Back to Journals » Neuropsychiatric Disease and Treatment » Volume 11
Intractable and highly active relapsing multiple sclerosis – role of alemtuzumab
Authors Dubey D, Cano C, Stuve O
Received 14 June 2015
Accepted for publication 13 August 2015
Published 18 September 2015 Volume 2015:11 Pages 2405—2414
DOI https://doi.org/10.2147/NDT.S90473
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Roger Pinder
Divyanshu Dubey,1 Christopher A Cano,1 Olaf Stuve1–3
1Department of Neurology and Neurotherapeutics, University of Texas Southwestern Medical Center, 2Neurology Section, VA North Texas Health Care System, Medical Service, Dallas, TX, USA; 3Department of Neurology, Klinikum rechts der Isar, Technische Universität München, Munich, Germany
Abstract: Alemtuzumab is a humanized recombinant monoclonal antibody that was recently approved by the US Food and Drug Administration and the European Medicines Agency for the management of relapsing forms of multiple sclerosis (MS). It has been utilized for the management of chronic lymphocytic leukemia, bone marrow and renal transplantation, or graft versus host disease. Because of its immunomodulatory properties, it was brought into clinical development in MS. One Phase II (CAMMS223) and two Phase III clinical trials (CARE-MSI and -II) have evaluated the safety and efficacy of alemtuzumab in patients with relapsing–remitting MS. Even though its efficacy profile and long-lasting effect have attracted much interest among physicians and patients, it has significant potential adverse effects that may limit its use to patients with active disease. Here, we review the history of drug development of alemtuzumab. Furthermore, we outline the postulated mechanisms of action, clinical evidence, and safety of alemtuzumab for its use as a disease-modifying agent in active and highly active MS.
Keywords: alemtuzumab, multiple sclerosis, monoclonal antibody, CD52, idiopathic thrombocytopenic purpura
Introduction
Even though the etiopathogenesis of multiple sclerosis (MS) has not been fully understood, our understanding of inflammation and neurodegeneration involved in this complex disease has increased significantly over the past years.1 Various antigens, including myelin oligodendrocyte glycoprotein, myelin basic protein, KiR4.1, and others, have been postulated to have a causal relation. Further research in this area will be required to establish them as the target of aberrant adaptive autoimmune responses.1,2
Approximately 8% of patients with MS experience more aggressive or hyperacute disease course.3 Some of these patients can be categorized as having highly active relapsing–remitting multiple sclerosis (HARRMS), although its definition remains debatable. European Medicines Agency (EMA) defined patients with HARRMS as treatment naïve patients with at least two disabling relapses in the last 1 year and at least one gadolinium-enhancing lesion or significant increase in T2-lesion load.4,5 Patients who have failed to respond to an adequate course of at least one disease-modifying therapy (DMT), presenting with at least one relapse in the previous year while on therapy and at least nine T2-hyperintense lesions or at least one gadolinium-enhancing lesion, were also characterized as having HARRMS.4–6 For these patients, most designated MS centers currently adopt an approach of rapid and effective immunomodulation in order to prevent aggressive disease progression and severe disability accumulation.7 Early initiation of effective immunotherapy is considered to be important in this group of patients due to a narrow therapeutic window for anti-inflammatory agents.7–9 Most of the conventional first-generation DMT may be ineffective in avoiding rapid accumulation of fixed disability.10–13
Development
Alemtuzumab is a humanized immunoglobulin (Ig) G kappa monoclonal antibody (mAb) that targets and depletes cells with CD52 surface antigen.14 CD52 is one of the most abundant membrane glycoproteins comprising ~5% of the cell surface.15 In humans, it is expressed on lymphocytes (except plasma cells), monocytes, eosinophils, and macrophages.16 This antigen is also found in the male reproductive tract, including epididymis, vas deferens, seminal vesicle, and spermatozoa.17 Despite its expression in the male reproductive system, adverse effects, such as infertility, have not been reported with the use of alemtuzumab.17 This may be due to lower concentration of this therapeutic antibody in the seminal fluid.15
Alemtuzumab was initially developed with the objective of depletion of T-cells for the management of graft versus host disease and lymphoid malignancies.15 During initial in vivo experiments, rat IgM antibody against CD52 surface antigen (CAMPATH-1M) was used for the management of lymphoid malignancies.18 Even though CAMPATH-1M coated the lymphocytes, there was no significant lymphocyte depletion. This was followed by the development of IgG2b anti-CD52 antibodies (CAMPATH-1G), which were able to bind to human Fc receptors and activate complement-mediated killing.15,19 CAMPATH-1G was successfully utilized for prevention of bone marrow transplant rejection.19 Subsequently, CAMPATH-1H, humanized IgG1 equivalent of CAMPATH-1G, was generated.20 This mAb was eventually approved for the management of chronic lymphocytic leukemia.
Mechanism of action
There are three mechanisms by which alemtuzumab mediates immune cell depletion (Figure 1): antibody-dependent cell-mediated cytotoxicity, complement-dependent cytotoxicity, and apoptosis.14,21 In vitro experiments showed that antibody-dependent cell-mediated cytotoxicity occurs at much lower concentrations of alemtuzumab (0.01 mg/mL) than the concentration required for complement-dependent cytotoxicity and apoptosis.22,23 The cellular CD52 epitope recognized by alemtuzumab is the C-terminal peptide and is part of the glycophosphatidylinositol anchor.15 Binding of the antibody to the epitope promotes deposition of activated complement molecules and facilitates cell-mediated killing.
