")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 12
Intracellular interactions of umeclidinium and vilanterol in human airway smooth muscle
Authors Shaikh N, Johnson M, Hall DA, Chung KF, Riley JH, Worsley S, Bhavsar PK
Received 10 February 2017
Accepted for publication 13 April 2017
Published 30 June 2017 Volume 2017:12 Pages 1903—1913
DOI https://doi.org/10.2147/COPD.S134420
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Russell
Nooreen Shaikh,1,2 Malcolm Johnson,3 David A Hall,4 Kian Fan Chung,1,2 John H Riley,3 Sally Worsley,5 Pankaj K Bhavsar1,2
1Experimental Studies, National Heart and Lung Institute, Imperial College London, 2Biomedical Research Unit, Royal Brompton and Harefield NHS Trust, London, 3Respiratory Global Franchise, GlaxoSmithKline, Uxbridge, 4Fibrosis and Lung Injury Development Planning Unit, GlaxoSmithKline, Stevenage, 5Respiratory Research & Development, GlaxoSmithKline, Uxbridge, UK
Background: Intracellular mechanisms of action of umeclidinium (UMEC), a long-acting muscarinic receptor antagonist, and vilanterol (VI), a long-acting β2-adrenoceptor (β2R) agonist, were investigated in target cells: human airway smooth-muscle cells (ASMCs).
Materials and methods: ASMCs from tracheas of healthy lung-transplant donors were treated with VI, UMEC, UMEC and VI combined, or control compounds (salmeterol, propranolol, ICI 118.551, or methacholine [MCh]). Cyclic adenosine monophosphate (cAMP) was measured using an enzyme-linked immunosorbent assay, intracellular free calcium ([Ca2+]i) using a fluorescence assay, and regulator of G-protein signaling 2 (RGS2) messenger RNA using real-time quantitative polymerase chain reaction.
Results: VI and salmeterol (10–12–10–6 M) induced cAMP production from ASMCs in a concentration-dependent manner, which was greater for VI at all concentrations. β2R antagonism by propranolol or ICI 118.551 (10–12–10–4 M) resulted in concentration-dependent inhibition of VI-induced cAMP production, and ICI 118.551 was more potent. MCh (5×10–6 M, 30 minutes) attenuated VI-induced cAMP production (P<0.05), whereas pretreatment with UMEC (10–8 M, 1 hour) restored the magnitude of VI-induced cAMP production. ASMC stimulation with MCh (10–11–5×10–6 M) resulted in a concentration-dependent increase in [Ca2+]i, which was attenuated with UMEC pretreatment. Reduction of MCh-induced [Ca2+]i release was greater with UMEC + VI versus UMEC. UMEC enhanced VI-induced RGS2 messenger RNA expression.
Conclusion: These data indicate that UMEC reverses cholinergic inhibition of VI-induced cAMP production, and is a more potent muscarinic receptor antagonist when in combination with VI versus either alone.
Keywords: COPD pharmacology, cough/mechanisms/pharmacology, drug reactions
Introduction
Bronchodilation is central to the pharmacological treatment of stable chronic obstructive pulmonary disease (COPD),1 and is mediated either directly by smooth-muscle (SM) relaxation via stimulation of the β2-adrenoreceptor (β2R) or indirectly by antagonism of the muscarinic receptor (MR) subtypes M2 and M3 in airway SM cells (ASMCs).2,3 Antagonism of the presynaptic M2 subtype leads to an increase in acetylcholine release from the vagal nerve ending, whereas antagonism of the postsynaptic M2 subtype increases the β2R-induced stimulation of adenylyl cyclase activity and subsequent airway SM relaxation.4 Antagonism of the M3 subtype inhibits bronchial contractility.5
The long-acting MR antagonist (LAMA) umeclidinium (UMEC) and the combination of UMEC with the long-acting β2R agonist (LABA) vilanterol (UMEC/VI) are approved maintenance treatments for COPD in the US, Canada, the EU, and several other countries.6–11 In a previous study, once-daily UMEC/VI 62.5/25 μg was shown to improve lung function compared with UMEC 62.5 μg, VI 25 μg, and placebo.12 Significant improvements in trough forced expiratory volume in 1 second (FEV1) were observed for UMEC/VI 62.5/25 μg compared with UMEC 62.5 μg (52 mL, P=0.004) and VI 25 μg (95 mL, P<0.001), in patients with moderate–severe COPD. In another 24-week trial, in patients with moderate–very severe COPD, an improvement of 90 mL (P=0.006) was observed for UMEC 62.5 μg added to VI 25 μg compared with VI 25 μg alone.13 Recently, the results of a trial demonstrated that the LAMA/LABA combination indacaterol/glycopyrronium was effective in preventing COPD exacerbations in patients with a history of exacerbations in the previous year.14
The mechanisms of action of LAMAs and LABAs alone are reasonably well understood,3,15 although the precise interactions between the two signaling pathways – β2R agonism and MR antagonism – are yet to be fully elucidated. Cyclic adenosine monophosphate (cAMP) is a key intracellular mediator in ASMCs for the activation of protein kinase A (PKA) (Figure 1). Stimulation of β2R increases the activity of adenylyl cyclase, resulting in increased production of intracellular cAMP.16 cAMP inhibits the release of intracellular free calcium ion ([Ca2+]i), reduces membrane [Ca2+]i entry, and removes Ca2+, which in turn leads to SM relaxation.17 Conversely, cholinergic activation of M2 receptors inhibits adenylyl cyclase and cAMP formation, and activation of M3 receptors results in an increase in inositol trisphosphate formation and [Ca2+]i release, leading to muscle contraction.3 These M2- and M3-receptor effects may be suppressed by MR antagonists, such as UMEC. In addition, stimulation of β2R increases the synthesis of regulator of regulator of G-protein signaling 2 (RGS2), a protein that downregulates signaling from multiple Gq-coupled receptors (including M1 and M3 receptors),18 and thus may enhance the therapeutic activity of a LAMA.19
