")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 14
Interstitial deletion within 7q31.1q31.3 in a woman with mild intellectual disability and schizophrenia
Authors Akahoshi K, Yamamoto T
Received 17 March 2018
Accepted for publication 9 May 2018
Published 5 July 2018 Volume 2018:14 Pages 1773—1778
DOI https://doi.org/10.2147/NDT.S168469
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Taro Kishi
Keiko Akahoshi,1 Toshiyuki Yamamoto2
1Department of Pediatrics, Tokyo Children’s Rehabilitation Hospital, Tokyo, Japan; 2Institute of Medical Genetics, Tokyo Women’s Medical University, Tokyo, Japan
Abstract: We report the case of a Japanese woman with an interstitial deletion within the 7q31.1q31.3 region, she presented with mild intellectual disability since infancy, and later developed characteristic psychiatric manifestations, including abnormal behavior, delusions, and hallucinations. She was diagnosed with paranoid schizophrenia (F20.0, International Statistical Classification of Diseases and Related Health Problems 10th Revision). Array comparative genomic hybridization examination revealed the deletion involving several important genes for neurodevelopment. Particularly, FOXP2, DOCK4, MET, and WNT2 in this region are suggested to be related to language impairment, autistic disorders, and cognitive disorders, via the WNT pathway. In addition, the WNT signal pathway has been suggested to be implicated in the pathogenesis of psychiatric disorders such as schizophrenia and bipolar disorder. However, there is no case report regarding schizophrenia associated with a 7q31 microdeletion. We suspect that the disruptions of these one or plural genes among the interstitial deletion of 7q31.1q31.3 may be involved in the development of schizophrenia in this woman. This is the first report on schizophrenia associated with a 7q31 microdeletion.
Keywords: chromosomal microarray, psychiatric disorder, autism spectrum disorder, ASD, Wnt pathway
Introduction
Schizophrenia is the most well-known chronic psychiatric disorder, characterized primarily by hallucinations and delusions and abnormal behaviors, predominantly developing in late puberty. Although the lifetime prevalence is approximately 0.5%–1.0% in the general population and a great amount of evidence has been accumulating on its cause, including responsible genes, it has not been clearly elucidated yet.1
The 7q31 chromosomal region is rich in genes. Within this region, there are several genes involved in neurodevelopment. These genes affected by the Wnt pathway may participate in the development of psychiatric disorders.
In this study, we describe the case of a 28-year-old Japanese woman with mild intellectual disability associated with an interstitial deletion in 7q31 region. This patient also showed the phenotypes of schizophrenia.
Patient report
Our case involved a 29-year-old female, who was described as cheerful and peaceful. Although her father and older brother are healthy, her mother succumbed to fungal pneumonia. The patient was born at 38 weeks of gestation via vaginal delivery with early membrane rupture. Her birth weight was 2,700 g. During pregnancy, maternal edema and proteinuria were observed. There was no birth asphyxia; however, the patient showed poor sucking and muscular hypotonia. Her development was delayed as evidenced by gaining head control at 6 months, rolling over at 18 months, and standing at 22 months. The diagnosis of intellectual disability was made at 7 years of age, because her intelligence quotient measured by the Binet–Tanaka test was 53. A neurological examination at that time confirmed the presence of psychomotor retardation. Some minor anomalies, such as a horizontal line in her palm, high-arched palate, and mandibular protrusion, were observed (Figure 1). After graduating from a special education school, she attended a workshop for the handicapped. She was active, helped her family well, and even participated in the Special Olympics.
