Back to Journals » Journal of Inflammation Research » Volume 15
Interrelationship and Sequencing of Interleukins4, 13, 31, and 33 – An Integrated Systematic Review: Dermatological and Multidisciplinary Perspectives
Authors Tatu AL ![]() , Nadasdy T
, Nadasdy T ![]() , Arbune A
, Arbune A ![]() , Chioncel V
, Chioncel V ![]() , Bobeica C
, Bobeica C ![]() , Niculet E
, Niculet E ![]() , Iancu AV
, Iancu AV ![]() , Dumitru C
, Dumitru C ![]() , Popa VT, Kluger N
, Popa VT, Kluger N ![]() , Clatici VG, Vasile CI
, Clatici VG, Vasile CI ![]() , Onisor C
, Onisor C ![]() , Nechifor A
, Nechifor A ![]()
Received 10 May 2022
Accepted for publication 13 August 2022
Published 8 September 2022 Volume 2022:15 Pages 5163—5184
DOI https://doi.org/10.2147/JIR.S374060
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Alin Laurentiu Tatu,1– 3,* Thomas Nadasdy,3,4 Anca Arbune,5,* Valentin Chioncel,6,* Carmen Bobeica,7 Elena Niculet,3,* Alina Viorica Iancu,7 Caterina Dumitru,8 Valentin Tudor Popa,3,9 Nicolas Kluger,10,11 Victor Gabriel Clatici,11 Claudiu Ionut Vasile,2 Cristian Onisor,7 Alexandru Nechifor2
1Dermatology Department, “Sf. Cuvioasa Parascheva” Clinical Hospital of Infectious Diseases, Galati, Romania; 2Clinical Medical Department, Faculty of Medicine and Pharmacy, “Dunarea de Jos” University, Galati, Romania; 3Multidisciplinary Integrated Center of Dermatological Interface Research (MIC-DIR) [Centrul Integrat Multi disciplinar de Cercetare de Interfata Dermatologica (CIM-CID)], Galați, Romania; 4Dermatology Department, Municipal Emergency Hospital, Timişoara, Romania; 5Neurology Department, Fundeni Clinical Institute, Bucharest, Romania; 6Neurology Department, “Bagdasar-Arseni” Emergency Clinical Hospital, Bucharest, Romania; 7Department of Morphological and Functional Sciences, Faculty of Medicine and Pharmacy, “Dunărea de Jos” University, Galați, Romania; 8Pharmaceutical Sciences Department, Faculty of Medicine and Pharmacy, “Dunarea de Jos” University, Galati, Romania; 9Dermatology Department, Center for the Morphologic Study of the Skin MORPHODERM, “Victor Babeș” University of Medicine and Pharmacy, Timișoara, Romania; 10Department of Dermatology, Allergology and Venereology, Helsinki University Central Hospital and University of Helsinki, Helsinki, Finland; 11Apolo Medical Center, Bucharest, Romania
*These authors contributed equally to this work
Correspondence: Anca Arbune, Neurology Department, Fundeni Clinical Institute, 258 Fundeni Street, Bucharest, 022328, Romania, Tel +40 21 275 0500, Email [email protected] Elena Niculet, Department of Morphological and Functional Sciences, Faculty of Medicine and Pharmacy, “Dunărea de Jos” University of Galați, 35 Alexandru Ioan Cuza Street, Galați, 800008, Romania, Tel +40 74 139 8895, Email [email protected]
Abstract: The interrelations and sequencing of interleukins are complex (inter)actions where each interleukin can stimulate the secretion of its preceding interleukin. In this paper, we attempt to summarize the currently known roles of IL-4, IL-13, IL-31, and IL-33 from a multi-disciplinary perspective. In order to conduct a comprehensive review of the current literature, a search was conducted using PubMed, Google Scholar, Medscape, UpToDate, and Key Elsevier for keywords. The results were compiled from case reports, case series, letters, and literature review papers, and analyzed by a panel of multi-disciplinary specialist physicians for relevance. Based on 173 results, we compiled the following review of interleukin signaling and its clinical significance across a multitude of medical specialties. Interleukins are at the bed rock of a multitude of pathologies across different organ systems and understanding their role will likely lead to novel treatments and better outcomes for our patients. New interleukins are being described, and the role of this inflammatory cascade is still coming to light. We hope this multi-discipline review on the role interleukins play in current pathology assists in this scope.
Keywords: interleukin cascade, IL-4, IL-13, IL-31, IL-33
Introduction
Interleukins are a diverse group of molecules exerting a plethora of autocrine and paracrine immune functions. They exhibit a pattern of interrelationing, where each interleukin can stimulate the secretion of its preceding interleukin. At present, 41 different members of this group of cytokines have been described, numbered from 1 to 41.1 With the most recent member of this group being discovered in 2019, it is evident we are still in the infancy of our understanding of how these molecules come to be and how they modulate intercellular signaling.2 In this paper, we attempt to summarize their current known clinical roles from a multi-disciplinary perspective. Classically, the interleukin cascade begins with a triggering stimulus, and the subsequent release of either damage-associated molecular patterns (DAMPs), or pathogen-associated molecular patterns (PAMPs), depending on the stimulus. In response to this trigger, dendritic, epithelial, fibroblast, and other resident immune cells that are present nearby, will secrete IL-1 as the initial step in immune activation leading to fever, upregulation of intracellular adhesion molecules, leukocyte stimulation and chemotaxis, and to a release of acute phase reactants. Release of subsequent interleukins will: act as polarizing cytokines for naïve T-cell differentiation (mainly IL-2, 4, 12, 22, and 23); lead to the activation of B-cells, and their differentiation into plasma cells (IL-4, 5, 6, 13, and 14); stimulate hematopoiesis in bone marrow stromal cells (IL-3 and 11); and further stimulate and promote chemotaxis of leukocytes into the damaged area, either directly, or by modifying local expression of adhesion molecules, chemokines, and vasoactive peptides (most other interleukins). The aforementioned cells, along with local stromal and epithelial cells, go on to secrete every other known interleukins.1,3 Thus, a positive feedback loop is created, with downstream interleukins acting to stimulate cells that release more of the same signaling molecules, and, in turn, perpetuate this inflammatory cascade (Figure 1, Table 1).
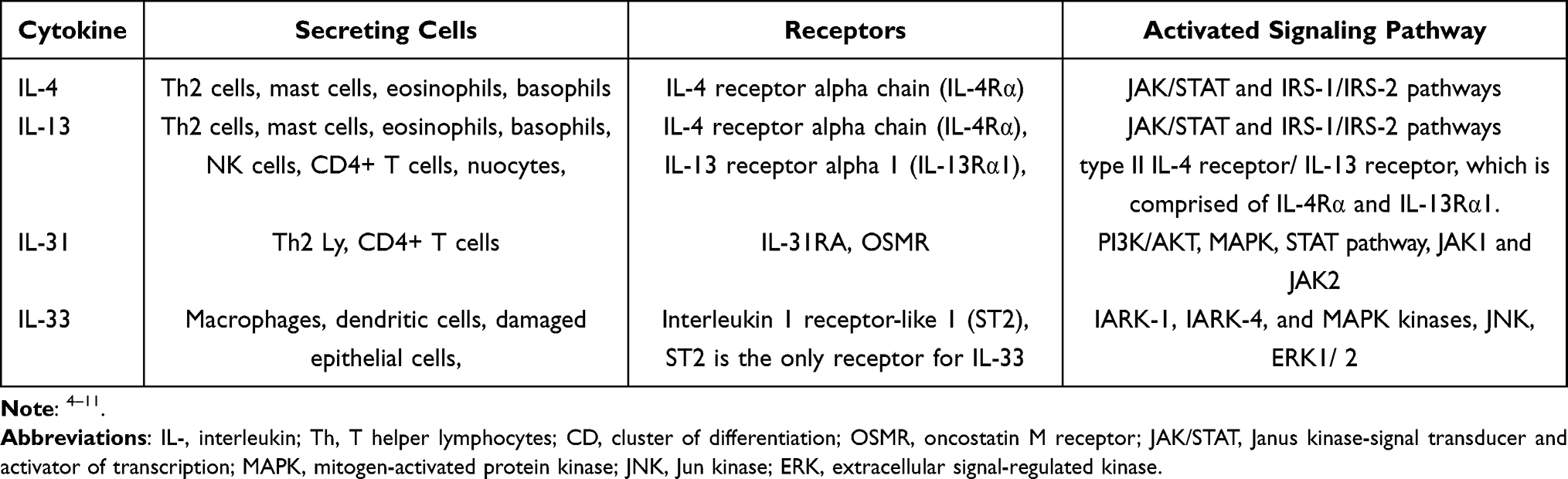 |
Table 1 IL-4, IL-13, IL-31 and IL-33-Secreting Cells, with the Respective IL Receptors and Signaling Pathways Which They Activate |
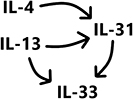 |
Figure 1 A snippet of the interleukin cascade. IL-4 and IL-13 are released under certain inflammatory stimuli. In turn they induce the release of IL-31, and IL-33; the latter is also induced by IL-31. |
Materials and Methods
In order to conduct a comprehensive descriptive review of the current literature, a search was conducted using PubMed, Medscape, UpToDate, and Key Elsevier for the terms “IL-4”, “IL-13”, “IL-31”, “IL-33” (OR) “Interleukin 4”, “Interleukin 13”, “Interleukin 31”, “Interleukin 33” in combination with the terms (AND) “Dermatology”, “Rheumatology”, “Cardiology”, “Neurology”, “Melanoma”, “Systemic Sclerosis”, “Atopic Dermatitis”, “Psoriasis”, “Rheumatoid arthritis”, “Ankylosing spondylitis”, “Systemic Lupus erythematosus”, “Memory impairment”, “Behcet”, “Neurodegenerative diseases”, “Alzheimer”, “Parkinson”, “Amyotrophic lateral sclerosis”, “Autoimmune encephalitis”, “Multiple Sclerosis”, “Toxoplasmosis”, “Neurocysticercosis”, “Zika”, “Intracerebral Hemorrhage”, “Epilepsy”, “Seizure”, “Psychiatric disorder”, “Pruritus”, “Schizophrenia”, “Depression”, “Autism”, “Cardiomyopathy”, “Myocarditis”, “Heart failure”, “Fibrosis”, “Kawasaki”, “Atherosclerosis”, “Ischemic heart disease”, “Infarction”. The results were compiled from original articles, case reports, case series, letters, and literature review papers, and analyzed by a panel of multi-disciplinary specialist physicians for relevance and pertinent information.
Results
Based on 173 relevant results, we compiled the following review of interleukin signaling and its clinical significance across a multitude of medical specialties (Figure 2).
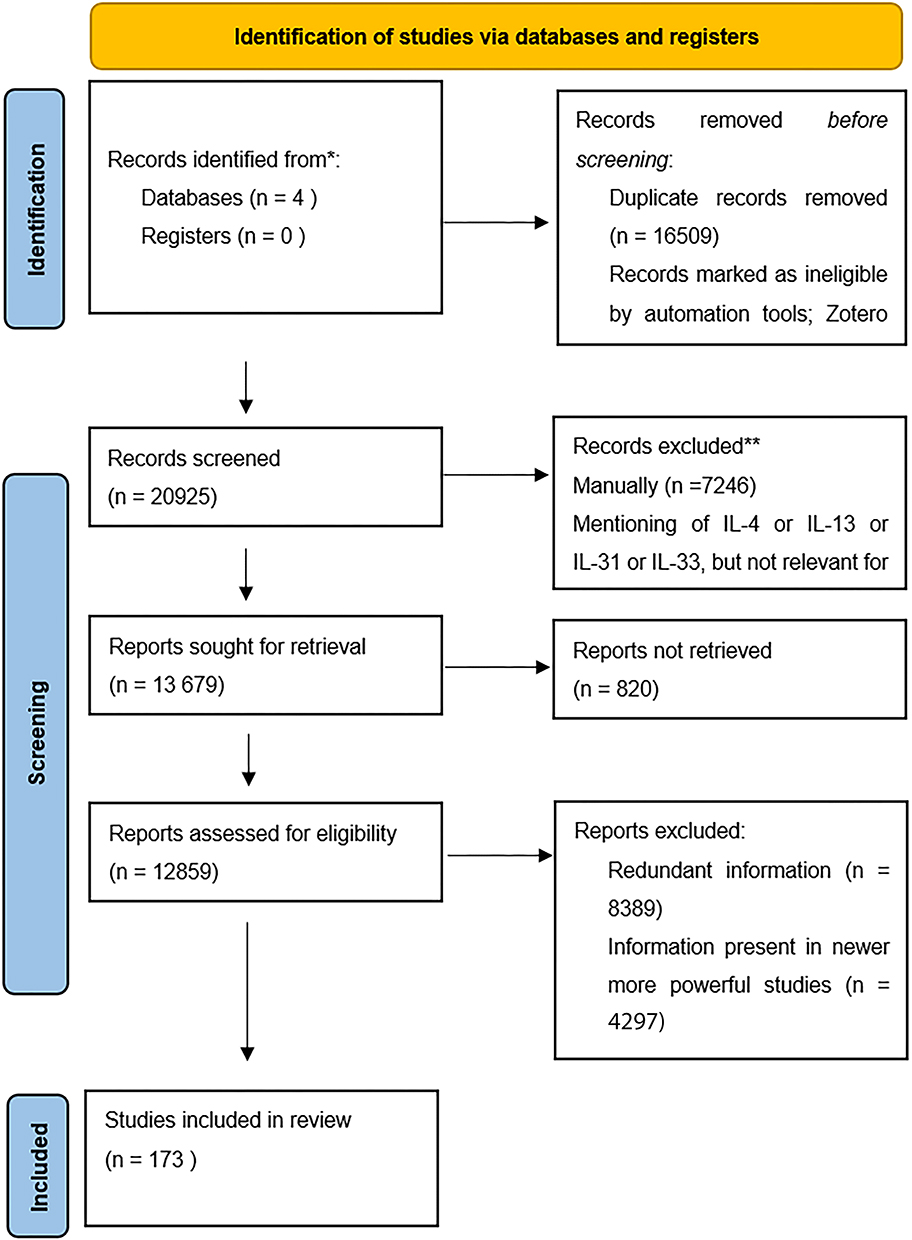 |
Figure 2 Compiling of results. Notes: PRISMA figure adapted from Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71. Creative Commons.191 For more information, visit: http://www.prisma-statement.org/. |
Discussion
Cardiology
Interleukin 4 (IL-4) is an anti-inflammatory and pro-fibrotic cytokine that regulates multiple biological functions and therefore has many implications in different pathologies.12,13 This cytokine stimulates the inflammatory response by activating the synthesis of collagen (types I and II) by fibroblasts, and also enhances the proliferation of fibroblasts, therefore contributing to the development and progression of fibrosis.14,15 IL-4 also augments platelet-derived growth factor (PDGF) and contributes to the increase of macrophages within developing regions of fibrosis. In this spirit, there are many cardiac diseases in which IL-4 is playing a major role. The patients with hypertensive cardiomyopathy had higher concentrations of IL-4, but it is still unknown whether chronically elevated IL-4 is sufficient to initiate a fibrotic response, leading to cardiac fibrosis and dysfunction.16 Inflammatory fibrosis (induced through overproduction and deposition of collagen by the fibroblasts) is also a typical feature of myocarditis and heart failure. Some studies in mice have already demonstrated a clear correlation between high levels of IL-4 and left ventricular enlargement (in response to sustained pressure overload), and depressed cardiac function (fibrotic cardiomyopathy).17,18 This important role of IL-4 in cardiac remodeling was also shown in human patients.19,20 Besides of the important role of IL-4 in upregulating fibrogenesis, it was shown that its neutralization leads to the attenuation of cardiac fibrosis, and administration of anti-IL-4 antibodies suppresses hypertension in rats.21,22 These findings are of great clinical interest and may serve as a potential target for future therapeutic interventions. IL-4 also has proinflammatory effects and, on one hand, determines the accelerated apoptosis of vascular endothelial cells, and, on the other hand, determines the increased endothelial oxidative stress, leading to atherogenesis.23 Through this effect, this cytokine induces early lung vascular inflammation, leading to the development of pulmonary hypertension and subsequent right ventricular hypertrophy and fibrosis.24,25 Another study on mice has shown that administration of IL-4 may augment cardiac M2-like macrophages and improve cardiac function after a myocardial infarction.26 Some pathological events were incriminated, such as: microvascular development in the viable myocardium and local formation of thicker connective tissue in the infarcted ventricular wall, helping in improving ventricular function. Thus, systemic administration (or maybe intracoronary injection while doing a percutaneous coronary intervention) of IL-4, may be a very promising therapeutic option for acute myocardial infarction. Another recent study demonstrated a close correlation between increased basophil count after myocardial infarction and IL-4 and, respectively, IL-13 levels, suggesting a contribution to tissue repair and infarct healing.27
Discovered by Minty and McKenzie in 1993, IL-13 is one of the powerful cytokines with broad functions, playing important roles in inflammation and immune responses.28,29 Widely expressed in most tissues, such as the heart, lung, liver and skin, IL-13 is not only involved in cardiac inflammatory diseases, such as myocarditis, but it is also relevant in the development of acute or chronic cardiovascular diseases with other pathogenic mechanisms, such as myocardial infarction and heart failure.30 Many sources suggest that IL-13 is associated with cardiac fibrosis, cardiomyocyte proliferation and myocardial hypertrophy. One study in aging mice models has shown that IL-13 is related to excessive collagen production, cardiac fibrosis and remodeling, leading to progressive cardiac dysfunction.31 Also, a higher serum level of IL-13 has been detected in vivo in patients with chronic heart failure and dilated cardiomyopathy, the authors concluding that in the near future, IL-13 might be a potential predictor or therapy target in these patients.32,33 Regarding atherosclerosis and ischemic heart disease, since 2013, Stanya et al have demonstrated that increased weight gain, hyperglycemia, and hepatic insulin resistance are commonly encountered in mice with IL-13 deficiency, indicating that IL-13 might inhibit atherogenesis.34 Many studies demonstrated that IL-13 is markedly expressed in myocardial infarction, and that it is also involved in cardiac wound healing and remodeling after an acute injury. Hofmann et al have shown the evolution of IL-13 concentration in acute myocardial infarction (registering an immediately increasing level, a peak at 3 days after infarction and a decline towards day 7), thus suggesting a close relation between this cytokine and cardiac function after infarction. In mice with IL-13 deficiency, significantly poor echocardiography parameters (within 7 days after myocardial infarction) suggest that IL-13 is involved in cardiac remodeling and might be a good prognostic marker in the very early phase of infarction.35 Another study evidenced that a reduction of IL-13 levels, registered at 3 months after primary angioplasty for acute myocardial infarction, is associated with a worse prognosis, so IL-13 level could be a novel prognostic marker in STEMI patients.36 On the other hand, IL-13 is an important regulatory factor involved in the pathogenesis of pulmonary vascular remodeling and in the development of pulmonary artery hypertension.37 Concerning the role of IL-13 in inflammatory heart diseases, it was demonstrated that IL-13 deficient mice develop severe myocarditis, having poor prognosis, and that experimental administration of IL-13 could significantly improve heart inflammation and cardiac injury induced by myocarditis.38,39 IL-13 is also associated with progression of valvular heart diseases. Rotter et Vianello have demonstrated a close correlation between higher IL-13 levels and severity of aortic valve diseases, suggesting that IL-13 might induce fibrosis, increased collagen depositing and valvular calcification.40,41 Taken together, it seems that further research revealing the role of IL-13 in cardiovascular diseases may lead to a promising immunotherapy in these patients.