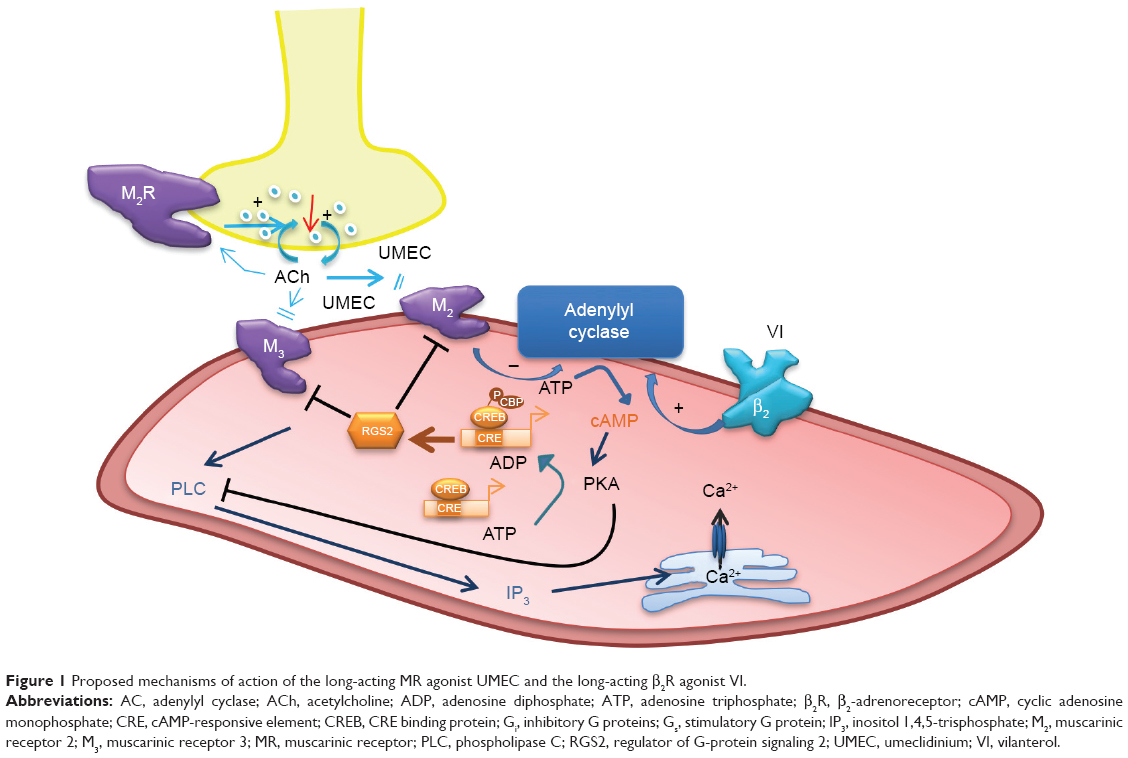 | Figure 1 Proposed mechanisms of action of the long-acting MR agonist UMEC and the long-acting β2R agonist VI. |
Although the pharmacological profiles of both UMEC monotherapy20 and VI monotherapy21,22 have been reported previously, these experiments were carried out on Chinese hamster ovary (CHO) cells. Here, we present results of the first study of either UMEC or VI on target cells – human ASMCs. This is the first report of pharmacological data for UMEC and VI acting together at the target cells. We investigated the ability of UMEC (with or without VI) to induce cAMP production in the presence and absence of cholinergic activation. We tested our hypothesis that UMEC would attenuate methacholine (MCh)-induced [Ca2+]i release (with or without VI), and that both UMEC and VI would induce RGS2.
Materials and methods
ASMC isolation and cell culture
ASMCs were isolated from healthy lung-transplant donor tracheas (n=8) and bronchoscopic biopsies (n=7) of healthy nonsmokers. All donors provided written informed consent. The study was approved by the Institution Ethics Committee (Royal Brompton and Harefield, and National Heart and Lung Institute, Imperial College London, London, UK [03-105]). Human biological samples were sourced ethically, and their research use was in accord with the terms of the informed consents.
ASMCs isolated from tracheas were transferred to a 25 cm2 culture flask. Cells were grown to confluence using Dulbecco’s Modified Eagle’s Medium (Thermo Fisher Scientific, Paisley, UK) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, 100 μg/mL streptomycin, and 1.5 μg/mL amphotericin B. For ASMCs isolated from bronchial biopsies, the biopsies were cut into small pieces (<1 mm2) and transferred to six-well culture plates. Once confluent, cells were harvested and split into larger flasks at each passage. Cultures were maintained in a humidified incubator at 37°C with 5% carbon dioxide, and medium were changed every 2–3 days. ASMCs were identified by the characteristic “hill-and-valley” morphology23,24 and expression of calponin, SM α-actin, and myosin heavy chains. By this definition, ASMCs comprised >95% of harvested cells.
After serum starvation, cells at passage five were plated into serum-deficient culture medium, and after 24 hours were treated with a range of concentrations of VI and UMEC (alone or in combination) or one of four control compounds: salmeterol (a β2R agonist), propranolol (a pan-β-receptor antagonist), ICI 118.551 (a β2R-specific antagonist), or MCh (an MR agonist).
cAMP-production experiments
The manufacturer’s protocol for the cAMP enzyme-linked immunosorbent-assay kit (Cayman Chemical Company, Ann Arbor, MI, US) was employed. Absorbance at 405–420 nm was measured using a colorimetric 96-well plate reader. Data are expressed as pmol/mL cAMP production into the culture medium. Background cAMP (ie, the concentration of cAMP in the absence of an agonist) was subtracted from the values of cAMP measured in the experiments.
cAMP production with β2R agonism
ASMCs were plated (4×104 cells/well) and treated with VI or salmeterol (both at 10−6 M). cAMP concentration in the resultant cell medium was analyzed at the following time points post-treatment: 10, 20, 30, 45, 60, and 90 minutes. In a separate repeat experiment, ASMCs were treated with VI or salmeterol at concentrations of 10−12–10−6 M for 60 minutes.
cAMP production with β2R antagonism
ASMCs were plated (4×104 cells/well) and pretreated with propranolol (10−6 M) or ICI 118.551 (10−6 M) for 30 minutes, followed by stimulation with VI (10−12–10−6 M) for 60 minutes.
Effect of UMEC on cAMP production
ASMCs were plated (4×104 cells/well) and pretreated with UMEC (10−8 M) for 1 hour, followed by MCh (5×10−6 M) for 30 min and VI over a range of concentrations (10−12–10−6 M) for a further 1 hour.
Calcium assay
ASMCs were plated (8×103 cells/well) and pre-treated for 2 minutes with UMEC (10−12–10−6 M), then stimulated with MCh (5×10−6 M) prior to measurement of [Ca2+]i. [Ca2+]i was measured using a Fluo-4 no-wash calcium assay. ASMC media were replaced with 100 μL Fluo-4 loading dye (Fluo-4 NW Calcium Assay Kits, Invitrogen, Paisley, UK) according to the manufacturer’s protocol. After 45 minutes’ incubation at 37°C, excitation/emission 494/516 nm was measured on a 96-well fluorescent plate reader (BMG GmbH, Ortenberg, Germany). Basal fluorescence was measured for 5 seconds, and after treatment with MCh, fluorescence was measured for a further 55 seconds. The following MCh concentrations were used: 10−11, 10−10, 10−8, 5×10−7, 10−6, and 5×10−6 M. Fluorescence was normalized to the maximal increase in fluorescence from baseline (=1) in response to MCh at 5×10−6 M. ASMCs were then pretreated with UMEC (10−10 M) for 2 minutes before measurement of [Ca2+]i, or pretreated for 2 minutes with UMEC (10−12–10−8 M) alone or in combination with VI (10−10, 10−8, 10−6 M) and then stimulated with MCh, before [Ca2+]i was determined.
