thank Rowshan Begum for her help in purifying proteins, and Drazen Raucher for providing the CPP-ELP expression vectors.
The abstract of this paper was presented at the 2015 Experimental Biology Conference as a poster presentation with interim findings. The poster’s abstract was published in “Poster Abstracts” in The Official Journal of the Federation of American Societies for Experimental Biology: http://www.fasebj.org/content/29/1_Supplement/834.2
Disclosure
The authors have no financial relationship with the organizations that sponsored the research. GLB is owner of Leflore Technologies, LLC, a private company working to develop ELP-based therapies for various diseases. The authors report no other conflicts of interest in this work.
References
Dhuria SV, Hanson LR, Frey WH 2nd. Intranasal delivery to the central nervous system: mechanisms and experimental considerations. J Pharm Sci. 2010;99(4):1654–1673. | ||
Xu J, Xiang Q, Su J, et al. Evaluation of the safety and brain-related tissues distribution characteristics of TAT-HaFGF via intranasal administration. Biol Pharm Bull. 2014;37(7):1149–1157. | ||
Falcone JA, Salameh TS, Yi X, et al. Intranasal administration as a route for drug delivery to the brain: evidence for a unique pathway for albumin. J Pharmacol Exp Ther. 2014;351(1):54–60. | ||
Yang JP, Liu HJ, Cheng SM, et al. Direct transport of VEGF from the nasal cavity to brain. Neurosci Lett. 2009;449(2):108–111. | ||
Casettari L, Illum L. Chitosan in nasal delivery systems for therapeutic drugs. J Control Release. 2014;190:189–200. | ||
Yang ZZ, Zhang YQ, Wang ZZ, Wu K, Lou JN, Qi XR. Enhanced brain distribution and pharmacodynamics of rivastigmine by liposomes following intranasal administration. Int J Pharm. 2013;452(1–2):344–354. | ||
Bidwell GL 3rd, Fokt I, Priebe W, Raucher D. Development of elastin-like polypeptide for thermally targeted delivery of doxorubicin. Biochem Pharmacol. 2007;73(5):620–631. | ||
George EM, Liu H, Robinson GG, Mahdi F, Perkins E, Bidwell GL 3rd. Growth factor purification and delivery systems (PADS) for therapeutic angiogenesis. Vasc Cell. 2015;7(1):1. | ||
Shamji MF, Whitlatch L, Friedman AH, Richardson WJ, Chilkoti A, Setton LA. An injectable and in situ-gelling biopolymer for sustained drug release following perineural administration. Spine (Phila Pa 1976). 2008;33(7):748–754. | ||
George EM, Liu H, Robinson GG, Bidwell GL. A polypeptide drug carrier for maternal delivery and prevention of fetal exposure. J Drug Target. 2014;22(10):935–947. | ||
Nicol A, Gowda C, Urry DW. Elastic protein-based polymers as cell attachment matrices. J Vasc Surg. 1991;13(5):746–748. | ||
Zhang YN, Avery RK, Vallmajo-Martin Q, et al. A highly elastic and rapidly crosslinkable elastin-like polypeptide-based hydrogel for biomedical applications. Adv Funct Mater. 2015;25(30):4814–4826. | ||
Moktan S, Perkins E, Kratz F, Raucher D. Thermal targeting of an acid-sensitive doxorubicin conjugate of elastin-like polypeptide enhances the therapeutic efficacy compared with the parent compound in vivo. Mol Cancer Ther. 2012;11(7):1547–1556. | ||
Liu W, Dreher MR, Chow DC, Zalutsky MR, Chilkoti A. Tracking the in vivo fate of recombinant polypeptides by isotopic labeling. J Control Release. 2006;114(2):184–192. | ||
Hearst SM, Shao Q, Lopez M, Raucher D, Vig PJS. The design and delivery of a PKA inhibitory polypeptide to treat SCA1. J Neurochem. 2014;131(1):101–114. | ||
Kamei N, Takeda-Morishita M. Brain delivery of insulin boosted by intranasal coadministration with cell-penetrating peptides. J Control Release. 2015;197:105–110. | ||
Khafagy el-S, Morishita M, Takayama K. The role of intermolecular interactions with penetratin and its analogue on the enhancement of absorption of nasal therapeutic peptides. Int J Pharm. 2010;388(1–2):209–212. | ||
