Back to Journals » Journal of Inflammation Research » Volume 19
Interleukin-Mediated Macrophage Polarization in Allergic Rhinitis Inflammation: A Systematic Review
Authors Luan H, Gao S, Ma Y, Yu Z, Li X
Received 5 March 2026
Accepted for publication 16 June 2026
Published 7 July 2026 Volume 2026:19 607167
DOI https://doi.org/10.2147/JIR.S607167
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Cynthia Koziol-White
Haihuan Luan1, Shouwei Gao2, Yuanyuan Ma3, Zeyu Yu3, Xiaonan Li1
1Department of Stomatology, The First Affiliated Hospital of Heilongjiang University of Chinese Medicine, Harbin, Heilongjiang, 150040, People’s Republic of China; 2Department of Rehabilitation, The Fourth Affiliated Hospital of Heilongjiang University of Chinese Medicine, Harbin, Heilongjiang, 150036, People’s Republic of China; 3Department of Pediatrics II, The First Affiliated Hospital of Heilongjiang University of Chinese Medicine, Harbin, Heilongjiang, 150040, People’s Republic of China
Correspondence: Xiaonan Li, Department of Stomatology, The First Affiliated Hospital of Heilongjiang University of Chinese Medicine, Harbin, Heilongjiang, 150040, People’s Republic of China, Email [email protected]
Objective: Allergic rhinitis (AR) is driven by interleukin (IL) network dysregulation and imbalanced macrophage polarization. This systematic review summarizes the molecular mechanisms by which ILs regulate macrophage phenotypic switching in AR inflammation.
Methods: We screened the PubMed and Embase databases from January 2010 to January 2026 to identify published studies. The search keywords used were as follows: [“interleukin” or “IL”], [“allergic rhinitis” or “AR”], [“macrophages”] and [“inflammation”]. A total of 223 peer-reviewed studies on human/animal models were included, and articles that did not meet the requirements were excluded.
Results: Pro-inflammatory ILs (IL-4/IL-13, IL-1β/IL-6/IL-17A) promote pathogenic M1/M2a macrophage polarization via STAT6, NF-κB and NLRP3 pathways, aggravating inflammatory responses. Anti-inflammatory ILs (IL-10, IL-27) induce M2c polarization and restore immune balance through STAT3/STAT1 signaling. Targeted interventions (dupilumab, MCC950, chlorogenic acid) effectively reverse polarization imbalance and alleviate AR symptoms. Clinically, AR patients present disrupted IL ratios, M1/M2 imbalance and impaired nasal mucosal barriers.
Conclusion: IL-mediated macrophage polarization is a core driver of AR inflammation. Targeting these regulatory pathways provides promising precision therapeutic strategies for AR.
Keywords: allergic rhinitis, interleukin, macrophage, inflammation, NLRP3
Introduction
Allergic rhinitis (AR), a globally prevalent chronic respiratory inflammatory disease, has seen a steady rise in its incidence worldwide. In China, the prevalence rate has reached 11.2% among adults1 and as high as 19.4% among the pediatric population,2 which severely impairs patients’ quality of life and imposes a heavy socioeconomic healthcare burden. Current therapeutic modalities have notable limitations. Intranasal glucocorticoids can rapidly alleviate clinical symptoms, but long-term administration may lead to adverse effects such as nasal mucosal atrophy and increased intraocular pressure.3 Antihistamines only target the early-phase response, with limited efficacy in ameliorating late-phase inflammation and nasal mucosal hyperresponsiveness.4 Allergen-Specific Immunotherapy (AIT) requires a long treatment course of 3–5 years, and the response rate is less than 60% in patients with moderate-to-severe disease.5 These limitations of symptomatic rather than etiological treatment urgently necessitates the exploration of novel therapeutic targets upstream of inflammatory pathogenesis. Current established first-line therapies for AR (intranasal glucocorticoids, antihistamines, leukotriene receptor antagonists, and allergen-specific immunotherapy) exert clinical efficacy partly by regulating interleukin networks and macrophage polarization, yet their precise immunological mechanisms remain to be integrated with novel targeted strategies.6
The core pathological feature of AR is chronic allergic inflammation driven by immune imbalance. As key effector cells of innate immunity, macrophages modulate the intensity and progression of inflammation directly via the balance of their polarization (ie, M1/M2 phenotypic switching).7 Under normal physiological conditions, M1 macrophages eliminate pathogens by secreting pro-inflammatory cytokines such as interleukin (IL)-6 and tumor necrosis factor-α (TNF-α), whereas M2 macrophages maintain immune tolerance through the release of IL-10 and transforming growth factor-β (TGF-β). The dynamic equilibrium between these two subsets constitutes the mucosal immune homeostasis.8 Interleukin network dysregulation disrupts this balance in AR. It forms a vicious cycle: abnormal interleukin secretion, disordered macrophage polarization, and amplified inflammatory signaling. Specifically, Th2-type interleukins (IL-4, IL-13) can induce the differentiation of macrophages into the pathogenic M2a subtype, while simultaneously promoting immunoglobulin E (IgE) production and eosinophil recruitment.9 In contrast, pro-inflammatory interleukins (IL-1β, IL-17) exacerbate the excessive activation of M1 macrophages, which release abundant inflammatory mediators and consequently lead to nasal mucosal epithelial damage.10
As key molecules bridging innate and adaptive immunity, interleukins precisely modulate macrophage phenotypic switching via specific receptor signaling, and thus serve as core targets for breaking the aforementioned vicious cycle.11 For instance, IL-10 can induce the polarization of macrophages toward the anti-inflammatory M2c subtype, thereby markedly suppressing nasal mucosal inflammation in AR mice.12 IL-27 modulates macrophages to exhibit a “balanced pro-inflammatory and anti-inflammatory phenotype” via the signal transducer and activator of transcription 1 (STAT1) pathway, and antagonizes Th2-type inflammatory responses.11 This regulatory axis of “interleukins and macrophage polarization” provides a novel direction for the precision treatment of AR by rectifying immune dysregulation at its source.
The complete regulatory network constituted by “interleukins, macrophage polarization, and AR inflammation” has not yet been systematically integrated in current research. International studies focus on the mechanistic elucidation of biologics, but lack sufficient attention to the individual differences in distinct polarization phenotypes.2 Domestic research has yielded fruitful achievements in the regulation of related pathways by natural products, yet most mechanistic investigations remain at the cellular level, with a paucity of clinical translational evidence.13 Additionally, the dual pro-/anti-inflammatory effects of some members of the interleukin family (eg, IL-32) remain controversial,14 and the impact of macrophage heterogeneity (eg, the role of the MDM3 subtype in nasal polyps15) on therapeutic strategies has not been clarified. Based on the current research status at home and abroad, this review systematically summarizes the molecular mechanisms by which interleukins regulate macrophage polarization to ameliorate AR inflammation, compares the characteristics of domestic and international studies, and thereby provides a comprehensive perspective for the research, development, and translation of precision therapies for AR.
Method
Search Strategy
We screened PubMed and Embase databases from January 2010 to January 2026 to search for published studies. The search keywords used are as follows: [“interleukin” or “IL”], [“allergic rhinitis” or “AR”], [“macrophages”] and [“inflammation”]. The retrieval process was completed independently by two investigators.
Inclusion and Exclusion Criteria
Inclusion Criteria
Study design: Original peer-reviewed articles, including clinical trials, animal experiments, and in vitro cell experiments;
Research topic: Directly focused on interleukin-mediated macrophage polarization in the regulation of AR inflammation;
Language: Publications in English or Chinese;
Publication time: From January 1, 2010, to January 1, 2026;
Data integrity: Complete full-text data and clear experimental results are available.
Exclusion Criteria
Non-original literature: meta-analyses, case reports, conference abstracts, letters, and guidelines;
Irrelevant studies: Not related to interleukins, macrophage polarization, or AR inflammation;
Unavailable literature: No full-text access, incomplete data, or duplicated publications;
Poor-quality literature: Studies with obvious data errors or unreported methodological details.
