Back to Journals » Infection and Drug Resistance » Volume 13
Intestinal Microbiota in Elderly Inpatients with Clostridioides difficile Infection
Authors Vakili B, Fateh A ![]() , Asadzadeh Aghdaei H
, Asadzadeh Aghdaei H ![]() , Sotoodehnejadnematalahi F
, Sotoodehnejadnematalahi F ![]() , Siadat SD
, Siadat SD
Received 9 May 2020
Accepted for publication 21 July 2020
Published 5 August 2020 Volume 2020:13 Pages 2723—2731
DOI https://doi.org/10.2147/IDR.S262019
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Bahareh Vakili,1 Abolfazl Fateh,2 Hamid Asadzadeh Aghdaei,3 Fattah Sotoodehnejadnematalahi,1 Seyed Davar Siadat2
1Department of Biology, School of Basic Science, Science and Research Branch, Islamic Azad University, Tehran, Iran; 2Department of Mycobacteriology and Pulmonary Research, Microbiology Research Center (MRC), Pasteur Institute of Iran, Tehran, Iran; 3Basic and Molecular Epidemiology of Gastrointestinal Disorders Research Center, Research Institute for Gastroenterology and Liver Diseases, Shahid Beheshti University of Medical Sciences, Tehran, Iran
Correspondence: Seyed Davar Siadat Tel +98 2164112282
Fax +98 2166465132
Email [email protected]
Purpose: The incidence of Clostridioides difficile infection (CDI) has been reported as 10-fold higher among the elderly population than in young adults. The aim of this study was to compare the targeted bacteria population in the fecal microbiota in two groups of hospitalized elderly, categorized according to CDI and non-CDI.
Patient and Methods: In this case–control study, 84 fecal samples of the 28 patients with CDI and 56 non-CDI patients (> 65 years) were studied. C. difficile isolates were characterized by anaerobic culture and multiplex PCR. Quantitative PCR was used to analyze the bacterial elements.
Results: CDI group differed significantly for a prolonged hospital stay, previous surgery, residence in nursing home and exposure to a range of antibiotics including quinolone, clindamycin and cephalosporin. CDI group had significantly fewer members of Bacteroides spp., Clostridium cluster IV, Bifidobacterium spp., Faecalibacterium prausnitzii, and Prevotella spp. in their fecal microbiota than the control group (P < 0.05). The abundances of Akkermansia muciniphila, Lactobacillus spp., Escherichia coli and Klebsiella spp. were higher in group CDI compared with the control group (P < 0.05).
Conclusion: CDI status is associated with the abundance of some bacterial populations. In this study, an increase in Akkermansia muciniphila, Lactobacillus spp., and Enterobacteriaceae genus was highlighted in CDI patients. A reduction in butyrate-producing bacteria was found in CDI patients. The differences in the composition of fecal microbiota can help to understand how antimicrobial agents impact on gut homeostasis and lead to loss of colonization resistance to C. difficile.
Keywords: Clostridioides difficile, risk factors, elderly, fecal microbiota
Introduction
Clostridioides difficile (C. difficile) is one of the frequent reasons of nosocomial diarrhea and being recognized as the leading cause of gastrointestinal infections worldwide, with 70–80% of C. difficile infections (CDIs) occurring in elderly hospitalized patients.1,2 The incidence of CDI has been reported as 10-fold higher among the elderly population than in young adults and death rates of CDI in patients over 80 years of age may exceed 20%.3,4 It can lead to readmission, significant increase in time, hospital stay and recurrences of infection.5,6 Elderly people are more common in hospitals and may be at an increased risk for CDI due to reduced immune status, experience more severe diseases and increased exposure to antibiotics.6,7 In fact, antibiotic treatment alters the composition of the gut microbiota that leads to the loss of colonization resistance to opportunistic bacteria, including C. difficile in this environment.3,8 The histopathologic damage is caused by C. difficile virulence factors, mainly toxins A (tcdA) and toxin B (tcdB) and the binary toxin (cdtA, and cdtB).1,9,10 Fecal microbiota transplantation (FMT) could be used as an effective therapy in managing patients with multiple recurrences of CDI and repeated antibiotic treatment.4,11 FMT is an emerging therapy for CDI that offers the potential for rapid and lasting elimination of CDI by the restoration of healthy microbiota especially in older patient populations.4 The elderly microbiota composition is distinct from that of young adults and marked by an unusual bacterial distribution associated with diet, age and hospital stay.3,8 Identification of the dominant bacterial species associated with CDI is important to show the disturbances of gut microbiota, which lead to C. difficile colonization and infection as the important cause of nosocomial diarrhea3,10 Several advanced molecular methods can be used to identify the microbial communities in the high-risk population of elderly hospitalized subjects.7,8,10 Next-generation sequencing technologies have provided invaluable data on the size and composition of intestine microbial. These sensitive methods are comparatively expensive and need skilled bioinformatics analysis for interpretation of the data.12,13 On the other side, quantitative polymerase chain reaction (qPCR) methods are fast cost-effective and reliable culture-independent techniques that have detected changes in the intestinal microbiome associated with antibiotic treatment or other diseases.13 This case–control study aimed to evaluate the targeted bacteria population in the fecal microbiota between two groups of hospitalized elderly, categorized according to CDI and non-CDI by using real-time qPCR. It is necessary for a better understanding of the alteration of the intestinal microbiome lead to CDI and providing potential targets for prevention and treatment.