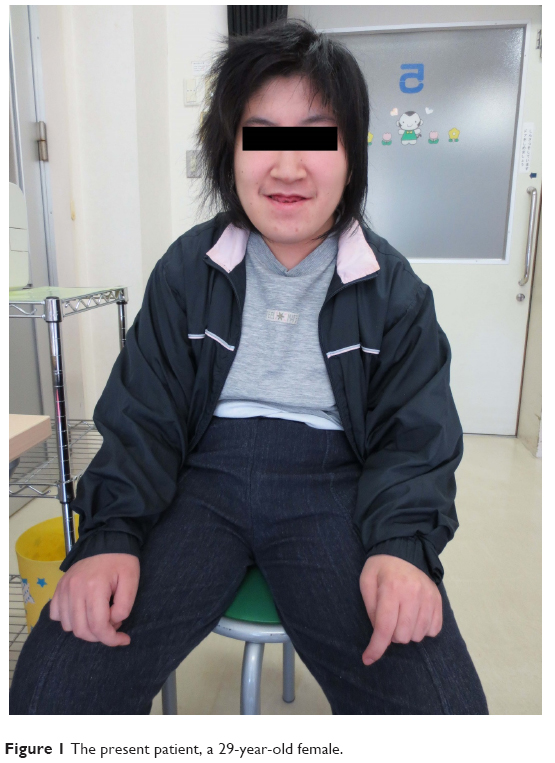 | Figure 1 The present patient, a 29-year-old female. |
At the age of 27 years, she suddenly became inactive and began neglecting her regular household chores. She refused to go to work and became withdrawn; she did not watch television, an activity she previously enjoyed. She was expressionless, was unkempt, and had stopped eating and bathing by herself. She suffered from insomnia, and her mood was unstable, irritable, and aggressive. She developed sluggish movements; talked to and laughed at herself; occasionally cried, shouted, and got irritated; and distressed her family.
Laboratory tests, including thyroid function, revealed no abnormalities with the exception of a chromosomal abnormality. Brain magnetic resonance imaging demonstrated no remarkable changes, and electroencephalography revealed no paroxysmal activity. She was prescribed psychotropic drugs, but she did not use them. Thereafter, she demonstrated obsessive-compulsive, repetitive, ritualized behavior, and she was no longer able to eat, bath, and excrete by herself. She also experienced hallucinations and delusions. Some of her complaints included “some people whisper in my ears,” “someone is chasing me,” “something moves my hand,” and “something touched my feet.” Eight months later, her family reported that she behaved as a completely different person. She was diagnosed with paranoid schizophrenia (F20.0, International Statistical Classification of Diseases and Related Health Problems 10th Revision).
With family cooperation, she started medication, and her hallucinations and delusions improved following the use of risperidone (2 mg/day) and duloxetine (20 mg/day). Six months later, she began to smile and her appetite and sleep normalized. One year later she was able to eat meals by herself. At present, she can excrete by herself. Although her hallucinations, delusions, and psychomotor sluggishness have improved with medication, she continues to talk to herself and exhibits obsessive-compulsive behavior. She often throws tantrums when the order of daily activities set by her gets disturbed.
Cytogenetic examination
Conventional chromosomal G-banding revealed an interstitial deletion in 7q, described as 46,XX,del(7)(q31.3q31.3) (data not shown). Permission to analyze parental karyotyping could not be obtained. To confirm the precise deletion region, chromosomal microarray testing was performed with an Agilent 60 K Human Genome Comparative Genomic Hybridization Microarray platform (Agilent Technologies, Santa Clara, CA, USA) in accordance with previously reported methods.2,3 For this purpose, a peripheral blood sample was collected after obtaining written informed consent. Genomic DNA was extracted with the QIA quick DNA extraction kit (Qiagen, Hilden, Germany). The results are described as arr[hg19] 7q31.1q31.31(111,105,151-119,120,843)X1.