Study Selection
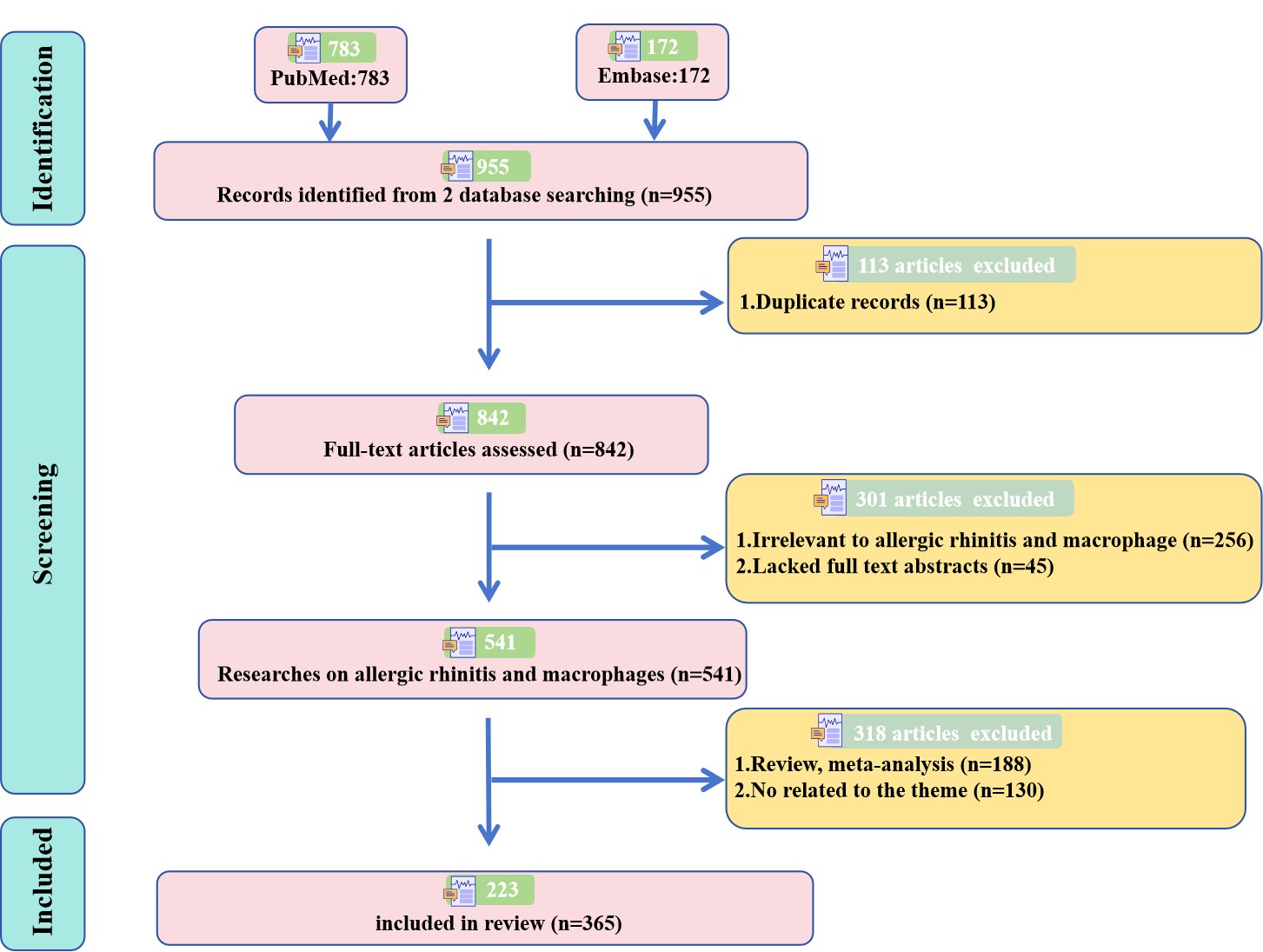
After removing 113 duplicate records, 842 records were initially identified. Two researchers independently screened titles and abstracts, and excluded 45 articles without full-text abstracts, 256 irrelevant articles, 188 reviews/meta-analyses, and 130 off-topic articles. Finally, 223 eligible studies were included. A clear PRISMA flow diagram is presented in Supplementary Figure 1 to show the complete screening process.
{kind=link}
Data Extraction
A standardized data extraction form was used to collect core information: interleukin family, study model, signaling pathway, key targets, mechanism, and outcomes. Discrepancies were resolved by discussion with a third investigator.
Risk of Bias Assessment
We performed standardized risk of bias assessment for all included studies:
For animal studies: The SYRCLE (Systematic Review Centre for Laboratory Animal Experimentation) risk of bias tool was used to evaluate selection, performance, detection, attrition, reporting, and other biases.
For clinical studies: The Cochrane Risk of Bias Tool was used to assess randomization, allocation concealment, blinding, outcome data, and selective reporting.
All assessments were completed independently by two researchers; disagreements were settled via consensus.
Overview of Interleukins and Macrophage Polarization Regulation in AR
Biological Characteristics of Interleukins and Their Secretory Regulation in AR
As key signaling molecules between immune cells and parenchymal cells, interleukins form a sophisticated regulatory network via shared receptor subunits or signaling pathways.11,16,17 Members of the IL-1 family (eg, IL-1β, IL-33) mostly exist in the form of alarmins and are highly expressed in the nasal mucosal epithelial cells of AR patients. When the nasal mucosa is subjected to allergen stimulation or tissue damage, IL-33 is released from the nucleus and binds to the ST2 receptor on the surface of macrophages, thereby initiating downstream inflammatory signaling.1,18,19 The IL-2 family (eg, IL-4, IL-13) transmits signals via the γc-chain receptor and is mainly secreted by T helper 2 (Th2) cells and group 2 innate lymphoid cells (ILC2s), serving as core factors that drive Th2-type inflammation in AR.2,20 The IL-6 family (eg, IL-6, IL-27) mediates signaling through the gp130 subunit. Among them, IL-6 exacerbates inflammatory amplification, while IL-27 exerts anti-inflammatory regulatory effects, thus forming a functional antagonism.11,21 The IL-10 family (eg, IL-10, IL-22) takes immune suppression as its core function and maintains immune homeostasis via activating the STAT3 pathway.12,22,23
The molecular pathogenesis of allergic rhinitis is illustrated in Figure 1. In the context of AR, the secretory regulation of interleukins is characterized by multi-cellular synergistic activation. For instance, upon stimulation by allergens such as house dust mites and pollen, nasal epithelial cells secrete IL-33 and thymic stromal lymphopoietin (TSLP) via the TLR4/NF-κB pathway,24 which further induces macrophages and mast cells to produce IL-4 and IL-1β.9,25 Th2 cells and macrophages amplify inflammatory signals through a “histamine-IL-4” positive feedback loop.9 The functional deficiency of regulatory T cells (Tregs) leads to insufficient IL-10 secretion, which fails to inhibit the excessive production of pro-inflammatory interleukins.5,26 Mendelian randomization studies have confirmed that elevated circulating levels of IL-18 and macrophage inflammatory protein-1α (MIP-1α) are significantly associated with an increased risk of AR onset, whereas elevated levels of TNF-related apoptosis-inducing ligand (TRAIL) exert a protective effect. These findings suggest that the dysregulated secretion of interleukins is a causal factor rather than a secondary effect in the pathogenesis of AR.27
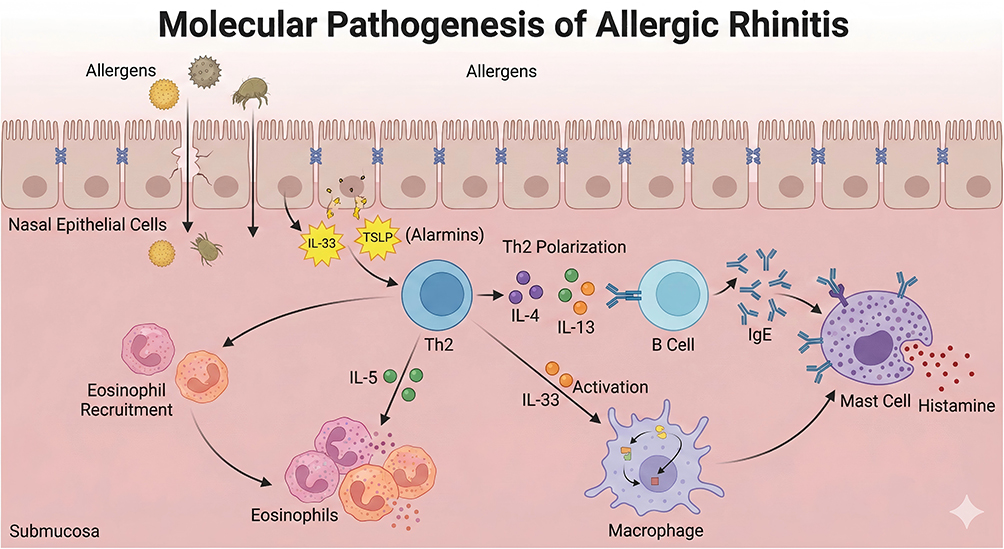 |
Figure 1 Molecular Pathogenesis of allergic rhinitis. This schematic depicts the stepwise cellular and molecular events driving allergic rhinitis. Inhaled allergens cross the nasal epithelial barrier, triggering nasal epithelial cells to release alarmins (IL-33, TSLP). These alarmins promote Th2 cell differentiation. Th2 cells secrete key cytokines. IL-4 and IL-13 drive B-cells to produce allergen-specific IgE antibodies, which bind to mast cells. IL-5 mediates eosinophil recruitment and activation in the submucosa. IL-33 also activates macrophages, amplifying inflammation. Mast cell-bound IgE cross-links upon repeat allergen exposure, triggering degranulation and release of histamine (and other mediators), which causes the classic symptoms of allergic rhinitis. |
Core Features of Inflammation and the Critical Role of Macrophage Polarization in AR
The core feature of AR inflammation is characterized by the trinity of “Th2-type inflammation predominance, mucosal barrier damage, and inflammatory cell infiltration”. Disruption of tight junctions in nasal mucosal epithelial cells leads to allergen translocation.28 A large number of inflammatory cells such as eosinophils, mast cells, and macrophages infiltrate the lesion site, releasing mediators including eosinophil cationic protein (ECP), histamine, and tryptase.8,29 The interaction between Th2 cells and B cells promotes IgE class switching, thereby inducing the formation of allergic memory.30,31 In this pathological process, macrophage polarization exerts a pivotal regulatory role. As the most abundant innate immune cells in the nasal mucosa, macrophages integrate interleukin signals through phenotypic switching, and then modulate the intensity of inflammation and the direction of immune responses.7
M1 macrophages are hyperactivated in AR. Upon activation by factors such as IL-1β and lipopolysaccharide (LPS), they secrete pro-inflammatory mediators including IL-6, TNF-α, and nitric oxide (NO) via the NF-κB and MAPK pathways, thereby exacerbating the apoptosis of nasal mucosal epithelial cells and the recruitment of inflammatory cells.8,32 Clinical sample analysis has shown that the expression of M1 markers, including inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2), is significantly upregulated in the nasal mucosa of AR patients, and positively correlates with the severity of nasal congestion and rhinorrhea symptoms.13
In contrast, M2 macrophages exhibit a characteristic of “functional differentiation”. Under normal physiological conditions, M2 macrophages inhibit inflammation and promote tissue repair by secreting IL-10 and TGF-β.33 However, in AR, M2 macrophages are induced by IL-4 and IL-13 into the pathogenic M2a subtype, which highly expresses CD206 and arginase 1 (Arg1), thereby promoting Th2 cell polarization and IgE production.7,34 The proportion of the M2a subtype (CD44+CCR2+CD64−) is increased in nasal polyp tissues, and positively correlates with the Lund-Mackay score, suggesting its involvement in the chronicization of AR.15
Disruption of macrophage polarization balance is a key driver of AR recurrence. The M1/M2 imbalance leads to sustained activation of inflammatory signaling, and repeated damage and repair of nasal mucosal epithelial cells induce tissue remodeling.35 Co-culture of macrophages with nasal epithelial cells can trigger epithelial-mesenchymal transition (EMT), thereby aggravating nasal mucosal fibrosis.7 Furthermore, efferocytotic dysfunction of macrophages results in impaired clearance of apoptotic cells, which further amplifies the inflammatory response. Knockdown of the myeloid cell-specific pyruvate kinase M2 (PKM2) gene was found to significantly suppress the pro-inflammatory activation of macrophages and reduce inflammatory infiltration in the nasal mucosa of AR mice, validating that disordered macrophage polarization is a core pathogenic driver of AR inflammation.36
Dual Roles of Interleukins in Regulating Macrophage Polarization in AR