Materials and Methods
Study Population
At the Internal Medicine and Critical Care Unit of Isfahan University Hospitals, Iran, we enrolled two groups of elderly (≥65 years old) patients hospitalized from August 2018 to June 2019. In this case–control study, the patients admitted to different wards have been included a wide range of acute diseases (Table 1). Each patient signed a written approved consent form before their stool being collected for use in this study. CDI group included 28 patients between 68 and 84 years old, exposed to antibiotic treatment during their hospitalization, and had been diagnosed with CDI at the moment of stool collection (Table 1). Non-CDI group was composed of 56 subjects without diarrhea or other symptoms of CDI, admitted for extra-intestinal illnesses and were undergoing antimicrobial therapy during their hospital stay. Faecal samples were routinely collected in plastic containers and shipped to the laboratory of Infectious Diseases Research Center within 24 h, where they have been immediately frozen at −80°C until further processing.
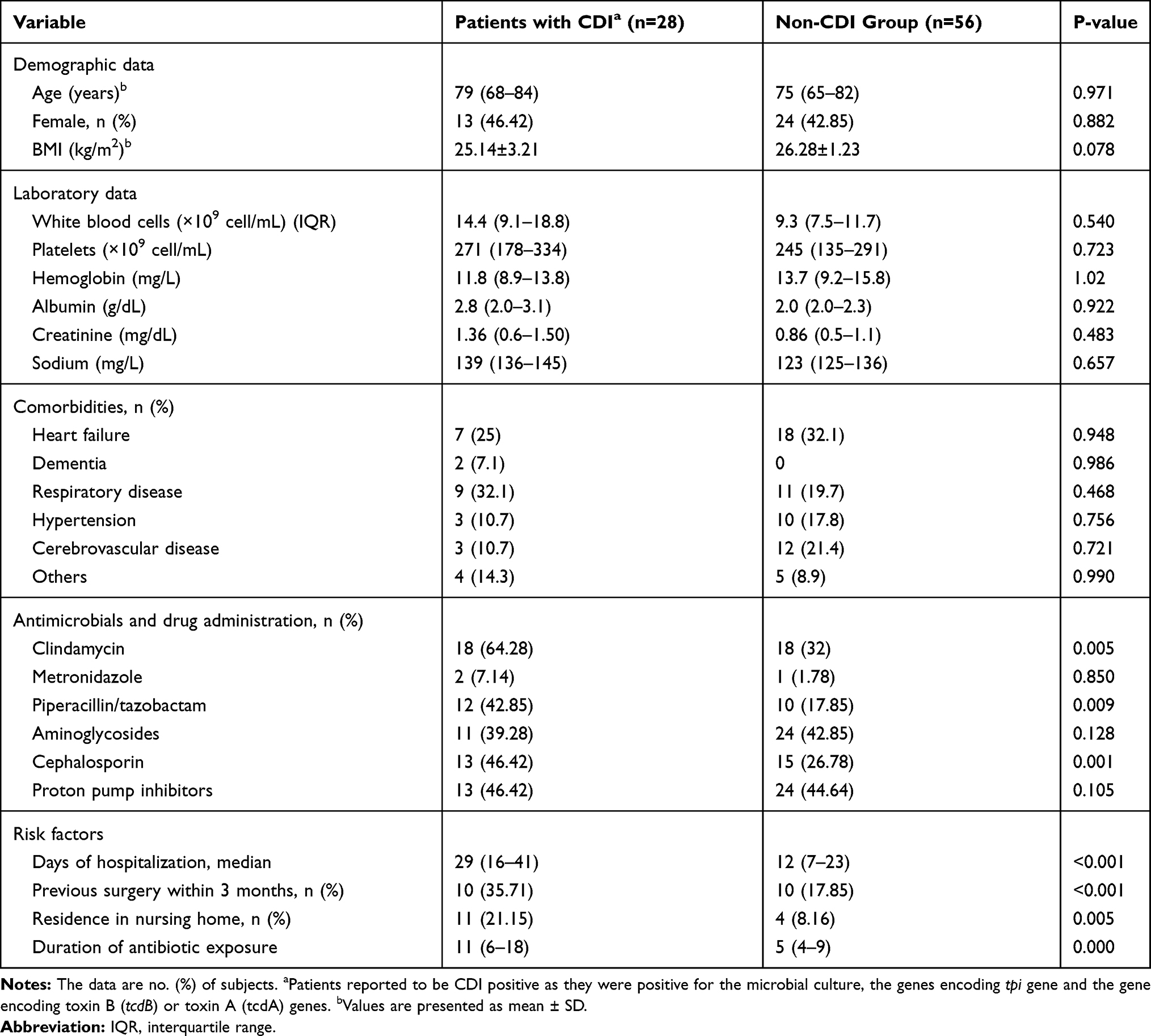 |
Table 1 Clinical Characteristics of Enrolled Subjects, Hospitalized Patients with CDI and Non-CDI Group |
Detection of Toxigenic C. difficile Isolates
Fecal samples from all subjects were inoculated into the selective medium C. difficile moxalactam norfloxacin (CDMN) broth and incubated in anaerobic jars for 48 hours at 37°C (80% N2, 10% CO2 and 10% H2). Alcohol shock treatment was performed and the pellet was inoculated into the CDMN agar and incubated anaerobically for 48 h at 37C°. Characteristic yellow colonies grown on this medium and L-proline aminopeptidase positive were identified as C. difficile.14,15
DNA extraction was performed with the procedure presented in the study by Pitcher et al.16,17 Polymerase chain reaction (PCR) targeting the species-specific triose phosphate isomerase gene, tpi to verify the presence of C. difficile in stool cultures. The presence of the genes encoding toxin A and B (tcd A and tcd B) and binary toxin (cdt A, cdt B) were performed using multiplex PCR method as described by Stubbs et al and Lemee et al.14,18 C. difficile, ribotype 027 was used as positive control in molecular and microbiological analysis.9
Antimicrobial Susceptibility Tests
Antimicrobial susceptibility testing to eight antibiotics was determined using disk diffusion method for isolated C. difficile strains. Antimicrobial disks were purchased from Padtan-teb Co. (Tehran, Iran) and Rosco Diagnostica A/S (Taastrup, Denmark) and interpretation of results were performed according to the guidelines of the Clinical and Laboratory Standards Institute (CLSI) 2012 and the European Committee on Antimicrobial Susceptibility Testing (EUCAST) (Table 2). In addition, E-tests (LiofilchemR, Italy) determined Minimum inhibitory concentrations (MICs) of vancomycin and metronidazole. Interpretation of the results and determination of the MICs were carried out according to the EUCAST guideline (Table 3). All tests were performed on Brucella Blood Agar containing vitamin K1, haemin, and defibrinated sheep red blood cells, and the antimicrobial agents tested were selected because of the emergence of reduced susceptibility.15 Streptococcus sp. MTCC 689 and Clostridium perfringens MTCC 13,124 strains were selected as controls.