Interleukin-31 (IL-31) is a proinflammatory cytokine from the gp130/IL-6 cytokine family, synthesized mainly by activated CD4+ T cells, being involved in different cardiac diseases.42,43 Some studies report a close relationship between a certain genotype of IL-31 (rs4758680 allele) and the susceptibility of developing dilated cardiomyopathy in Han Chinese people; patients with this IL-31 genotype (rs4758680) had a higher chance of developing a poorer outcome.44 The researchers also discovered that IL-31 acted through the receptor complex of IL-31RA, therefore inducing left ventricular hypertrophy and a loss of sarcomeres and cardiac fibroblasts, finally affecting myocardial contraction.45–47 Another cardiac pathology in which IL-31 is involved is Kawasaki disease. Pro-inflammatory cytokine IL-31 triggered release from eosinophils might be the key mechanism that explains this association.48 Some authors suggest that IL-31 may also be a predictor of coronary lesion development in patients with Kawasaki disease.49
IL-33 (a recently identified member of IL-1 family) is a complex immunomodulatory cytokine, acting like a pro- and anti-inflammatory factor, depending on the disease.50–53 This cytokine was identified on endothelial and epithelial cells, smooth muscle cells, keratinocytes, fibroblasts, activated macrophages. It acts through the ST2 receptor just like an alarm factor. Its release is promoted by stressful conditions, inducing protective measures in neighboring cells.54–58 In the human heart, IL-33 is mainly present in endothelial cells, cardiomyocytes and fibroblasts, functioning as a cardioprotective cytokine. It is also an indicator of cardiac stress in pressure overload situations (hypertension, heart failure, aortic stenosis and cardiomyopathies), and it favorably regulates the cardiac remodeling process.59–61 In chronic heart failure, the levels of sST2 are elevated and they are strongly associated with heart failure severity and with adverse outcomes.62 As such, IL-33/sST2 might become a powerful prognostic biomarker in chronic heart failure (with a reduced ejection fraction) and also a very good stratification marker (by being unaffected by body mass index, renal function, or age, as opposed to natriuretic peptides). Like a biomarker of cardiac mechanical strain, IL-33/sST2 may also have prognostic value in hypertension.63 It was demonstrated that higher sST2 levels are associated with alterations of left ventricular geometry and with an increased systolic blood pressure.64–66 In the same spirit, there is evidence about a close correlation between pulmonary arterial hypertension and sST2 release, secondary to an increased right ventricular afterload and a high myocardial stretch.67,68 Some authors pointed out that elevated sST2 was associated with systolic dysfunction and right ventricular dilatation and it independently predicts 1-year mortality in PAH patients.69 IL-33 is playing an important role in atherosclerosis and in ischemic heart disease. It may be used as an alarm signal and a diagnostic marker, as suggested by a research study wherein patients with acute coronary syndrome showed a significantly higher serum concentration of IL-33 (attributed to the myocardial damage) in comparison with the healthy control group.70 There is much evidence regarding the protective role of IL-33 in atherosclerosis. It was demonstrated that the administration of recombinant IL-33 in mice may reduce the number of cardiomyocyte deaths, it might decrease plaque burden and secondary infarct size, and it might reduce cardiac remodeling.71–73 Also, in men, the IL-33/sST2 pathway may inhibit the development of atherosclerosis, and sST2 administration may improve systolic function.74,75 Thus, the clinical use of IL-33/sST2 may be soon a very useful diagnostic and prognostic biomarker in many cardiovascular diseases. Starting from the suggested benefit of IL-33 in some cardiovascular pathologies further work is needed to confirm these results in order to be able to use this cytokine as a valuable instrument for the prevention and treatment of cardiac diseases.76
The cardiovascular system is markedly influenced by IL-4, IL-13 and IL-31, these cytokines playing various roles (sometimes at the opposite poles – anti-inflammatory and pro-inflammatory) with (pro)fibrotic activities which in turn influence the cardiac remodeling processes; IL-4 increases the number of fibroblasts and macrophages in a damaged site and is involved in hypertension (systemic and pulmonary), cardiac dysfunction and ventricular enlargement; IL-13 inhibits atherogenesis, is involved in cardiac wound healing, remodeling and hypertension (pulmonary), while IL-31 plays a role in (left) ventricular hypertrophy and Kawasaki disease, regulating in a positive way the remodeling of the heart.
Dermatology
The role of interleukins in dermatology is vast, having relevance in a large number of pathologies, extending from the neoplastic ones to immunological ones. We can witness the dysregulation of inflammation manifesting as dermatoses that come in all shapes and sizes; sometimes, this can be highlighted by different dermatoses appearing in the same patient, on a similar inflammatory substrate.77 Local inflammation may also lower the threshold for the development of a lesion, a good example being what it is seen in the Koebner phenomenon.78 Treatments aimed at interrupting this inflammation are often employed successfully in such diseases.79 Likewise, inflammation is a substrate for the appearance of various types of skin cancer.80,81 Melanoma is one of the most severe neoplastic diseases encountered in dermatologic practice, and it often poses a diagnostic challenge due to possible similarities with benign lesions, usually forcing excision for proper diagnosis.82 Still, the therapeutic approach in this disease is often an even greater challenge. Below we explore the key roles interleukins play in these diseases, and what possible therapeutic options they might create. IL-4 may have a positive immunomodulatory role in melanoma, with overall protective effects. Direct activation of IL-4 receptors in cultured melanoma cells demonstrated significant reduction in the size of the cell-cultured tumor, along with changes in tumor antigen expression.83 Melanoma has some of the highest concentrations of IL-4-α receptor when compared to other solid tumors. The in vitro stimulation of this receptor with recombinant human IL-4 (rhIL-4) inhibits cell growth in a dose-dependent manner. Furthermore, IL-4 overexpressing transgenic mice were injected with B16F10 melanoma cells and were compared to non-transgenic mice with the same tumor cell line. The overexpressing mice were observed to have decreased tumor volumes and weights as compared to the non-transgenic mice, showing that IL-4 stimulation has a protective effect in vivo as well. The proposed mechanism for IL-4ʹs role in melanoma is highlighted through: the upregulation of p53 and p21; the activation of the JAK/STAT6 pathway; upregulation of proapoptotic proteins, including bax, caspases −3, −8, and −9; and through the downregulation of cell survival protein bcl-2.84 While this beneficial effect of IL-4Rα activation appears to show promise in opening the way for new and innovative treatments for melanoma, a Phase 2 clinical trial using rhIL-4 to directly treat human subjects was unable to demonstrate a significant benefit in terms of survival and tumor growth. Thirty-four eligible patients were treated with 5 μg/kg/day rhIL-4 for two 28-day cycles with a 7 day rest period in between cycles. Complete reduction was witnessed in one patient, 27 had progressive disease, 2 remained stable, 1 had a premature death related to melanoma, and 3 were lost during the assessment. While the response rate was limited, the study mentions that the administering route may have been inadequate for delivering IL-4 at the local level of the tumor. Direct treatment with rhIL-4 was poorly tolerated by the subjects, leading to 4 patients completely dropping out of the study, while 11 had to receive dose reductions. The one patient who did benefit, seemed to have a long-lasting remission at 16 months, when contact was lost.85 Further experimental studies using different administering routes need to be undertaken, before IL-4 therapy can be completely dismissed as a possible treatment in melanoma.
IL-13 seems to play a role in the increased proliferation of certain tumors, including gliomas, melanoma, thyroid, breast, pancreatic, and ovarian cancers.86 Certain subtypes of these tumors express an alternate form of interleukin-13 receptor (IL-13Rα2) which is associated with increased tumor proliferation, immune escape, and poorer prognosis. In the case of melanoma, the frequency of this mutation appears to be around 7.5%, as estimated by Okahamoto et al, in a study using the immunohistochemical staining of 187 primary melanomas. It was well preserved in the metastases of these tumors, with 90% of metastases being positive in the one studied patient identified with IL-13Rα2+ variant melanoma. This mutation appears to increase tumor size by promoting neovascularization of tumor tissue, possibly through the upregulation of amphiregulin.87 The receptor is relatively specific to malignant tumors, as it is not expressed in healthy tissues outside of spermatocytes, and thus it is an excellent target for future oncologic therapy. Some authors have observed that inflammation can induce IL13Rα2 expression, but this appears to be a pathologic manifestation related to certain diseases.88 IL-13 plays an important role in immune regulation, and its direct inhibition would likely be undesirable. Targeted therapy against the mutant receptor, however, remains an option. Many of the following treatments are still in their experimental phases and their application in the treatment of melanoma has not yet been tested, but the rationale for their use in melanoma parallels that of other solid tumors that express IL13Rα2, such as glioblastoma multiforme, where its association with a poor prognosis is better established. These therapies include: anti-IL13Rα2 antibodies, which could bind to the mutant receptor and activate the complement system, deliver pseudomonas exotoxin into the tumor, or bind titanium oxide microdiscs which shear the tumor through mechanical forces when exposed to an alternating magnetic field; autologous dendritic cell vaccines that have been exposed to IL-13Rα2; and chimeric antigen receptor-modified cytotoxic T cells.89
Interleukin-31 is secreted mainly by activated CD4+ Th2 cells and mast cells. Only one receptor is known (IL-31R) which is present on a diverse group of both immune and non-immune cells. Activation of this receptor causes actions exerted downstream via the JAK/STAT, ERK/MAPK and PI3K/AKT pathways. IL-31 induces the release of chemokines CXCL 1 and 3, and CCL2, 7, 13, and 15; upregulates matrix metalloproteinases 1, 3, 7, and 25; upregulates IL-6, 16, and 32; and causes the release of VEGF and EGF. It has been studied for its involvement in maintaining pruritus, dermatitis, and psoriasis. Mouse models have demonstrated that upregulation of IL-31 produces an atopic dermatitis-like phenotype.89 IL-31 and IL-4 levels are significantly increased in patients with psoriasis and atopic dermatitis who present pruritus when compared to healthy control groups. This appears to be a unique cytokine pattern related to pruritus. Different patterns were observed in the patient groups depending on their respective pathology, with IL13/33 being decreased in psoriasis patients, while the inverse was true for atopic patients.90 The interleukin 4/13 pattern appears to be central to the pathogenesis of atopic dermatitis. Indeed, IL-4 and IL-13 share a common receptor chain and overlap in their signaling pathways where they can both activate type 2 IL-4R. Their individual effects are primarily due to the ratio of expression of type 1:type 2 IL-4aR complexes on their target cells. Non-hematopoietic cells primarily possess type 2 receptors, while type 1 is preferentially expressed in leukocytes. Myeloid cells contain both type 1 and type 2 receptors and are therefore responsive to both interleukins.91 In atopic dermatitis, IL-13 appears to stimulate epidermal hyperplasia, while IL-4 induces chemotaxis of eosinophils. Both IL-13 and IL-4 must work together synergistically to increase lymphocyte and macrophage proliferation. Knock-out mice studies have shown that while IL-13 is the main contributor to the pathogenesis of atopic dermatitis, IL-4 is necessary for upstream polarization of Th2 cells. In its absence, IL-13-dependent effects are dampened. However, it seems that activation of alternative IL-13 induction pathways via IL-33, IL-25, and TSLP will produce a similar clinical phenotype.92,93 This opens up the possibility of IL-13 targeting, such as with tralokinumab, as treatment for atopic dermatitis. This is a recent medication, but it has shown promising results in clinical trials for the treatment of atopic dermatitis.94 Dupilumab, an IgG monoclonal antibody that targets the alpha subunit of the IL-4 receptor, and thus inhibiting both IL-4 and IL-13 pathways, has been approved for use in the treatment of severe atopic dermatitis. There is strong evidence that it decreases the impact of atopic dermatitis, with significant improvements seen in objective clinical severity scores, quality of life, reductions in pruritus, and histological studies.95 IL-4 and IL-13 binding to IL-4α leads to phosphorylation of Janus Kinases 1 and 3, which, through a cascade of downstream tyrosine protein phosphorylation events, activate signal transducer and activator 6. This upregulates cellular transcription in those immune cells, responsible for the pathologic events seen in atopic dermatitis. This pathway is probably centrally responsible for the manifestations seen in atopic dermatitis. Inhibitors of the JAK/STAT6 pathway, such as leflunomide and ruxolitinib, may be employed with some success in treatment.96 Ruxolitinib even has the advantage of being formulated for topical administration, and may even be more effective than some topical corticosteroids in the treatment of atopic dermatitis, further highlighting what an integral part the IL-4/IL-13 pathway plays in this disease.97 IL-32 agonism may also have a role in the treatment of atopic dermatitis. IL-32 appears to control inflammation in atopic dermatitis by reducing inflammatory cell chemotaxis, inhibiting miR-205 expression, suppressing NF-κB activation, and decreasing TNFα, IL1, and IL-6 signaling. Mice induced to express an atopic dermatitis phenotype with the help of phthalic anhydride, were treated with recombinant human IL-32γ. The mice demonstrated histologic improvement at 4 weeks with decreased epidermal thickening and reduced leukocytic infiltration.98 It is therefore important to recognize the nuance that inflammatory cytokines play in dermatological diseases, with a potential to act as both aggravating and relieving factors. Future therapies that interrupt or accentuate these pathways will probably see development in the coming years. There is an increasing demand for newer therapeutic agents which help fight various diseases through these cytokines: drugs which employ IL-4 (and possibly make use of its receptors) may fight against melanoma, inhibiting its growth; tumor growth inhibition could also be obtained by using anti-IL-13 agents, in tumors such as gliomas, melanoma and cancers of the breast, thyroid, pancreas and ovaries; not only anti-IL-13 medication could be beneficial for patients suffering from atopic dermatitis, but the entire IL-4/IL-13 axis; IL-31, being responsible for pruritus, psoriasis and other skin diseases is a future target for medication.