The regulation of macrophage polarization by interleukins exhibits a “bidirectional regulation” feature, encompassing both “pathological polarization” induced by pro-inflammatory interleukins and “homeostatic repair” mediated by anti-inflammatory interleukins. Pro-inflammatory interleukins drive macrophages toward pro-inflammatory phenotypes through specific signaling pathways. IL-4 and IL-13 bind to macrophage IL-4Rα. They activate the STAT6 pathway, induce M2a polarization, and promote IL-13 and CCL17 secretion.9,37 IL-1βactivates the NLR family pyrin domain containing 3 (NLRP3) inflammasome, promotes macrophage pyroptosis, and triggers the release of IL-18, thereby further amplifying M1-type inflammation.8,38 IL-17A enhances macrophage M1 polarization through the NF-κB pathway and synergizes with IL-4 to exacerbate Th2-type inflammation.39,40 Clinical studies have confirmed that the expression levels of IL-4 and IL-17A in the nasal mucosa of AR patients exhibit a significant positive correlation with the expression of M2a and M1 markers, respectively.10,39
Anti-inflammatory interleukins maintain macrophage polarization balance through dual mechanisms of “direct inhibition and phenotypic switching”. IL-10 binds to the IL-10 receptor (IL-10R) on macrophages, activates the STAT3 pathway, inhibits NF-κB activity, and induces M2c polarization along with the secretion of anti-inflammatory factors.12,41 IL-27 downregulates the expression of the M2 marker CD206 via the STAT1 pathway, while promoting the secretion of interferon-gamma (IFN-γ) to antagonize Th2-type inflammation.11,42 IL-35 inhibits the excessive activation of T cells by inducing macrophages to express programmed death-ligand 1 (PD-L1).5,43 Bioactive components of traditional Chinese medicine (TCM), such as Xiaoqinglong Decoction and Melastoma dodecandrum polysaccharide, can remodel the M1/M2 balance of macrophages by upregulating IL-10 and downregulating IL-4 expression,13,44 confirming that anti-inflammatory interleukin-mediated polarization repair is an effective therapeutic approach for AR.
Controversies persist in the polarization-regulating effects of certain interleukins. In human AR patient samples (limited human observational data), IL-32 is highly expressed in the nasal mucosa.45 In in vitro cellular models (preclinical mechanistic data), IL-32 promotes macrophage pro-inflammatory activation by activating caspase-1; however, the anti-inflammatory effect of the IL-32γ isoform is only observed in tumor models (non-AR preclinical evidence) and represents an emerging, unconfirmed hypothesis in AR.14,43 No in vivo animal studies have validated the function of IL-32 in AR, so its definitive regulatory mechanism remains to be established.
For IL-33, it can induce M2 polarization to exacerbate inflammation on the one hand, and alleviate inflammation by promoting regulatory T cells (Tregs) to secrete IL-10 on the other hand.46 This dual role and contradictory conclusions may stem from three key factors. First, IL-33 exhibits concentration-dependent effects (low concentrations promote M2 polarization, high concentrations activate M1 polarization), which are rarely controlled consistently across studies. Second, methodological gaps exist: most evidence is derived from murine AR models, while few human nasal mucosal ex vivo studies are available, and animal strain differences lead to heterogeneous immune responses. Third, the relative levels of other cytokines (eg, IL-10, TNF-α) in the local inflammatory microenvironment differ significantly between studies, resulting in opposite polarization outcomes.
Mechanistic Specificity of Different Interleukins in Regulating Macrophages to Ameliorate AR Inflammation
IL-1 Family: Dual Regulation of Inflammation Initiation and Polarization Orientation
As a classic pro-inflammatory cytokine, IL-1β binds to IL-1 receptor type 1 (IL-1R1) on macrophages to activate the NF-κB pathway, thereby promoting M1 polarization and the secretion of IL-6 and TNF-α, which exacerbates inflammatory cell infiltration and tissue damage in the nasal mucosa.8,10,47 Clinical sample studies have shown that IL-1β expression in the nasal mucosa of AR patients is significantly correlated with NLRP3 inflammasome activation; inhibiting NLRP3 can reduce IL-1β release and reverse macrophage M1 polarization.48 IL-33 regulates macrophage polarization balance via the ST2 receptor: low concentrations of IL-33 induce M2 polarization and the secretion of IL-4 and IL-13, while high concentrations activate the NF-κB pathway to promote M1 activation.18,49 Serum IL-33 levels are significantly elevated in AR patients and positively correlate with the expression of the M2 marker CD206 in the nasal mucosa. The clinical potential of anti-IL-33 antibodies demonstrated in asthma and chronic obstructive pulmonary disease (COPD) suggests that IL-33 may serve as a novel therapeutic target for AR.
IL-18 drives the production of IL-1β and granulocyte-macrophage colony-stimulating factor (GM-CSF) by macrophages, enhances their antigen-presenting capacity, and amplifies Th2-type inflammatory responses.50 Mendelian randomization studies have confirmed that genetically predicted elevated IL-18 levels are associated with an increased risk of AR onset,27 while IL-18 binding protein (IL-18BP) can inhibit macrophage activation by neutralizing IL-18.50 As an IL-1 receptor antagonist (IL-1Ra), IL-1Ra competitively binds to IL-1 receptors, suppresses the pro-inflammatory activation of macrophages, and reduces the release of pro-inflammatory cytokines, thereby providing a “receptor blockade” strategy for AR treatment.51
IL-2 Family (γc Chain Family): Core Regulation of Th2 Inflammation Dominance and Polarization Balance
IL-4, a key factor inducing macrophage M2 polarization, upregulates the expression of CD206 and arginase 1 (Arg1) via the IL-4Rα/STAT6 pathway, promotes the secretion of IL-13 and C-C motif chemokine ligand 2 (CCL2), and recruits Th2 cells and eosinophils.9,52 Clinical data confirm positive correlations between IL-4/IL-13 levels and M2a abundance in AR nasal mucosa.7,53 Knockdown of IL-4 or blockade of IL-13Rα1 (via miR-31/miR-143) effectively inhibits M2a-mediated EMT and inflammatory amplification.54,55
IL-2 inhibits macrophage M2 polarization by activating the STAT5 pathway, promotes IFN-γ secretion, and antagonizes Th2-type inflammatory responses.5,56 Immunostimulatory sequence oligodeoxynucleotide (ISS-ODN) can enhance anti-inflammatory effects by inducing macrophages to produce IL-2, and its efficacy is superior to that of dexamethasone57 Although IL-5 does not directly regulate macrophage polarization, it can enhance the chemotactic and activating capacities of macrophages toward eosinophils, and amplify Th2-type inflammation via paracrine effects.58 Montelukast can reduce inflammatory infiltration by inhibiting IL-5-mediated macrophage-eosinophil interaction.59
IL-6 Family: Functional Antagonism Between Pro-Inflammatory Amplification and Anti-Inflammatory Regulation
IL-6 promotes macrophage M1 polarization via the gp130/JAK/STAT3 pathway, induces the secretion of TNF-α and IL-1β, and exacerbates nasal mucosal inflammatory damage.60 The expression of IL-6 in the nasal mucosa of AR patients is positively correlated with the M1 marker inducible nitric oxide synthase (iNOS). Chlorogenic acid can block IL-6-mediated macrophage activation by inhibiting the TLR4/MAPK/NF-κB pathway.32 As an anti-inflammatory member of this family, in animal and in vitro preclinical models (established preclinical mechanism), IL-27 inhibits macrophage M2 polarization by activating the STAT1 pathway, downregulates the expression of IL-4 and IL-13, and simultaneously promotes IFN-γ secretion.11 Preclinical studies (AR mouse model) have confirmed that IL-27 pretreatment significantly reduces nasal mucosal inflammatory infiltration (animal data only). In human AR patients, the immunomodulatory role of IL-27 remains an emerging hypothesis with no clinical or ex vivo human data to validate it.