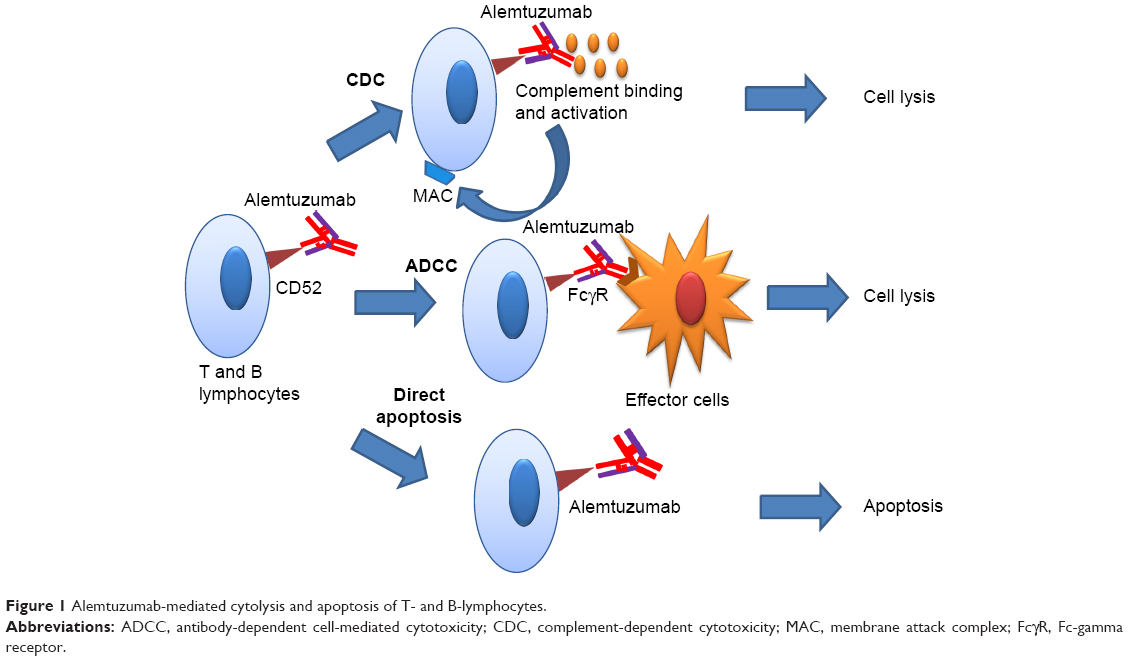 | Figure 1 Alemtuzumab-mediated cytolysis and apoptosis of T- and B-lymphocytes. |
Numerous studies have evaluated the mechanism of growth arrest and apoptosis of B- and T-cells by CD52 antibody, but the exact mechanism remains unclear.24 The biological effects may be related to immunoprecipitation of cell signaling pathways by antibodies, leading to increase in tyrosine phosphorylation, which appears to correlate with the degree of growth inhibition. Decline in growth inhibitions seems to be also dependent on the density of CD52 receptors of target cells.24 Another study demonstrated that alemtuzumab, especially in combination with a cross-linking anti-Fc antibody, leads to activation of caspase 3 and 8.25 Caspase 8 has been shown to initiate cell death in response to extracellular apoptosis-inducing ligands. Caspase 8 signaling is further amplified by caspase 3.26
Potential neuroprotective role of alemtuzumab was highlighted by a post hoc analysis conducted by Jones et al.27 Among patients in CAMMS223 trial with no clinical disease activity immediately before receiving treatment, there was significant improvement in disability in the alemtuzumab-treated group compared to those receiving interferon β1a (INF-β1a). This study also showed that peripheral blood mononuclear cell cultures when specifically stimulated with myelin basic protein, following treatment with alemtuzumab, produced an increased concentration of brain-derived neurotrophic factor, platelet-derived growth factor, and ciliary neurotrophic factor. T-cells played the most prominent role in the production of these neurotrophins. Post-alemtuzumab peripheral blood culture media promoted increased survival of rat neurons. These effects were reversed to some extent by the use of brain-derived neurotrophic factor and ciliary neurotrophic factor neutralizing antibodies. Peripheral blood mononuclear cell-conditioned media also enhanced survival and maturation of oligodendrocyte precursor cells.
Repopulation of leukocytes
CD19 cells (B-lymphocytes) are the first leukocyte subset to repopulate following alemtuzumab treatment, reaching the 25th percentile of pretreatment levels within 3–6 months.21,28 This is followed by CD8+ cells (cytotoxic T-lymphocytes), which reach the 25th percentile of pretreatment levels by 6–9 months. Helper T-cells (CD4+ T-cells) repopulate at slower rate, taking >2 years to return to the 25th percentile pretreatment numbers. Pattern of leukocyte repopulation has been hypothesized to contribute to immune-mediated adverse effects of this therapeutic mAb.21,29
Administration/dosing
In patients with MS, alemtuzumab is administered intravenously at a dose of 12 mg/d for five consecutive days, with a further 3-day course 12 months later.30 This dose is significantly lower than the dose used in patients with chronic lymphocytic leukemia.31 To prevent inflammatory response due to excessive release of cytokines, patients are premedicated with corticosteroids (methylprednisolone 1,000 mg or equivalent) for the first 3 days, immediately prior to alemtuzumab administration.21 Antihistamines and/or antipyretics are also used in some patients. Owing to high prevalence of herpes infection among patients receiving alemtuzumab, antiviral prophylaxis is initiated on the first day of treatment.29 The antiviral prophylaxis is continued for at least 2 months after completion of alemtuzumab and until CD4+ lymphocyte count is ≥200/mm3.
Efficacy
Phase II trial
CAMMS223 was a Phase II, randomized, and single-blinded trial (Table 1), in which 334 previously untreated, early relapsing–remitting MS (RRMS) patients were randomized (1:1:1 ratio) to receive either annual cycles of intravenous alemtuzumab at 12 mg/d or 24 mg/d, or three times per week subcutaneous INF-β1a at a dose of 44 μg.32 The two coprimary measures of efficacy were rate of relapse and time to 6-month sustained accumulation of disability (SAD). The study was designed to last 36 months, but treatment with alemtuzumab was suspended prematurely after three patients developed immune thrombocytopenic purpura (ITP) and one of them died. This prevented two patients (1%) from receiving their second cycle of alemtuzumab at 12 months and 155 patients (75%) from receiving their third cycle of alemtuzumab at 24 months. The INF-β1a-treatment group completed the study unabated.