Discussion
In this study, an interstitial deletion in the 7q31 region was identified in a female patient with intellectual disability. In the deletion region, 25 registered RefSeq genes were identified. The identified genes included FOXP2, DOCK4, MET, and WNT2. These genes play an important role in the function of central nervous system while affecting each other via the Wnt/β-catenin signaling pathway. Several studies have investigated the association between Wnt/β-catenin signaling pathway and autism spectrum disorders (ASD), and 7q31 has been considered a “hotspot” for ASD.4 The canonical Wnt pathway has been indicated to play a role in normal brain development and synaptic function and to affect dendrite growth and activity.5,6 Reportedly, Wnt and its receptors are involved in the embryonic development of central nervous system and are also highly expressed in adult brains, suggesting that their expression may affect higher brain functions and that Wnt signaling may be involved in higher brain functions. Therefore, genes affected by the Wnt pathway may be involved in the development of psychiatric disorders, such as schizophrenia or bipolar disorder.7,8
Meanwhile, several previous studies have reported deletion cases similar to that reported here (Figure 2). Among them, cases with FOXP2 deletion have mild-to-moderate developmental delays (Table 1).2,9–16 FOXP2 encodes a transcription factor comprising a polyglutamine tract and a forkhead DNA-binding domain.17 FOXP2 protein may regulate the expression of other genes in the cortico-basal ganglia and cerebellar circuits of the brain.18 Disruptions in FOXP2 are associated with language, autistic, and cognitive disorders.19,20 Therefore, developmental delay in such patients is likely to be derived from FOXP2 haploinsufficiency. Interestingly, there is a report that FOXP2 may also regulate DISC1, which has attracted the most attention as a candidate gene for schizophrenia.21 However, there are both positive and negative reports on the association between FOXP 2 and schizophrenia.22–25 Further, in several previous studies as stated above including FOXP2 (Figure 2), schizophrenia has not been reported. DOCK4, a guanine nucleotide exchange factor (GEF) for Rac, is a component of the β-catenin destruction complex. Dock4 is expressed in the rat hippocampal neurons and plays an important role in supine formation.26 It has also been reported as candidate gene for autism, dyslexia, and schizophrenia.27–29 MET is the receptor for hepatocyte growth factor (HGF). HGF-MET signaling contributes to neuronal differentiation, cerebral cortex and cerebellum development, and axon growth. MET transcription is regulated by FOXP2,30 and DOCK4 and MET are related to the Wnt pathway through β-catenin.4 Finally, WNT2 is a homologue of WNT1, and Wnt1 has been suggested to regulate genetic networks in midbrain dopaminergic neuron development.31 Wnt-1 protein has increased expression in the brains of individuals with schizophrenia.32 Similarly, WNT2 may be associated with the development of schizophrenia.
 | Figure 2 Genome map around 7q31 region depicting reported deletion regions. |
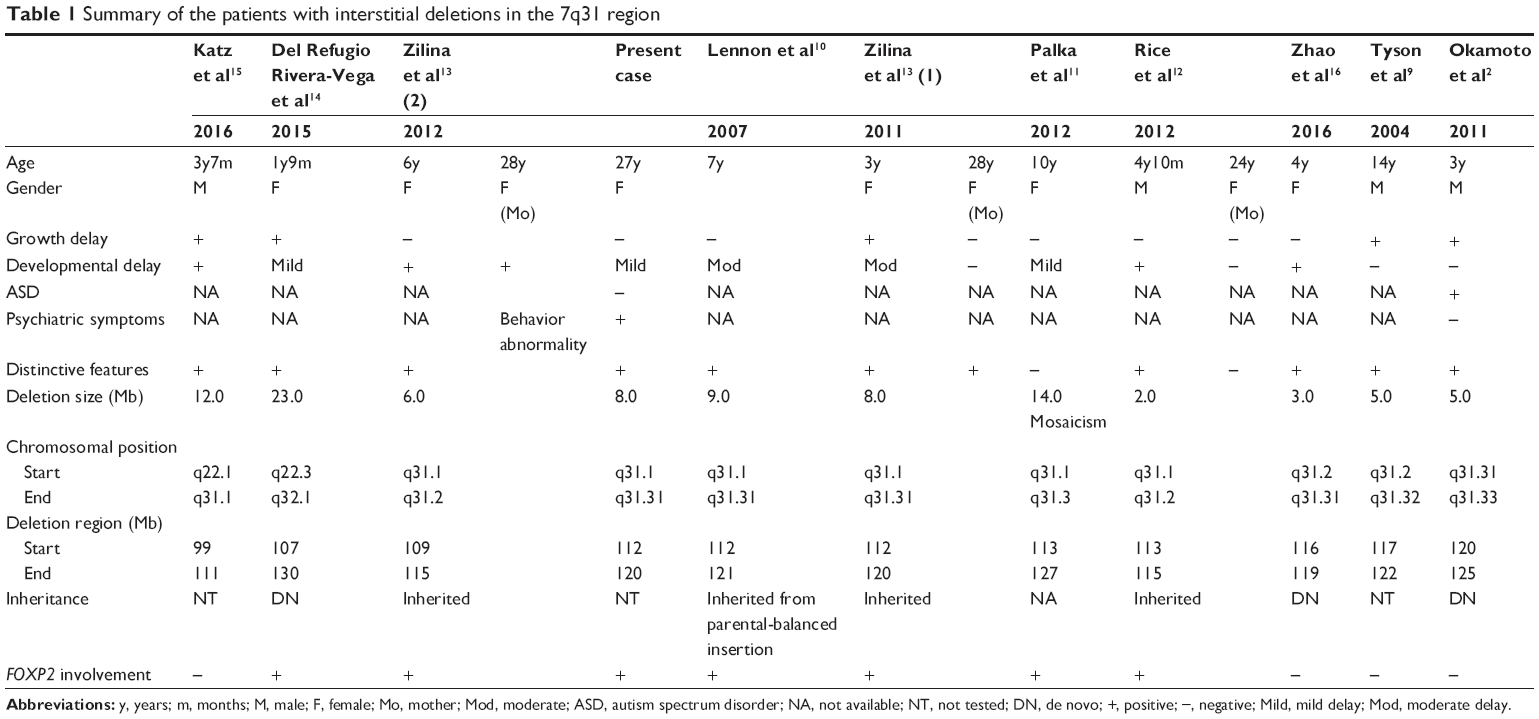 | Table 1 Summary of the patients with interstitial deletions in the 7q31 region |
FOXP2, DOCK4, MET, and WNT2 are heterozygous in this case. We suspected that one possibility is that mutation or epigenetic change occurs somewhere in the contralateral allele and the disruptions of these plural genes affect each other and may be involved in the onset of schizophrenia in this patient.