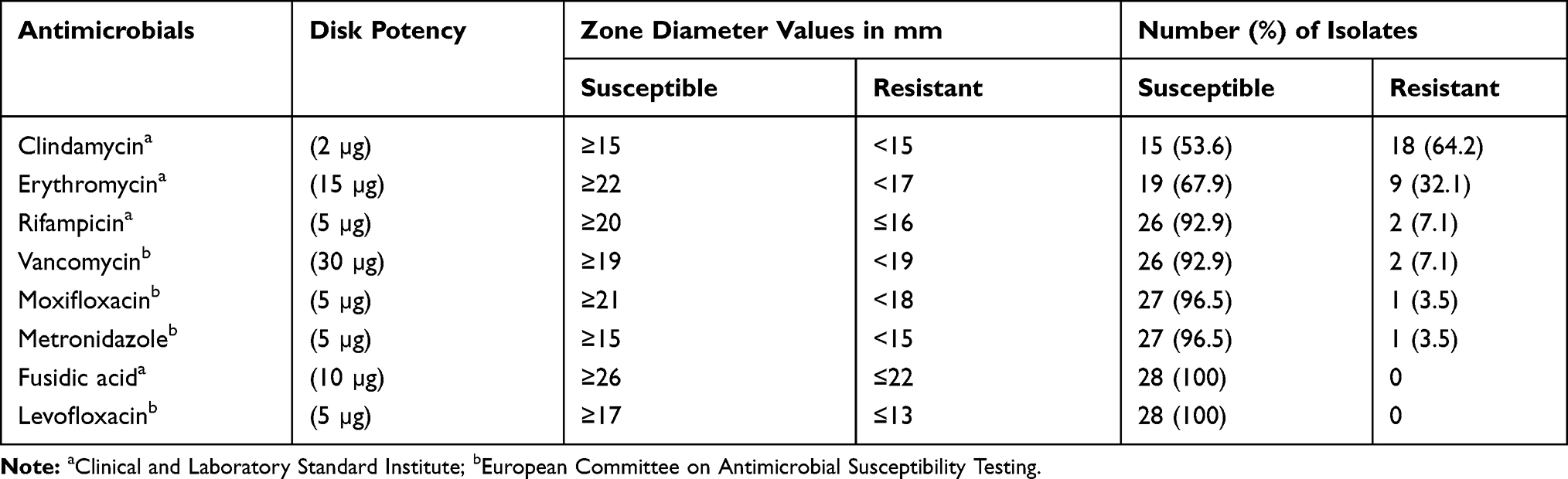 |
Table 2 Antimicrobial Susceptibility of 28 Toxigenic C. difficile Isolates |
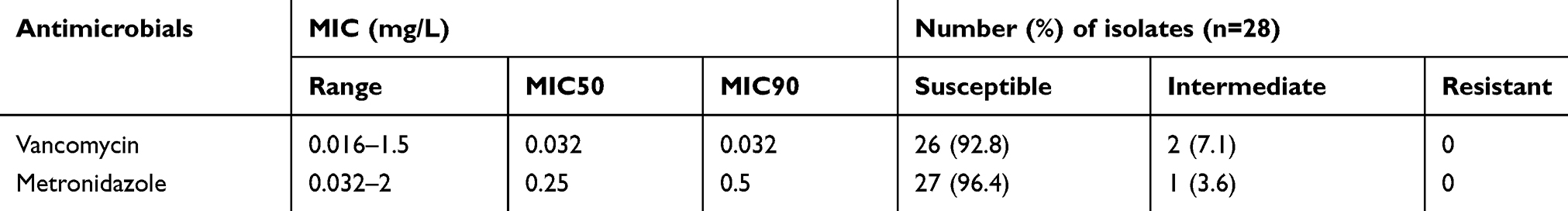 |
Table 3 Minimum Inhibitory Concentrations of 28 Toxigenic Isolates to Vancomycin and Metronidazole |
DNA Extraction
Briefly, 220 mg of frozen stool sample was treated with ASL buffer and heated to 95°C for 5 min to obtain bacterial lysis. DNA was extracted with the QIAamp DNA Stool Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions.12 DNA concentration was quantified using a QubitTM 4Florometer (Life Technologies, Invitrogen, Singapore). Fecal DNA samples were adjusted to equal concentrations and 50 ng of faecal DNA was used for qPCR analysis.