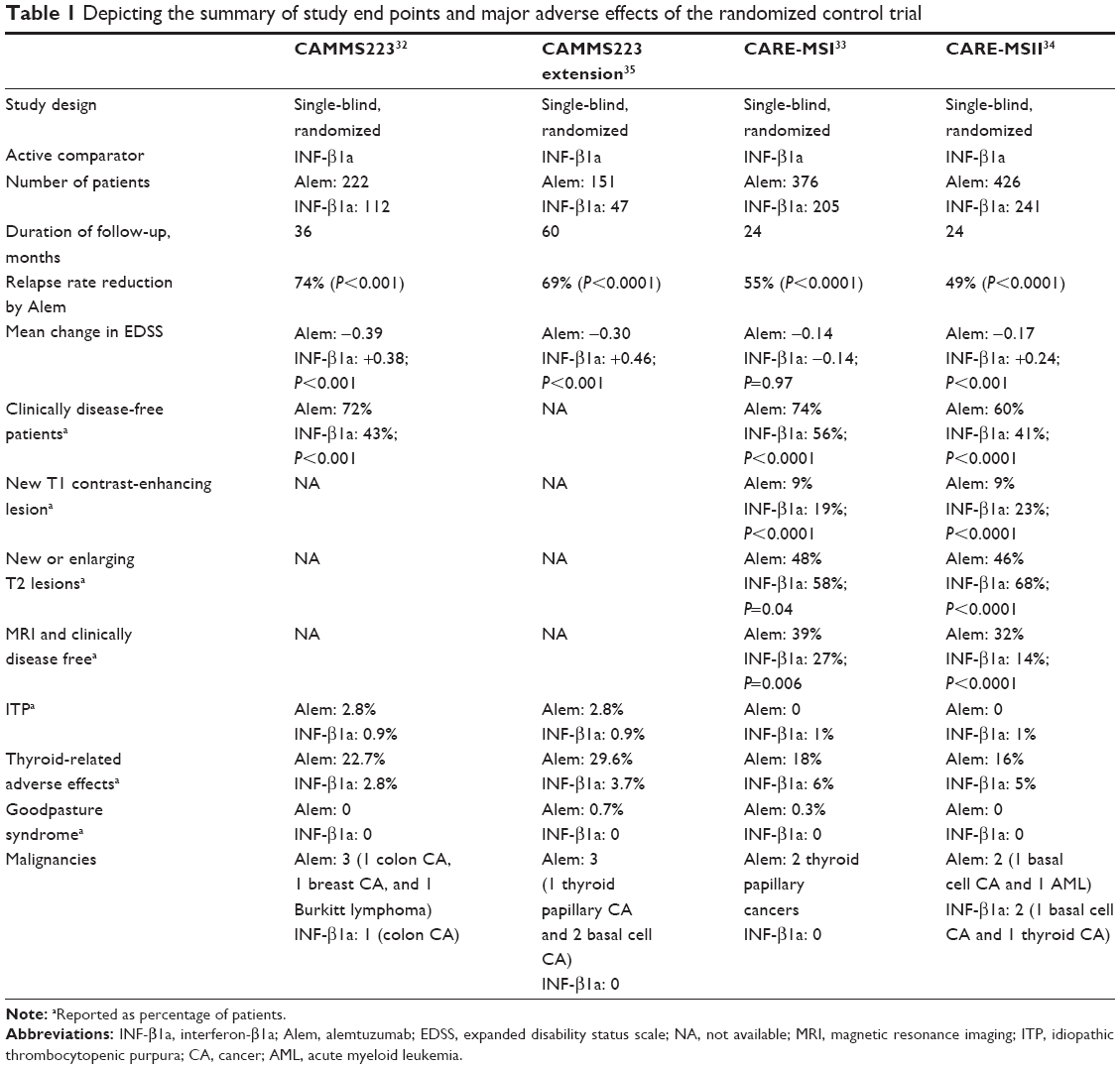 | Table 1 Depicting the summary of study end points and major adverse effects of the randomized control trial |
The two alemtuzumab dosing groups were both statistically equally efficacious, and their data were combined to demonstrate a 74% reduction in relapse rate and 71% reduction in risk of 6-month SAD when compared with INF-β1a group. Thus, both coprimary measures of efficacy were met.
Phase III trials
CARE-MSI and CARE-MSII were Phase III, randomized, and single-blinded trials (Table 1) that were designed similar to the CAMMS223 trial. These trials also compared efficacy of alemtuzumab to INF-β1a for the treatment of RRMS by measuring the rate of relapses and the time to 6-month SAD.32–34 Both CARE-MS studies enrolled patients with active MS who fulfilled 2005 McDonald criteria and had ≥2 relapses in the prior 2 years and ≥1 relapse in the prior year.33,34 The CARE-MSI and CARE-MSII trials differed in that the CARE-MSI trial enrolled treatment naïve patients with early RRMS, like the CAMMS223 trial, whereas the CARE-MSII trial enrolled previously treated patients with RRMS. The CARE-MSII patient cohort had experienced relapses after being on a DMT for a minimum of 6 months. These patients had more advanced disease than those in the CARE-MSI trial as they were allowed to have a higher expanded disability status scale (EDSS) score (≤5 versus ≤3) and a longer disease duration (<10 years versus <5 years since the first clinical manifestation).
The CARE-MSI trial enrolled 581 patients who were randomized in a 2:1 ratio to receive either annual cycles of intravenous alemtuzumab at 12 mg/d or three times per week subcutaneous INF-β1a at 44 μg that was followed for 24 months. The alemtuzumab-treated patient group had 55% reduction in relapse rate compared to the INF-β1a-treated group. However, there was no significant difference in the risk of SAD.