RGS2 messenger RNA
ASMCs were plated (1.6×105 cells/well) and treated with VI or UMEC alone (both at 10−8 M) or in combination (both at 10−9 M) over a 4-hour time course. Total RNA was isolated from cells using an RNeasy minikit (Qiagen, Crawley, UK) and converted to complementary DNA using random primers and avian myeloblastosis virus reverse transcriptase (Promega, Southampton, UK). Messenger RNA (mRNA) expression was determined by real-time quantitative polymerase chain reaction (Rotor-Gene 3000; Corbett Research, St Neots, UK) using SYBR Green PCR Mix Reagent (Qiagen) and normalized to 18S RNA expression.
Statistics
GraphPad Prism version 5.03 (GraphPad Software, La Jolla, CA, US) software was used for statistical analysis. Concentration-dependent responses were examined using one-way-analysis of variance (ANOVA) (Kruskal–Wallis test) followed by Dunn’s multiple comparison test. Mann–Whitney test comparison compared results between the groups. Negative logarithm of the inhibition constant (pKi) data were determined from the inhibitor concentration causing a 50% reduction in response (IC50) using the Cheng–Prusoff correction.25 Data in all figures are plotted as mean ± standard error of the mean. Each replicate was performed on a separate sample of cells from a different donor. P<0.05 was taken as statistically significant.
Results
cAMP production following β2R agonism
Both VI (10−6 M) and salmeterol (10−6 M) induced cAMP production over a 90-minute time course; however, the magnitude of induction was greater with VI compared with salmeterol (Figure 2A). The greatest difference occurred at 30 minutes poststimulation, where VI induced a cAMP production of 21.65±1.86 pmol/mL compared with 3.45±2.09 pmol/mL for salmeterol.
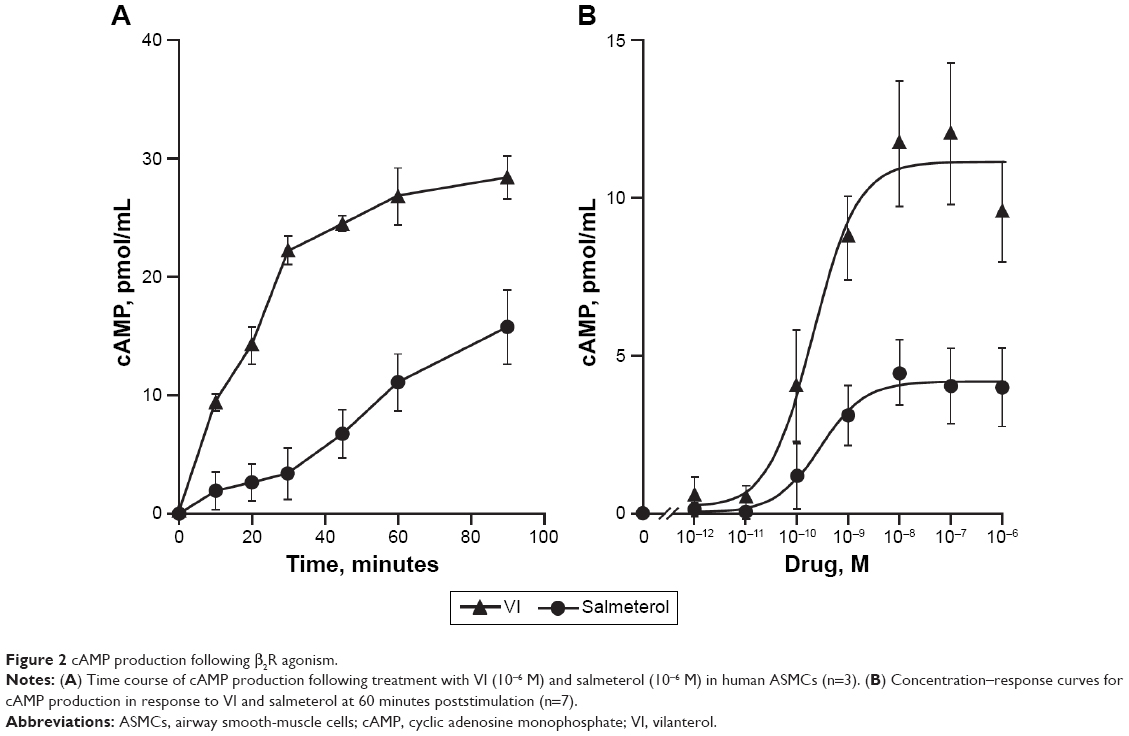 | Figure 2 cAMP production following β2R agonism. |
The time to 50% of the maximal recorded cAMP concentration for VI and salmeterol was 20 versus 38 minutes, respectively. cAMP production in response to a range of concentrations (10−12–10−6 M) of VI or salmeterol at 60 minutes poststimulation was subsequently examined. cAMP production was found to be concentration-dependent for both VI and salmeterol (Figure 2B), and greater for VI versus salmeterol at all concentrations. The maximal cAMP production for VI (10−7 M) and salmeterol (10−7 M) was 12.03±2.18 pmol/mL and 4.46±1.16 pmol/mL (P<0.01, Kruskal–Wallis, n=7), respectively (Figure 2B). The concentration of VI and salmeterol required to stimulate half the maximal response (EC50) was 3.5×10−10±1.1×10−10 M and 2.1×10−8±1.3×10−8 M, respectively.
cAMP production following β2R antagonism
Propranolol effectively inhibited VI-induced cAMP production, causing a substantial decrease in its potency while having a limited effect on maximal response (as expected for a competitive antagonist) (Figure 3A). In contrast, ICI 118.551 effectively inhibited VI-induced cAMP production over all concentrations examined (Figure 3A). The EC50 for VI in the presence of propranolol and ICI 118.551 was 7×10−8±3.4×10−8 M and 6×10−10±3.5×10−10 M, respectively.