SYBR Green q-PCR
The SYBR® Green-assay was carried out in a Rotor-Gene 6000 real-time PCR cycler (Qiagen Corbett, Hilden, Germany) using the QuantiTect SYBR® Green PCR kit. The genes encoding 16S rRNA from specific bacterial groups were amplified using qPCR master mix (Yekta Tajhiz Azma, Tehran, Iran). All primers and annealing temperatures are detailed in Supplementary Table S1.
Samples were run in duplicate in a volume of 25 μL containing 1 × SYBR green qPCR master mix, 0.5 μM of each primer and 50 ng of purified fecal DNA. The PCR program for bacteria consisted of one cycle at 95 °C for 15 min, 40 cycles of denaturation at 95 °C for 1 min, 30 s at the appropriate annealing temperature (Table 2), and 72°C for 1 min followed by a final extension step at 72 °C for 5 min.
Following the amplification, a melting temperature analysis of PCR products was performed to determine the specificity of the PCR.
Quantification was done using melt curve analysis obtained from continuous fluorescence measurement along with slow heating at 0.1 °C/s from 72 °C to 95 °C. The copy number of 16SrRNA gene operons of targeted bacteria in crude DNA templates was determined against serially diluted plasmid DNA standards. The appropriate set of standards melting curve analysis of the PCR products was conducted following each assay to confirm that the fluorescence signal originated from specific PCR products and not from primer-dimers or other artifacts. In each run, a non-amplification control (NTC) which did not contain any DNA template. The standard curve was constructed for assessment of the number of different bacterial groups present in each sample using serially diluted bacterial genomic DNA extracted from the reference strains of a pure culture of targeted bacterial groups. The bacterial concentration from each fecal sample was calculated from the threshold cycle values (Ct) acquired from the standard curve and expressed as the number of bacteria per gram feces.19–21
Statistical Analysis
Statistical analysis was performed using SPSS version 21.0 software (SPSS Inc. Chicago, IL, USA). Clinical characteristics of subjects were expressed as the means ± SD. The qPCR results were graphically presented by box plots. To compare the means of different variables between the study groups, an independent sample t-test was used.
For continuous variables (eg, age and weight), one-way analysis of variance was utilized. For categorical variables, Pearson’s chi-square was performed. P <0.05 was regarded as statistically significant.
Results
Eighthly four fecal samples of the 28 patients with CDI and 56 healthy controls were studied. All specimens introduced in this study were from subjects older than 65 years, with the same proportion of females and males in both the healthy (average of 75 years of age) and CDI groups (average of 79 years of age), to minimize differences due to age or gender. Table 1 summarizes the clinical characteristics of these subjects; correlations between both groups were analyzed using the Mann–Whitney U-test. There were no significant differences between age, gender, BMI, metronidazole, aminoglycosides or proton pump inhibitors (PPI) treatment. They differed significantly for a prolonged hospital stay, previous surgery, residence in nursing home and exposure to a range of antibiotics including quinolone, clindamycin or cephalosporin. Nearly all of the C. difficile strains were susceptible to metronidazole, vancomycin, fusidic acid and levofloxacin. Clindamycin and erythromycin were the most resistant antibiotics (64.4% and 31.2%, respectively) (Tables 2 and 3).