The CARE-MSII trial enrolled 840 patients who were followed for 24 months and randomized at a 2:2:1 ratio to receive either annual cycles of intravenous alemtuzumab at 12 mg/d or 24 mg/d or three times per week subcutaneous INF-β1a at 44 μg. The intravenous alemtuzumab at 24 mg/d group was discontinued to aid in the recruitment of patients into the other two groups, and data collected were utilized to assess safety but not efficacy. The two coprimary end points in this trial were met with a 49% reduction in relapse rate and 42% reduction in the risk of SAD in the alemtuzumab-treated group compared to the INF-β1a group at 24 months.
Long-term extension studies of alemtuzumab efficacy
The CAMMS223 extension study followed 68% of the alemtuzumab-treated and 42% of the INF-β1a-treated patients from the original study.35 At 5 years, there was a continued reduction in relapse rate by 69% and reduction in SAD by 72% in the alemtuzumab-treated group compared to the INF-β1a-treated group. Interestingly, there was a 56% reduction in relapse rate in the alemtuzumab-treated group compared to the INF-β1a-treated group from years 3 to 5 of the study, which did not reach statistical significance.
Tuohy et al conducted an observational cohort study of 87 RRMS patients who received alemtuzumab. These patients had experienced ≥1 relapse in the preceding year, displayed an EDSS score <6.0, had a disease duration <10 years, and had not been exposed to any experimental therapy.36 The median period for follow-up was 7 years (33–144 months). The majority of patients (52%) required just two cycles of alemtuzumab, except for the patients who experienced relapses and required additional treatment cycles: three cycles (36%), four cycles (8%), or five cycles (1%). Compared to their baseline EDSS scores, 68% patients had an improved or stable level of neurological disability. By the area under the curve analysis, 60% of patients had an overall improvement or stabilization of disability. There was also a reduction in the mean annualized relapse rate (ARR) after treatment with alemtuzumab from 1.78 (SD 0.82) to 0.16 (SD 0.26).
The CARE-MSI and CARE-MSII extension studies are currently ongoing and will follow patients up to 10 years to assess efficacy and safety of alemtuzumab.21
Management of highly active relapsing MS
Hirst et al reported an open-label experience from three regional centers in Europe using alemtuzumab as a therapeutic agent for the management of HARRMS in a consecutive series of 39 patients.7 The patients were followed up for a mean of 1.89 years. The mean ARR fell from 2.48 to 0.19 following treatment with alemtuzumab. Nearly one-third (29%) of the relapses were observed within the first 12 weeks following the infusion. This represents a reduction in the ARRs by 87% in the first year and 92% overall. There was also a significant change in the mean EDSS scores: −0.36 overall and −0.15 in those patients who completed ≥1 year of follow-up. Eighty-three percent of patients had stable or improved disability following treatment.
Perumal et al published two cases with HARRMS who were treated with subcutaneous alemtuzumab.37 There was a significant reduction in the number of relapses and an improvement in the disability in these two patients. Subcutaneous alemtuzumab was well tolerated with no major adverse effects.
In another study, 16 patients with aggressive MS (eight patients with RRMS and eight patients with secondary progressive MS [SPMS]) who were previously treated with mitoxantrone were administered alemtuzumab.38 Assessment of rate of disability progression and relapses were prospectively performed at 12 months and 6 months before treatment, at baseline, and then every 3 months following alemtuzumab administration. Among the SPMS patients, two remained disease free up to the last evaluation (4.7 years and 8 years), five improved or stabilized, one patient worsened. Among eight RRMS patients, seven had improvement in disability, four of whom remained disease free up to the last visit, and one patient progressively worsened.
Other therapies for management of HARRMS
Natalizumab
Seventy patients with HARRMS were treated with natalizumab and assessed for freedom from clinical, radiological, and disease activities (clinical and radiological).8 Fifty-two patients completed 2 years of treatment; of them 48% had freedom from disease activity. There was significant reduction in ARR from 2.49 to 0.47 at the end of the first year and to 0.34 at the end of the second year. There was also a significant reduction in the percentage of patients with contrast-enhancing lesions from 21 to 5.7 at the end of the first year and to 5.8 during the second year.
Another post hoc analyses of AFFIRM and SENTINEL trials showed that among treatment naïve patients who met the criteria for HARRMS; natalizumab reduced the relapse rate by 81% and the risk of disability progression by 64% compared to placebo.39 Among patients with HARRMS who were on INF-β1a, natalizumab reduced relapse rate and disability progression by 76% and 58%, respectively.
Fingolimod
Derfuss et al performed a post hoc analysis (FREEDOMS and FREEDOMS II trials) to evaluate the efficacy of fingolimod versus placebo in patients with HARRMS.40 Based on the inclusion criteria for HARRMS, 249 and 257 patients were allocated to fingolimod and placebo groups, respectively. There was a significant reduction in ARR (48%) in the fingolimod group compared to placebo. There was also a significant reduction in 3-month (34%) and 6-month (45%) confirmed disability progression and brain volume loss (46%) among patients receiving fingolimod versus placebo. The number of contrast-enhancing lesions and new or enlarging T2 lesions was also reduced significantly by 65% and 69%, respectively.
Daclizumab
A post hoc analysis of patient with HARRMS in SELECT trial was performed evaluating the effects of daclizumab high-yield process (DAC HYP) versus placebo.41 In this study, patients with daclizumab were defined as patients with ≥2 relapses in the year before randomization and ≥1 gadolinium-enhancing lesion at baseline. Among the patients in the DAC HYP group, there was a significant reduction in ARR by 50% compared to placebo. New or newly enlarging T2 lesion and gadolinium-enhancing lesions were also significantly reduced by 76% and 89%, respectively, in the DAC HYP group versus placebo. Sustained disability progression over 6 months was reduced by 88% among patients receiving DAC HYP compared to placebo, but this difference did not reach statistical significance (P=0.057).