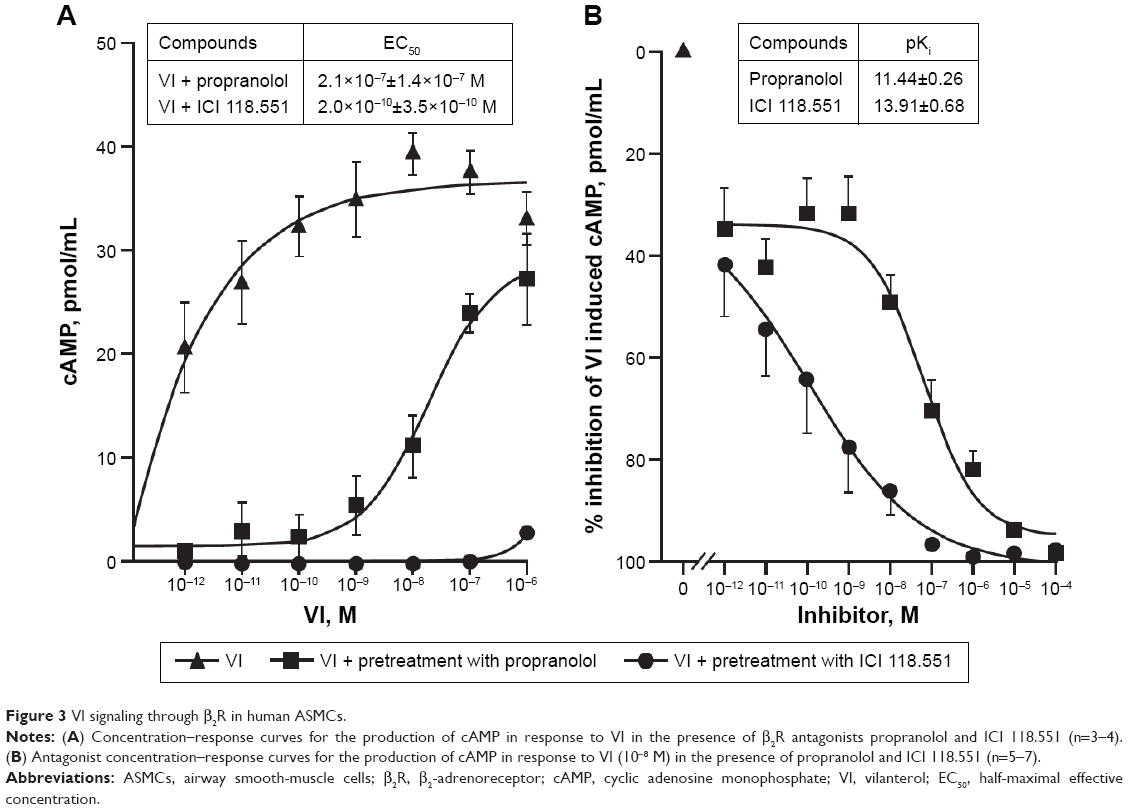 | Figure 3 VI signaling through β2R in human ASMCs. |
Further analysis across a range of ICs showed that both propranolol and ICI 118.551 inhibited VI-induced cAMP accumulation in a concentration-dependent manner (Figure 3B). The pKi for ICI 118.551 was greater than that of propranolol (pKi±SEM: 13.91±0.68 versus 11.44±0.26, respectively).
Effect of cholinergic stimulation in presence and absence of UMEC on VI-induced cAMP production
For all concentrations of VI, pretreatment of ASMCs with MCh (5×10−6 M) for 30 minutes attenuated VI-induced cAMP production (Figure 4A). At the lowest concentration of VI (10−12 M), cAMP production was reduced from 6.95±1.56 pmol/mL to 4.25±1.37 pmol/mL, and at the highest concentration of VI (10−6 M) cAMP production was reduced from 12.09±1.8 pmol/mL to 9.14±1.34 pmol/mL.
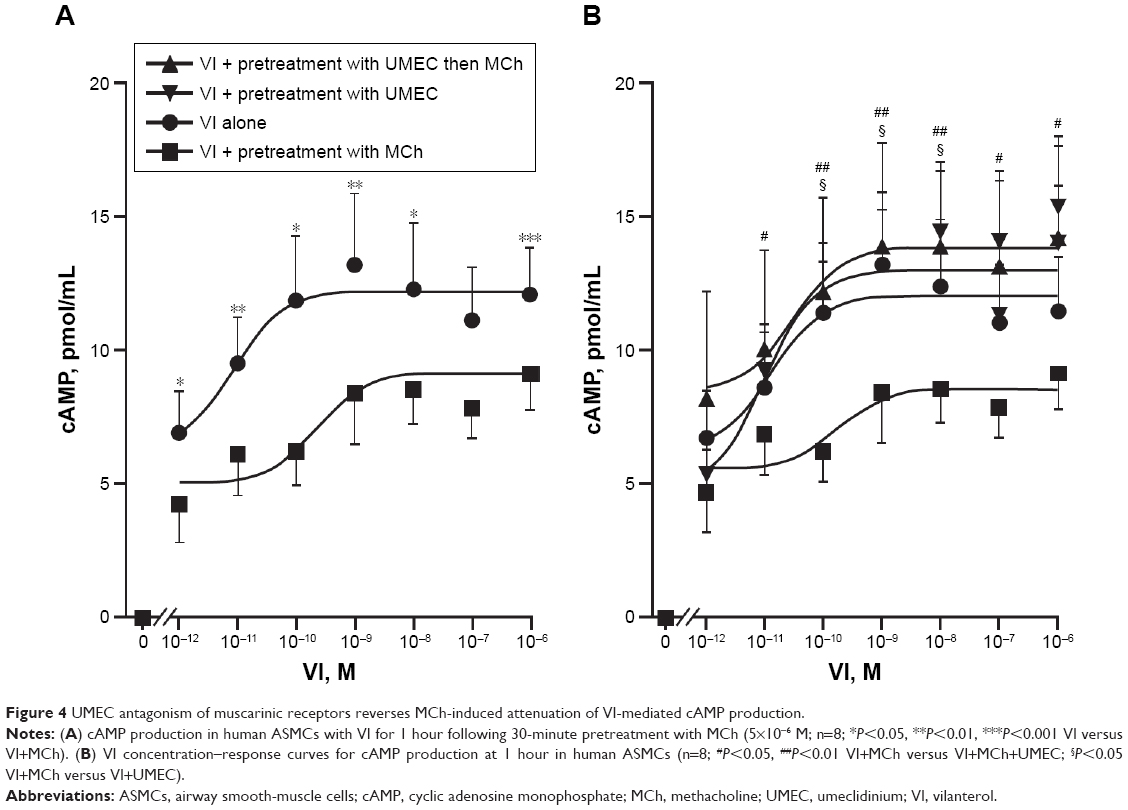 | Figure 4 UMEC antagonism of muscarinic receptors reverses MCh-induced attenuation of VI-mediated cAMP production. |
Pretreatment with UMEC (10−8 M) for 1 hour restored the magnitude of the VI-induced cAMP production at all concentrations of VI in the presence of MCh (Figure 4B). The EC50 and the maximum effect (Emax) elicited by the agonist for VI alone was 7.6×10−10 M and 12.1±1.8 pmol/mL, respectively; for UMEC + VI, EC50 and Emax were 2.6×10−10 M and 14.2±3.8 pmol/mL, respectively; for UMEC + VI + MCh, EC50 and Emax were 2×10−10 M and 12.4±2.04 pmol/mL, respectively. In the absence of MCh, pretreatment with UMEC did not enhance VI-induced cAMP production, and importantly did not result in a reduction in cAMP production. Neither UMEC nor MCh alone induced a detectable difference in cAMP production (data not shown).
Effect of M2- and M3-receptor antagonism (UMEC) on MCh-induced Ca2+ release
Stimulation of ASMCs with MCh (10−11–5×10−6 M) resulted in a concentration-dependent increase in [Ca2+]i (P<0.001, Kruskal–Wallis, Figure 5A). UMEC attenuated MCh-induced [Ca2+]i release in a concentration-dependent manner, with a maximum suppressive effect of 50% at a concentration of 10−6 M (Figure 5B). Attenuation of [Ca2+]i release was observed over the full range of MCh concentrations used (Figure 5C), reaching significance at 5×10−7 M (P<0.05) and 5×10−6 M (P<0.01).