In 2020, IL-31ʹs receptor targeting, which was considered a key element in (moderate-to-severe) prurigo nodularis treatment, encountered clinical success in a randomized clinical trial: patients received subcutaneous nemolizumab (a human monoclonal antibody) and after 4 weeks their pruritus score improved (as compared to placebo) and after 12 weeks, some lesions subsided.99 Pruritus in prurigo nodularis and IL-31 have long been considered for therapy targeting; also, IL-31 is involved in the development of pruritus not only in the latter disease, but also in other dermatological diseases which present with pruritus, such as: atopic dermatitis or psoriasis. Patients suffering from pruritic diseases have been found to have elevated serum levels of IL-31 and its respective receptor.100
Rheumatology
The accumulation of proinflammatory cytokines plays a central role in the pathogenic chain of systemic sclerosis (SSc).101 SSc is an autoimmune disease with an inflammatory substrate that evolves into extensive multiorgan fibrosis.102 The pathogenesis of SSc involves an immunological alteration followed by vasculopathy and cutaneous and visceral fibrosis.106 Endothelial lesions appear to be the origin of “cascading” events, which end with the accumulation of cytokines and chemokines with a proinflammatory potential, along with a number of growth factors that promote fibrogenesis. The mechanisms of the fibrotic process are not clear, but the development of fibrotic lesions seems to be conditioned by the chronic inflammation present in SSc.101 Inflammation is maintained by the marked expression of cytokines: IL-17A, IL-4, IL-25, transforming growth factor (TGF) β1.102 Activated inflammatory cells accumulate in the damaged vascular endothelium and turn into myofibroblasts that expose α-actin smooth muscle (α-SMA).101 At the origin of this disease one finds an endothelial cell lesion, activated in the context of overexpression of surface selectin E and adhesion molecules: ELAM-1 (endothelial leukocyte adhesion molecule-1), VCAM-1 (vascular cell adhesion molecule-1) and ICAM-1 (intercellular adhesion molecule-1). Leukocytes are chemotactically activated by activated endothelial cells. Through diapedesis, CD4 + Th lymphocytes pass into the perivascular space and constitute inflammatory infiltrates.103,104 Leukocytes attach to the endothelial wall, activated by a ligand, P-selectin. Monocytes and macrophages adhere to endothelial cells via L-selectin and integrin α-2 molecules present on their surface. Transforming growth factor β (TGFβ), produced in excess by mast cells, activates dermal fibroblasts.105 The substrate of endothelial alteration is based on the production of anti-endothelial antibodies and on the existence of anti-ICAM-1 antibodies.106 Hyperactivated endothelial cells provide connective tissue growth factors and interleukins such as: IL-1, IL-4, IL-6, IL-8, TGFβ, TNFα. All of these cytokines, and especially TGFβ, induce hyperactivation of the fibroblast responsible for excessive fibrosis. PEDF (pigment epithelium-derived factor), normally present in the epidermis in the germ layer, has been identified both perivascular and in resident fibroblasts at the base of the reticular dermis.107,108 IL-4 is responsible for promoting proliferation, extracellular matrix deposition, and chemotaxis in fibroblasts. It is elevated in the blood of patients with systemic sclerosis. In particular, an alternative variant, IL-4δ2, seems to be related to this disease state. This RNA variant is normally present in other tissues including the thymus, lung, gut, and placenta. An increased ratio of IL-4 to IL-4δ2 mRNA is, however, associated with a variety of rheumatic pathologies, including SSc, rheumatoid arthritis, and possibly asthma. The transcribed IL-4δ2 protein may act as an antagonist of IL-4 via competitive inhibition on its receptors in a variety of immune cells, including multiple subtypes of lymphocytes and of macrocytes. In contrast, it seems to have an analogous role to IL-4 in fibroblasts.109 This, therefore, may be a key mediator to the aberrant deposition of collagen seen in SSc, having both immunomodulatory and fibrogenic activities. Anti-IL-4 therapy has been proposed in SSc. Neutralizing IL-4 antibodies when given to tight-skin mice inhibit the deposition of dermal collagen.110 Further human studies would be required to clarify whether IL-4 could be used in the treatment of SSc. Studies have also noted the involvement of IL-13 in the pathogenic chain of SSc. In this regard, a group of researchers have identified the molecular mechanism by which CD8 + T lymphocytes produce IL-13. They analyzed the confocal images of blood samples, and those of hardened skin. The thickened skin samples were pre-frozen, then stained by immunofluorescence techniques. Subsequently, CD8 + T lymphocytes were isolated from the fibrous skin and blood samples. In the early stages of SSc, fibrous skin contains elevated levels of CD8+ T lymphocytes that produce IL-13. CD4+ T lymphocytes also synthesize IL-13, but to a lesser extent. For these reasons, it was observed that IL-13 generating CD8+ T lymphocytes play a key role in excessive tissue fibrosis.111 Moreover, it has been observed that elevated levels of IL-13 producing CD8+ T lymphocytes in fibrotic tissue are associated with a high level of GATA-3 transcription factor. In other words, these lymphocytes, which overexpress the transcription factor, produce an excess of IL-13 that precedes extensive fibrosis. Experimental deletion of small interfering RNA (siRNA) was followed by IL-13 synthesis inhibition. The authors analyzed the molecular process in depth and noted that the T-bet transcription factor modulates the manifestation of GATA-3 factor. In turn, GATA-3 regulates the differentiation of T helper cells and modulates the expression of several cytokines: IL-4, IL-5, IL-13.111 Although the authors believed that the T-bet factor does not regulate GATA-3, later in 2018, they observed that the T-bet transcription factor canceled GATA-3, and through IFNγ transcription, is able to block the synthesis of IL-13. They concluded that in fibrous skin and in the blood of patients with the diffuse subset of SSc, CD8 + T lymphocytes overexpress GATA-3 transcription factor and induce a high level of T-bet transcription factor.111 Early endothelial injury induces microvasculopathy by reducing the number of capillaries. Neoangiogenesis and a faulty vascular repair mechanism affect both blood capillaries and lymphatic vessels, leading to edema.112 Newer studies have shown that angiogenesis is promoted by high levels of endothelin-1 (ET-1). Also, it was observed that microangiopathy is associated with a high level of vascular endothelial growth factor A (VEGF-A). Excessive fibrosis is subsequent to vasculopathy and is induced by fibroblast activation. Therefore, the fibroblast initiates an aberrant production of type I collagen that deposits in the skin and viscera.109 Excessive collagen deposition is responsible for the hardening of the skin.113 IL-31 plays a role in SSc by direct stimulation of a variety of skin cells, including macrophages, fibroblasts, vascular endothelial cells, and mesenchymal fat cells. Increased levels of this interleukin have been observed in both skin biopsies and blood samples of SSc patients. When stimulated by IL-31, fibroblasts undergo a phenotypic change that increases their proliferation independent of the TGFβ pathway. Collagen protein deposition is also increased via the STAT3 pathway. IL-31 also appears to mediate vascular inflammation and contribute to the aforementioned vasculopathy.114 IL-31Ra seems to be upregulated in surrounding tissue cells during the SSc disease state, showing that this axis is likely stimulated in an aberrant manner.115 IL-33 has been studied as a disease marker in SSc. It follows the disease state of SSc tightly, with increases in affected individuals, and whose drops correlate well with treatment efficacy.116 Certain gene polymorphisms, such as the rs7044343 that encode for IL-33 may lead to more severe diseases.117
Ankylosing spondylitis (AS) is an immune-mediated chronic inflammatory rheumatic disease that strongly associates with the HLA-B27 allele. The role of HLA-B27 in the pathogenesis of AS is not very clear. One of the theories issued, the theory of arthritogenic peptides, supports that HLA-B27 has a key role in directing joint-specific peptide fragments to cytotoxic CD8+ T lymphocytes. HLA-B27 binds specific peptides and presents them to activating CD8+ T lymphocytes. This theory cannot be fully supported because arthritogenic peptides have not been identified in the joints, as is the case in reactive arthritis caused by Chlamydia. Most likely, HLA-B27 modifies the person’s microbiome.118 IL-4 attenuates the manifestation of ankylosing spondylitis. Arthritis has been observed to be relieved by IL-4 agonism, whereas anti-IL-4 antibodies appear to worsen symptoms.119 The reasons why, are less clear. When IL-4 is injected intra-articularly in proteoglycan-induced arthritis mice models, there is a decrease in both clinical and histologic changes associated with arthritis. It’s possible that monocyte/macrophage polarization plays a role in this, as IL-4 appears to promote the phenotypic shift from M1 to M2 macrophages both in vitro and in mouse models.120 While the clinical utility of IL-4 agonism is still under investigation, IL-4 may not play a key pathogenic role in the development of AS, as levels of IL-4 levels appear to not be significantly increased in these patients, as compared to those of healthy controls.121 IL-33 may also play relevant roles in AS. Both the concentrations of IL-33 and of its decoy ligand, sST2, were markedly higher in serum samples of patients with AS, as compared to controls when measured by ELISA (P < 0.0001).122 The significance of IL-33 elevations in AS is less well established. It may act to promote angiogenesis and sustain inflammation locally. One possible clinical utility would be in the differential diagnosis with psoriatic arthritis, which is often difficult as both diseases affect the axial skeleton and are associated with the HLA-B27 genotype. Here, elevated serum IL-33 could be used to plead for the occurrence of ankylosing spondylitis.123
Similar to SSc, the pathogenesis of SLE is heterogeneous. Stimulation of the immune system under the action of antigens (environmental factors or infectious agents) activates B lymphocytes and T lymphocytes. Antigen presenting cells recruit these antigens and expose them to T lymphocytes. Therefore, T lymphocytes induce the synthesis of cytokines (IL-6, IL-10, IL-12, IL-23) which leads to an increase in the number of monocytes and lymphocytes. On the other hand, high levels of IL-17 and IL-21 induce an overproduction of T lymphocytes. At the same time, the increase of IFNγ in SLE induces defective T lymphocytes. High concentrations of interleukins activate B lymphocytes to plasma cells that produce elevated autoantibody titers characteristic of SLE. The presence of autoantibodies causes the deposition of circulating immune complexes and complement fractions in various organs, causing damage. B lymphocytes also function as antigen presenting cells to the T lymphocytes they activate. In other words, the activation of T and B cells is reciprocal, self-sustaining and amplifies the autoimmune process. And in the case of SLE, interleukins maintain the inflammatory process and activate autoimmune mechanisms.124 Overall, serum levels of IL-4 are generally reduced in lupus patients. However, increases of IL-4 expressing Th lymphocytes are seen in the dermal and epidermal biopsies of SLE patients.119 This incongruence has led to some speculation as to what IL-4ʹs mechanism is in SLE. It may act to promote tissue fibrosis, synovial proliferation, osteoclastic bone resorption, the production of autoantibodies from B-cells, and a decrease the immunomodulatory role of T-cells. One interesting observation has been that neutralizing IL-4 with antibodies leads to delays in end-stage renal disease in patients with glomerulosclerosis, as a consequence of lupus nephritis.125,126 It is therefore reasonable to conclude that localized IL-4 elevations may still contribute to the end-organ damage seen in SLE, but this is not reflected in the serum levels of patients. At the same time, a study from 2002, by Sugimoto et al, found that a cytokine profile which favored a higher number of IL-4 producing cells was registered in patients who also suffered from SLE renal involvement.127 A more recent research developed on Wild-type/IL-4 knockout BALB/c mice found that IL-4 has a central role in developing lupus-like disease and IL-4 inhibition in human lupus might improve the clinical aspects of this disease.128 IL-13 is a clearer contributor to the inflammation seen in SLE, with marked elevations seen in affected patients, especially in those with lupus nephropathy.129 An activating genotype of IL-13, rs20541, also carries with it an increased risk of developing SLE.130,131 Similarly activating IL-31 mutations rs4758680 G/T and rs7977932 C/G also carry an increased risk of developing SLE. This is possibly due to IL-31ʹs ability to increase inflammatory IL-6 release from inflammatory cells.130,131 Increasing evidence, such as the 2011 study by Yang et al, supported by Moreau et al in 2021, has revealed that IL-33 plays an important role in the development of SLE (in the acute phase), being significantly increased in these patients’ serum.132,133 However, IL-33 acts as a double-edged sword, having also protective properties in what concerns the development of SLE, as demonstrated in a young mice population study from 2020; further research needs to be done in order to explore this protective role.134
The pathogenesis of rheumatoid arthritis (RA) takes place in stages and is triggered by a series of environmental factors that can act on a predisposing genetic field. Following the citrullination process, proteins behave like an antigen and the immune system loses its tolerance. “Self” becomes “non-self”, and as a result, B lymphocytes are activated and synthesize an increased titer of cyclic citrullinate peptide (CCP) antibodies. The inflammatory infiltrate comprises many macrophages, mast cells, natural killer (NK) cells, T lymphocytes, B lymphocytes, plasma cells and dendritic cells. Macrophages, T lymphocytes, and B lymphocytes release chemokines, matrix metalloproteinases (MMPs) and cytokines (TNFα, IL-6, IL-1, IL-15, IL-17) that promote chronic inflammation of the synovial membrane in the joints.135 IL-4 acts as an anti-inflammatory cytokine in rheumatoid arthritis. Mouse models show that knockout of IL-4 leads to increased arthritic changes due to loss of regulatory control over proinflammatory mediators IL-1, IL-12, TNFα, IFNγ, and VEGF.119,136 This joint inflammation is reduced when recombinant IL-4 is administered, possibly due to a switch of Th1-type response towards a predominantly Th2-type that acts as a positive feedback loop sustaining increased IL-4 signaling.137 IL-4 also has an inhibitory effect on B cell development, and thus can decrease the citrullination process described above.119 In a phase two clinical trial, patients treated with methotrexate were compared to a treatment group receiving a low dose combination of IL-4, IL-10, and anti-IL-1 antibodies. Disease activity was measured using standardized clinical scales, alongside with the assessment of inflammatory markers (ESR, CRP), and subjective pain in both groups, over the course of year. No statistical significance was found between either group, in any of the categories, showing that these anti-inflammatory signaling molecules may be effective in the treatment of RA.138 Contrary to what has been discussed so far, increased serum and bronchial fluid levels of IL-4 have been associated with lung fibrosis in RA. However, these IL-4 levels were significantly increased in those with pulmonary fibrosis, as compared to both patients with RA, but without lung fibrosis and controls, and there was no difference in IL-4 levels between those with RA without fibrosis and the controls. IL-4 may therefore be a marker of this disease, but likely results as a response to the uncontrolled inflammatory response rather than from its pathogenesis.139 IL-13 has an analogous anti-inflammatory effect to IL-4 in RA, but whose increased level is associated closely to the disease state. Decreases in IL-13 levels are also concurrent with an improved clinical response to TNFα inhibitor medication in these patients.140 This cytokine therefore has the potential to become a marker for disease activity. One study even suggests a cutoff value of 10.73 pg/mL as an accurate diagnostic indicator of early RA, and finds it more accurate at predicting disease activity than either CRP, ESR, or anti-CCP antibodies alone.141 Increased cytokine signaling via the IL-33 axis has been implicated in RA as well. IL-33ʹs ligand, ST2 is primarily located on mast cells, Treg cells, and eosinophils, where it tends to stimulate these cells to increase protein synthesis, multiply, or release other cytokines. This proinflammatory activity may contribute to the pathogenesis of RA. High levels of IL-33 are associated with increased RA severity, whereas administration of ST2 inhibitors reduces articular inflammatory infiltrates, bone erosions, and synovial hyperplasia.141 Indeed, some authors have even suggested that this cytokine, through its alarming role, may be the triggering mechanism for the development of RA.143 An interesting observation has been demonstrated with Rituximab and Tocilizumab, where higher pretreatment serum levels of IL-33 may predict a positive response. This result has not been observed with TNFα inhibitors.117
IL-4 is involved in SSc (in the excessive fibrotic skin response), in AS (by inhibiting the clinical manifestations of the disease), in SLE, where it promotes fibrosis, synovial proliferation and osteoclastic bone resorption; in RA it seems to have anti-inflammatory properties. IL-13 is also involved in SSc, in the pro-fibrotic response, in SLE where patients have increased serum values corresponding to the disease activity (with high titers in the acute phase of the disease), and in RA, with effects analogous to those of IL-4.