Acknowledgments
We would like to thank the patient’s family for their written informed consent to publish this case report and photographs. We also acknowledge the DECIPHER database for checking overlapping deletions. We are grateful to the technicians from our laboratories, including Ms Yumiko Ondo for her skillful help. This research was supported by the Practical Research Project for Rare/Intractable Diseases from the Japan Agency for Medical Research and Development (AMED), and the Japan Society for the Promotion of Science (JSPS) KAKENHI grant number 15K09631 (TY).
Disclosure
The authors report no conflicts of interest in this work.
References
Freedman R. Schizophrenia. N Engl J Med. 2003;349(18):1738–1749. | ||
Okamoto N, Hatsukawa Y, Shimojima K, Yamamoto T. Submicroscopic deletion in 7q31 encompassing CADPS2 and TSPAN12 in a child with autism spectrum disorder and PHPV. Am J Med Genet A. 2011;155A(7):1568–1573. | ||
Sangu N, Shimojima K, Takahashi Y, Ohashi T, Tohyama J, Yamamoto T. A 7q31.33q32.1 microdeletion including LRRC4 and GRM8 is associated with severe intellectual disability and characteristics of autism. Hum Genome Var. 2017;4:17001. | ||
Kalkman HO. A review of the evidence for the canonical Wnt pathway in autism spectrum disorders. Molecular Autism. 2012;3(1):10. | ||
Lie DC, Colamarino SA, Song HJ, et al. Wnt signalling regulates adult hippocampal neurogenesis. Nature. 2005;437(7063):1370–1375. | ||
Hiester BG, Galati DF, Salinas PC, Jones KR. Neurotrophin and Wnt signaling cooperatively regulate dendritic spine formation. Mol Cell Neurosci. 2013;56:115–127. | ||
Wilson SW, Houart C. Early steps in the development of the forebrain. Dev Cell. 2004;6(2):167–181. | ||
Ciani L, Salinas PC. WNTs in the vertebrate nervous system: from patterning to neuronal connectivity. Nat Rev Neurosci. 2005;6(5):351–362. | ||
Tyson C, McGillivray B, Chijiwa C, Rajcan-Separovic E. Elucidation of a cryptic interstitial 7q31.3 deletion in a patient with a language disorder and mild mental retardation by array-CGH. Am J Med Genet A. 2004;129A(3):254–260. | ||
Lennon PA, Cooper ML, Peiffer DA, et al. Deletion of 7q31.1 supports involvement of FOXP2 in language impairment: clinical report and review. Am J Med Genet A. 2007;143A(8):791–798. | ||
Palka C, Alfonsi M, Mohn A, et al. Mosaic 7q31 deletion involving FOXP2 gene associated with language impairment. Pediatrics 2012;129(1):e183–e188. | ||
Rice GM, Raca G, Jakielski KJ, et al. Phenotype of FOXP2 haploinsufficiency in a mother and son. Am J Med Genet A. 2012;158A(1):174–181. | ||
Zilina O, Reimand T, Zjablovskaja P, et al. Maternally and paternally inherited deletion of 7q31 involving the FOXP2 gene in two families. Am J Med Genet A. 2012;158A(1):254–256. | ||
Del Refugio Rivera-Vega M, Gomez-Del Angel LA, Valdes-Miranda JM, et al. A Novel 23.1 Mb interstitial deletion involving 7q22.3q32.1 in a girl with short stature, motor delay, and craniofacial dysmorphism. Cytogenet Genome Res. 2015;145(1):1–145. | ||
Katz OL, Krantz ID, Noon SE. Interstitial deletion of 7q22.1q31.1 in a boy with structural brain abnormality, cardiac defect, developmental delay, and dysmorphic features. Am J Med Genet C Semin Med Genet. 2016;172(2):92–101. | ||