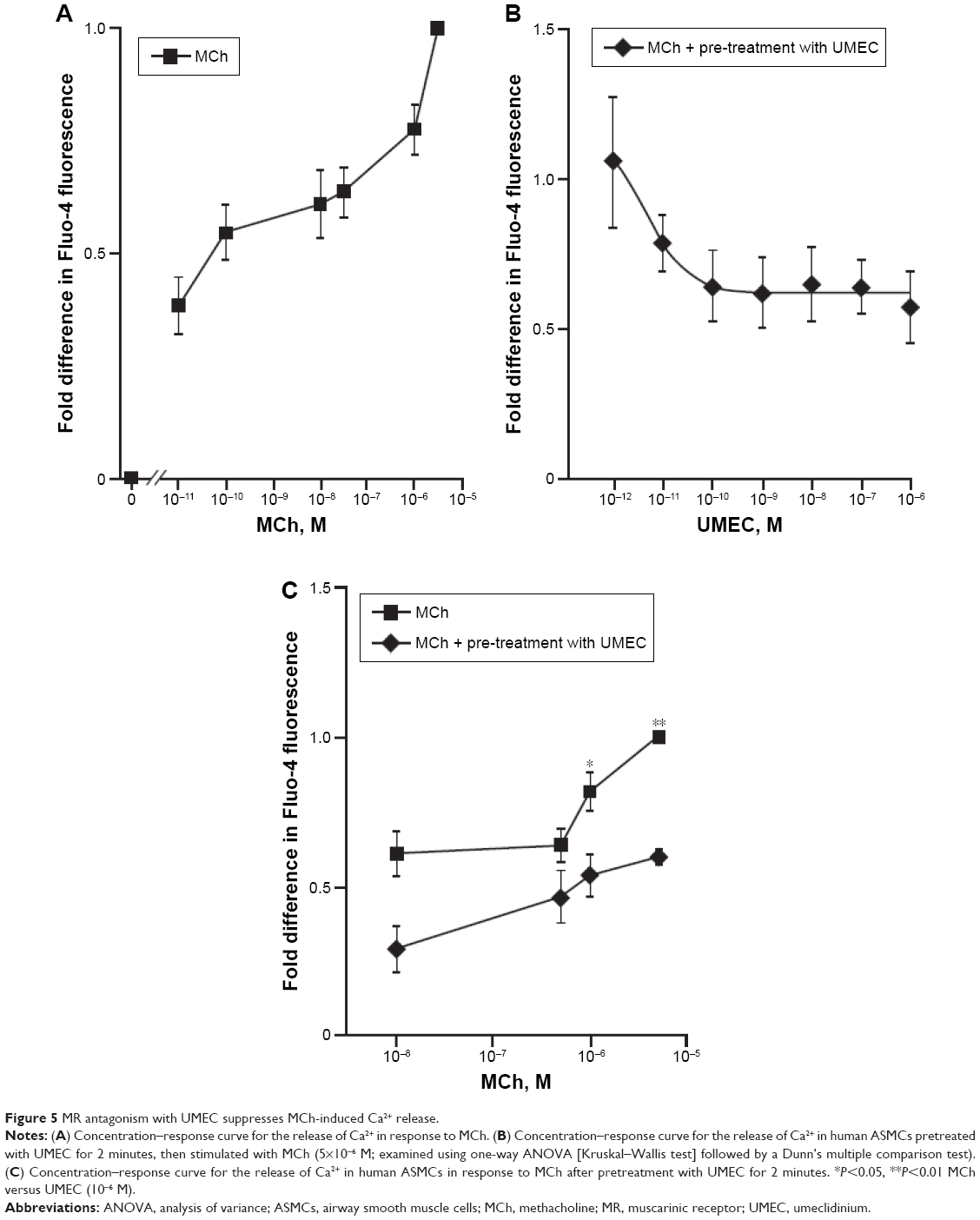 | Figure 5 MR antagonism with UMEC suppresses MCh-induced Ca2+ release. |
Effect of β2R activation (VI) on M2- and M3-receptor antagonist (UMEC)-mediated attenuation of MCh-activated Ca2+ release
Following the observation of the suppressive effect of M2- and M3-receptor antagonism on MCh-activated [Ca2+]i release, potential cross talk between the muscarinic and β2R-signaling pathways on attenuation of [Ca2+]i was investigated. UMEC (10−12–10−8 M) in combination with VI (10−10, 10−8, 10−6 M) reduced the [Ca2+]i release elicited by MCh to a greater extent than UMEC alone (Figure 6A and B). The inhibitory effect of UMEC alone at 10−10 M was achieved with a 100-fold reduction in concentration of UMEC (10−12 M) in the presence of even the lowest concentration of VI (10−10 M). The greatest enhancement in the suppressive effect of UMEC was observed at 10−6 M VI (P<0.01) (Figure 6B).
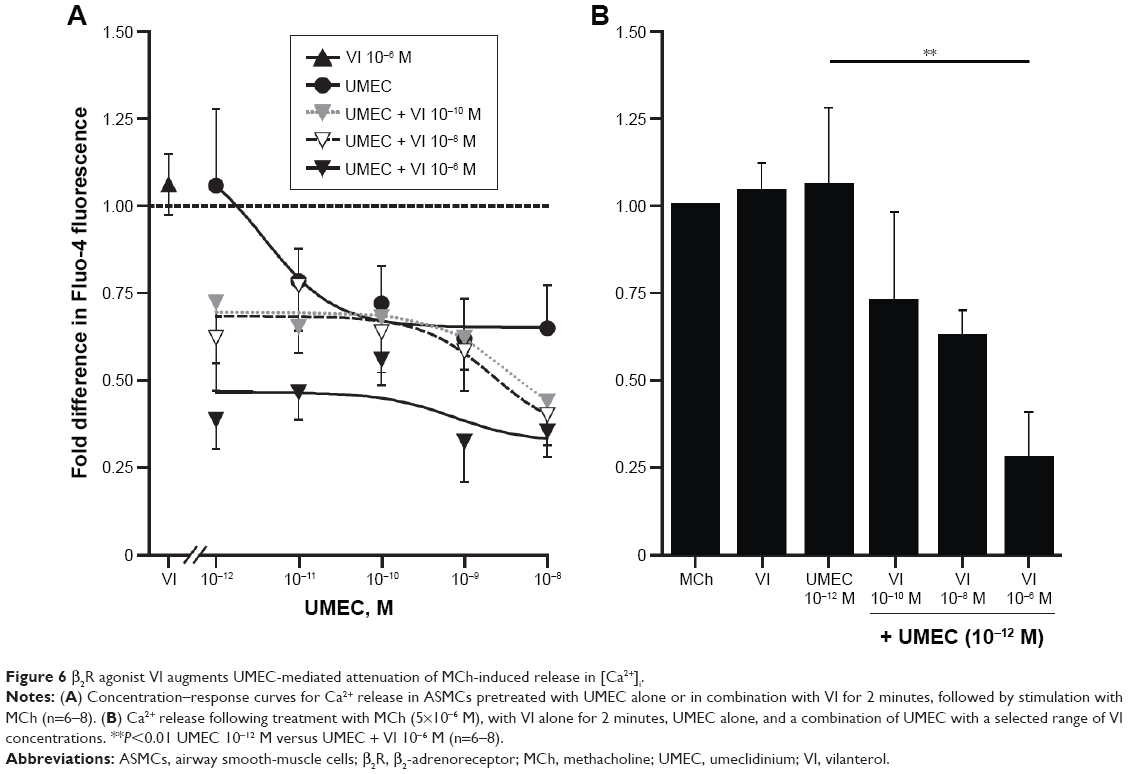 | Figure 6 β2R agonist VI augments UMEC-mediated attenuation of MCh-induced release in [Ca2+]i. |
RGS2 marker for possible β2R and MR interaction
VI induced expression of RGS2 mRNA, with maximal expression reached at approximately 60 minutes. This was maintained to 2 hours postinduction, but expression then declined to 50% of the maximum value at 4 hours (Figure 7). UMEC alone had no observed effect on RGS2 mRNA expression. However, the presence of UMEC-enhanced VI-induced RGS2 mRNA expression across all time points, reaching significance at 4 hours compared with VI alone (8.5±1.3- versus 3.2±0.7-fold increase compared with baseline, respectively; P<0.05).