IL-31 acts as a stimulating agent in SSc, with effects on macrophages, fibroblasts, endothelial and adipose cells, effectively promoting excessive fibrosis. IL-33 is involved in SSc, acting as a disease marker with values evolving parallel to the disease, in AS, where it sustains local inflammation and promotes angiogenesis, and in RA, where it act as a pro-inflammatory factor, with high levels reflecting the disease severity.
Neurology
A large number of cytokines are known to play an important role in neuroinflammation and in an array of neurological diseases. While IL-4 and IL-13 have been intensively studied for their role in the immune regulation of the CNS, more recently discovered IL-31 and IL-33 emerge as potential biomarkers and therapeutic targets in CNS afflictions. We further summarize the current available evidence on the implications of these cytokines in specific neurological conditions.
IL-4 is one of the most important cytokines in neuroinflammation and it can exert either pro- or anti-inflammatory effects in specific neuronal populations. Overall, low levels or absence of IL-4 seem to increase vulnerability to neuroinflammation.5 A very intriguing study by King et al found that patients with mild cognitive impairment (MCI) preceding Alzheimer’s disease (AD) and dementia with Lewy bodies (DLB) have high levels of peripheral IL-4, while patients with established dementia showed no difference to controls.144 Furthermore, the study evidenced that lower IL-4 levels correlated with a more severe disease course. These findings indicate that neuroinflammation, with IL-4 as one of the major mediators, plays an important role in early cognitive neurological diseases’ pathogenesis. Multiple studies have identified that mice with absent or suppressed adaptive immunity exhibit cognitive impairment and decreased learning abilities, as well as reduced neurogenesis. After performing specific cognitive tasks, these mice exhibited increased IL-4 production in meningeal T-cells. Interestingly, the cognitive defects have been shown to be reduced by restoration of the T-cell population.144,145 Moreover, aging mice with cognitive impairment or AD models demonstrate improvement in neuronal connection strength, memory and learning tasks, after IL-4 administration, either direct or as genes via a viral vector.144 A study from 2020, by Dawling et al, supports the positive role that IL-4 injections have on cognitive improvement in AD mice models by activating microglial changes in such neurodegenerative diseases.146
IL-13 and IL-4 share a common alpha chain complex receptor, formed through heterodimerization of IL-4R alpha chain (IL-4Rα), with IL-13 receptor alpha 1 chain (IL-13Rα1), thus the role of IL-13 in memory and cognitive impairment having also been studied. The study of Brombacher et al demonstrated that IL-13-deficient mice have significant working memory impairment and reduced reference memory.147,148 The authors also suggest that both IL-4 and IL-13 can stimulate astrocytes from the meninges and hippocampus during complex cognitive tasks, thus exhibiting important targets for dementia disorders treatment. A more recent study from the same group showed that IL-4 and IL-13 deficient mice have significantly impaired learning and reference memory, while IL-4 alpha receptor deficiency only impairs reference memory, with sparing of spatial learning abilities.149 IL-33 is a more recently discovered cytokine pertaining to the IL-1 family with multiple roles in neurological diseases’ pathology. A study by Xiong et al identified through immunohistochemical studies that IL-33 and its receptor, ST2, are overly expressed in the vicinity of amyloid plaques and neurofibrillary tangles in the glial cells of patients with AD, as compared to controls.150 Later, serum and CSF IL-33 levels have been found to be lower in patients with mild cognitive impairment and Alzheimer’s disease. Moreover, the ST2 receptor levels are higher in the same patients.151–154 Genetic studies in the Chinese Hunan Han population have identified that a specific single nucleotide polymorphisms (SNPs) in the IL-33 gene was associated with late-onset AD, while a larger study in a Caucasian population further identified two SNPs associated with increased AD risk.154,155 In animal models, similar observations were confirmed and studies established that IL-33 expression in astrocytes increases with age and aging. IL-33 deficient mice display a disease course similar to AD, with increased neurodegeneration and abnormal tau deposition. Interestingly, the administration of IL-33 in animal models improved memory and learning impairments, increased amyloid plaque clearance and reversed synaptic plasticity deficiencies.151,153–156 A more recent study by Reverchon et al observed that IL-33 injections into the hippocampus of mice induced increased inflammatory responses and spatial memory impairment.5 This induced overexpression of IL-1β in glial cells was registered within the first two days after injection, but IL-1αβ deficient mice were resistant to cognitive decline.156 All the aforementioned findings highlight the versatile modulating role of IL-33 in the brain, with positive or negative influence on cognitive impairment and neurodegeneration.
A recent study by Yan et al identified significantly higher levels of Th2 cells producing IL-4 and a significant decrease in naive B cells levels in patients with Parkinson’s disease (PD) as compared with controls.157 Previous studies have suggested the cytotoxic effects of IL-4 and IL-13 on dopaminergic neurons in the brain, with increased survival upon administration of IL-4 antibodies.148,158,159 This can be partially explained by the increased expression of IL-13Rα1 receptor, a component of the common alpha chain complex receptor mediating both IL-4 and IL-13 functions, in the substantia nigra and ventral tegmental area neurons, which are preferentially affected in PD. Moreover, mice with IL-13Rα1 deficiency demonstrated resistance to dopaminergic neuron loss during chronic inflammation, further supporting previous evidence.148,158,160 In humans, genetic studies have placed the genes encoding this receptor on the X chromosome, within the PARK12 region, known to be a risk factor to PD development.148,160,161 More recently, Aguirre et al identified in humans two rare SNPs in IL-13 and IL13RA1 genes that were related to early onset PD and increased the cytotoxicity of IL-13 on human neurons in vitro.161 This data highlights the potential role of both IL-4 and IL-13 as therapeutic targets in Parkinson’s disease. Additionally, IL-33 was identified as a possible contributor to neuroinflammation in PD development, in a few in vitro studies. Moreover, increased expression of IL-33 in the midbrain and striatum was found in patients with PD as compared to controls.153
In a mouse model of amyotrophic lateral sclerosis (ALS), IL-4 administration induced microglia proliferation with a skew towards embryogenic traits, resulting in improved locomotion and slower disease progression. However, no improved motor neuron or overall survival was observed, suggesting a more complex interplay of cytokines in ALS.162 Multiple studies have identified that serum IL-33 levels are significantly reduced, and that the ST2 receptor levels are increased in patients with ALS, in contrast to healthy controls.151,153,154 Interestingly, long-term IL-33 administration to transgenic mice with G93A-superoxide dismutase-1 mutation, specific for ALS development, induced delayed disease onset only in females.163
Current available data suggest that IL-4 plays a neuroprotective and anti-inflammatory role in models of experimental autoimmune encephalomyelitis (EAE).5,164,165 Some studies showed that IL-4 deficit in the CNS of mice led to increased neuroinflammation and a more severe clinical course of the disease.164,165
IL-13 was successfully identified in the active lesions in the spinal cord of multiple sclerosis (MS) patients and a positive correlation between IL-13 levels and EDSS scores in relapsing-remitting MS was established.166 Other studies observed increased IL-13 production in patients with glatiramer acetate treatment. In animal models, it was demonstrated to have anti-inflammatory effects in EAE mice alone, or in combination with IL-4. A few studies on mice also suggest that IL-13 expression and function might be influenced by gender, with possible implications in the higher female prevalence of MS.148 Studies have identified high levels of IL-33 and ST2 receptor in the plasma and in the brain and spinal cord lesions (both acute and chronic) of MS patients, as compared to controls. The suggested function of IL-33 was to modulate both the immune response as well as CNS myelination.149,151,154,167 In MS patients with IFN-β-1a treatment plasma IL-33 levels were reduced.155 In some animal models, higher endogenous IL-33 production delayed EAE onset and reduced disease severity.151,154 In other studies on mice, IL-33 antibody administration produced a similar effect, while treatment with IL-33 increased neuroinflammation and worsened the disease course of EAE.153,154 The exact effect of IL-33 on the remyelination of MS lesions is also controversial, some studies indicating inhibition of CNS myelination, while others indicate favoring of myelin repair.153–155,168
A few studies have identified higher expression of IL-33 in the serum and CSF of patients with neuro-Behcet disease (NBD) (in comparison with patients suffering from non-inflammatory neurological diseases), as well as an association with higher levels of specific chemokines.153,154 However, the exact role of IL-33 in NBD needs further clarifications.
A recent study by Ygberg et al demonstrated that the immunological response to a viral infection matures during childhood and adolescence.156 In this study, CSF expression of IL-4 and IL-13 was found to increase with age in response to viral infections, suggesting their importance in the risk of developing of long-term sequelae.168 In their study, Silva et al found that IL-4 knockout mice with experimentally induced neurocysticercosis had lower cerebral edema, lower inflammatory infiltrates and prolonged survival of the parasites in the brain.169 This indicates that IL-4 is preferentially expressed in response to infections in an attempt to reduce brain damage, possibly explaining the previous observations that asymptomatic patients from endemic areas exhibit increased production of IL-4 and IL-13.
IL-33 has been more extensively studied in different types of infections.151,153,154,170–173 In cerebral malaria models, IL-33 levels are increased and IL-33 treatment reduced the parasitic load in the early phase of the infection.151,154 Moreover, ST2 receptor deficient mice did not develop the disease, had a longer survival and displayed no neurological signs or no cognitive impairment in comparison with wild type mice.171,173 Multiple studies on ST2 receptor deficient mice infected with Toxoplasma gondii observed an increased susceptibility to infection, increased parasite load and encephalitis symptoms.138,140,142 These findings suggest that IL-33 induces a more severe neuroinflammatory response to parasitic infections and leads to the emergence of brain lesions and neurological signs. One of the proposed mechanisms for IL-33 attenuation by parasitic or viral-induced encephalitis is through the downregulation of the inducible nitric oxide synthase (iNOS) expression in the brain.172 In the presence of bacterial endotoxins, IL-33 production is increased, while deficits fail to stimulate microglia to produce a proper neuroinflammatory response. In other studies, IL-33 was positively correlated with IL-1b expression, with implications in the pro-inflammatory response to viral infections such as Zika or HIV.153
Early studies of IL-4 and IL-13 have discovered increased production in the microglia of the mouse models in response to experimental ischemic lesions, in an attempt to restrict the neuroinflammatory response and neuronal damage.148,174 An interesting study by Kolosowska et al studied the effects of exogenous IL-13 administration in mice with middle cerebral artery occlusion during the acute phase. The authors conclude that early IL-13 treatment determined a reduced inflammatory infiltration in the ischemic area and reduced lesion volumes, as well as a neuroprotective effect in the peri-ischemic area, with overall improvement of clinical symptoms at 14 days post-occlusion.174 However, another study demonstrated that IL-13 and IL-4 play no major role in the ischemic stroke outcome in mice with combined genetic deletion of IL-4, IL-5, IL-9, and IL-13.175 Studies on acute ischemic stroke patients have identified an increase in serum IL-33 and ST2 receptor and further established a negative correlation with lesion volume and patient outcome. In animal models, ST2 receptor deficiency resulted in larger ischemic lesions and more severe behavioral and neurological deficits on the long term. Consequently, many studies on exogenous IL-33 administration in mice found a strong anti-inflammatory effect after experimental ischemia, with reduced neuronal damage and reduced neurological deficits. Other studies even highlight a neuroprotective effect of IL-33 in atherosclerosis pathogenesis in mice. However, IL-33 does not directly exert its action on neurons or glial cells, but through the stimulation of IL-4 production in T-cells in the brain.151,153,154 These findings indicate that IL-33 can become a reliable diagnostic and prognostic marker in stroke patients, as well as an early treatment option to reduce long-term effects.