IL-31 enhances the release of pro-inflammatory cytokines by activating the MAPK pathway in macrophages, thereby aggravating pruritus-associated inflammatory responses of the nasal mucosa.61 Serum IL-31 levels in AR patients are positively correlated with nasal pruritus symptom scores. The success of IL-31-targeted biologics in atopic dermatitis (AD) provides a cross-disease reference for AR treatment.
IL-10 Family: Core Mediators of Anti-Inflammatory Polarization and Immune Tolerance
IL-10 is the primary anti-inflammatory interleukin governing M2c polarization, upregulates the secretion of IL-10 and TGF-β, and inhibits the activation of NF-κB and the NLRP3 inflammasome.8,62 The peroxisome proliferator-activated receptor gamma (PPAR-γ) agonist ciglitazone can inhibit M2 polarization and alleviate nasal symptoms in AR mice by inducing macrophages to secrete IL-10.12 IL-22 synergizes with IL-10 to enhance the tissue repair capacity of macrophages, suppress M1 polarization, and protect the integrity of the nasal mucosal epithelial barrier.63 IL-22 expression is downregulated in the nasal mucosa of AR patients, and exogenous supplementation of IL-22 can significantly improve the tight junction function of the nasal mucosa.28
Other Key Interleukins: Complementary Mechanisms of Inflammatory Amplification and Regulation
The IL-17 family (including IL-17A and IL-17F) promotes macrophage M1 polarization by activating NF-κB pathway, enhances the release of IL-6 and TNF-α, and recruits neutrophil infiltration.10,39 AR symptoms are significantly alleviated in IL-17A-deficient mice. Melastoma dodecandrum polysaccharide can inhibit macrophage pyroptosis by regulating the NLRP3/IL-17 axis.44 IL-32 promotes the pro-inflammatory activation of macrophages by activating caspase-1 and upregulates the expression of IL-1β and TNF-α.45,64 Bamboo salt can alleviate macrophage-mediated inflammatory responses by inhibiting IL-32 signaling.64 GM-CSF promotes macrophage M2 polarization, enhances antigen-presenting capacity, and synergizes with IL-5 to maintain eosinophil survival.65 Specific immunotherapy can reduce GM-CSF expression in nasal polyp tissues and alleviate macrophage-mediated inflammation.66 We have summarized the research on the regulation of macrophages by different interleukins to improve allergic rhinitis inflammation, as shown in Table 1.
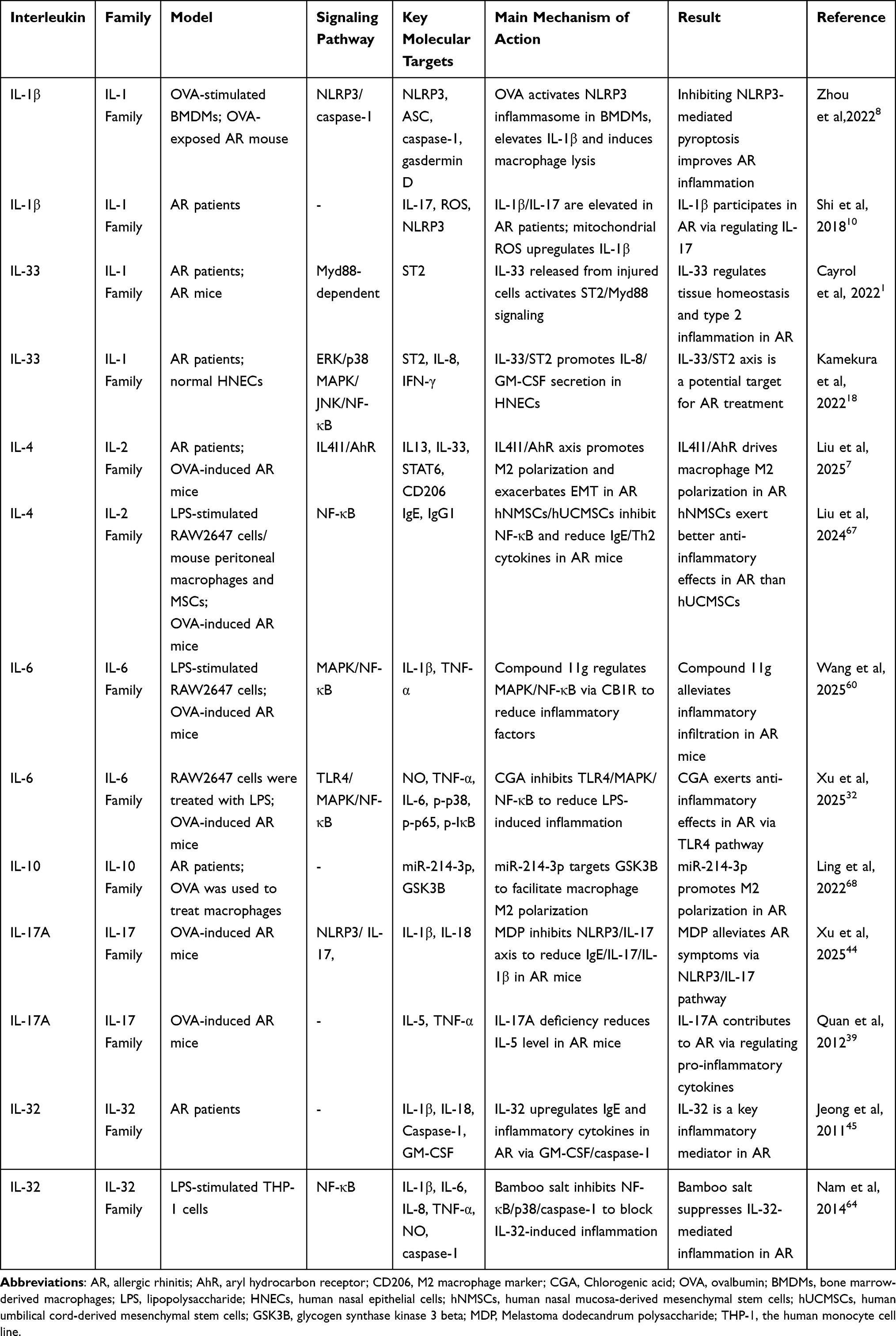 |
Table 1 Summary of Studies on Different Interleukins Regulating Macrophages to Improve Allergic Rhinitis Inflammation |
Interaction Mechanisms Between Aberrant Macrophage Polarization and the Interleukin Network in AR
Polarization Characteristics and Functional Imbalance of Macrophages in AR
In the context of AR, allergens (eg, house dust mites and pollen) activate macrophages via the TLR4/NF-κB pathway, inducing their polarization toward the M1 phenotype32 Meanwhile, pro-inflammatory cytokines such as IL-1β and TNF-α form a positive feedback loop that sustains the persistent activation of M1 macrophages8 By secreting mediators including IL-6, IL-12, and NO, M1 macrophages directly impair the tight junctions of nasal mucosal epithelial cells (as evidenced by the downregulated expression of ZO-1 and occludin), and recruit neutrophils and Th17 cells via the secretion of chemokines such as CCL2 and macrophage inflammatory protein-2 (MIP-2)28,39 Clinical sample analyses have shown that the proportion of M1 macrophages in the nasal mucosa of AR patients is 23-fold higher than that in healthy individuals, and positively correlates with nasal mucosal thickness and the number of infiltrating inflammatory cells69 In vitro experiments have confirmed that M1-conditioned medium can significantly promote the apoptosis of nasal epithelial cells, whereas blockade of IL-6 and TNF-α can reverse this effect, suggesting that M1 macrophage hyperactivation is a direct cause of nasal mucosal damage.32
Under normal physiological conditions, M2 macrophages inhibit inflammatory responses and promote nasal mucosal epithelial repair by secreting IL-10 and transforming growth factor-β (TGF-β) Whereas in AR, M2 macrophages exhibit a feature of pathogenic activation: they are induced into the M2a subtype by IL-4 and IL-13, highly express CD206 and arginase 1 (Arg1), and promote Th2 cell polarization and IgE production via the secretion of IL-4 and IL-13. Meanwhile, co-culture of M2a macrophages with nasal epithelial cells can induce epithelial–mesenchymal transition (EMT), thereby facilitating nasal mucosal fibrosis and tissue remodeling.7 In nasal polyp tissues, the proportion of the M2a subtype (CD44+CCR2+CD64−) is significantly increased, and positively correlates with the number of infiltrating eosinophils as well as IL-5 expression.15 In SENP3-deficient mice, enhanced M2 polarization elevates the risk of nasal polyp formation, confirming that pathogenic M2 macrophages are a key driver of AR chronicization.35
As an anti-inflammatory member of the M2 subset, the M2c subtype exhibits functional deficiency in AR. In the nasal mucosa of AR patients, the expression of the M2c marker CD163 is downregulated, accompanied by insufficient IL-10 secretion, which fails to inhibit the hyperactivation of M1 and M2a macrophages. The functional impairment of regulatory Tregs serves as a critical factor contributing to insufficient M2c activation. Allergen immunotherapy (AIT) can promote M2c polarization by restoring Treg function.5
Disruption of M1/M2 polarization balance contributes to the persistent and recurrent features of AR inflammation: M1 hyperactivation triggers acute inflammatory responses (eg, sneezing, nasal pruritus), while pathogenic differentiation of M2a sustains chronic inflammation (eg, nasal congestion, nasal mucosal hypersecretion). Their synergistic effect induces nasal mucosal hyperresponsiveness, characterized by excessive responses to non-specific stimuli such as cold air and odors.69 Long-term polarization imbalance also leads to the formation of immune memory. M2a macrophages enhance antigen-presenting capacity to promote the formation of Th2 cell memory, resulting in AR patients triggering inflammatory responses even upon exposure to low-dose allergens. Clinical studies have confirmed that AR patients have a high recurrence rate after discontinuing medication after drug withdrawal, and the M1/M2 ratio in their nasal mucosa remains significantly imbalanced, suggesting that the failure to restore polarization balance is the core cause of recurrence.3,70 AR patients have a high recurrence rate after discontinuing medication.