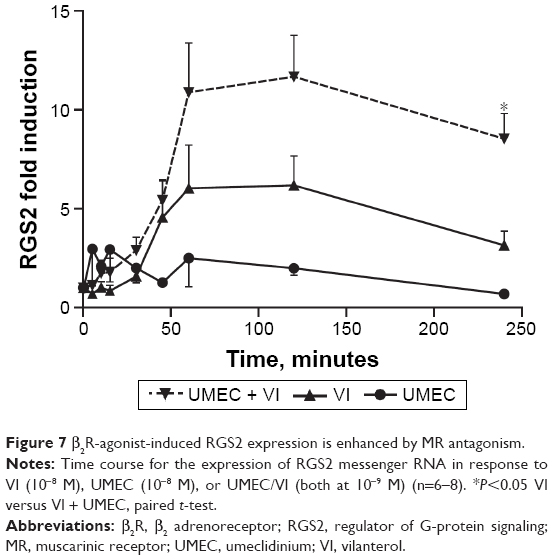 | Figure 7 β2R-agonist-induced RGS2 expression is enhanced by MR antagonism. |
Discussion
We investigated the intracellular effects of UMEC and VI at their sites of action in healthy human ASMCs to provide an insight into the possible mechanisms underlying improvement in lung function observed in patients with COPD receiving UMEC/VI treatment compared with its components UMEC and VI. Overall, results demonstrated that VI is a more effective inducer of cAMP production than salmeterol over the range of time points and concentrations studied, thus supporting previous research conducted in CHO cells.21 Antagonism of β2R suppressed the ability of VI to induce cAMP expression. We also demonstrated that MCh stimulation of ASMCs resulted in a concentration-dependent increase in [Ca2+]i, which was attenuated by pretreatment with UMEC. A major finding of this study was that attenuation of VI-induced cAMP production by MCh was counteracted by UMEC. Further, although pretreatment of ASMCs with VI alone had no effect on inhibiting MCh-induced [Ca2+]i release, pretreatment with UMEC in the presence of VI improved both the potency of UMEC (between 10- and 100-fold), and increased maximal inhibition. VI-induced RGS2 mRNA expression was also enhanced by UMEC, though UMEC alone did not affect RGS2 expression. Together, these data show that UMEC is a more potent inhibitor of the effects of MCh when in combination with VI versus UMEC monotherapy, and suggest that dual bronchodilators may work together to elicit bronchodilation via the upregulation of RGS2 and the augmentation of cAMP production.
VI is a LABA with a fast onset of activity and a long duration of action,26 resulting in bronchodilation for at least 24 hours.21 In human lung slices, VI has been shown to induce rapid reversal of cholinergic bronchoconstriction in vitro, reaching maximal bronchodilation in less than 15 minutes.21 This was quicker than salmeterol, supporting the results presented here, where VI induced more rapid cAMP production than salmeterol (Figure 2A).
While MR activation attenuated VI-induced cAMP production, pretreatment with UMEC restored the magnitude of the VI-induced cAMP production. This indicates that blocking of MR with UMEC prevents interaction with β2R, thus potentially enabling VI in the presence of MCh to elicit a biological response. VI-induced cAMP production was blocked by a specific β2R antagonist (ICI 118.551), indicating that VI acts through the β2R on ASMCs. This corresponds with a recent in vitro study, which demonstrated the high selectivity for β2R versus β1R of VI.27 Clinically, this finding would suggest that any transfer of VI from the lung into the circulatory system is unlikely to have negative side effects, eg, in the heart (cardiac myocytes), where the β1R predominates.28,29 This suggestion is supported by a previous randomized trial, in which no dose-related effects on blood pressure or pulse rate with inhaled VI were reported.30
The therapeutic benefit of such LAMAs as UMEC lies in their ability to counteract bronchoconstriction in response to vagal reflexes triggered by stimulation of airway sensory nerve endings and the subsequent release of acetylcholine (Figure 1).31,32 Studies in COPD also report heightened levels of acetylcholine release in patients with COPD; this is correlated with disease severity and an increase in M3-receptor expression and signaling, which enhance the bronchoconstrictive drive. The major mechanism by which MR antagonists, such as UMEC, affect bronchodilation is through blocking the action of acetylcholine at the M2 and M3 receptors on ASMCs, inhibiting bronchoconstriction and thereby leading to SM relaxation.
A key finding in this study was that the reduction in [Ca2+]i release elicited by MCh was greater with the UMEC–VI combination compared with UMEC alone. These data provide strong evidence for an interaction among the two signaling pathways, β2R agonism, and MR antagonism, and that β2R agonists have an effect on MR agonist-induced [Ca2+]i increase, but only in the presence of UMEC. Of note, the reduction in [Ca2+]i release was observed down to 10−12 M of UMEC.