IL-4 and IL-13 are not considered major players in traumatic brain injury recovery.162 However, more recent studies on mice with TBI who received intranasal IL-4 or IL-13 found reduced neuronal tissue loss, improved white matter integrity and improved long-term functional outcomes, suggesting a more complex interplay than previously regarded and opening new treatment possibilities.176,177,178 In trauma animal models, IL-33 was found to be immediately released by damaged oligodendrocytes, with consequent influence on local astrocytes and microglia chemokine production. Moreover, IL-33 deficient mice demonstrated larger lesions and impaired recovery after TBI, whereas IL-33 treatment, after experimental TBI in mice, reduced myeloid cell infiltrates, tissue damage and demyelination, with an additional overall improved recovery by inducing Th2 responses and activating meningeal ILC2s. Thus, IL-33 is considered one of the alarms released after injury to activate both CNS and immune cells to promote recovery. In animal models with intracerebral hemorrhage (ICH), IL-33 was found to be predominantly produced in astrocytes and microglia around an intracerebral hematoma. Similarly to cerebral ischemia, IL-33 exerts a neuroprotective effect in mice with ICH, reducing neuronal death, brain edema and pro-inflammatory responses when exogenously administered.151,153,154 In a recent study on patients with spontaneous ICH, Miao et al found that higher levels of IL-33 were negatively correlated with initial NIHSS severity scores and hematoma volume, characterized better outcomes for patients and even established a cut-off level for poor prognosis.179 In contrast, in subarachnoid hemorrhage (SAH) models, studies identified high levels of both IL-33 and ST2 receptor in the CSF, along with increased levels of other pro-inflammatory cytokines, suggesting a potential neurotoxic effect of IL-33.154
A few studies have identified a relationship between IL-33/ST2 pathway and epilepsy.153,180–182 Plasma ST2 levels were found higher in patients with increased epileptiform abnormalities after SAH, as well as patients with focal temporal lobe epilepsy.153,180 Moreover, oxidative stress index, which is significantly higher in patients not responding to antiepileptic drug treatment, was positively correlated with IL-33 levels.182 Memory performance was also correlated with IL-33 levels in temporal lobe epilepsy.180 In an animal model of recurrent neonatal seizures, IL-33 administration improved learning, memory, and behavior deficits, as well as body weight gain secondary to seizures by inhibition of apoptosis, endoplasmic reticulum stress, and neuroinflammation.181 Thus, IL-33 seems to be a new, promising treatment option for reducing the negative impact of recurrent seizures.
IL-31 is a novel cytokine with the special function of connecting the immune system with the skin and the peripheral nervous system, by inducing pruritus behavior, but not directly skin inflammation. Interestingly, the infliction of pain mediated by the Y neuropeptide interneurons determines a reduction in itch perception induced by IL-31 in patients with atopic dermatitis.183,184 Studies on neurons from the dorsal root ganglions suggest that IL-31 might be involved in neuronal proliferation, survival, and metabolism.173 However, its importance in neurological conditions needs further research. IL-33/ST2 signaling has been observed to exert a stimulatory effect on sensory neurons, with an increase in the levels of both cytokine and receptor in inflammatory spinal cord lesions in mice. Studies have identified an important role of IL-33 in hyperalgesia induction and neuropathic pain through the modulation of TNF-α, IL-1β in animal models. Moreover, IL-33 is considered one of the inductors of inflammation responsible for cancer-related pain, making it a potential target for palliative treatments development.151,153,154
An interesting inverse relationship between allergies and glioblastoma multiforme (GBM) led to the investigation of IL-4 and IL-13. Cell cultures from patients with GBM seem to have a high expression of receptors for IL-4 (IL4Rα) and IL-13 (IL-13Rα2), the latter being found only in glioblastoma cells and presenting as a promising GBM biomarker. Other studies have identified the crucial role of IL-4 in tumor control through the mediation of angiogenesis and eosinophil recruitment, specific polymorphisms being correlated with better survival.5,148 Studies on IL-33 have deemed it a diagnostic and prognostic marker in specific types of cancer, including gliomas. Multiple studies have identified that the IL-33 and ST2 receptor levels are elevated in tumor tissues in comparison with normal brain tissue, and correlate with tumor progression and decreased survival.151,153,154 Other studies have identified that IL-33 recruit and activate a large array of immune cells, creating a favorable environment for tumor proliferation.154,185 A recent study identified that IL-33 significantly reduces the efficacy of temozolomide, an important chemotherapy agent for CNS tumors.186 Thus, the development of treatments targeting the blockage of IL-33/ST2 signaling pathway might improve glioma survival.
A very intriguing study by Thurmann et al determined that elevated IL-13 levels during pregnancy increased the risk of children to later develop behavioral problems, especially hyperactive/inattentive disorder. However, the elevated levels of IL-13 were found to occur secondary to an insufficient supply of polyunsaturated fatty acids.187 More studies are required to elucidate the importance of IL-13 levels during pregnancy. In addition to the previously described roles, IL-33 seems to promote the normal development of the nervous system. Some studies have identified that IL-33 is required for normal synapse numbers and circuit function in the thalamus and spinal cord, while others have highlighted its importance in oligodendrocyte maturation and myelination during the early postnatal period. The IL-33/ST2 pathway also seems to play an important part in sustaining the blood–brain barrier integrity and function. Interestingly, the IL-33 gene is one of the five genes that promote astrocyte differentiation in the human forebrain. In addition, a study on the behavior of IL-33 knockout mice revealed reduced anxiety behaviors and impaired social recognition, despite intact sociability, suggesting the involvement of IL-33 in the normal maturation of the forebrain.153,188,189
A recent review of the current literature by Pandolfo et al concluded that elevated IL-33 levels, in humans and animal models, correlate with a change of the phase of specific psychiatric diseases. The authors suggest that when a stressor arises in an individual with genetic susceptibility, an abnormal increase in IL-33 occurs and determines acute symptoms in schizophrenia and affective disorders, as well as the appearance of a new recurrence in major depression disorder.190 In patients with schizophrenia, IL-33 and ST2 levels have been positively correlated with cognitive performance, but there was difference in the levels of each molecule between patients and healthy controls. Genetic studies have revealed that specific IL-33 polymorphisms can determine a decreased risk of susceptibility to developing schizophrenia, as well as an increased risk of developing major depressive disorder.151,153,154 Previous studies on IL-33 deficient mice highlighted the appearance of anxiety and abnormal social behaviors, but the exact implications of IL-33/ST2 signaling pathway in schizophrenia or autism spectrum disorder remain unknown.151,188 We are still at the beginning of our understanding of the complex relationship between cytokines, neuroinflammation and neurological disorders, but the aforementioned studies have shed light on some pieces of the puzzle. IL-4, IL-13 and IL-33 seem to be a few of the important therapeutic targets of CNS diseases that need to be considered in further research.
IL-4 is a cytokine with pro- and anti-inflammatory effects in the CNS; high serum levels of IL-4 can be found in patients suffering from MCI, AD, DLB, PD. In ALS and EAE, IL-4 has positive effects (such as improving symptoms or a protective role). IL-4 and IL-13 both help in the immune regulation of CNS and are seen as targets for dementia treatment. IL-13 alone is linked to cognitive impairment and reduced reference memory when in low levels. In MS, IL-13 is involved in active lesions, while elevated levels during pregnancy are linked to behavioral problems in children; it also has neuroprotective effects in the peri-ischemic area of the brain in mouse models. IL-31 creates a favorable microenvironment for tumor proliferation. At last, IL-33 has low levels in the serum and CSF of AD patients, is increased in NBD, PD and MS patients. IL-33 is also involved in the maturation of brain tissue, and it activates immune cells for recovery in traumatic lesions.189,190
Conclusions
As we can see, interleukins are at the bedrock of a multitude of pathologies across different organ systems and understanding their role will likely lead to novel treatments and better outcomes for our patients. IL-4 promotes fibrosis, atherosclerosis, remodeling of tissues, as well as cognition. IL-13 promotes fibrosis, tissue remodeling, cognition, modulates neuroinflammation, and inhibits atherosclerosis. IL-31 promotes angiogenesis, pruritus, and tissue remodeling. IL-33 acts as an alarm in response to acute stress. The tables below (Tables 2 and 3) summarize the activities these interleukins play and their role in the aforementioned pathologies. As the interleukins rely on their preceding signaling molecules to accentuate their own signal, so too can we draw upon the works and knowledge of preceding researchers in order to further our own practice. New interleukins are being described, and the role of this inflammatory cascade is still coming to light. We hope this multi-discipline review on the role interleukins play in current pathology assists in this scope.
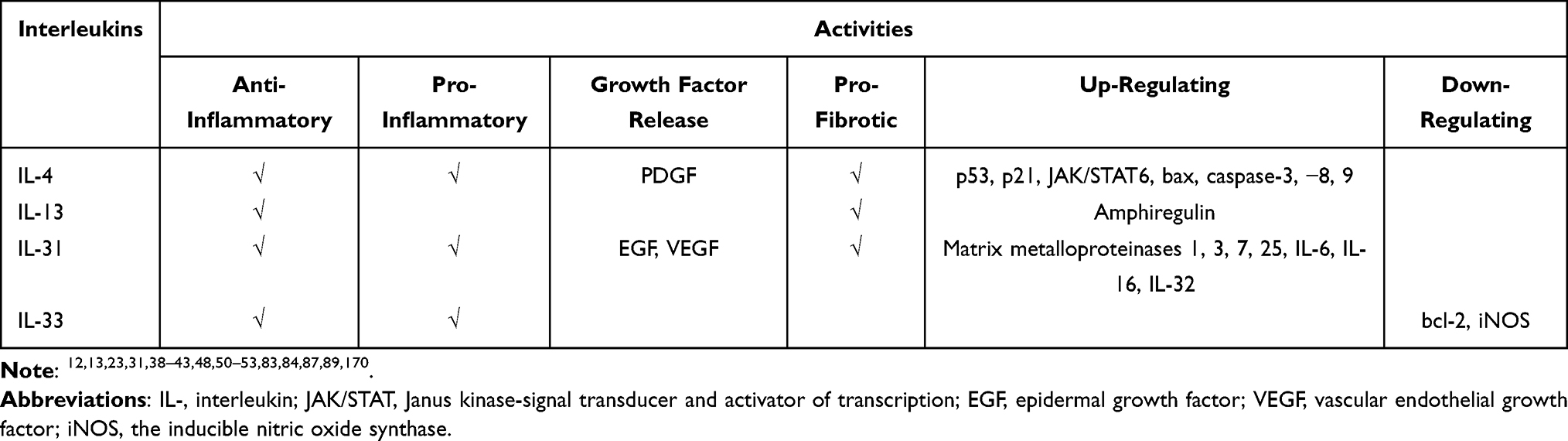 |
Table 2 Overview of the Activities of IL-4, IL-13, IL-31 and IL-33 |
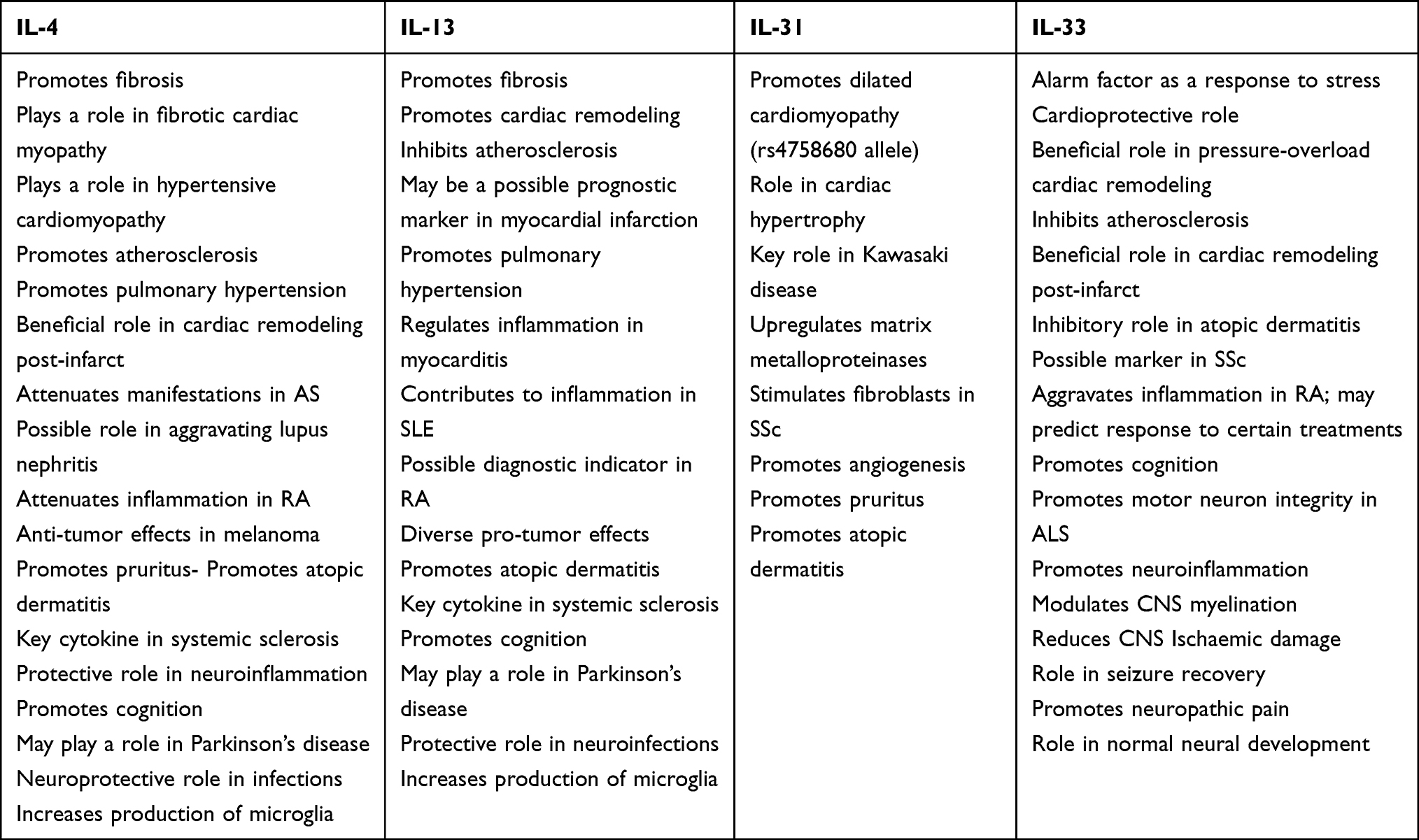 |
Table 3 Summary of Key Findings for IL-4, IL-13, IL-31, and IL-33 |
Acknowledgments
The current work was academically supported by the ‘Dunarea de Jos’ University of Galati, Romania, through the research center – Multidisciplinary Integrated Center of Dermatological Interface Research (MIC-DIC) [Centrul Integrat Multi-disciplinar de Cercetare de Interfata Dermatologica (CIM-CID)].
Funding
The article publishing charge was paid by the “Dunarea de Jos” University of Galati, Romania, grant number RF3668/01.10.2021.
Disclosure
Dr Nicolas Kluger reports personal fees from Novartis Finland, during the conduct of the study. The authors report no other conflicts of interest in this work.