Skotland T, Iversen T, Torgersen M, Sandvig K. Cell-penetrating peptides: possibilities and challenges for drug delivery in vitro and in vivo. Molecules. 2015;20(7):13313–13323. | ||
Kanazawa T, Akiyama F, Kakizaki S, Takashima Y, Seta Y. Delivery of siRNA to the brain using a combination of nose-to-brain delivery and cell-penetrating peptide-modified nano-micelles. Biomaterials. 2013;34(36):9220–9226. | ||
Silhol M, Tyagi M, Giacca M, Lebleu B, Vivès E. Different mechanisms for cellular internalization of the HIV-1 Tat-derived cell penetrating peptide and recombinant proteins fused to Tat. Eur J Biochem. 2002; 269(2):494–501. | ||
Rousselle C, Clair P, Lefauconnier JM, Kaczorek M, Scherrmann JM, Temsamani J. New advances in the transport of doxorubicin through the blood-brain barrier by a peptide vector-mediated strategy. Mol Pharmacol. 2000;57(4):679–686. | ||
Bidwell GL 3rd, Davis AN, Raucher D. Targeting a c-Myc inhibitory polypeptide to specific intracellular compartments using cell penetrating peptides. J Control Release. 2009;135(1):2–10. | ||
Walker LR, Perkins E, McNally LR, Raucher D. Fusion of cell-penetrating peptides to thermally responsive biopolymer improves tumor accumulation of p21 peptide in a mouse model of pancreatic cancer. Drug Des Devel Ther. 2014;8:1649–1658. | ||
Wang Z, Chen Y, Liu E, Gong J, Shin MC, Huang Y. CPP-mediated protein delivery in a noncovalent form: proof-of-concept for percutaneous and intranasal delivery. Protein Pept Lett. 2014;21(11):1129–1136. | ||
Lyons DF, Le VH, Kramer WH, et al. The effect of basic cell penetrating peptides on the structural, thermodynamic and hydrodynamic properties of a novel drug delivery vector: ELP [V5G3A2-150]. Biochemistry. 2014;53(6):1081–1091. | ||
Bidwell GL, Perkins E, Hughes J, Khan M, James JR, Raucher D. Thermally targeted delivery of a c-Myc inhibitory polypeptide inhibits tumor progression and extends survival in a rat glioma model. PLoS One. 2013;8(1):e55104. | ||
Sela H, Cohen H, Elia P, Zach R, Karpas Z, Zeiri Y. Spontaneous penetration of gold nanoparticles through the blood brain barrier (BBB). J Nanobiotechnology. 2015;13:71. | ||
Stockwell J, Abdi N, Lu X, Maheshwari O, Taghibiglou C. Novel central nervous system drug delivery systems. Chem Biol Drug Des. 2014;83(5):507–520. | ||
Kim JS, Yoon TJ, Yu KN, et al. Toxicity and tissue distribution of magnetic nanoparticles in mice. Toxicol Sci. 2006;89(1):338–347. | ||
Lovisolo D, Gilardino A, Ruffinatti F. When neurons encounter nanoobjects: spotlight on calcium signalling. Int J Environ Res Public Health. 2014;11(9):9621–9637. | ||
Ruigrok MJR, de Lange ECM. Emerging insights for translational pharmacokinetic and pharmacokinetic-pharmacodynamic studies: towards prediction of nose-to-brain transport in humans. AAPS J. 2015;17(3):493–505. | ||
Craft S, Baker LD, Montine TJ, et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment. Arch Neurol. 2012;69(1):29–38. | ||
Chade AR, Tullos NA, Harvey TW, Mahdi F, Bidwell GL 3rd. Renal therapeutic angiogenesis using a bioengineered polymer-stabilized vascular endothelial growth factor construct. J Am Soc Nephrol. 2016;27(6):1741–1752. | ||
Vig PJS, Subramony SH, D’Souza DR, Wei J, Lopez ME. Intranasal administration of IGF-I improves behavior and Purkinje cell pathology in SCA1 mice. Brain Res Bull. 2006;69(5):573–579. | ||
Vig PJ, Hearst S, Shao Q, Lopez ME, Murphy HA 2nd, Safaya E. Glial S100B protein modulates mutant ataxin-1 aggregation and toxicity: TRTK12 peptide, a potential candidate for SCA1 therapy. Cerebellum. 2011;10(2):254–266. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.