In this study, UMEC was shown to enhance VI-induced RGS2 mRNA expression, while UMEC alone did not affect RGS2 expression. Acetylcholine is known to activate MAPK signaling,33 which influences gene transcription. UMEC may enhance the effects of β2R activation by inhibiting the activation of MR through reversing the effects of acetylcholine produced by ASMCs.34,35 Regulators of G-protein signaling proteins are GTPase-activating proteins that attenuate signaling by heterotrimeric Gq-coupled G-protein-coupled receptors, including muscarinic M1, M2, and M3 receptors.36,37 RGS2 is a highly potent and relatively selective inhibitor of Gαq function,38 which is coupled to both the M2 and M3 receptors. RGS2 activation/induction is known to block M3 receptor-mediated activation of phospholipase C and PKA, and hence airway SM contraction,36,39 and inhibit M2-receptor signaling.40 The latter could also reduce the negative impact of cholinergic activation of the M2 receptor on β2R-stimulated adenylyl cyclase activity (Figure 1). The RGS2 proximal gene promoter contains a conserved cAMP-response element that is critical for CREB binding and RGS2-promoter activation,41 and RGS2 gene transcription has been shown to be cAMP-dependent in ASMC.19 Moreover, LABAs and glucocorticoids can synergistically enhance RGS2 expression;42 evidence from our group shows VI can enhance the anti-inflammatory effects of the inhaled corticosteroid fluticasone furoate. In blood cells from patients with COPD and asthma, VI may act as a steroid-sparing agent (Khorasani et al, personal communication, 2016). Overall, these studies suggest the potential for triple therapy, where the addition of low-dose fluticasone furoate could improve the bronchodilatory effects of UMEC/VI by synergistically increasing VI-induced RGS2 expression. Future studies need to test the effect of adding an inhaled corticosteroid to a LAMA/LABA combination in ASMCs. Data also suggest that the potential synergy between UMEC and VI could allow for reduced therapeutic doses of UMEC/VI compared with corresponding doses of monotherapy, thereby reducing the risk of side effects associated with long-term usage of long-acting bronchodilators.
Limitations of this study include the different cell batches used, which may have introduced variations in the study data. Additionally, experiments in this study were conducted in ASMCs from healthy donor lungs, in order to provide a surrogate for studying tissue of patients affected by COPD or asthma. However, the use of healthy lung donor tissue may be perceived as a possible limitation, and it would be informative for future studies to be conducted in ASMCs from patients with COPD or asthma. A study by Lo et al (personal communication, 2016) showed that salmeterol can reduce the proliferation, myofibroblastic differentiation, and CC-chemokine receptor 7 expression of fibrocytes from healthy subjects and patients with nonsevere asthma, but not from patients with severe asthma. Moreover, fibrocytes from patients with severe asthma had lower baseline surface β2R expression, but were not insensitive to the direct effects of exogenous cAMP (8-Br-cAMP). This finding suggests an impairment at the level of β2R itself. It is possible that impairments in β2R or β2R signaling may lead to increased use of bronchodilator therapy in patients with severe COPD or asthma, though further studies would need to test this hypothesis.
With regard to the mechanisms involved in RGS2 expression, further experiments would be useful in confirming our hypothesis that UMEC+VI enhances RGS2 expression compared with VI alone. These experiments include Fluo4 fluorescence studies in RGS2-knockout ASMCs to determine whether the absence of RGS2 expression affects [Ca2+]i release, and real-time examination of the effect of UMEC, VI and UMEC/VI on [Ca2+]i release and RGS2 expression/activity in the presence and absence of β2R antagonists over the time frame of the cAMP-monitoring experiments (90 minutes). This would provide further evidence to determine whether RGS2 expression is directly affected by UMEC and VI. These experiments were beyond the scope of the current investigation, but should be considered for future studies.
In summary, we showed that in human ASMCs: 1) VI induces cAMP production from β2R more effectively than salmeterol, 2) cholinergic activation attenuates VI-induced cAMP production, 3) attenuation of VI-induced cAMP production by MCh is counteracted by UMEC, 4) the combination of UMEC+VI attenuated cholinergic agonist-induced [Ca2+]i release to a greater extent than UMEC alone, and 5) UMEC enhanced VI-induced RGS2 mRNA expression. The mechanism for the enhanced [Ca2+]i release with UMEC+VI compared with UMEC alone could lie in the increase in RGS2 mRNA expression, which inhibits Gq-coupled receptors. We thus provided some insight into the mechanisms of and rationale for the use of dual (LAMA/LABA) bronchodilators over their monotherapy components.
Acknowledgments
This study was funded by GlaxoSmithKline. Malcolm Johnson was a GlaxoSmithKline employee at the time of the study, but is now retired. Editorial assistance was provided by Matthew Robinson, DPhil, of Fishawack Indicia Ltd, UK.
Author contributions
All authors contributed toward drafting and revising the paper. NS, MJ, KFC, JHR, SW and PKB designed the research. NS and PKB performed the research. NS, MJ, DAH, JHR, SW and PKB analyzed the data. All authors agree to be accountable for all aspects of the work.
Disclosure
NS reports no conflicts of interest in this work. PKB received research support from GlaxoSmithKline. KFC has been on advisory boards for GlaxoSmithKline, Merck, and Gilead, and received research support from the National Institute for Health Research, Medical Research Council UK, Asthma UK, and the Wellcome Trust. DAH, JHR, and SW are employees of GlaxoSmithKline and hold GlaxoSmithKline stocks/shares. MJ is an ex-employee of GlaxoSmithKline and is now retired; he holds GlaxoSmithKline stocks/shares.
References
Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management and prevention of COPD. 2017. Available from: http://goldcopd.org/gold-2017-global-strategy-diagnosis-management-prevention-copd. Accessed April 7, 2017. | ||
Cazzola M, Segreti A, Matera MG. New developments in the combination treatment of COPD: focus on umeclidinium/vilanterol. Drug Des Devel Ther. 2013;7:1201–1208. | ||
Brusasco V. Reducing cholinergic constriction: the major reversible mechanism in COPD. Eur Respir Rev. 2006;15(99):32–36. | ||
Montuschi P, Ciabattoni G. Bronchodilating drugs for chronic obstructive pulmonary disease: current status and future trends. J Med Chem. 2015;58(10):4131-4164. | ||
van Nieuwstadt RA, Henricks PA, Hajer R, van der Meer WA, Breukink HJ, Nijkamp FP. Characterization of muscarinic receptors in equine tracheal smooth muscle in vitro. Vet Q. 1997;19(2):54–57. | ||