Mitoxantrone
Mitoxantrone has been utilized in patients with aggressive MS with some success, but its adverse effect profile limits the use.42,43 Additionally, a recent study showed that patients who had continued disease progression despite being on mitoxantrone showed some clinical and radiological benefits by transitioning to alemtuzumab.38
Autologous hematopoietic stem cell transplantation
Autologous hematopoietic stem cell transplantation (AHSCT) is another therapeutic strategy currently being evaluated.44 In a Phase II trial, AHSCT was compared to mitoxantrone for the management of aggressive forms of RRMS or SPMS.45 The primary end point of the study was cumulative number of new T2 lesions. The secondary end points included number of gadolinium-enhancing lesions, ARR, and disability progression. There was a significant reduction in the number of T2 lesions in the AHSCT group compared to mitoxantrone. There were no gadolinium-enhancing lesions over a period of 4 years among patients who underwent AHSCT, whereas 56% of patients receiving mitoxantrone had new gadolinium-enhancing lesions. There was also significant reduction in relapse rate following AHSCT versus mitoxantrone. The results of this trial are promising, but more data points will be required to evaluate this novel therapeutic strategy. Profound immunosuppression associated with ASHCT puts patients at risk of opportunistic infections.46 This needs to further be evaluated to ascertain that the benefits of this therapy outweigh the risks.
Thus far, there are no head-to-head trials comparing the efficacy of abovementioned therapeutic strategies versus alemtuzumab for the management of HARRMS. It is not clearly evident if choosing one DMT (natalizumab, fingolimod, daclizumab, or alemtuzumab) over another would be beneficial. Therefore, clinicians should inform their patients about available treatment options. Side-effect profile, contraindications, route of administration, and frequency of dosing of each agent should be evaluated before deciding preferred therapeutic agent, for example, in a patient with positive JC virus serology, DMTs other than natalizumab should be considered.
Adverse events
There were elevated rates of infusion reactions, infections, and autoimmunity in patients who received alemtuzumab for RRMS in the Phase II and Phase III trials compared to controls.32–34 Interestingly, the number of patients dropping out of these studies because of treatment-related adverse effects was lower in patients treated with alemtuzumab (1.3%–3%) than those treated with INF-β1a (5.8%–12.1%). Even in the study evaluating the effect of alemtuzumab in HARRMS, infusion reaction was the most common adverse effect, occurring in 90%–99% of patients. These reactions were generally mild (fever, fatigue, nausea, headache, rash, and pruritus).7 Transient worsening of preexisting neurological deficits during infusion was also observed in three patients.
Infections occurred in 77% of patients treated with alemtuzumab 12 mg and were predominantly mild to moderate in severity and primarily affected the upper respiratory and urinary tracts.32–34 There was a relatively high incidence (16%) of herpes infections in alemtuzumab-treated patients, which was alleviated by prophylactic treatment with acyclovir (or its equivalent). Spirochetal gingivitis, pyogenic granuloma, and listeria meningitis were the only infections observed in the open-label studies that may have been attributable to immunosuppression.21,32,35 Progressive multifocal leukoencephalopathy was not reported. There were no infection-related deaths in alemtuzumab-treated patients. Low rate of opportunistic infection could be due to preservation of innate immune system.29,30 This is likely explained by the fact that alemtuzumab depletes predominantly naïve T-cells and naïve B-cells, whereas memory T-cells and memory B-cells are relatively spared.29,47,48
An increased risk of autoimmunity (Table 1) among patients receiving alemtuzumab was also identified.30 This side effect is thought to be due to the pattern of repopulation of lymphocytes.49 The mechanisms through which lymphocytes repopulate include generation of new T-cells in the thymus or homeostatic proliferation of mature lymphocytes that escaped depletion by alemtuzumab.28,49 Latter mechanism seems to be more common among patients having more significant autoimmune adverse effects.
The most common autoimmune adverse event in the Phase III trials was thyroid autoimmunity (29.6%) in alemtuzumab-treated patients.33,34 In the CAMMS223 5-year follow-up study, 39% of patients treated with alemtuzumab 12 mg and 29% of those who received the 24 mg dose had autoimmune thyroid dysfunction.35 The onset of thyroid autoimmunity ranged from 6 months to 61 months post initiation of alemtuzumab treatment, and patients generally responded well to thyroid hormone replacement therapy.21,33,34 The rate of thyroid autoimmunity was even higher (41%) in the long-term follow-up study.36 The rate of autoimmune thyroid disease did not seem to be associated with the number of treatment courses; most probably due to longer follow-up, more cases were indentified.
In the abovementioned clinical trials, ITP was seen at higher frequency in alemtuzumab-treated group compared to the INF-β1a-treated group.32–34 ITP associated with alemtuzumab differs from other drug-induced immune thrombocytopenia, which typically occurs within days of exposure to the offending agent and resolves within days of its discontinuation. There seems to be significant delay in the presentation of alemtuzumab-associated ITP (median 24.5 months) following initial drug administration.50 This delay may point toward the potential role of lymphocyte repopulation in the manifestation of ITP.29,50 Seven patients were diagnosed with ITP in CAMMS223.32 Six were in alemtuzumab-treatment group, whereas one was in the INF-β1a-treatment group. In June 2005, one of the six patients died secondary to intracranial hemorrhage. Following this fatality, investigators were instructed to perform monthly complete blood counts (CBC) to identify thrombocytopenia or other hematologic abnormalities and contact patients and advise them to report abnormal bleeding, bruising, or petechial rash. Administration of alemtuzumab was transiently suspended following development of mucocutaneous bleeding in two additional patients, who were diagnosed with ITP. Dosing of alemtuzumab was resumed in 2008 following amendments to the study protocol to include a formal monitoring plan to ensure timely detection and treatment of ITP.