References
1. Catalan-Dibene J, McIntyre LL, Zlotnik A. Interleukin 30 to interleukin 40. J Interferon Cytokine Res. 2018;38:423–439. doi:10.1089/jir.2018.0089
2. Onuora S. Novel cytokine, IL-41, linked with PsA. Nat Rev Rheumatol. 2019;15:636.
3. Justiz Vaillant AA, Qurie A. Interleukin. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2022.
4. Maier E, Werner D, Duschl A, Bohle B, Horejs-Hoeck J. Human Th2 but not Th9 cells release IL-31 in a STAT6/NF-κB-dependent way. J Immunol. 2014;193(2):645–654. doi:10.4049/jimmunol.1301836
5. Macedo RBV, Kakehasi AM, Melo de Andrade MV. IL33 in rheumatoid arthritis: potential contribution to pathogenesis. Rev Bras Reumatol. 2016;56:451–457. doi:10.1016/j.rbr.2016.01.006
6. Interleukin 13. Available from: https://en.wikipedia.org/wiki/Interleukin_13#:~:text=IL%2D13%20is%20a%20cytokine,fibrosis%20and%20chitinase%20up%2Dregulation.
7. Zhang Q, Putheti P, Zhou Q, Liu Q, Gao W. Structures and biological functions of IL-31 and IL-31 receptors. Cytokine Growth Factor Rev. 2008;19(5–6):347–356. doi:10.1016/j.cytogfr.2008.08.003
8. Furukawa S, Moriyama M, Miyake K, et al. Interleukin-33 produced by M2 macrophages and other immune cells contributes to Th2 immune reaction of IgG4-related disease. Sci Rep. 2017;7:42413. doi:10.1038/srep42413
9. Chan BCL, Lam CWK, Tam LS, Wong CK. IL33: roles in allergic inflammation and therapeutic perspectives. Front Immunol. 2019;10:364. doi:10.3389/fimmu.2019.00364
10. Di Salvo E, Ventura-Spagnolo E, Casciaro M, Navarra M, Gangemi S. IL-33/IL-31 axis: a potential inflammatory pathway. Mediators Inflamm. 2018;2018:3858032. doi:10.1155/2018/3858032
11. Hershey GK. IL-13 receptors and signaling pathways: an evolving web. J Allergy Clin Immunol. 2003;111(4):677–90, quiz 691. doi:10.1067/mai.2003.1333
12. Jenkins SJ, Ruckerl D, Cook PC, et al. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science. 2011;332:1284–1288. doi:10.1126/science.1204351
13. Ho I-C, Miaw S-C. Regulation of IL-4 expression in immunity and diseases. Adv Exp Med Biol. 2016;941:31–77. doi:10.1007/978-94-024-0921-5_3
14. Sempowski GD, Beckmann MP, Derdak S, Phipps RP. Subsets of murine lung fibroblasts express membrane-bound and soluble IL-4 receptors. role of IL-4 in enhancing fibroblast proliferation and collagen synthesis. J Immunol. 1994;152:3606–3614.
15. Sommer M, Eismann U, Gerth J, Stein G. Interleukin 4 co-stimulates the PDGF-BB- and BFGF-mediated proliferation of mesangial cells and myofibroblasts. Nephron. 2002;92:
16. Levick SP, McLarty JL, Murray DB, Freeman RM, Carver WE, Brower GL. Cardiac mast cells mediate left ventricular fibrosis in the hypertensive rat heart. Hypertens. 2009;53(6):1041–1047. doi:10.1161/HYPERTENSIONAHA.108.123158
17. Yu Q, Horak K, Larson DF. Role of T lymphocytes in hypertension-induced cardiac extracellular matrix remodeling. Hypertens. 2006;48(1):98–104. doi:10.1161/01.HYP.0000227247.27111.b2
18. Peng H, Yang X-P, Carretero OA, et al. Angiotensin II-induced dilated cardiomyopathy in Balb/c but not C57BL/6J mice. Exp Physiol. 2011;96:756–764. doi:10.1113/expphysiol.2011.057612
19. Roselló-Lletí E, Rivera M, Bertomeu V, Cortés R, Jordán A, González-Molina A. Interleukin-4 and cardiac fibrosis in patients with heart failure. Rev Esp Cardiol. 2007;60:777–780. doi:10.1157/13108284
20. Catapano G, Pedone C, Nunziata E, Zizzo A, Passantino A, Incalzi RA. Nutrient intake and serum cytokine pattern in elderly people with heart failure. Eur J Heart Fail. 2008;10:428–434. doi:10.1016/j.ejheart.2008.02.016
21. Kanellakis P, Ditiatkovski M, Kostolias G, Bobik A. A pro-fibrotic role for interleukin-4 in cardiac pressure overload. Cardiovasc Res. 2012;95:77–85. doi:10.1093/cvr/cvs142
22. van Heuven-Nolsen D, De Kimpe SJ, Muis T, et al. Opposite role of interferon-gamma and interleukin-4 on the regulation of blood pressure in mice. Biochem Biophys Res Commun. 1999;254:816–820. doi:10.1006/bbrc.1998.8742
23. Lee YW, Kühn H, Kaiser S, Hennig B, Daugherty A, Toborek M. Interleukin 4 induces transcription of the 15-lipoxygenase I gene in human endothelial cells. J Lipid Res. 2001;42:783–791. doi:10.1016/S0022-2275(20)31641-2
24. Yamaji-Kegan K, Su Q, Angelini DJ, Myers AC, Cheadle C, Johns RA. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELMalpha) increases lung inflammation and activates pulmonary microvascular endothelial cells via an IL-4-dependent mechanism. J Immunol. 2010;185:5539–5548. doi:10.4049/jimmunol.0904021
25. Yamaji-Kegan K, Takimoto E, Zhang A, et al. Hypoxia-induced mitogenic factor (FIZZ1/RELMα) induces endothelial cell apoptosis and subsequent interleukin-4-dependent pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2014;306:L1090–1103. doi:10.1152/ajplung.00279.2013
26. Shintani Y, Ito T, Fields L, et al. IL-4 as a repurposed biological drug for myocardial infarction through augmentation of reparative cardiac macrophages: proof-of-concept data in mice. Sci Rep. 2017;7:6877. doi:10.1038/s41598-017-07328-z
27. Sicklinger F, Meyer IS, Li X, et al. Basophils balance healing after myocardial infarction via IL-4/IL-13. J Clin Invest. 2021;131:136778. doi:10.1172/JCI136778
28. Minty A, Chalon P, Derocq JM, et al. Interleukin-13 is a new human lymphokine regulating inflammatory and immune responses. Nature. 1993;362:248–250. doi:10.1038/362248a0
29. McKenzie AN, Culpepper JA, de Waal Malefyt R, et al. Interleukin 13, a T-cell-derived cytokine that regulates human monocyte and B-cell function. Proc Natl Acad Sci USA. 1993;90:3735–3739. doi:10.1073/pnas.90.8.3735
30. Akaiwa M, Yu B, Umeshita-Suyama R, et al. Localization of human interleukin 13 receptor in non-haematopoietic cells. Cytokine. 2001;13:75–84. doi:10.1006/cyto.2000.0814
31. Cieslik KA, Taffet GE, Carlson S, Hermosillo J, Trial J, Entman ML. Immune-inflammatory dysregulation modulates the incidence of progressive fibrosis and diastolic stiffness in the aging heart. J Mol Cell Cardiol. 2011;50:248–256. doi:10.1016/j.yjmcc.2010.10.019
32. Amir O, Spivak I, Lavi I, Rahat MA. Changes in the monocytic subsets CD14dimCD16+ and CD14++CD16− in chronic systolic heart failure patients. Mediators Inflamm. 2012;2012:616384. doi:10.1155/2012/616384
33. Ohtsuka T, Inoue K, Hara Y, et al. Serum markers of angiogenesis and myocardial ultrasonic tissue characterization in patients with dilated cardiomyopathy. Eur J Heart Fail. 2005;7:689–695. doi:10.1016/j.ejheart.2004.09.011
34. Stanya KJ, Jacobi D, Liu S, et al. Direct control of hepatic glucose production by interleukin-13 in mice. J Clin Invest. 2013;123:261–271. doi:10.1172/JCI64941
35. Hofmann U, Knorr S, Vogel B, et al. Interleukin-13 deficiency aggravates healing and remodeling in male mice after experimental myocardial infarction. Circ Heart Fail. 2014;7:822–830. doi:10.1161/CIRCHEARTFAILURE.113.001020
36. Parisi V, Cabaro S, D’Esposito V, et al. Epicardial adipose tissue and IL-13 response to myocardial injury drives left ventricular remodeling after ST elevation myocardial infarction. Front Physiol. 2020;11:575181. doi:10.3389/fphys.2020.575181
37. Yuan S-M. Interleukin-13 in the pathogenesis of pulmonary artery hypertension. J Lab Med. 2019;43:5–11. doi:10.1515/labmed-2018-0323
38. Cihakova D, Barin JG, Afanasyev M, et al. Interleukin-13 protects against experimental autoimmune myocarditis by regulating macrophage differentiation. Am J Pathol. 2008;172:1195–1208. doi:10.2353/ajpath.2008.070207
39. Yang H, Chen Y, Gao C. Interleukin-13 reduces cardiac injury and prevents heart dysfunction in viral myocarditis via enhanced M2 macrophage polarization. Oncotarget. 2017;8:99495–99503. doi:10.18632/oncotarget.20111
40. Rotter Sopasakis V, Sandstedt J, Johansson M, et al. Toll-like receptor-mediated inflammation markers are strongly induced in heart tissue in patients with cardiac disease under both ischemic and non-ischemic conditions. Int J Cardiol. 2019;293:238–247. doi:10.1016/j.ijcard.2019.06.033
41. Vianello E, Marrocco-Trischitta Massimiliano M, Dozio E, et al. Correlational study on altered epicardial adipose tissue as a stratification risk factor for valve disease progression through IL-13 signaling. J Mol Cell Cardiol. 2019;132:210–218. doi:10.1016/j.yjmcc.2019.05.012
42. Dillon SR, Sprecher C, Hammond A, et al. Interleukin 31, a cytokine produced by activated T cells, induces dermatitis in mice. Nat Immunol. 2004;5:752–760. doi:10.1038/ni1084
43. Pflanz S, Hibbert L, Mattson J, et al. WSX-1 and glycoprotein 130 constitute a signal-transducing receptor for IL-27. J Immunol. 2004;172:2225–2231. doi:10.4049/jimmunol.172.4.2225
44. Song H, Peng Y, Zhou B, et al. Associations between interleukin-31 gene polymorphisms and dilated cardiomyopathy in a Chinese population. Dis Markers. 2017;2017:4191365. doi:10.1155/2017/4191365
45. Saito M, Yoshida K, Hibi M, Taga T, Kishimoto T. Molecular cloning of a murine IL-6 receptor-associated signal transducer, Gp130, and its regulated expression in vivo. J Immunol. 1992;148:4066–4071.
46. Lafontant PJ, Burns AR, Donnachie E, Haudek SB, Smith CW, Entman ML. Oncostatin M differentially regulates CXC chemokines in mouse cardiac fibroblasts. Am J Physiol Cell Physiol. 2006;291:C18–26. doi:10.1152/ajpcell.00322.2005
47. Kunsleben N, Rüdrich U, Gehring M, Novak N, Kapp A, Raap U. IL-31 induces chemotaxis, calcium mobilization, release of reactive oxygen species, and CCL26 in eosinophils, which are capable to release IL-31. J Invest Dermatol. 2015;135:1908–1911. doi:10.1038/jid.2015.106
48. Cheung PF-Y, Wong C-K, Ho AW-Y, Hu S, Chen D-P, Lam CW-K. Activation of human eosinophils and epidermal keratinocytes by Th2 Cytokine IL-31: implication for the immunopathogenesis of atopic dermatitis. Int Immunol. 2010;22:453–467. doi:10.1093/intimm/dxq027
49. Tseng W-N, Lo M-H, Guo MM-H, Hsieh K-S, Chang W-C, Kuo H-C. IL-31 associated with coronary artery lesion formation in Kawasaki disease. PLoS One. 2014;9:e105195. doi:10.1371/journal.pone.0105195
50. Miller AM. Role of IL-33 in inflammation and disease. J Inflamm. 2011;8:22. doi:10.1186/1476-9255-8-22
51. Pascual-Reguant A, BayatSarmadi J, Baumann C, et al. TH17 cells express ST2 and are controlled by the alarmin IL-33 in the small intestine. Mucosal Immunol. 2017;10:1431–1442. doi:10.1038/mi.2017.5
52. Jha HC, Divya A, Prasad J, Mittal A. Plasma circulatory markers in male and female patients with coronary artery disease. Heart Lung J Crit Care. 2010;39:296–303. doi:10.1016/j.hrtlng.2009.10.005
53. Demyanets S, Speidl WS, Tentzeris I, et al. Soluble ST2 and interleukin-33 levels in coronary artery disease: relation to disease activity and adverse outcome. PLoS One. 2014;9:e95055. doi:10.1371/journal.pone.0095055
54. Yanaba K, Yoshizaki A, Asano Y, Kadono T, Sato S. Serum IL-33 levels are raised in patients with systemic sclerosis: association with extent of skin sclerosis and severity of pulmonary fibrosis. Clin Rheumatol. 2011;30:825–830. doi:10.1007/s10067-011-1686-5
55. Komine M, Sato A, Adachi A, et al. Nuclear expression of IL-33 in epidermal keratinocyte is up-regulated in differentiated cells, while suppressed in proliferating cells. J Dermatol Sci. 2016;84:e43. doi:10.1016/j.jdermsci.2016.08.138
56. Travers J, Rochman M, Wen T, Rothenberg ME. IL-33 is selectively expressed by esophageal basal layer epithelial cells during allergic inflammation. J Allergy Clin Immunol. 2016;137:AB228. doi:10.1016/j.jaci.2015.12.879
57. Oshikawa K, Yanagisawa K, Tominaga S, Sugiyama Y. Expression and function of the ST2 gene in a murine model of allergic airway inflammation. Clin Exp Allergy J. 2002;32:1520–1526. doi:10.1046/j.1365-2745.2002.01494.x
58. Griesenauer B, Paczesny S. The ST2/IL-33 axis in immune cells during inflammatory diseases. Front Immunol. 2017;8:475. doi:10.3389/fimmu.2017.00475
59. Sanada S, Hakuno D, Higgins LJ, Schreiter ER, McKenzie ANJ, Lee RT. IL-33 and ST2 comprise a critical biomechanically induced and cardioprotective signaling system. J Clin Invest. 2007;117:1538–1549. doi:10.1172/JCI30634
60. Chen W-Y, Hong J, Gannon J, Kakkar R, Lee RT. Myocardial pressure overload induces systemic inflammation through endothelial cell IL-33. Proc Natl Acad Sci USA. 2015;112:7249–7254. doi:10.1073/pnas.1424236112
61. Veeraveedu PT, Sanada S, Okuda K, et al. Ablation of IL-33 gene exacerbate myocardial remodeling in mice with heart failure induced by mechanical stress. Biochem Pharmacol. 2017;138:73–80. doi:10.1016/j.bcp.2017.04.022
62. Dieplinger B, Mueller T. Soluble ST2 in heart failure. Clin Chim Acta Int J Clin Chem. 2015;443:57–70. doi:10.1016/j.cca.2014.09.021
63. Ojji DB, Opie LH, Lecour S, Lacerda L, Adeyemi OM, Sliwa K. The effect of left ventricular remodelling on soluble ST2 in a cohort of hypertensive subjects. J Hum Hypertens. 2014;28:432–437. doi:10.1038/jhh.2013.130
64. Ho JE, Larson MG, Ghorbani A, et al. Soluble ST2 predicts elevated SBP in the community. J Hypertens. 2013;31:
65. Coglianese EE, Larson MG, Vasan RS, et al. Distribution and clinical correlates of the interleukin receptor family member soluble ST2 in the Framingham heart study. Clin Chem. 2012;58:1673–1681. doi:10.1373/clinchem.2012.192153
66. Zheng Y-G, Yang T, He J-G, et al. Plasma soluble ST2 levels correlate with disease severity and predict clinical worsening in patients with pulmonary arterial hypertension. Clin Cardiol. 2014;37:365–370. doi:10.1002/clc.22262
67. Agoston-Coldea L, Lupu S, Hicea S, Paradis A, Mocan T. Serum levels of the soluble IL-1 receptor family member ST2 and right ventricular dysfunction. Biomark Med. 2014;8:95–106. doi:10.2217/bmm.13.116
68. Daniels LB, Clopton P, Iqbal N, Tran K, Maisel AS. Association of ST2 levels with cardiac structure and function and mortality in outpatients. Am Heart J. 2010;160:721–728. doi:10.1016/j.ahj.2010.06.033
69. Carlomagno G, Messalli G, Melillo RM, et al. Serum soluble ST2 and interleukin-33 levels in patients with pulmonary arterial hypertension. Int J Cardiol. 2013;168:1545–1547. doi:10.1016/j.ijcard.2012.12.031