Regulatory Effects of the Interleukin Network on Macrophage Polarization
Th2-type interleukins (IL-4 and IL-13) serve as the core signals inducing macrophage polarization toward the M2a subtype. IL-4 binds to IL-4Rα on the macrophage surface, activates the STAT6 pathway, and upregulates the expression of M2a markers such as CD206 and arginase 1 (Arg1).7 IL-13 acts via the IL-13 receptor alpha 1 (IL-13Rα1)/STAT6 pathway, synergizes with IL-4 to enhance M2a polarization, and simultaneously promotes the expression of mucus-related genes.54 The expression levels of IL-4 and IL-13 in the nasal mucosa of AR patients are 3.1-fold and 2.7-fold higher than those in healthy individuals, respectively, and positively correlate with the proportion of M2a macrophages.55 MicroRNA-143 (miR-143) can block IL-13-mediated M2a polarization by inhibiting IL-13Rα1 expression, confirming that Th2-type interleukins are key drivers of M2a polarization.55
Pro-inflammatory interleukins (IL-1β, IL-6, IL-17) synergistically induce macrophage polarization toward the M1 phenotype. IL-1β promotes macrophage pyroptosis and triggers the release of IL-18 by activating the NLRP3 inflammasome, thereby further enhancing M1 activation.8 IL-6 upregulates the expression of iNOS and COX-2 via the JAK/STAT3 pathway, strengthening the pro-inflammatory function of M1 macrophages.32 IL-17A synergizes with IL-4 through the NF-κB pathway to not only promote M1 polarization but also augment Th2-type inflammation.39 These interleukins exert a cascade amplification effect: IL-1β induces macrophages to secrete IL-6 and IL-17, which in turn recruit Th17 cells to secrete IL-17, exacerbating M1 polarization.7
The IL-10 family restores macrophage polarization balance via dual mechanisms of direct inhibition + phenotypic switching. IL-10 binds to the IL-10R on macrophages, activates the STAT3 pathway, inhibits NF-κB activity, downregulates the expression of M1 markers, and simultaneously induces M2c polarization.12 IL-10 expression is downregulated in the nasal mucosa of AR patients, and exogenous supplementation of IL-10 can significantly reduce the M1/M2a ratio and alleviate inflammatory symptoms.12 IL-27 induces macrophages to exhibit an “M1/M2 balanced phenotype” by activating the STAT1 pathway, which not only reduces the secretion of IL-6 and TNF-α but also decreases the expression of CD206 and IL-13.11 IL-22 synergizes with IL-10 to enhance the tissue repair capacity of macrophages and protect the integrity of the nasal mucosal epithelial barrier.14
Other anti-inflammatory mediators (eg, TRAIL, IL-35) are also involved in polarization regulation: TRAIL reduces the secretion of pro-inflammatory cytokines by macrophages via inhibiting the NF-κB pathway.27 IL-35 indirectly restores polarization balance by inducing macrophages to express PD-L1 and inhibiting excessive T cell activation.5
Vicious Cycle Between Macrophage Polarization and Interleukins
A closed loop of “aberrant macrophage polarization-interleukin imbalance” is formed in AR. Allergen stimulation induces M1/M2 imbalance in macrophages: M1 macrophages secrete IL-1β and IL-6, while M2a macrophages secrete IL-4 and IL-13, and these interleukins further exacerbate the disorder of macrophage polarization.9 Meanwhile, macrophages with abnormal polarization enhance their antigen-presenting capacity to promote Th2 cell activation and interleukin secretion, amplifying inflammatory signals. For instance, Th2 cells and macrophages mutually activate each other through a histamine-IL-4 loop. Histamine secreted by macrophages promotes Th2 cells to secrete IL-4 via the histamine H4 receptor, and IL-4 further induces M2a polarization of macrophages and histamine release.9 Activation of the NLRP3 inflammasome leads to macrophage pyroptosis, and the released IL-1β induces Th17 cells to secrete IL-17, which in turn exacerbates M1 polarization.8
This vicious cycle leads to sustained amplification of inflammatory signals (as shown in Figure 2). Damage to nasal mucosal epithelial cells induces allergen translocation, which further activates macrophages and the interleukin network.24 Inflammatory cell infiltration exacerbates tissue damage, forming a “damage-inflammation-re-damage” cycle.28 Ultimately, this drives AR to progress from acute inflammation to chronic inflammation, and may even develop into complications such as asthma and nasal polyps.2
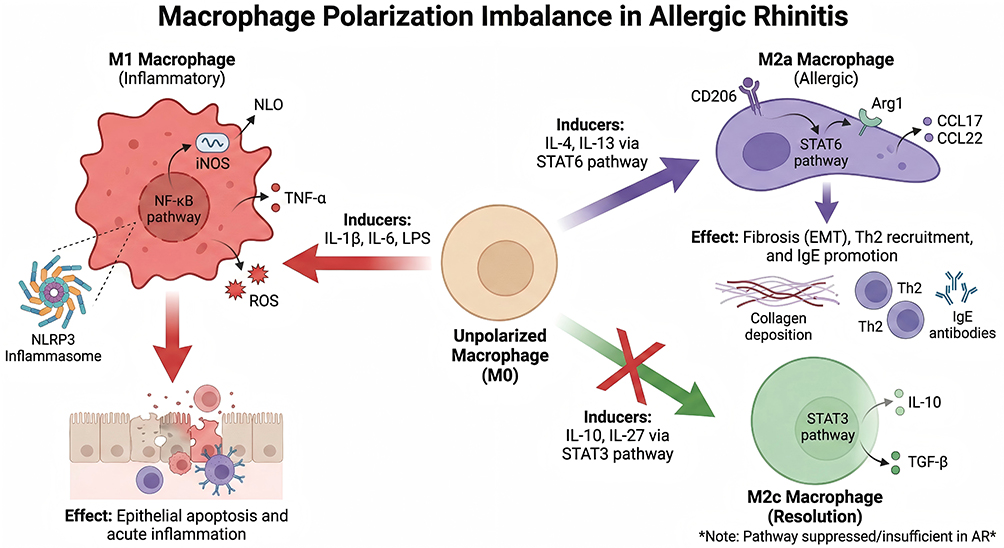 |
Figure 2 The polarization of macrophages in the nasal mucosa of allergic rhinitis is imbalanced. This schematic illustrates the dysregulation of macrophage polarization in allergic rhinitis (AR), depicting how unpolarized (M0) macrophages differentiate into functionally distinct subsets and their contributions to allergic inflammation. M1 (Pro-Inflammatory) Macrophages: Induced by triggers including IL-1β, IL-6, and LPS via the NF-κB pathway, these cells produce pro-inflammatory mediators (TNF-α, reactive oxygen species [ROS], iNOS, and NLO) and interact with the NLRP3 inflammasome. This drives epithelial cell apoptosis and acute mucosal inflammation. M2a (Allergic/Type 2) Macrophages: Polarized by IL-4 and IL-13 through the STAT6 signaling cascade, M2a macrophages express markers CD206 and Arg1, and secrete chemokines CCL17/CCL22. They promote allergic pathology by driving collagen deposition (fibrosis/epithelial-mesenchymal transition, EMT), Th2 cell recruitment, and IgE antibody production. M2c (Anti-Inflammatory/Pro-Resolution) Macrophages: Normally induced by IL-10 and IL-27 via the STAT3 pathway to produce anti-inflammatory cytokines IL-10 and TGF-β, this homeostatic subset is suppressed or insufficient in AR. |
Core Mechanisms of Interleukin-Regulated Macrophage Polarization for Ameliorating AR Inflammation
Targeting Pro-Inflammatory Interleukins: Blocking Abnormal Polarization-Inducing Signals
As the core inducing signals for M2a polarization, IL-4/IL-13 and their receptor IL-4Rα represent important therapeutic targets.71 Dupilumab, an anti-IL-4Rα monoclonal antibody, has been confirmed in phase III clinical trials to significantly reduce the expression of the M2a marker CD206 in the nasal mucosa of AR patients, decrease IL-4/IL-13 levels, and improve nasal mucosal hyperresponsiveness.2,72 Semi-quantitative synthesis shows a consistent effect trend: By regulating CD206/TLR2, eosinophil infiltration was reduced by ≥55% and AR pathology in mice was effectively improved.73 Animal experiments have shown that anti-IL-13 antibodies can inhibit macrophage M2a polarization and reduce eosinophil recruitment.54 In addition, miRNA-based regulatory strategies have also shown promising potential. miR-31 and miR-143 can block IL-13-mediated macrophage activation by inhibiting IL-13Rα1 expression.74,75 Intranasal delivery of miR-31 mimics can significantly alleviate symptoms in AR mice.55
IL-1β-mediated M1 polarization and macrophage pyroptosis serve as crucial initiating events in AR inflammation.76 MCC950, a selective NLRP3 inhibitor, can suppress macrophage pyroptosis, reduce IL-1β release, reverse M1 polarization, and alleviate nasal mucosal inflammation in AR mice.8,77 Melastoma dodecandrum polysaccharide inhibits macrophage pyroptosis and IL-17 secretion by downregulating the expression of NLRP3 and gasdermin D (GSDMD).44,78 Canakinumab, a monoclonal antibody against IL-1β, has been confirmed in AR animal models to inhibit M1 macrophage activation and reduce pro-inflammatory cytokine release, thereby providing a solid basis for clinical translation.10,79
IL-17A synergistically promotes M1 polarization and Th2-type inflammation, and therapeutic strategies targeting IL-17 have demonstrated efficacy. In IL-17A-deficient mice, AR symptoms are significantly alleviated, with reduced levels of M1 markers and Th2 cytokines in the nasal mucosa.39,80 Traditional Chinese medicine (TCM) components (eg, bamboo salt, Nanhuzhu herb) can reduce the pro-inflammatory activation of macrophages by inhibiting the IL-32/IL-17 axis.64,81
Enhancing Anti-Inflammatory Interleukins: Activating Polarization-Balancing Signals