In the Phase III CARE-MS trial program, the rate of ITP was ~1%; three patients in CARE-MSI and seven patients in CARE-MSII were involved in the program.33,34 Three of these patients needed to be initiated on rituximab, and one patient in CARE-MS studies also had to undergo splenectomy. The rate of occurrence of ITP was even higher (3.5%) in the Cambridge long-term study.36
The ITP monitoring protocol includes monthly questionnaire for signs and symptoms of ITP offset by 2 weeks from the monthly CBC and peripheral blood smears.32–35 Patients and investigators are educated about these cutaneous lesions through visual depictions of petechiae and ecchymoses.50 Patients are advised to continue safety monitoring for at least 4 years after their last dose.
Alemtuzumab has also been associated with the development of nephropathy.29 In a study by Meyer et al two patients developed antiglomerular basement membrane (anti-GBM) disease and had to undergo renal transplantation.51 In CAMMS223 and CARE-MS trials, four cases of glomerulonephritis were reported among patients treated with alemtuzumab (0.3%), including two cases of membranous glomerulonephritis (MGN) and two cases of anti-GBM disease.32–35 Patients with MGN responded well to medical treatment, including diuretics and/or lisinopril. Two cases of anti-GBM disease showed improvement after treatment with plasmapheresis, cyclophosphamide, and glucocorticosteroids.
The onset of renal dysfunction following alemtuzumab infusion ranges from 4 months to 39 months. Clinical manifestations may include elevation of serum creatinine, hematuria, and/or proteinuria. Regular monitoring of serum creatinine and urinalysis have been recommended for the first 48 months.52 Any detection of worsening renal function (serum creatinine), unexplained hematuria, and/or proteinuria should prompt evaluation for possible nephropathy, especially MGN or anti-GBM disease.
Outlook
The criteria for enrollment of patients in the CARE-MS studies may help guide clinicians to select patients with active disease (defined as ≥1 relapse in the past year and ≥2 relapses in the past 2 years, plus the presence of magnetic resonance imaging lesions). Both treatment naïve patients with active RRMS based on CARE-MSI and those refractory to treatment with other DMTs based on CARE-MSII demonstrated benefits with alemtuzumab. Therefore, among patients who meet criteria for active disease, consideration of initiation of alemtuzumab is warranted.
Additionally, the efficacy of alemtuzumab in challenging cases with HARRMS points toward its potential role in patients requiring early and aggressive immunomodulation to prevent severe disability accumulation. HARRMS criteria as suggested by European Medicines Agency can further help in the selection of treatment naïve patients for initiation of alemtuzumab as first-line DMT.4,5
Most treated patients show significant disease control following two cycles of alemtuzumab.35,36 As more long-term data are made available to clinicians, there will perhaps be a better understanding of its therapeutic potential in patients receiving it for more than two infusion cycles. Even though the pattern of repopulation of immune cells has been attributed to be the cause of heightened risk of autoimmunity, clear understanding of precise mechanism of these adverse effects is still lacking.
The prescription of alemtuzumab is restricted under the Risk Evaluation and Mitigation Strategy program because of the high risk of infusion reactions, autoimmunity, and malignancy.52 The Risk Evaluation Mitigation Strategy program requires physicians, patients, pharmacies, and health care facilities all to enroll in order for alemtuzumab to be prescribed, distributed, and, finally, infused. Physicians, in particular, must complete a training course before prescribing alemtuzumab. A safety measure has been introduced with monthly monitoring of laboratory tests, including serum creatinine, CBC with differential, urinalysis with microscopy, and three monthly monitoring of thyroid function tests until 48 months after receiving last alemtuzumab infusion.21,29,30
Lemtrada (alemtuzumab) was introduced in USA in December 2014 for the treatment of RRMS.53 Alemtuzumab entered a competitive market, which includes eleven other DMT options (five have been released in the last 5 years).54 Restrictions over prescription and monthly laboratory monitoring may impact the sales of alemtuzumab, compared to other disease-modifying agents. To our knowledge, there are currently no publicly available data that detail the prescription numbers for Lemtrada in the USA or Europe. Genzyme, Lemtrada’s manufacturer, predicted physician adoption rates of 5% 1 year after introduction to market and 18%–20% 5 years after introduction to market.55 Genzyme predicted that physicians principally would prescribe Lemtrada as a second- and third-line medication and that the drug would be most popular among treatment-experienced patients.
Acknowledgments
Olaf Stuve has received grant support from Teva Pharmaceuticals and Opexa Therapeutics and has been funded by a merit grant from the US Department of Veterans Affairs. This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Disclosure
Olaf Stuve serves on the editorial boards of JAMA Neurology, Multiple Sclerosis Journal, and Therapeutic Advances in Neurological Disorders. He has served on data monitoring committees of Pfizer and Sanofi-Aventis without monetary compensation. He has served on an advisory board for Sanofi Genzyme and Genentech and has provided advices for Huron Life Sciences. The other authors report no conflicts of interest in this work.