Zhao J, Noon SE, Krantz ID, Wu Y. A de novo interstitial deletion of 7q31.2q31.31 identified in a girl with developmental delay and hearing loss. Am J Med Genet C Semin Med Genet. 2016;172(2):102–108. | ||
Lai CS, Fisher SE, Hurst JA, Vargha-Khadem F, Monaco AP. A forkhead-domain gene is mutated in a severe speech and language disorder. Nature. 2001;413(6855):519–523. | ||
Enard W. FOXP2 and the role of cortico-basal ganglia circuits in speech and language evolution. Curr Opin Neurobiol. 2011;21(3):415–424. | ||
Reuter MS, Riess A, Moog U, et al. FOXP2 variants in 14 individuals with developmental speech and language disorders broaden the mutational and clinical spectrum. J Med Genet. 2017;54:64–72. | ||
Vernes SC, Spiteri E, Nicod J, et al. High-throughput analysis of promoter occupancy reveals direct neural targets of FOXP2, a gene mutated in speech and language disorders. Am J Hum Genet. 2007;81(6):1232–1250. | ||
Walker RM, Hill AE, Newman AC, et al. The DISC1 promoter: characterization and regulation by FOXP2. Hum Mol Genet. 2012;21(13):2862–2872. | ||
Sanjuán J, Tolosa A, González JC, et al. Association between FOXP2 polymorphisms and schizophrenia with auditory hallucinations. Psychiatr Genet. 2006;16(2):67–72. | ||
Tolosa A, Sanjuán J, Dagnall AM, et al. FOXP2 gene and language impairment in schizophrenia: association and epigenetic studies. BMC Med Genet. 2010;22(11):114. | ||
Rao W, Du X, Zhang Y, et al. Association between forkhead-box P2 gene polymorphism and clinical symptoms in chronic schizophrenia in a Chinese population. J Neural Transm (Vienna). 2017;124:891–897. | ||
Yin J, Jia N, Liu Y, et al. No association between FOXP2 rs10447760 and schizophrenia in a replication study of the Chinese Han population. Psychiatr Genet. 2018;28(2):19–23. | ||
Ueda S, Negishi M, Katoh H. Rac GEF Dock4 interacts with cortactin to regulate dendritic spine formation. Mol Biol Cell. 2013;24(10):1602–1613. | ||
Pagnamenta AT, Bacchelli E, de Jonge MV, et al. Characterization of a family with rare deletions in CNTNAP5 and DOCK4 suggests novel risk loci for autism and dyslexia. Biol Psychiatry. 2010;68(4):320–328. | ||
Maestrini E, Pagnamenta AT, Lamb JA, et al. High-density SNP association study and copy number variation analysis of the AUTS1 and AUTS5 loci implicate the IMMP2L-DOCK4 gene region in autism susceptibility. Mol Psychiatry. 2010;15(9):954–968. | ||
Alkelai A, Lupoli S, Greenbaum L, et al. DOCK4 and CEACAM21 as novel schizophrenia candidate genes in the Jewish population. Int J Neuropsychopharmacol. 2012;15(4):459–469. | ||
Mukamel Z, Konopka G, Wexler EL, et al. Regulation of MET by FOXP2, genes implicated in higher cognitive dysfunction and autism risk. J Neurosci. 2011;31(32):11437–11442. | ||
Wurst W, Prakash N. Wnt1-regulated genetic networks in midbrain dopaminergic neuron development. J Mol Cell Biol. 2014;6(1):34–41. | ||
Miyaoka T, Seno H, Ishino H. Increased expression of Wnt-1 in schizophrenic brains. Schizophr Res. 1999;38(1):1–6. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.