IL-10-induced M2c polarization is key to restoring immune homeostasis.82 Ciglitazone, a peroxisome proliferator-activated receptor gamma (PPAR-γ) agonist, can inhibit M2a polarization and alleviate symptoms in AR mice by inducing macrophages to secrete IL-10.12,83 Mesenchymal stem cell-derived exosomes (MSC-Exos) carry miR-146a-5p, which can upregulate IL-10 expression in macrophages and promote M2c polarization.84 IL-35 indirectly promotes anti-inflammatory polarization of macrophages by enhancing regulatory T cell (Treg) function, and its mimetics have shown favorable safety profiles in preclinical studies.5
IL-27-mediated balanced polarization exhibits unique advantages.85 It not only inhibits M2a polarization but also does not exacerbate M1 activation; instead, it induces macrophages to adopt an anti-inflammatory phenotype. Intranasal administration of IL-27 analogs can significantly reduce IL-4 and IL-6 levels in the nasal mucosa of AR mice and ameliorate their symptoms.11,86 Natural products (eg, chlorogenic acid) can regulate macrophage polarization by upregulating IL-27 expression, which provides insights into the development of multi-targeted therapy.32
Downstream Anti-Inflammatory Effects: Ameliorating Core Pathological Features of AR
Modulation of macrophage polarization significantly reduces the recruitment of inflammatory cells such as eosinophils and mast cells: inhibition of M2a polarization decreases the secretion of CCL17 and CCL22, thereby reducing the infiltration of Th2 cells and eosinophils.7 Activation of M2c polarization suppresses mast cell degranulation via IL-10, leading to a reduction in histamine release.12
Animal experiments have shown that Xiaoqinglong Decoction markedly decreases the numbers of eosinophils and macrophages in the nasal lavage fluid of AR mice by inhibiting M1 polarization and the transient receptor potential vanilloid 1 (TRPV1) pathway.13 MP-AzeFlu (azelastine + fluticasone propionate) synergistically inhibits the secretion of pro-inflammatory cytokines by macrophages and reduces eosinophil viability.87
Restoration of polarization balance can promote the recovery of nasal mucosal epithelial barrier function. TGF-β secreted by M2c macrophages can facilitate epithelial cell proliferation and tight junction reconstruction.12,88 IL-22 synergistically enhances the expression of the tight junction proteins ZO-1 and occludin in epithelial cells together with IL-10.28,89 Asarum sieboldii oil (ASO) improves tight junction function of the nasal mucosa in AR mice and reduces allergen translocation by modulating macrophage polarization.28 Psoralen inhibits macrophage inflammation and mucus production in nasal epithelial cells via activating the SIRT1/Nrf2 pathway.90
The core mechanism of interleukin-regulated macrophage polarization to improve AR inflammation is shown in Figure 3. Modulation of macrophage polarization can indirectly restore Treg/Th2 balance. IL-10 secreted by M2c macrophages can promote Treg proliferation and functional activation.5 Inhibition of M2a polarization reduces Th2 cell activation and IgE production. Allergen immunotherapy (AIT) restores immune tolerance by decreasing GM-CSF and IL-5 expression and reshaping macrophage polarization balance.66 Immunostimulatory sequence oligodeoxynucleotides (ISS-ODN) induce macrophages to produce IFN-γ, antagonize Th2-type inflammation, and exert a superior efficacy over dexamethasone.57
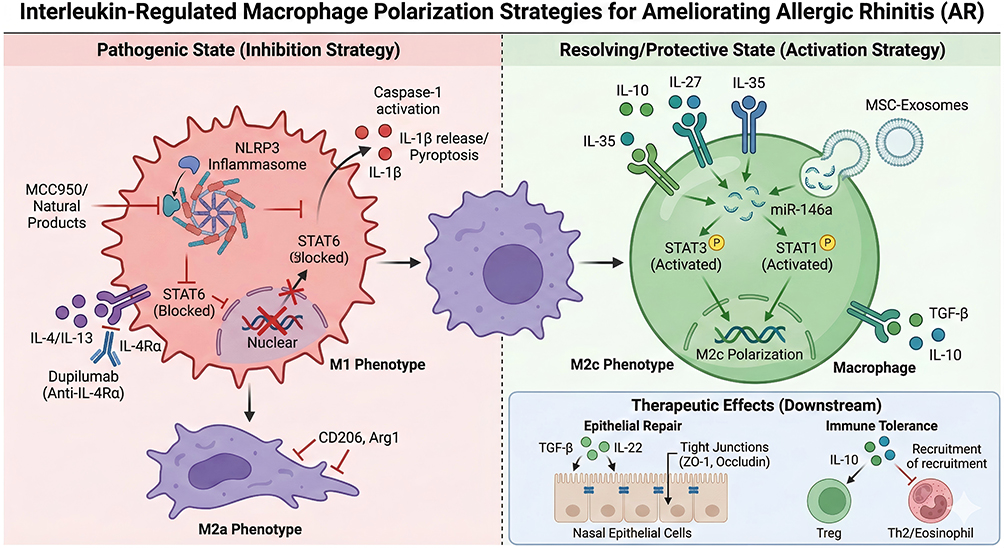 |
Figure 3 Molecular mechanism of Interleukin-targeted therapies regulating Macrophage Polarization in AR. This diagram outlines macrophage-targeted therapies for AR. The left panel depicts inhibition of pathogenic M1 (via NLRP3 inflammasome blockade) and M2a (via IL-4/IL-13/STAT6 inhibition) macrophage phenotypes. The right panel shows activation of pro-resolving M2c macrophages by IL-10, IL-27, IL-35, or MSC-exosomes (via STAT3/STAT1 signaling), which drive nasal epithelial barrier repair and immune tolerance. |
Current Status and Characteristic Comparison of Domestic and International Research
International Research Progress: In-Depth Mechanism Elucidation and Biologic Agent Development
International research has focused on the in-depth elucidation of interleukin-macrophage polarization pathway mechanisms, with particular emphasis on signaling pathway regulation and molecular target mining. It has been clarified that IL-4/IL-13 regulate Arg1 expression in macrophages via the JAK1/STAT6 pathway, IL-10 inhibits the transcription of M1-associated genes through the STAT3 pathway,12 and activation of the NLRP3 inflammasome is a critical step in macrophage pyroptosis.8,91 The development of biologic agents represents the core direction of international research. The anti-IL-4Rα monoclonal antibody (dupilumab) has been approved for the treatment of AR, and phase III clinical trials have demonstrated that it can significantly ameliorate symptoms in patients with moderate-to-severe AR.2 Anti-IL-33 antibodies have shown clinical potential in asthma and chronic obstructive pulmonary disease (COPD), and their therapeutic efficacy for AR is currently being validated in clinical trials.1,33 IL-10 fusion proteins have exhibited favorable anti-inflammatory effects in preclinical studies by targeting M2c polarization of macrophages.12
Exploration of precision medicine constitutes another distinctive feature of international research. Personalized treatment regimens have been formulated based on the macrophage polarization phenotypes of AR patients, namely M1-dominant, M2a-dominant, and mixed types.2 Heterogeneous subtypes of nasal mucosal macrophages (eg, MDM3) have been identified via single-cell RNA sequencing (scRNA-seq), providing novel targets for targeted therapy.15 Mendelian randomization studies have clarified that IL-18 and MIP-1α are causal risk factors for AR, offering clear directions for drug development.27
Domestic Research Progress: Regulation by Natural Products and Features of Clinical Translation
Domestic research has achieved remarkable outcomes in the regulation of the interleukin-macrophage polarization pathway by natural products (including traditional Chinese medicine [TCM] and plant extracts). TCM formulae (eg, Xiaoqinglong Decoction and modified Yupingfeng Powder) can reshape the macrophage M1/M2 ratio by regulating the IL-6/IL-10 balance, thereby ameliorating symptoms in AR patients without significant side effects.13,46
Active TCM components (eg, Melastoma dodecandrum polysaccharide, chlorogenic acid, and psoralen) inhibit the pro-inflammatory activation of macrophages by suppressing pathways such as NLRP3 and NF-κB.32,44,90 Ethnic medicinal materials (eg, bamboo salt and Nanhuzhu herb) alleviate macrophage-mediated inflammatory responses via inhibiting IL-32 signaling.64,81
Clinical translation and population-specific research represent the advantages of domestic studies. Cohort studies on macrophage polarization phenotypes of AR patients in the Chinese population have been conducted, which clarified that IL-17 and IL-33 act as drivers of aberrant macrophage polarization in domestic AR patients.39,46 Derivatives of natural products (eg, astragalus polysaccharide nanoparticles) have been developed to enhance the targeting efficiency toward macrophages.92
The gut microbiota-interleukin-macrophage polarization axis has been explored, and it was found that probiotics can improve macrophage function in AR by upregulating IL-10 expression.84 In addition, research on cell therapy has been gradually advanced: human nasal mucosa-derived mesenchymal stem cells (hNMSCs) suppress macrophage inflammation via the NF-κB pathway, and their anti-inflammatory efficacy is superior to that of human umbilical cord-derived mesenchymal stem cells.67 Mesenchymal stem cell-derived exosomes carry miR-146a-5p, which can modulate macrophage polarization.84
Challenges and Future Perspectives
Core Challenges in Clinical Translation