To quantify the total bacterial DNA targeted bacterial sub-populations in all subjects, qPCR was used to analyze the original DNA samples using bacterial species-specific primers. qPCR data were expressed as copies per gram total microbial DNA as seen in Table 4 and Figure 1. CDI group had significantly fewer members of Bacteroides spp., Clostridium cluster IV, Bifidobacterium spp., Faecalibacterium prausnitzii, and Prevotella spp. in their fecal microbiota than the control group (P < 0.05). The abundances of Akkermansia muciniphila, Lactobacillus spp., Escherichia coli, and Klebsiella spp. were higher in group CDI compared with the control group (P < 0.05) (Figure 2). The differences of intestinal bacterial genera in CDI Patients were represented in Supplementary Figure S1.
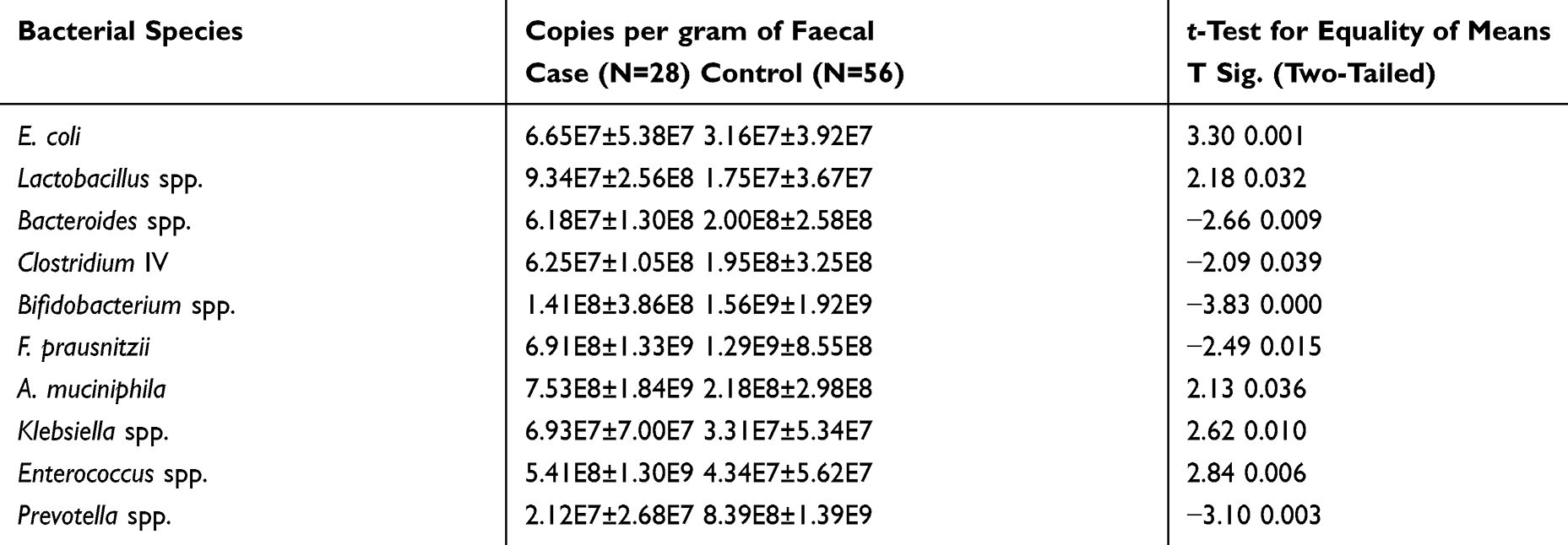 |
Table 4 Comparison of Population Numbers of Selected Microbial Groups in Case (CDI Patients) and Control (Hospitalized Patients without Diarrhea) Studied Groups Participated in This Study |
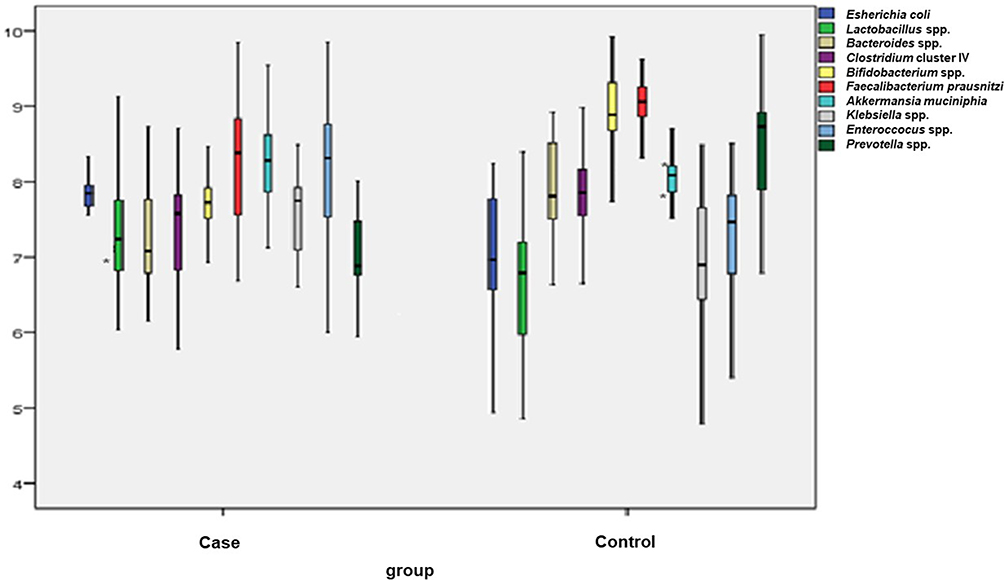 |
Figure 1 Box-and-whisker plots of bacterial groups quantified by qPCR. Notes: Bacterial groups quantified by SYBR Green qPCR and expressed as Log10 bacteria per gram stool in hospitalized patients with CDI and non-CDI group. Outlier point were shown by *. |
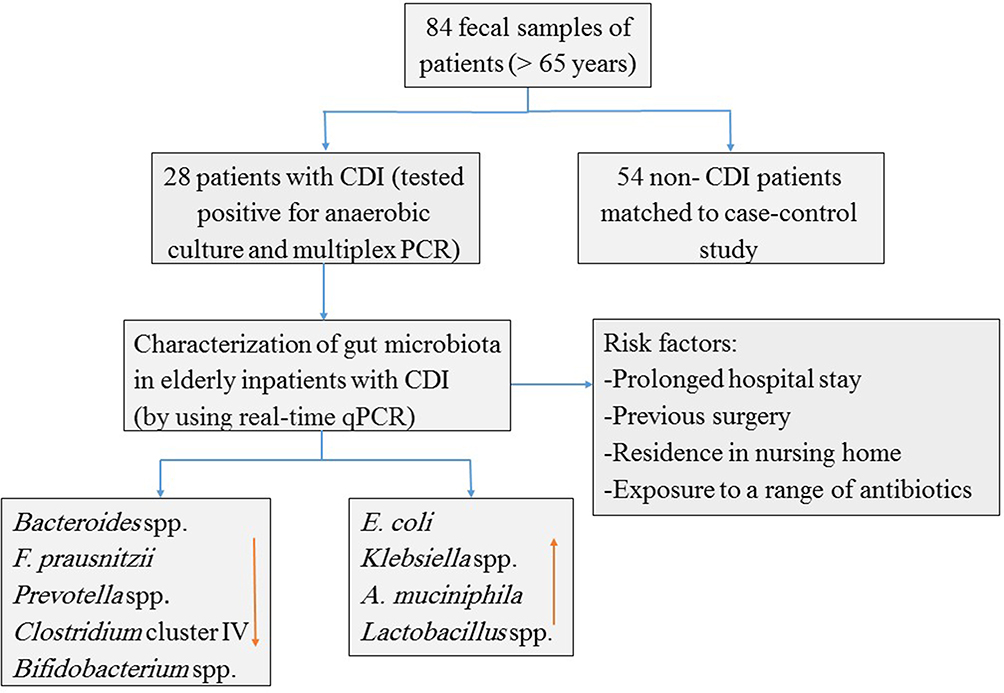 |
Figure 2 Flow diagram illustrating the number of patients and their gut microbiota patent. |
Discussion
CDI is regarded as a major health problem throughout the world because of its critical role in increased morbidity and mortality. A recent study in Iran showed that significant risk factors for CDI were advanced age, undergoing chemotherapy, previous surgery, and residence in the nursing home.9 C. difficile colonization has been reported as 10 times higher in elderly patients than in the general population living outside long-term care facilities.22 We have applied targeted qPCR method for assessment of each predominant genus of commensal and pathogenic organisms in gut microbiota separately. A previous study on elderly and CDI showed that there was little difference regarding the microbiota composition between CDI subjects and asymptomatic C. difficile carriers and a minor bacterial taxon showed a statistical difference between CDI patients and non-CDI individuals.7
The 16S rRNA profiling has been performed on a limited case-study of C. difficile negative and positive individuals. In accordance with previous studies, we found that in these elderly patients, C. difficile colonization was associated with more significant differences in microbiota between both groups.7,8
We could focus on previous antibiotic use during the survey and our results showed that prolonged hospital stay, previous surgery, residence in the nursing home, and exposure to a range of antibiotics (quinolone, clindamycin, or cephalosporin) were clinical risk factors for CDI. However, other similar studies revealed that these risk factors did not significantly differ between CDI and non-CDI patients with diarrhea.23–25 The discriminatory power of stool testing and the limited number of studied populations may be the cause of this difference.