ANORO ELLIPTA™ [summary of product characteristics]. 2017. Available from: http://www.medicines.org.uk/emc/medicine/28949. Accessed April 7, 2017. | ||
ANORO ELLIPTA™ [highlights of prescribing information]. 2017. Available from: https://www.gsksource.com/pharma/content/dam/GlaxoSmithKline/US/en/Prescribing_Information/Anoro_Ellipta/pdf/ANORO-ELLIPTA-PI-MG.PDF. Accessed April 7, 2017. | ||
INCRUSE ELLIPTA™ [highlights of prescribing information]. 2016. Available from: https://www.gsksource.com/pharma/content/dam/GlaxoSmithKline/US/en/Prescribing_Information/Incruse_Ellipta/pdf/INCRUSE-ELLIPTA-PI-PIL.PDF. Accessed April 7, 2017. | ||
INCRUSE™ [summary of product characteristics]. 2017. Available from: https://www.medicines.org.uk/emc/medicine/29394. Accessed April 7, 2017. | ||
ANORO ELLIPTA™ [product monograph]. 2017. Available from: http://ca.gsk.com/media/588409/anoro-ellipta.pdf. Accessed April 7, 2017. | ||
INCRUSE ELLIPTA™ [product monograph]. 2017. Available from: http://ca.gsk.com/media/591190/incruse_ellipta.pdf. Accessed April 7, 2017. | ||
Donohue JF, Maleki-Yazdi MR, Kilbride S, Mehta R, Kalberg C, Church A. Efficacy and safety of once-daily umeclidinium/vilanterol 62.5/25 mcg in COPD. Respir Med. 2013;107(10):1538–1546. | ||
Decramer M, Anzueto A, Kerwin E, et al. Efficacy and safety of umeclidinium plus vilanterol versus tiotropium, vilanterol, or umeclidinium monotherapies over 24 weeks in patients with chronic obstructive pulmonary disease: results from two multicentre, blinded, randomised controlled trials. Lancet Respir Med. 2014;2(6):472–486. | ||
Wedzicha JA, Banerji D, Chapman KR, et al. Indacaterol-glycopyrronium versus salmeterol-fluticasone for COPD. N Engl J Med. 2016;374(23):2222–2234. | ||
Johnson M. Molecular mechanisms of β2-adrenergic receptor function, response, and regulation. J Allergy Clin Immunol. 2006;117(1):18–24. | ||
Widdop S, Daykin K, Hall IP. Expression of muscarinic M2 receptors in cultured human airway smooth muscle cells. Am J Respir Cell Mol Biol. 1993;9(5):541–546. | ||
Canning BJ, Fischer A. Neural regulation of airway smooth muscle tone. Respir Physiol. 2001;125(1–2):113–127. | ||
Heximer SP. RGS2-mediated regulation of Gqα. Methods Enzymol. 2004;390:65–82. | ||
Holden NS, Bell MJ, Rider CF, et al. β2-Adrenoceptor agonist-induced RGS2 expression is a genomic mechanism of bronchoprotection that is enhanced by glucocorticoids. Proc Natl Acad Sci U S A. 2011;108(49):19713–19718. | ||
Salmon M, Luttmann MA, Foley JJ, et al. Pharmacological characterization of GSK573719 (umeclidinium): a novel, long-acting, inhaled antagonist of the muscarinic cholinergic receptors for treatment of pulmonary diseases. J Pharmacol Exp Ther. 2013;345(2):260–270. | ||
Slack RJ, Barrett VJ, Morrison VS, et al. In vitro pharmacological characterization of vilanterol, a novel long-acting β2-adrenoceptor agonist with 24-hour duration of action. J Pharmacol Exp Ther. 2013;344(1):218–230. | ||
Malerba M, Radaeli A, Montuschi P, Morjaria JB. Vilanterol trifenatate for the treatment of COPD. Expert Rev Respir Med. 2016;10(7):719–731. | ||
Eskin SG, Sybers HD, Lester JW, Navarro LT, Gotto AM Jr, DeBakey ME. Human smooth muscle cells cultured from atherosclerotic plaques and uninvolved vessel wall. In Vitro. 1981;17(8):713–718. | ||
Kang HS, Moon YJ, Kim YY, et al. Smooth-muscle progenitor cells isolated from patients with moyamoya disease: novel experimental cell model. J Neurosurg. 2014;120(2):415–425. | ||
Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22(23):3099–3108. | ||
Procopiou PA, Barrett VJ, Bevan NJ, et al. Synthesis and structure-activity relationships of long-acting β2 adrenergic receptor agonists incorporating metabolic inactivation: an antedrug approach. J Med Chem. 2010;53(11):4522–4530. | ||
Aparici M, Gavaldà A, Ramos I, et al. In vitro and in vivo preclinical profile of abediterol (LAS100977), an inhaled long-acting β2-adrenoceptor agonist, compared with indacaterol, olodaterol and vilanterol. Eur J Pharmacol. 2016;770:61–69 | ||
Capote LA, Mendez Perez R, Lymperopoulos A. GPCR signaling and cardiac function. Eur J Pharmacol. 2015;763(Pt B):143–148. | ||
Lymperopoulos A, Rengo G, Koch WJ. Adrenergic nervous system in heart failure: pathophysiology and therapy. Circ Res. 2013;113(6):739–753. | ||
Hanania NA, Feldman G, Zachgo W, et al. The efficacy and safety of the novel long-acting β2 agonist vilanterol in patients with COPD: a randomized placebo-controlled trial. Chest. 2012;142(1):119–127. | ||
Coleridge HM, Coleridge JC, Schultz HD. Afferent pathways involved in reflex regulation of airway smooth muscle. Pharmacol Ther. 1989;42(1):1–63. | ||
Groneberg DA, Quarcoo D, Frossard N, Fischer A. Neurogenic mechanisms in bronchial inflammatory diseases. Allergy. 2004;59(11):1139–1152. | ||
Jiménez E, Gámez MI, Bragado MJ, Montiel M. Muscarinic activation of mitogen-activated protein kinase in rat thyroid epithelial cells. Cell Signal. 2002;14(8):665–672. | ||
Profita M, Albano GD, Riccobono L, et al. Increased levels of Th17 cells are associated with non-neuronal acetylcholine in COPD patients. Immunobiology. 2014;219(5):392–401. | ||
Pera T, Penn RB. Crosstalk between β-2-adrenoceptor and muscarinic acetylcholine receptors in the airway. Curr Opin Pharmacol. 2014;16:72–81. | ||
Tovey SC, Willars GB. Single-cell imaging of intracellular Ca2+ and phospholipase C activity reveals that RGS 2, 3, and 4 differentially regulate signaling via the Gαq/11-linked muscarinic M3 receptor. Mol Pharmacol. 2004;66(6):1453–1464. | ||
Bernstein LS, Ramineni S, Hague C, et al. RGS2 binds directly and selectively to the M1 muscarinic acetylcholine receptor third intracellular loop to modulate Gq/11α signaling. J Biol Chem. 2004;279(20):21248–21256. | ||
Heximer SP, Watson N, Linder ME, Blumer KJ, Hepler JR. RGS2/G0S8 is a selective inhibitor of Gqαfunction. Proc Natl Acad Sci U S A. 1997;94(26):14389–14393. | ||
Tovey SC, Taylor CW. Cyclic AMP directs inositol (1,4,5)-trisphosphate-evoked Ca2+ signalling to different intracellular Ca2+ stores. J Cell Sci. 2013;126(Pt 10):2305–2313. | ||
Mark MD, Wittemann S, Herlitze S. G protein modulation of recombinant P/Q-type calcium channels by regulators of G protein signalling proteins. J Physiol. 2000;528(Pt 1):65–77. | ||
Xie Z, Liu D, Liu S, et al. Identification of a cAMP-response element in the regulator of G-protein signaling-2 (RGS2) promoter as a key Cis-regulatory element for RGS2 transcriptional regulation by angiotensin II in cultured vascular smooth muscles. J Biol Chem. 2011;286(52):44646–44658. | ||
Holden NS, George T, Rider CF, et al. Induction of regulator of G-protein signaling 2 expression by long-acting β2-adrenoceptor agonists and glucocorticoids in human airway epithelial cells. J Pharmacol Exp Ther. 2014;348(1):12–24. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.