70. Mohsen K. Serum levels of interleukin (IL)-33 in patients with ischemic heart disease. MOJ Immunol. 2018;6(2):29–32.
71. McLaren JE, Michael DR, Salter RC, et al. IL-33 reduces macrophage foam cell formation. J Immunol. 2010;185:1222–1229. doi:10.4049/jimmunol.1000520
72. Seki K, Sanada S, Kudinova AY, et al. Interleukin-33 prevents apoptosis and improves survival after experimental myocardial infarction through ST2 signaling. Circ Heart Fail. 2009;2:684–691. doi:10.1161/CIRCHEARTFAILURE.109.873240
73. Ruisong M, Xiaorong H, Gangying H, et al. The protective role of interleukin-33 in myocardial ischemia and reperfusion is associated with decreased HMGB1 expression and up-regulation of the P38 MAPK signaling pathway. PLoS One. 2015;10:e0143064. doi:10.1371/journal.pone.0143064
74. Miller AM, Xu D, Asquith DL, et al. IL-33 reduces the development of atherosclerosis. J Exp Med. 2008;205:339–346. doi:10.1084/jem.20071868
75. Abston ED, Barin JG, Cihakova D, et al. IL-33 independently induces eosinophilic pericarditis and cardiac dilation: ST2 improves cardiac function. Circ Heart Fail. 2012;5:366–375. doi:10.1161/CIRCHEARTFAILURE.111.963769
76. Miller AM, Liew FY. The IL-33/ST2 pathway–a new therapeutic target in cardiovascular disease. Pharmacol Ther. 2011;131:179–186. doi:10.1016/j.pharmthera.2011.02.005
77. Tatu AL, Ionescu MA. Multiple autoimmune syndrome type 3- thyroiditis, vitiligo and alopecia areata. Acta Endocrinol Buchar. 2017;13:124–125. doi:10.4183/aeb.2017.124
78. Nwabudike LC, Tatu AL, Happle R, et al. Koebner’s sheep in wolf’s clothing: does the isotopic response exists as a distinct phenomenon? J Eur Acad Dermatol Venereol. 2018;32:e336–e337. doi:10.1111/jdv.14900
79. Tatu AL, Nwabudike LC. The treatment options of male genital lichen sclerosus et atrophicus. In:
80. Niculet E, Chioncel V, Elisei AM, et al. Multifactorial expression of IL-6 with update on COVID-19 and the therapeutic strategies of its blockade (review). Exp Ther Med. 2021;21:263. doi:10.3892/etm.2021.9693
81. Elisei A, Nwabudike LC, Buzia OD, Miulescu M, Tatu AL. Statins. A review on structural perspectives, adverse reactions and relations with non-melanoma skin cancer. Rev Chim. 2018;69:2557–2562.
82. Tatu AL. Umbilicated blue-black lesion on the lateral thorax. J Cutan Med Surg. 2017;21:252. doi:10.1177/1203475417694859
83. Hoon DS, Banez M, Okun E, Morton DL, Irie RF. Modulation of human melanoma cells by interleukin-4 and in combination with gamma-interferon or alpha-tumor necrosis factor. Cancer Res. 1991;51:2002–2008.
84. Lee HL, Park MH, Song JK, et al. Tumor growth suppressive effect of IL-4 through P21-mediated activation of STAT6 in IL-4Rα overexpressed melanoma models. Oncotarget. 2016;7:23425–23438. doi:10.18632/oncotarget.8111
85. Whitehead RP, Unger JM, Goodwin JW, et al. Phase II trial of recombinant human interleukin-4 in patients with disseminated malignant melanoma: a southwest oncology group study. J Immunother. 1998;21(6):440–446. doi:10.1097/00002371-199811000-00006
86. Chong ST, Tan KM, Kok CYL, et al. IL13RA2 is differentially regulated in papillary thyroid carcinoma vs follicular thyroid carcinoma. J Clin Endocrinol Metab. 2019;104:5573–5584. doi:10.1210/jc.2019-00040
87. Okamoto H, Yoshimatsu Y, Tomizawa T, et al. Interleukin-13 receptor Α2 is a novel marker and potential therapeutic target for human melanoma. Sci Rep. 2019;9:1281. doi:10.1038/s41598-019-39018-3
88. Fichtner-Feigl S, Strober W, Kawakami K, Puri RK, Kitani A. IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 production and fibrosis. Nat Med. 2006;12:99–106. doi:10.1038/nm1332
89. Castellani ML, Felaco P, Galzio RJ, et al. IL-31 a Th2 cytokine involved in immunity and inflammation. Int J Immunopathol Pharmacol. 2010;23:709–713. doi:10.1177/039463201002300304
90. Bodoor K, Al-Qarqaz F, Heis LA, et al. IL-33/13 axis and IL-4/31 axis play distinct roles in inflammatory process and itch in psoriasis and atopic dermatitis. Clin Cosmet Investig Dermatol. 2020;13:419–424. doi:10.2147/CCID.S257647
91. Junttila IS. Tuning the cytokine responses: an update on interleukin (IL)-4 and IL-13 receptor complexes. Front Immunol. 2018;9:888. doi:10.3389/fimmu.2018.00888
92. Bitton A, Avlas S, Reichman H, et al. A key role for IL-13 signaling via the type 2 IL-4 receptor in experimental atopic dermatitis. Sci Immunol. 2020;5:eaaw2938. doi:10.1126/sciimmunol.aaw2938
93. Bieber T. Interleukin-13: targeting an underestimated cytokine in atopic dermatitis. Allergy. 2020;75:54–62. doi:10.1111/all.13954
94. Wollenberg A, Blauvelt A, Guttman-Yassky E, et al. Tralokinumab for moderate-to-severe atopic dermatitis: results from two 52-week, randomized, double-blind, multicentre, placebo-controlled phase III trials (ECZTRA 1 and ECZTRA 2). Br J Dermatol. 2021;184:437–449. doi:10.1111/bjd.19574
95. TameezUd Din A, Malik I, Arshad D, TameezUd Din A. Dupilumab for atopic dermatitis: the silver bullet we have been searching for? Cureus. 2020;12:e7565. doi:10.7759/cureus.7565
96. Wozel G, Vitéz L, Pfeiffer C. Severe atopic dermatitis and leflunomide: first clinical experience and highlights of pertinent experimental data. Dermatol Online J. 2006;12:6.
97. Kim BS, Howell MD, Sun K, et al. Treatment of atopic dermatitis with ruxolitinib cream (JAK1/JAK2 inhibitor) or triamcinolone cream. J Allergy Clin Immunol. 2020;145:572–582. doi:10.1016/j.jaci.2019.08.042
98. Lee YS, Han S-B, Ham HJ, et al. IL-32γ suppressed atopic dermatitis through inhibition of MiR-205 expression via inactivation of nuclear factor-kappa B. J Allergy Clin Immunol. 2020;146:156–168. doi:10.1016/j.jaci.2019.12.905
99. IL-31 drug succeeds for Prurigo Nodularis — less itching and twice as many lesions resolved with investigational agent. MedpageToday. Available from: https://www.medpagetoday.com/dermatology/generaldermatology/84974.
100. Kabashima K, Irie H. Interleukin-31 as a clinical target for pruritus treatment. Front Med. 2021;8:638325. doi:10.3389/fmed.2021.638325
101. Asano Y. Recent advances in animal models of systemic sclerosis. J Dermatol. 2016;43:19–28. doi:10.1111/1346-8138.13185
102. Guggino G, Lo Pizzo M, Di Liberto D, et al. Interleukin-9 over-expression and T helper 9 polarization in systemic sclerosis patients. Clin Exp Immunol. 2017;190:208–216. doi:10.1111/cei.13009
103. Denton CP. Advances in pathogenesis and treatment of systemic sclerosis. Clin Med. 2015;15(6):s58–63. doi:10.7861/clinmedicine.15-6-s58
104. Sierra-Sepúlveda A, Esquinca-González A, Benavides-Suárez SA, et al. Systemic sclerosis pathogenesis and emerging therapies, beyond the fibroblast. BioMed Res Int. 2019;2019:4569826. doi:10.1155/2019/4569826
105. Asano Y. Systemic sclerosis. J Dermatol. 2018;45:128–138. doi:10.1111/1346-8138.14153
106. Altorok N, Wang Y, Kahaleh B. Endothelial dysfunction in systemic sclerosis. Curr Opin Rheumatol. 2014;26:615–620. doi:10.1097/BOR.0000000000000112
107. Matucci-Cerinic M, Steen V, Nash P, Hachulla E. The complexity of managing systemic sclerosis: screening and diagnosis. Rheumatol Oxf. 2009;48(3):iii8–13. doi:10.1093/rheumatology/ken482
108. Liakouli V, Elies J, El-Sherbiny YM, et al. Scleroderma fibroblasts suppress angiogenesis via tgf-β/caveolin-1 dependent secretion of pigment epithelium-derived factor. Ann Rheum Dis. 2018;77:431–440. doi:10.1136/annrheumdis-2017-212120
109. Atamas SP, White B. Interleukin 4 in systemic sclerosis: not just an increase. Clin Diagn Lab Immunol. 1999;6:658–659. doi:10.1128/CDLI.6.5.658-659.1999
110. Gasparini G, Cozzani E, Parodi A. Interleukin-4 and interleukin-13 as possible therapeutic targets in systemic sclerosis. Cytokine. 2020;125:154799. doi:10.1016/j.cyto.2019.154799
111. Cascio S, Medsger TA, Hawse WF, et al. 14-3-3z sequesters cytosolic T-Bet, upregulating IL-13 levels in TC2 and CD8+ lymphocytes from patients with scleroderma. J Allergy Clin Immunol. 2018;142:109–119.e6. doi:10.1016/j.jaci.2017.10.029
112. Manetti M, Pratesi S, Romano E, et al. Decreased circulating lymphatic endothelial progenitor cells in digital ulcer-complicated systemic sclerosis. Ann Rheum Dis. 2019;78:575–577. doi:10.1136/annrheumdis-2018-214240
113. Yamashita T, Lakota K, Taniguchi T, et al. An orally-active adiponectin receptor agonist mitigates cutaneous fibrosis, inflammation and microvascular pathology in a murine model of systemic sclerosis. Sci Rep. 2018;8:11843. doi:10.1038/s41598-018-29901-w
114. Yaseen B, Lopez H, Taki Z, et al. Interleukin-31 promotes pathogenic mechanisms underlying skin and lung fibrosis in scleroderma. Rheumatol Oxf. 2020;59:2625–2636. doi:10.1093/rheumatology/keaa195
115. Kuzumi A, Yoshizaki A, Matsuda KM, et al. Interleukin-31 promotes fibrosis and T helper 2 polarization in systemic sclerosis. Nat Commun. 2021;12:5947. doi:10.1038/s41467-021-26099-w
116. Wagner A, Köhm M, Nordin A, Svenungsson E, Pfeilschifter JM, Radeke HH. Increased serum levels of the IL-33 neutralizing SST2 in limited cutaneous systemic sclerosis. Scand J Immunol. 2015;82:269–274. doi:10.1111/sji.12317
117. Murdaca G, Greco M, Tonacci A, et al. IL-33/IL-31 axis in immune-mediated and allergic diseases. Int J Mol Sci. 2019;20:5856. doi:10.3390/ijms20235856
118. Simone D, Al Mossawi MH, Bowness P. Progress in our understanding of the pathogenesis of ankylosing spondylitis. Rheumatol Oxf. 2018;57:vi4–vi9. doi:10.1093/rheumatology/key001
119. Dong C, Fu T, Ji J, Li Z, Gu Z. The role of interleukin-4 in rheumatic diseases. Clin Exp Pharmacol Physiol. 2018;45:747–754. doi:10.1111/1440-1681.12946
120. Lin S, Qiu M, Chen J. IL-4 modulates macrophage polarization in ankylosing spondylitis. Cell Physiol Biochem. 2015;35:2213–2222. doi:10.1159/000374026
121. Wang J, Zhao Q, Wang G, et al. Circulating levels of Th1 and Th2 chemokines in patients with ankylosing spondylitis. Cytokine. 2016;81:10–14. doi:10.1016/j.cyto.2016.01.012
122. Li X, Lin T, Qi C, et al. Elevated serum level of IL-33 and SST2 in patients with ankylosing spondylitis: associated with disease activity and vascular endothelial growth factor. J Investig Med. 2013;61:848–851. doi:10.2310/JIM.0b013e31828deed2
123. Talabot-Ayer D, McKee T, Gindre P, et al. Distinct serum and synovial fluid interleukin (IL)-33 levels in rheumatoid arthritis, psoriatic arthritis and osteoarthritis. Joint Bone Spine. 2012;79:32–37. doi:10.1016/j.jbspin.2011.02.011
124. Justiz Vaillant AA, Goyal A, Bansal P, Varacallo M. Systemic lupus erythematosus. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2022.
125. Singh RR. IL-4 and many roads to lupus-like autoimmunity. Clin Immunol. 2003;108:73–79. doi:10.1016/S1521-6616(03)00145-1
126. Nakajima A, Hirose S, Yagita H, Okumura K. Roles of IL-4 and IL-12 in the development of lupus in NZB/W F1 mice. J Immunol. 1997;158:1466–1472.