Nearly all identified C. difficile strains in our study showed susceptibility to vancomycin and metronidazole. Full susceptibility of C. difficile isolates to these antibiotics has been reported in other studies.9,26
In the current study, C. difficile isolates showed the highest antibiotic resistance rates to clindamycin and erythromycin. Lower susceptibility to metronidazole in recent studies may be due to differences in the number of investigated isolates and excessive use of this antibiotic to treat CDI.27
Circulating strains in different regions have the potential to extend worldwide and genotyping of strains is important in the identification of epidemic, hypervirulent genotypes, and the relations between them.9 Our findings were in accordance with similar studies in Asia revealed that toxigenic C. difficile strains were susceptible to vancomycin and metronidazole as the most common choices for CDI treatment.9
Antibiotics induce short- and long-term changes in the composition of gut microbiota which persisted 6 months to 2 years after treatment.10,11 Systemic antimicrobial therapy caused a decrease in specific taxa of gut microbiota diversity and disturbances regardless of the research technique used including a decrease of the Bacteroides and Firmicutes phylae together with an overgrowth of Enterobacteriaceae.3,10,11 Following the use of quinolones, recovering the initial composition of microbiota diversity can be incomplete. Clindamycin, penicillins, cephalosporins, and fluoroquinolones are the classes that are more frequently associated with CDI, such as a Cephalosporin which leads to a decrease in Clostridia, Lactobacilli, and Bifidobacteria.11 Clindamycin leads to a decrease in total anaerobic bacteria, Bifidobacterium, Clostridium, and some species in the Bacteroides genera.8,11,28 Studies suggest a probable association between fluoroquinolone use, age over 65 years, and PPI use with a higher risk of CDI.1 The link between using PPI and CDI was not found in our study; however, a recent metagenomics study has compared the gut microbiota of patients who had been using PPI for more than 5 years revealed a decrease in Bacteroidetes and an increase in Firmicutes phylae while in another study by Faleck et al, no changes in gut diversity were observed.29,30 A significant alteration of gut microbiota was described during CDI along with a decreased number of bacterial genera, ie, Bactericides, Prevotella, and Bifidobacteria, with increased numbers of facultative species, ie, Clostridium and Lactobacillus spp. and our results were similar to these findings. We also observed decreased numbers of Clostridium cluster IV in patients with CDI that were in agreement with Antharam et al study.6,22,31
In a model of CDI in aged mice compared to young mice, the severity of the disease, relapse, and mortality was increased and recovery from infection delayed.5 Various animal models showed more complexes including changes in immune responses and gut microbiota.32 These aged mice generated lower serum levels of anti–C. difficile toxin A immunoglobulin M and immunoglobulin G, and had altered microbiota structure5 The specific alterations in the gut microbiota of the treated mice with antibiotics have been shown the loss of colonization resistance to C. difficile, an increase in the Firmicutes and Proteobacteria, and more specifically in the Lactobacillaceae and Pseudomonadaceae families.32
Some other bacterial groups could be associated with a CDI or non-CDI status, which depends on the other microorganisms present in the microbiota.2 Loss of Bacteroidales may be a biomarker for C. difficile positive status and worsening the clinical prognosis.25 Patients with CDI revealed higher abundances of Lactobacillus spp., Escherichia coli, and Klebsiella spp. which were in agreement with those of other studies in which patients with CDI revealed an enriched Lactobacillus profile in the gut and the overrepresentation of opportunistic pathogens, such as Klebsiella and members of Enterobacteriaceae, may clearly express a blooming phenomenon, because of a reduced ecological niche competition.3,8 Our subjects had high rates of prior antimicrobial treatment which resulted in an abundance of the members of Enterobacteriaceae family, including Escherichia and Klebsiella. These common opportunistic pathogens become dominant when the normal gut microbiota is disturbed by antibiotics. The link between Akkermansia and C. difficile infection was found within some of the CDI samples. The relative overrepresentation of Akkermansia spp. may reflect gut inflammation with increased enteric mucous production.8,33
CDI group had significantly fewer members of Bacteroides spp., Clostridium cluster IV, Bifidobacterium spp., F. prausnitzii, and Prevotella spp. in their fecal microbiota than non-CDI group.