References
Frohman EM, Racke MK, Raine CS. Multiple sclerosis – the plaque and its pathogenesis. N Engl J Med. 2006;354:942–955. | ||
Brickshawana A, Hinson SR, Romero MF, et al. Investigation of the KIR4.1 potassium channel as a putative antigen in patients with multiple sclerosis: a comparative study. Lancet Neurol. 2014;13:795–806. | ||
Confavreux C, Aimard G, Devic M. Course and prognosis of multiple sclerosis assessed by the computerized data processing of 349 patients. Brain. 1980;103:281–300. | ||
European Medicines Agency. Gilenya Summary of Product Characteristics; 2011. Available from http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002202/WC500104528.pdf. Accessed August 1, 2015. | ||
Fazekas F, Bajenaru O, Berger T, et al. How does fingolimod (gilenya((R))) fit in the treatment algorithm for highly active relapsing-remitting multiple sclerosis? Front Neurol. 2013;4:10. | ||
Devonshire V, Havrdova E, Radue EW, et al; FREEDOMS study group. Relapse and disability outcomes in patients with multiple sclerosis treated with fingolimod: subgroup analyses of the double-blind, randomised, placebo-controlled FREEDOMS study. Lancet Neurol. 2012;11:420–428. | ||
Hirst CL, Pace A, Pickersgill TP, et al. Campath 1-H treatment in patients with aggressive relapsing remitting multiple sclerosis. J Neurol. 2008;255:231–238. | ||
Cobo-Calvo Á, Bau L, Matas E, et al. Effectiveness of natalizumab in patients with highly active relapsing remitting multiple sclerosis. Eur Neurol. 2015;73:220–229. | ||
Ostberg A, Pittas F, Taylor B. Use of low-dose mitozantrone to treat aggressive multiple sclerosis: a single-centre open-label study using patient self-assessment and clinical measures of multiple sclerosis status. Intern Med J. 2005;35:382–387. | ||
Randomised double-blind placebo-controlled study of interferon beta-1a in relapsing/remitting multiple sclerosis. PRISMS (Prevention of Relapses and Disability by Interferon beta-1a Subcutaneously in Multiple Sclerosis) Study Group. Lancet. 1998;352:1498–1504. | ||
IFNB Multiple Sclerosis Study Group. Interferon beta-lb is effective in relapsing-remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. The IFNB Multiple Sclerosis Study Group. Neurology. 1993;43(4):655–661. | ||
Jacobs LD, Cookfair DL, Rudick RA, et al. Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. The Multiple Sclerosis Collaborative Research Group (MSCRG). Ann Neurol. 1996;39:285–294. | ||
The IFNB Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. Interferon beta-1b in the treatment of multiple sclerosis: final outcome of the randomized controlled trial.Neurology. 1995;45(7):1277–1285. | ||
Dubey D, Kieseier BC, Hartung HP, Hemmer B, Miller-Little WA, Stuve O. Clinical management of multiple sclerosis and neuromyelitis optica with therapeutic monoclonal antibodies: approved therapies and emerging candidates. Expert Rev Clin Immunol. 2015;11:93–108. | ||
Hale G. The CD52 antigen and development of the CAMPATH antibodies. Cytotherapy. 2001;3:137–143. | ||
Elsner J, Hochstetter R, Spiekermann K, Kapp A. Surface and mRNA expression of the CD52 antigen by human eosinophils but not by neutrophils. Blood. 1996;88:4684–4693. | ||
Hale G, Rye PD, Warford A, Lauder I, Brito-Babapulle A. The glycosylphosphatidylinositol-anchored lymphocyte antigen CDw52 is associated with the epididymal maturation of human spermatozoa. J Reprod Immunol. 1993;23:189–205. | ||
Hale G, Zhang MJ, Bunjes D, et al. Improving the outcome of bone marrow transplantation by using CD52 monoclonal antibodies to prevent graft-versus-host disease and graft rejection. Blood. 1998;92: 4581–4590. | ||
Oakhill A, Pamphilon DH, Potter MN, et al. Unrelated donor bone marrow transplantation for children with relapsed acute lymphoblastic leukaemia in second complete remission. Br J Haematol. 1996;94:574–578. | ||
Phillips J, Drumm A, Harrison P, et al. Manufacture and quality control of CAMPATH-1 antibodies for clinical trials. Cytotherapy. 2001;3:233–242. | ||
Menge T, Stuve O, Kieseier BC, Hartung HP. Alemtuzumab: the advantages and challenges of a novel therapy in MS. Neurology. 2014; 83:87–97. | ||
Mone AP, Cheney C, Banks AL, et al. Alemtuzumab induces caspase-independent cell death in human chronic lymphocytic leukemia cells through a lipid raft-dependent mechanism. Leukemia. 2006;20: 272–279. | ||
Riechmann L, Clark M, Waldmann H, Winter G. Reshaping human antibodies for therapy. Nature. 1988;332:323–327. | ||
Rowan W, Tite J, Topley P, Brett SJ. Cross-linking of the CAMPATH-1 antigen (CD52) mediates growth inhibition in human B- and T-lymphoma cell lines, and subsequent emergence of CD52-deficient cells. Immunology. 1998;95:427–436. | ||
Nuckel H, Frey UH, Roth A, Duhrsen U, Siffert W. Alemtuzumab induces enhanced apoptosis in vitro in B-cells from patients with chronic lymphocytic leukemia by antibody-dependent cellular cytotoxicity. Eur J Pharmacol. 2005;514:217–224. | ||
Jones DT, Ganeshaguru K, Virchis AE, et al. Caspase 8 activation independent of Fas (CD95/APO-1) signaling may mediate killing of B-chronic lymphocytic leukemia cells by cytotoxic drugs or gamma radiation. Blood. 2001;98:2800–2807. | ||
Jones JL, Anderson JM, Phuah CL, et al. Improvement in disability after alemtuzumab treatment of multiple sclerosis is associated with neuroprotective autoimmunity. Brain. 2010;133:2232–2247. | ||