127. Sugimoto K, Morimoto S, Kaneko H, et al. Decreased IL-4 producing CD4+ T cells in patients with active systemic lupus erythematosus-relation to IL-12R expression. Autoimmunity. 2002;35(6):381–387. doi:10.1080/0891693021000008535
128. Reséndiz-Mora A, Wong-Baeza C, Nevárez-Lechuga I, et al. Interleukin 4 deficiency limits the development of a lupus-like disease in mice triggered by phospholipids in a non-bilayer arrangement. Scand J Immunol. 2021;93(3):e13002. doi:10.1111/sji.13002
129. Morimoto S, Tokano Y, Kaneko H, Nozawa K, Amano H, Hashimoto H. The increased interleukin-13 in patients with systemic lupus erythematosus: relations to other Th1-, Th2-related cytokines and clinical findings. Autoimmunity. 2001;34:19–25. doi:10.3109/08916930108994122
130. Wang R, Lu Y-L, Huang H-T, et al. Association of interleukin 13 gene polymorphisms and plasma IL 13 level with risk of systemic lupus erythematosus. Cytokine. 2018;104:92–97. doi:10.1016/j.cyto.2017.09.034
131. Huang H-T, Chen J-M, Guo J, Lan Y, Wei Y-S. The association of interleukin-31 polymorphisms with interleukin-31 serum levels and risk of systemic lupus erythematosus. Rheumatol Int. 2016;36:799–805. doi:10.1007/s00296-016-3422-6
132. Yang Z, Liang Y, Xi W, Li C, Zhong R. Association of increased serum IL-33 levels with clinical and laboratory characteristics of systemic lupus erythematosus in Chinese population. Clin Exp Med. 2011;11(2):75–80. doi:10.1007/s10238-010-0115-4
133. Moreau A, Nicaise C, Awada A, Soyfoo MS. Soluble ST2 is increased in systemic lupus erythematous and is a potential marker of lupus nephritis. Clin Exp Rheumatol. 2021;2021:34128798.
134. Mohd Jaya FN, Liu Z, Chan GC. Early treatment of interleukin-33 can attenuate lupus development in young NZB/W F1 mice. Cells. 2020;9(11):2448. doi:10.3390/cells9112448
135. Firestein G, McInnes IB. Immunopathogenesis of rheumatoid arthritis. Immunity. 2017;46:183–196. doi:10.1016/j.immuni.2017.02.006
136. Finnegan A, Grusby MJ, Kaplan CD, et al. IL-4 and IL-12 regulate proteoglycan-induced arthritis through stat-dependent mechanisms. J Immunol Baltim. 2002;169:3345–3352. doi:10.4049/jimmunol.169.6.3345
137. Finnegan A, Mikecz K, Tao P, Glant TT. Proteoglycan (Aggrecan)-induced arthritis in BALB/c mice Is a Th1-type disease regulated by Th2 cytokines. J Immunol Baltim. 1999;163:5383–5390.
138. Martin-Martin LS, Giovannangeli F, Bizzi E, et al. An open randomized active-controlled clinical trial with low-dose SKA cytokines versus DMARDs evaluating low disease activity maintenance in patients with rheumatoid arthritis. Drug Des Devel Ther. 2017;11:985–994. doi:10.2147/DDDT.S118298
139. Shen H, Xia L, Lu J. Interleukin-4 in rheumatoid arthritis patients with interstitial lung disease: a pilot study. Indian J Med Res. 2013;138:919–921.
140. Tokayer A, Carsons SE, Chokshi B, Santiago-Schwarz F. High levels of interleukin 13 in rheumatoid arthritis sera are modulated by tumor necrosis factor antagonist therapy: association with dendritic cell growth activity. J Rheumatol. 2002;29:454–461.
141. Siloşi I, Boldeanu MV, Cojocaru M, et al.. The relationship of cytokines IL-13 and IL-17 with autoantibodies profile in early rheumatoid arthritis. J Immunol Res. 2016;3109135. doi:10.1155/2016/3109135
142. Duan L, Chen J, Gong F, Shi G. The role of IL-33 in rheumatic diseases. Clin Dev Immunol. 2013;924363. doi:10.1155/2013/924363
143. Gadani SP, Cronk JC, Norris GT, Kipnis J. IL-4 in the brain: a cytokine to remember. J Immunol Baltim. 2012;189:4213–4219. doi:10.4049/jimmunol.1202246
144. King E, O’Brien JT, Donaghy P, et al. Peripheral inflammation in prodromal Alzheimer’s and Lewy body dementias. J Neurol Neurosurg Psychiatry. 2018;89:339–345. doi:10.1136/jnnp-2017-317134
145. Derecki NC, Cardani AN, Yang CH, et al. Regulation of learning and memory by meningeal immunity: a key role for IL-4. J Exp Med. 2010;207:1067–1080. doi:10.1084/jem.20091419
146. Dionisio-Santos DA, Behrouzi A, Olschowka JA, O’Banion MK. Evaluating the effect of interleukin-4 in the 3xTg mouse model of Alzheimer’s disease. Front Neurosci. 2020;14:441. doi:10.3389/fnins.2020.00441
147. Brombacher TM, Nono JK, De Gouveia KS, et al. IL-13-mediated regulation of learning and memory. J Immunol Baltim. 2017;198:2681–2688. doi:10.4049/jimmunol.1601546
148. Mori S, Maher P, Conti B. Neuroimmunology of the interleukins 13 and 4. Brain Sci. 2016;6:E18. doi:10.3390/brainsci6020018
149. Brombacher TM, Berkiks I, Pillay S, Scibiorek M, Moses BO, Brombacher F. IL-4R alpha deficiency influences hippocampal-BDNF signaling pathway to impair reference memory. Sci Rep. 2020;10:16506. doi:10.1038/s41598-020-73574-3
150. Xiong Z, Thangavel R, Kempuraj D, Yang E, Zaheer S, Zaheer A. Alzheimer’s disease: evidence for the expression of interleukin-33 and its receptor ST2 in the brain. J Alzheimers Dis. 2014;40:297–308. doi:10.3233/JAD-132081
151. Fairlie-Clarke K, Barbour M, Wilson C, Hridi SU, Allan D, Jiang H-R. Expression and function of IL-33/ST2 axis in the central nervous system under normal and diseased conditions. Front Immunol. 2018;9:2596. doi:10.3389/fimmu.2018.02596
152. Saresella M, Marventano I, Piancone F, et al. IL-33 and its decoy SST2 in patients with Alzheimer’s disease and mild cognitive impairment. J Neuroinflammation. 2020;17:174. doi:10.1186/s12974-020-01806-4
153. Sun Y, Wen Y, Wang L, et al. Therapeutic opportunities of interleukin-33 in the central nervous system. Front Immunol. 2021;12:654626. doi:10.3389/fimmu.2021.654626
154. Du L-X, Wang Y-Q, Hua G-Q, Mi W-L. IL-33/ST2 pathway as a rational therapeutic target for CNS diseases. Neuroscience. 2018;369:222–230. doi:10.1016/j.neuroscience.2017.11.028
155. Abd RachmanIsnadi MF, Chin VK, Abd Majid R, et al. Critical roles of IL-33/ST2 pathway in neurological disorders. Mediators Inflamm. 2018;2018:5346413. doi:10.1155/2018/5346413
156. Reverchon F, de Concini V, Larrigaldie V, et al. Hippocampal interleukin-33 mediates neuroinflammation-induced cognitive impairments. J Neuroinflammation. 2020;17:268. doi:10.1186/s12974-020-01939-6
157. Yan Z, Yang W, Wei H, et al. Dysregulation of the adaptive immune system in patients with early-stage Parkinson disease. Neurol Neuroimmunol Neuroinflammation. 2021;8:e1036. doi:10.1212/NXI.0000000000001036
158. Morrison BE, Marcondes MCG, Nomura DK, et al. Cutting edge: IL-13Rα1 expression in dopaminergic neurons contributes to their oxidative stress-mediated loss following chronic peripheral treatment with lipopolysaccharide. J Immunol Baltim. 2012;189:5498–5502. doi:10.4049/jimmunol.1102150
159. Bok E, Cho EJ, Chung ES, Shin W-H, Jin BK. Interleukin-4 contributes to degeneration of dopamine neurons in the lipopolysaccharide-treated substantia nigra in vivo. Exp Neurobiol. 2018;27:309–319. doi:10.5607/en.2018.27.4.309
160. Mori S, Sugama S, Nguyen W, et al. Lack of interleukin-13 receptor Α1 delays the loss of dopaminergic neurons during chronic stress. J Neuroinflammation. 2017;14:88. doi:10.1186/s12974-017-0862-1
161. Aguirre CA, Concetta Morale M, Peng Q, et al. Two single nucleotide polymorphisms in IL13 and IL13RA1 from individuals with idiopathic Parkinson’s disease increase cellular susceptibility to oxidative stress. Brain Behav Immun. 2020;88:920–924. doi:10.1016/j.bbi.2020.04.007
162. Rossi C, Cusimano M, Zambito M, et al. Interleukin 4 modulates microglia homeostasis and attenuates the early slowly progressive phase of amyotrophic lateral sclerosis. Cell Death Dis. 2018;9:250. doi:10.1038/s41419-018-0288-4
163. Korhonen P, Pollari E, Kanninen KM, et al. Long-term interleukin-33 treatment delays disease onset and alleviates astrocytic activation in a transgenic mouse model of amyotrophic lateral sclerosis. IBRO Rep. 2019;6:74–86. doi:10.1016/j.ibror.2019.01.005
164. Falcone M, Rajan AJ, Bloom BR, Brosnan CF. A critical role for IL-4 in regulating disease severity in experimental allergic encephalomyelitis as demonstrated in IL-4-deficient C57BL/6 mice and BALB/c mice. J Immunol Baltim. 1998;160:4822–4830.
165. Ponomarev ED, Maresz K, Tan Y, Dittel BN. CNS-derived interleukin-4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. J Neurosci. 2007;27:10714–10721. doi:10.1523/JNEUROSCI.1922-07.2007
166. Ghezzi L, Cantoni C, Cignarella F, et al. T cells producing GM-CSF and IL-13 are enriched in the cerebrospinal fluid of relapsing MS patients. Mult Scler. 2020;26:1172–1186. doi:10.1177/1352458519852092
167. Allan D, Fairlie-Clarke KJ, Elliott C, et al. Role of IL-33 and ST2 signalling pathway in multiple sclerosis: expression by oligodendrocytes and inhibition of myelination in central nervous system. Acta Neuropathol Commun. 2016;4:75. doi:10.1186/s40478-016-0344-1
168. Ygberg S, Fowler Å, Wickström R. Age-related changes in the inflammatory responses to viral infections in the central nervous system during childhood. Pediatr Res. 2022;91(1):204–208. doi:10.1038/s41390-021-01423-8
169. Silva HM, Vinaud MC, de Lino RS. Experimental neurocysticercosis: absence of IL-4 induces lower encephalitis. Arq Neuropsiquiatr. 2017;75:96–102. doi:10.1590/0004-282x20160194
170. Peng H, Sun R, Zhang Q, et al. Interleukin 33 mediates type 2 immunity and inflammation in the central nervous system of mice infected with angiostrongylus cantonensis. J Infect Dis. 2013;207:860–869. doi:10.1093/infdis/jis682
171. Palomo J, Reverchon F, Piotet J, et al. Critical role of IL-33 receptor ST2 in experimental cerebral malaria development. Eur J Immunol. 2015;45:1354–1365. doi:10.1002/eji.201445206
172. Franca RFO, Costa RS, Silva JR, et al. IL-33 signaling is essential to attenuate viral-induced encephalitis development by downregulating INOS expression in the central nervous system. J Neuroinflammation. 2016;13:159. doi:10.1186/s12974-016-0628-1
173. Reverchon F, Mortaud S, Sivoyon M, et al. IL-33 receptor ST2 regulates the cognitive impairments associated with experimental cerebral malaria. PLoS Pathog. 2017;13:e1006322.
174. Kolosowska N, Keuters MH, Wojciechowski S, et al. Peripheral administration of IL-13 induces anti-inflammatory microglial/macrophage responses and provides neuroprotection in ischemic stroke. Neurotherapeutics. 2019;16:1304–1319. doi:10.1007/s13311-019-00761-0
175. Perego C, Fumagalli S, Miteva K, Kallikourdis M, De Simoni M-G. Combined genetic deletion of IL (Interleukin)-4, IL-5, IL-9, and IL-13 does not affect ischemic brain injury in mice. Stroke. 2019;50:2207–2215. doi:10.1161/STROKEAHA.119.025196
176. Woodcock T, Morganti-Kossmann MC. The role of markers of inflammation in traumatic brain injury. Front Neurol. 2013;4:18. doi:10.3389/fneur.2013.00018
177. Pu H, Ma C, Zhao Y, et al. Intranasal delivery of interleukin-4 attenuates chronic cognitive deficits via beneficial microglial responses in experimental traumatic brain injury. J Cereb Blood Flow Metab. 2021;41(11):2870–2886. doi:10.1177/0271678X211028680
178. Miao W, Zhao Y, Huang Y, et al. IL-13 ameliorates neuroinflammation and promotes functional recovery after traumatic brain injury. J Immunol Baltim. 2020;204:1486–1498. doi:10.4049/jimmunol.1900909
179. Miao Y, Zhang Z-X, Feng X, Sun W-M. IL-33 as a novel serum prognostic marker of intracerebral hemorrhage. Oxid Med Cell Longev. 2021;2021:5597790. doi:10.1155/2021/5597790
180. de Toscano ECB, Lessa JMK, de Oliveira GN, et al. Peripheral levels of SST2 are increased in patients with temporal lobe epilepsy: additional evidence of low-grade inflammation. Epilepsy Behav. 2020;112:107351. doi:10.1016/j.yebeh.2020.107351
181. Gao Y, Luo C, Yao Y, et al. IL-33 alleviated brain damage via anti-apoptosis, endoplasmic reticulum stress, and inflammation after epilepsy. Front Neurosci. 2020;14:898. doi:10.3389/fnins.2020.00898
182. Ethemoglu O, Calık M, Koyuncu I, et al. Interleukin-33 and oxidative stress in epilepsy patients. Epilepsy Res. 2021;176:106738. doi:10.1016/j.eplepsyres.2021.106738
183. Takamori A, Nambu A, Sato K, et al. IL-31 is crucial for induction of pruritus, but not inflammation, in contact hypersensitivity. Sci Rep. 2018;8:6639. doi:10.1038/s41598-018-25094-4
184. Nemmer JM, Kuchner M, Datsi A, et al. Interleukin-31 signaling bridges the gap between immune cells, the nervous system and epithelial tissues. Front Med. 2021;8:639097. doi:10.3389/fmed.2021.639097
185. De Boeck A, Ahn BY, D’Mello C, et al. Glioma-derived IL-33 orchestrates an inflammatory brain tumor microenvironment that accelerates glioma progression. Nat Commun. 2020;11:4997. doi:10.1038/s41467-020-18569-4
186. Lin L, Li Y, Liu M, Li Q, Liu Q, Li R. The interleukin-33/ST2 axis promotes glioma mesenchymal transition, stemness and TMZ resistance via JNK activation. Aging. 2020;12(2):1685–1703. doi:10.18632/aging.102707
187. Thürmann L, Herberth G, Rolle-Kampczyk U, et al. Elevated gestational IL-13 during fetal development is associated with hyperactivity and inattention in eight-year-old children. Front Immunol. 2019;10:1658. doi:10.3389/fimmu.2019.01658
188. Dohi E, Choi EY, Rose IVL, et al. Behavioral changes in mice lacking interleukin-33. eNeuro. 2017;4(6):
189. Vainchtein ID, Chin G, Cho FS, et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science. 2018;359:1269–1273. doi:10.1126/science.aal3589
190. Pandolfo G, Genovese G, Casciaro M, et al. IL-33 in mental disorders. Medicina. 2021;57(4):315. doi:10.3390/medicina57040315
191. Page MJ, McKenzie JE, Bossuyt PM, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71. doi:10.1136/bmj.n71
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
IL-4 and IL-13 in Cardiovascular Disease: From Immune Modulation to Therapeutic Possibilities – A Narrative Review
Kılıç AT, Bora R, Toprak B
Journal of Inflammation Research 2025, 18:10669-10679
Published Date: 8 August 2025