Besides, in the present study, the levels of F. prausnitzii were lower in the CDI group than the control group. The levels of F. prausnitzii and Bifidobacterium are reduced in patients with chronic gut inflammation, and F. prausnitzii, in particular, has been shown to exert anti-inflammatory effects on inflammatory bowel disease models of colitis.34 Microbiome disturbance, such as prolonged antibiotic treatment could overwhelm the homeostasis maintenance and protection against CDI. Specific dysbiosis in the microbiota of the elderly populations resulting in increased susceptibility to opportunistic pathogens like C. difficile.8
The relatively low number of patients, dietary regimen, and the clinical complexity of patients were the major limitations of this study which can certainly influence comparisons between CDI and non-CDI groups. Further studies on the elderly population needed to improve our knowledge of the CDI. To our knowledge, this is the first study exploring the composition of fecal microbiota using a qPCR approach in a hospitalized elderly population with CDI.
Conclusions
Changes were observed in C. difficile positive individuals in the abundance of some bacterial populations, including Akkermansia muciniphila, Lactobacillus spp., Escherichia coli, and Klebsiella spp. were higher in CDI patients than in non-CDI patients. A decrease of the members of Bacteroides spp., Clostridium cluster IV, Bifidobacterium spp., Faecalibacterium prausnitzii, and Prevotella spp. was revealed in the CDI group. This may indicate that the effects of antibiotic treatment and CDI leading to a significant difference between the intestinal microbiota of these two groups, but further studies are still needed for better understanding the alteration of intestinal microbiome lead to CDI and providing potential targets for prevention and treatment.
Data Sharing Statement
Data generated or analyzed during this study are available from the corresponding author and some are included in this article. (Gel pictures of different PCRs are available).
Ethical Approval and Consent to Participate
The present study was approved by the human research ethics committee of Isfahan University of Medical Sciences (grant number 293347). All individuals agreed to participate in the study and gave their informed consent. All assessments were performed following the principles of the Declaration of Helsinki.
Acknowledgments
The authors would like to express their gratitude to Dr Parisa Shoaei and Dr Bahram Bagherpour for their special cooperation in our study. We are thankful to the staff of Isfahan University Training Hospitals and Infectious Disease Research Center, Isfahan, University of Medical Sciences, Isfahan, Iran for their great help in performing tests.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors have no conflicts of interest to declare regarding this paper.
References
1. Skraban J, Dzeroski S, Zenko B, Mongus D, Gangl S, Rupnik M. Gut microbiota patterns associated with colonization of different Clostridium difficile ribotypes. PLoS One. 2013;8(2):e58005. doi:10.1371/journal.pone.0058005
2. Asempa TE, Nicolau DP. Clostridium difficile infection in the elderly: an update on management. Clin Interv Aging. 2017;12:1799. doi:10.2147/CIA.S149089
3. Barbosa TF, Secco DA, Peixoto RS, et al. Bacterial fecal microbiota in healthy subjects and inpatients with Clostridium difficile infection. Adv Microbiol. 2017;7(01):10. doi:10.4236/aim.2017.71002
4. Tauxe WM, Haydek JP, Rebolledo PA, et al. Fecal microbiota transplant for Clostridium difficile infection in older adults. Therap Adv Gastroenterol. 2016;9(3):273–281. doi:10.1177/1756283X15622600
5. Fischer N, Relman DA. Clostridium Difficile, Aging, and the Gut: Can Microbiome Rejuvenation Keep Us Young and Healthy? Oxford University Press US; 2018.
6. Rea MC, O’Sullivan O, Shanahan F, et al. Clostridium difficile carriage in elderly subjects and associated changes in the intestinal microbiota. J Clin Microbiol. 2012;50(3):867–875. doi:10.1128/JCM.05176-11
7. Rodriguez C, Taminiau B, Korsak N, et al. Longitudinal survey of Clostridium difficile presence and gut microbiota composition in a Belgian nursing home. BMC Microbiol. 2016;16(1):229. doi:10.1186/s12866-016-0848-7
8. Milani C, Ticinesi A, Gerritsen J, et al. Gut microbiota composition and Clostridium difficile infection in hospitalized elderly individuals: a metagenomic study. Sci Rep. 2016;6(1):1–12. doi:10.1038/srep25945
9. Shoaei P, Shojaei H, Khorvash F, et al. Molecular epidemiology of Clostridium difficile infection in Iranian hospitals. Antimicrob Resist Infect Control. 2019;8(1):12. doi:10.1186/s13756-018-0454-6
10. Samarkos M, Mastrogianni E, Kampouropoulou O. The role of gut microbiota in Clostridium difficile infection. Eur J Intern Med. 2018;50:28–32. doi:10.1016/j.ejim.2018.02.006
11. Lagier J-C. Gut microbiota and Clostridium difficile infections. Human Microbiome J. 2016;2:10–14. doi:10.1016/j.humic.2016.10.003
12. Vakili B, Fateh A, Asadzadeh Aghdaei H, Sotoodehnejadnematalahi F, Siadat SD. Characterization of gut microbiota in hospitalized patients with Clostridioides difficile infection. Curr Microbiol. 2020;15:1–8.
13. Jimeno R, Brailey PM, Barral P. Quantitative polymerase chain reaction-based analyses of murine intestinal microbiota after oral antibiotic treatment. JoVE. 2018;141:e58481.