Macrophage polarization phenotypes in AR patients exhibit substantial interindividual heterogeneity. Approximately 35% of patients present with M1-dominant inflammation, 42% with M2a-dominant inflammation, and 23% with a mixed phenotype, resulting in limited response rates to single interleukin-targeted drugs. For instance, anti-IL-4Rα monoclonal antibodies exert significant therapeutic efficacy in M2a-dominant patients but yield limited improvement in M1-dominant patients.2 NLRP3 inhibitors are effective in M1-dominant patients but show poor efficacy in M2a-dominant patients.8 In addition, factors such as age, allergen type, and comorbidities (eg, asthma, nasal polyps) of AR patients can also affect macrophage polarization phenotypes, further increasing the difficulty of treatment.
Existing biologic agents are unable to specifically act on local macrophages in the nasal mucosa. Monoclonal antibodies such as anti-IL-4Rα and anti-IL-1β are administered systemically, which may impair the function of peripheral macrophages and thereby increase the risk of infection.2
Although nasal preparations (eg, glucocorticoids) can exert local effects, they lack targeting ability toward macrophages, resulting in relatively limited efficacy in restoring polarization balance.3 How to achieve targeted delivery characterized by nasal mucosal locality and macrophage specificity is the key to improving therapeutic efficacy and reducing adverse effects.
Although domestic research on natural products is abundant, it is confronted with problems such as inadequate in-depth mechanism elucidation and insufficient standardization. The exact targets of action for most active TCM components remain unclear; it is only known that they regulate pathways such as NF-κB and NLRP3, whereas their specific binding sites and molecular interactions are yet to be identified.93
TCM formulae have complex compositions and lack unified quality control standards, which leads to poor reproducibility of clinical efficacy.13 In addition, the pharmacokinetic properties of natural products (eg, low bioavailability and rapid metabolism) also limit their clinical application.90
Most studies remain at the cellular and animal model levels, lacking high-quality clinical data. Domestic research on natural products is mostly limited to small-sample clinical observations, with a shortage of large-sample, long-term follow-up phase III clinical trials.13 Real-world treatment considerations further restrict translational value, including variable patient adherence to nasal preparations, comorbidity interactions (asthma, conjunctivitis), economic burden of biologics, and regional differences in clinical practice, which are rarely combined with interleukin-macrophage targeted strategies in current research.94,95 While international biologic agents have been tested in clinical trials, subgroup analyses targeting patients with different polarization phenotypes are insufficient.2 In addition, clinical detection methods for macrophage polarization-related biomarkers (eg, CD206, iNOS, IL-10) have not been standardized, which prevents their application in therapeutic efficacy monitoring.5
Future Research Directions
Combination therapeutic strategies should be formulated based on polarization phenotypes. For patients with M1-dominant inflammation, “anti-IL-1β monoclonal antibody in combination with IL-10 mimetic” is recommended, which not only inhibits M1 activation but also induces M2c polarization.8,12 For patients with M2a-dominant inflammation, “anti-IL-4Rα monoclonal antibody in combination with IL-27 analog” is proposed to block M2a polarization and restore a balanced phenotype.2,11 For patients with the mixed phenotype, “multi-targeted nanocarriers” can be employed to simultaneously deliver IL-4Rα inhibitors and NLRP3 inhibitors.84 Machine learning algorithms can be used to integrate patients’ polarization phenotypes, genetic backgrounds, and clinical characteristics to construct therapeutic efficacy prediction models, thus enabling personalized treatment.96
Construct nasal macrophage-targeted delivery systems. Liposomes and nanoparticles modified with macrophage-targeting peptides (eg., CD206 ligands) can be used to load interleukin inhibitors or natural products, achieving targeted delivery to local nasal mucosal macrophages.84
Develop stimuli-responsive nanocarriers (eg., pH-sensitive and ROS-sensitive ones) that specifically release drugs in the inflammatory microenvironment to enhance targeting precision.90 Intranasal administration not only avoids systemic adverse effects but also acts directly on nasal mucosal macrophages, thereby improving therapeutic efficacy.28
Multi-omics technologies (transcriptomics, proteomics, metabolomics) should be applied to screen the core targets of active TCM components. The binding mode of chlorogenic acid to TLR4 needs to be clarified,32 and the molecular mechanism by which dried ginger in Xiaoqinglong Decoction inhibits TRPV1 should be elucidated.13 Standardized extraction processes and quality control standards for active TCM components should be established to ensure the stability of clinical efficacy.93 Structural modifications (eg., esterification and glycosylation) can be performed to improve the bioavailability and metabolic stability of natural products.90
Multi-center, large-sample clinical trials of natural products and biologic agents should be conducted, with a focus on therapeutic efficacy differences among subgroups with distinct polarization phenotypes.13 Detection kits for macrophage polarization-related biomarkers (eg., serum IL-10/IL-4 ratio, nasal mucosal CD206/iNOS expression ratio) should be developed for patient stratification and therapeutic efficacy monitoring.5 Early intervention strategies should be explored, targeting populations with allergic diathesis (eg., children), to prevent the onset of AR or delay disease progression by modulating the interleukin-macrophage polarization pathway.96
The relationship between macrophage heterogeneity and AR should be further explored. It is necessary to clarify the role of subtypes such as MDM3 in nasal polyp formation and identify novel therapeutic targets.15 The epigenetic mechanisms underlying interleukin-mediated regulation of macrophage polarization (eg., DNA methylation and non-coding RNA) should be investigated.97 The regulatory effect of the gut-nasal mucosa axis on macrophage polarization should be explored to provide new insights into AR treatment.98
Conclusions
The core pathological mechanism of allergic rhinitis (AR) revolves around interleukin (IL)-mediated immune dysregulation and mucosal inflammatory cascades, driven by disrupted network balance between pro-inflammatory and anti-inflammatory ILs rather than a single factor. This review systematically summarizes the key regulatory roles of the IL network in AR, providing a mechanistic basis for understanding AR pathogenesis and developing targeted therapies.
Th2-type ILs (IL-4, IL-5, IL-13) act as pivotal pathogenic factors: IL-4 promotes IgE production and mast cell degranulation; IL-5 drives eosinophilic infiltration; IL-13 disrupts nasal mucosal tight junctions. Anti-inflammatory ILs (IL-10, IL-35) maintain immune homeostasis by suppressing Th2 inflammation and pro-inflammatory cytokine release, while IL-6/IL-17 amplifies inflammatory cascades via immune subset regulation.
This review systematically clarifies the synergistic mechanisms of IL network-mediated immune dysregulation and mucosal barrier injury, complementing previous single-factor studies and providing an integrated view of AR pathogenesis. Clinically, it supports the translational potential of IL-targeted agents (IL-4/IL-13 receptor antagonists, anti-IL-5 biologics) and links these novel strategies to established AR therapies (intranasal glucocorticoids, antihistamines, allergen-specific immunotherapy). Real-world challenges including patient phenotypic heterogeneity, administration routes, cost accessibility, and long-term safety should be integrated into future therapeutic development to overcome the limitations of low efficacy and high recurrence of conventional treatments. This review provides a mechanistic foundation for AR precision therapy and supports the optimization of clinical treatment strategies.
Data Sharing Statement
No new data was generated for this study.