Cossburn MD, Harding K, Ingram G, et al. Clinical relevance of differential lymphocyte recovery after alemtuzumab therapy for multiple sclerosis. Neurology. 2013;80:55–61. | ||
Havrdova E, Horakova D, Kovarova I. Alemtuzumab in the treatment of multiple sclerosis: key clinical trial results and considerations for use. Ther Adv Neurol Disord. 2015;8:31–45. | ||
Willis MD, Robertson NP. Alemtuzumab for the treatment of multiple sclerosis. Ther Clin Risk Manag. 2015;11:525–534. | ||
Elter T, Molnar I, Kuhlmann J, Hallek M, Wendtner C. Pharmacokinetics of alemtuzumab and the relevance in clinical practice. Leuk Lymphoma. 2008;49:2256–2262. | ||
CAMMS223 Trial Investigators; Coles AJ, Compston DA, Selmaj KW, et al. Alemtuzumab vs. interferon beta-1a in early multiple sclerosis. N Engl J Med. 2008;359:1786–1801. | ||
Cohen JA, Coles AJ, Arnold DL, et al; CARE-MS I investigators. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet. 2012;380:1819–1828. | ||
Coles AJ, Twyman CL, Arnold DL, et al; CARE-MS II investigators. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet. 2012;380:1829–1839. | ||
Coles AJ, Fox E, Vladic A, et al. Alemtuzumab more effective than interferon beta-1a at 5-year follow-up of CAMMS223 clinical trial. Neurology. 2012;78:1069–1078. | ||
Tuohy O, Costelloe L, Hill-Cawthorne G, et al. Alemtuzumab treatment of multiple sclerosis: long-term safety and efficacy. J Neurol Neurosurg Psychiatry. 2015;86:208–215. | ||
Perumal JS, Foo F, Cook P, Khan O. Subcutaneous administration of alemtuzumab in patients with highly active multiple sclerosis. Mult Scler. 2012;18:1197–1199. | ||
Le PE, Deburghgraeve V, Lester MA, Cardiet I, Leray E, Edan G. Alemtuzumab as rescue therapy in a cohort of 16 aggressive multiple sclerosis patients previously treated by Mitoxantrone: an observational study. J Neurol. 2015;262:1024–1034. | ||
Hutchinson M, Kappos L, Calabresi PA, et al; AFFIRM and SENTINEL Investigators. The efficacy of natalizumab in patients with relapsing multiple sclerosis: subgroup analyses of AFFIRM and SENTINEL. J Neurol. 2009;256:405–415. | ||
Derfuss T, Bergvall NK, Sfikas N, Tomic DL. Efficacy of fingolimod in patients with highly active relapsing-remitting multiple sclerosis. Curr Med Res Opin. Epub 2015 August 24. | ||
Giovannoni G, Radue EW, Havrdova E, et al. Effect of daclizumab high-yield process in patients with highly active relapsing-remitting multiple sclerosis. J Neurol. 2014;261:316–323. | ||
Correale J, Rush C, Amengual A, Goicochea MT. Mitoxantrone as rescue therapy in worsening relapsing-remitting MS patients receiving IFN-beta. J Neuroimmunol. 2005;162:173–183. | ||
Fox EJ. Management of worsening multiple sclerosis with mitoxantrone: a review. Clin Ther. 2006;28:461–474. | ||
Saccardi R, Mancardi GL, Solari A, et al. Autologous HSCT for severe progressive multiple sclerosis in a multicenter trial: impact on disease activity and quality of life. Blood. 2005;105:2601–2607. | ||
Mancardi GL, Sormani MP, Gualandi F, et al; ASTIMS Haemato-Neurological Collaborative Group, On behalf of the Autoimmune Disease Working Party (ADWP) of the European Group for Blood and Marrow Transplantation (EBMT); ASTIMS Haemato-Neurological Collaborative Group On behalf of the Autoimmune Disease Working Party ADWP of the European Group for Blood and Marrow Transplantation EBMT. Autologous hematopoietic stem cell transplantation in multiple sclerosis: a phase II trial. Neurology. 2015;84: 981–988. | ||
Daikeler T, Tichelli A, Passweg J. Complications of autologous hematopoietic stem cell transplantation for patients with autoimmune diseases. Pediatr Res. 2012;71:439–444. | ||
Hale G, Bright S, Chumbley G, et al. Removal of T cells from bone marrow for transplantation: a monoclonal antilymphocyte antibody that fixes human complement. Blood. 1983;62:873–882. | ||
Jacobs P, Wood L, Fullard L, Waldmann H, Hale G. T cell depletion by exposure to Campath-1G in vitro prevents graft-versus-host disease. Bone Marrow Transplant. 1994;13:763–769. | ||
Jones JL, Phuah CL, Cox AL, et al. IL-21 drives secondary autoimmunity in patients with multiple sclerosis, following therapeutic lymphocyte depletion with alemtuzumab (Campath-1H). J Clin Invest. 2009;119:2052–2061. | ||
Cuker A, Coles AJ, Sullivan H, et al. A distinctive form of immune thrombocytopenia in a phase 2 study of alemtuzumab for the treatment of relapsing-remitting multiple sclerosis. Blood. 2011;118:6299–6305. | ||
Meyer D, Coles A, Oyuela P, Purvis A, Margolin DH. Case report of anti-glomerular basement membrane disease following alemtuzumab treatment of relapsing-remitting multiple sclerosis. Mult Scler Relat Disord. 2013;2:60–63. | ||
LEMTRADA REMS program overview. Available from https://www.lemtradarems.com/Docs/Pdf/LEMTRADA_REMS_Program_Overview.pdf. Accessed August 1, 2015. | ||
FDA Approves Lemtrada™ (alemtuzumab) for Relapsing MS – UPDATE; 2014. Available from http://www.nationalmssociety.org/About-the-Society/News/FDA-Approves-Lemtrada%E2%84%A2-(alemtuzumab)-for-Relapsing. Accessed August 1, 2015. | ||
Disease Modification; 2015. Available from http://www.nationalmssociety.org/For-Professionals/Clinical-Care/Managing-MS/Disease-Modification#section-1. Accessed August 1, 2015. | ||
Genzyme Details Market Potential of Alemtuzumab for MS; 2014. Available from http://www.worldpharmanews.com/genzyme/1529-genzyme-details-market-potential-of-alemtuzumab-for-ms. Accessed August 1, 2015. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.