14. Lemee L, Dhalluin A, Pestel-Caron M, Lemeland J-F, Pons J-L. Multilocus sequence typing analysis of human and animal Clostridium difficile isolates of various toxigenic types. J Clin Microbiol. 2004;42(6):2609–2617. doi:10.1128/JCM.42.6.2609-2617.2004
15. Shoaei P, Shojaei H, Jalali M, et al. Clostridium difficile isolated from faecal samples in patients with ulcerative colitis. BMC Infect Dis. 2019;19(1):361. doi:10.1186/s12879-019-3965-8
16. Pitcher D, Saunders N, Owen R. Rapid extraction of bacterial genomic DNA with guanidium thiocyanate. Lett Appl Microbiol. 1989;8(4):151–156. doi:10.1111/j.1472-765X.1989.tb00262.x
17. Shoaei P, Shojaei H, Khorvash F, et al. Clostridium difficile infection in cancer patients with hospital acquired diarrhea at the teaching hospitals in Iran: multilocus sequence typing analysis (MLST) and Antimicrobial resistance pattern. Ann Ig. 2019;31(4):365–373. doi:10.7416/ai.2019.2298
18. Stubbs S, Rupnik M, Gibert M, Brazier J, Duerden B, Popoff M. Production of actin-specific ADP-ribosyltransferase (binary toxin) by strains of Clostridium difficile. FEMS Microbiol Lett. 2000;186(2):307–312. doi:10.1111/j.1574-6968.2000.tb09122.x
19. Carroll IM, Chang Y-H, Park J, Sartor RB, Ringel Y. Luminal and mucosal-associated intestinal microbiota in patients with diarrhea-predominant irritable bowel syndrome. Gut Pathog. 2010;2(1):19. doi:10.1186/1757-4749-2-19
20. Mello CS, Do Carmo Rodrigues MS, de Araújo Filho HB, et al. Fecal microbiota analysis of children with small intestinal bacterial overgrowth among residents of an urban slum in Brazil. Jornal De Pediatria (Versão Em Português). 2018;94(5):483–490. doi:10.1016/j.jpedp.2017.10.012
21. Nabizadeh E, Jazani NH, Bagheri M, Shahabi S. Association of altered gut microbiota composition with chronic urticaria. Ann Allergy Asthma Immunol. 2017;119(1):48–53. doi:10.1016/j.anai.2017.05.006
22. Arvand M, Moser V, Schwehn C, Bettge-Weller G, Hensgens MP, Kuijper J. High prevalence of Clostridium difficile colonization among nursing home residents in Hesse, Germany. PLoS One. 2012;7(1):e30183. doi:10.1371/journal.pone.0030183
23. Hegarty JP, Sangster W, Harris III LR, Stewart DB. Proton pump inhibitors induce changes in colonocyte gene expression that may affect Clostridium difficile infection. Surgery. 2014;156(4):972–978. doi:10.1016/j.surg.2014.06.074
24. Regnault H, Bourrier A, Lalande V, et al. Prevalence and risk factors of Clostridium difficile infection in patients hospitalized for flare of inflammatory bowel disease: a retrospective assessment. Dig Liver Dis. 2014;46(12):1086–1092. doi:10.1016/j.dld.2014.09.003
25. Sangster W, Hegarty JP, Schieffer KM, et al. Bacterial and fungal microbiota changes distinguish C. difficile infection from other forms of diarrhea: results of a prospective inpatient study. Front Microbiol. 2016;7:789. doi:10.3389/fmicb.2016.00789
26. Shoaei P, Shojaei H, Shirani K. Phenotypic and genotypic characteristics of Clostridium difficile isolates in patients with type 2 diabetes in Iran. Infect Drug Resist. 2020;13:683. doi:10.2147/IDR.S225829
27. Alimolaei M, Rahimi H-R, Ezatkhah M, Bafti MS, Afzali S. Prevalence, characteristics and antimicrobial susceptibility patterns of Clostridioides difficile isolated from hospitals in Iran. J Glob Antimicrob Resist. 2019;19:22–27. doi:10.1016/j.jgar.2019.02.013
28. Lagier J-C, Million M, Hugon P, Armougom F, Raoult D. Human gut microbiota: repertoire and variations. Front Cell Infect Microbiol. 2012;2:136. doi:10.3389/fcimb.2012.00136
29. Clooney A, Bernstein C, Leslie W, et al. A comparison of the gut microbiome between long‐term users and non‐users of proton pump inhibitors. Aliment Pharmacol Ther. 2016;43(9):974–984. doi:10.1111/apt.13568
30. Faleck DM, Salmasian H, Furuya EY, Larson EL, Abrams JA, Freedberg DE. Proton pump inhibitors do not affect risk for Clostridium difficile infection in the intensive care unit. Am J Gastroenterol. 2016;111(11):1641. doi:10.1038/ajg.2016.343
31. Antharam VC, Li EC, Ishmael A, et al. Intestinal dysbiosis and depletion of butyrogenic bacteria in Clostridium difficile infection and nosocomial diarrhea. J Clin Microbiol. 2013;51(9):2884–2892. doi:10.1128/JCM.00845-13
32. Kachrimanidou M, Tsintarakis E. Insights into the role of human gut microbiota in Clostridioides difficile infection. Microorganisms. 2020;8(2):200. doi:10.3390/microorganisms8020200
33. Hernández M, de Frutos M, Rodríguez-Lázaro D, López-Urrutia L, Quijada NM, Eiros JM. Fecal microbiota of toxigenic Clostridioides difficile-associated diarrhea. Front Microbiol. 2019;9:3331. doi:10.3389/fmicb.2018.03331
34. Gu S, Chen Y, Zhang X, et al. Identification of key taxa that favor intestinal colonization of Clostridium difficile in an adult Chinese population. Microbes Infect. 2016;18(1):30–38. doi:10.1016/j.micinf.2015.09.008
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.