Author Contributions
Haihuan Luan: Writing - review & editing, Methodology, Conceptualization. Shouwei Gao: Writing - review & editing, Methodology, Conceptualization. Yuanyuan Ma: Writing - review & editing, Investigation, Conceptualization. Zeyu Yu: Writing - original draft, Formal analysis. Xiaonan Li: Writing - review & editing, Project administration, Methodology, Conceptualization. All authors gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agreed to be accountable for all aspects of the work.
Funding
The authors declare that no funding was received for this study.
Disclosure
All authors declare no conflicts of interest for this study.
References
1. Cayrol C, Girard JP. Interleukin-33 (IL-33): a critical review of its biology and the mechanisms involved in its release as a potent extracellular cytokine. Cytokine. 2022;156:155891. doi:10.1016/j.cyto.2022.155891
2. Ogulur I, Mitamura Y, Yazici D, et al. Type 2 immunity in allergic diseases. Cell Mol Immunol. 2025;22:211–19. doi:10.1038/s41423-025-01261-2
3. Fokkens WJ, Godthelp T, Holm AF, Blom H, Klein-Jan A. Allergic rhinitis and inflammation: the effect of nasal corticosteroid therapy. Allergy. 1997;52:29–32. doi:10.1111/j.1398-9995.1997.tb04819.x
4. Hasala H, Janka-Junttila M, Moilanen E, Kankaanranta H. Levocetirizine and cytokine production and apoptosis of human eosinophils. Allergy Asthma Proc. 2007;28:582–591. doi:10.2500/aap2007.28.3045
5. Zissler UM, Schmidt-Weber CB. Predicting success of allergen-specific immunotherapy. Front Immunol. 2020;11:1826. doi:10.3389/fimmu.2020.01826
6. Wise SK, Damask C, Roland LT, et al. International consensus statement on allergy and rhinology: allergic rhinitis - 2023. Int Forum Allergy Rhinol. 2023;13:293–859.
7. Liu Q, Deng G, Jiang X, et al. Macrophage-mediated activation of the IL4I1/AhR axis is a key player in allergic rhinitis. Int Immunopharmacol. 2025;152:114439. doi:10.1016/j.intimp.2025.114439
8. Zhou H, Zhang W, Qin D, et al. Activation of NLRP3 inflammasome contributes to the inflammatory response to allergic rhinitis via macrophage pyroptosis. Int Immunopharmacol. 2022;110:109012. doi:10.1016/j.intimp.2022.109012
9. Iwasaki N, Terawaki S, Shimizu K, et al. Th2 cells and macrophages cooperatively induce allergic inflammation through histamine signaling. PLoS One. 2021; 16:e0248158.
10. Shi Q, Lei Z, Cheng G, et al. Mitochondrial ROS activate interleukin-1β expression in allergic rhinitis. Oncol Lett. 2018;16:3193–3200. doi:10.3892/ol.2018.8984
11. Kato A. Group 2 innate lymphoid cells in airway diseases. Chest. 2019;156:141–149. doi:10.1016/j.chest.2019.04.101
12. Fukui N, Honda K, Ito E, Ishikawa K. Peroxisome proliferator-activated receptor gamma negatively regulates allergic rhinitis in mice. Allergol Int. 2009;58:247–253. doi:10.2332/allergolint.08-OA-0047
13. Zhou L, Huang L, He G, Li H, Liu M, Xu J. Xiao-Qing-Long-Tang alleviates allergic rhinitis by inhibiting M1 macrophage polarization and modulating the TRPV1 channel. Phytomedicine. 2025;150:157732. doi:10.1016/j.phymed.2025.157732
14. Boreika R, Sitkauskiene B. Interleukin-32 in pathogenesis of atopic diseases: proinflammatory or anti-inflammatory role? J Interferon Cytokine Res. 2021;41:235–243. doi:10.1089/jir.2020.0230
15. Zhang Y, Yang M, Li Y, et al. Role of CD44(+)CCR2(+)CD64(-)monocyte-derived macrophage in chronic rhinosinusitis with nasal polyps. Cell Immunol. 2025;411–412:104953. doi:10.1016/j.cellimm.2025.104953
16. Teufel LU, Taks EJM, van Gemert J, et al. Interleukin 38 reduces antigen-presentation capacity and antibody production after vaccination. Vaccine. 2024;42:126396. doi:10.1016/j.vaccine.2024.126396
17. Liu G, Ma N, Cheng K, et al. Bacteria-derived nanovesicles enhance tumour vaccination by trained immunity. Nat Nanotechnol. 2024;19:387–398. doi:10.1038/s41565-023-01553-6
18. Kamekura R, Kojima T, Takano K, Go M, Sawada N, Himi T. The role of IL-33 and its receptor ST2 in human nasal epithelium with allergic rhinitis. Clin Exp Allergy. 2012;42:218–228. doi:10.1111/j.1365-2222.2011.03867.x
19. Kuo CJ, Chen CY, Lo HR, et al. Helicobacter pylori induces IL-33 production and recruits ST-2 to lipid rafts to exacerbate inflammation. Cells. 2019;9:8. doi:10.3390/cells9010008
20. Hou Y, Li FF, Kong JW, et al. Adipose tissue-derived mesenchymal stem cells regulate Th17/Treg cells in a rat model of allergic rhinitis by activating IL-2/JAK-3/STAT-5 signaling pathway. Int Arch Allergy Immunol. 2025;186:811–823. doi:10.1159/000543758
21. Shi C, Bopp T, Lo HW, Tkaczuk K, Lin J. Bazedoxifene as a potential cancer therapeutic agent targeting IL-6/GP130 signaling. Curr Oncol. 2024;31:5737–5751. doi:10.3390/curroncol31100426
22. Jiang Y, Nguyen TV, Jin J, Yu ZN, Song CH, Chai OH. Bergapten ameliorates combined allergic rhinitis and asthma syndrome after PM2.5 exposure by balancing Treg/Th17 expression and suppressing STAT3 and MAPK activation in a mouse model. Biomed Pharmacother. 2023;164:114959. doi:10.1016/j.biopha.2023.114959
23. Wang J, Shen Y, Li C, et al. IL-37 attenuates allergic process via STAT6/STAT3 pathways in murine allergic rhinitis. Int Immunopharmacol. 2019;69:27–33. doi:10.1016/j.intimp.2019.01.013
24. Jing Y, Xie J, Huang M, Zhao W, Long S, Zhao S. MiR-193b-3p regulates TLR4 expression to inhibit inflammation by targeting ETS1 in allergic rhinitis. Int Arch Allergy Immunol. 2023;184:727–735. doi:10.1159/000528393
25. Fu Y, Gong T, Loughran PA, et al. Roles of TLR4 in macrophage immunity and macrophage-pulmonary vascular/lymphatic endothelial cell interactions in sepsis. Commun Biol. 2025;8:469. doi:10.1038/s42003-025-07921-3
26. Zhang ZT, Zhang DY, Xie K, Wang CJ, Xu F. Luteolin activates Tregs to promote IL-10 expression and alleviating caspase-11-dependent pyroptosis in sepsis-induced lung injury. Int Immunopharmacol. 2021;99:107914. doi:10.1016/j.intimp.2021.107914
27. Zhang X, Wang P, Dang Q, Huang X, Xiao Y, Guan B. Inflammatory cytokines and risk of allergic rhinitis: a Mendelian randomization study. Cytokine. 2024;177:156547. doi:10.1016/j.cyto.2024.156547
28. Choi YY, Jin SC, Yi S, Yang WM. The essential oils from Asarum sieboldii Miq. Alleviate allergic rhinitis by regulating tight junction and inflammation; Network analysis and preclinical validation. J Ethnopharmacol. 2025;338:119032. doi:10.1016/j.jep.2024.119032
29. Ogulur I, Pat Y, Ardicli O, et al. Advances and highlights in biomarkers of allergic diseases. Allergy. 2021;76:3659–3686. doi:10.1111/all.15089
30. Gao YD, Wang ZJ, Ogulur I, et al. The evolution, immunopathogenesis and biomarkers of type 2 inflammation in common allergic disorders. Allergy. 2025;80:1848–1877. doi:10.1111/all.16620
31. Kappen JH, Durham SR, Veen HI, Shamji MH. Applications and mechanisms of immunotherapy in allergic rhinitis and asthma. Ther Adv Respir Dis. 2017;11:73–86. doi:10.1177/1753465816669662
32. Xu X, Wang L, Wu G, Li X. Therapeutic effects of chlorogenic acid on allergic rhinitis through TLR4/MAPK/NF-κB pathway modulation. Biomol Biomed. 2025;25:1571–1580. doi:10.17305/bb.2024.11582
33. Singh D, Guller P, Reid F, et al. A phase 2a trial of the IL-33 monoclonal antibody tozorakimab in patients with COPD: FRONTIER-4. Eur Respir J. 2025;66.
34. Hong JY, Chung Y, Steenrod J, et al. Macrophage activation state determines the response to rhinovirus infection in a mouse model of allergic asthma. Respir Res. 2014;15:63. doi:10.1186/1465-9921-15-63
35. Bao X, Liu B, Jiang Y, et al. Loss of SENP3 mediated the formation of nasal polyps in nasal mucosal inflammation by increasing alternative activated macrophage. Immun Inflamm Dis. 2023; 11:e781.
36. Guo D, Zhou H, Wu X, Xu Y. Dysregulation of PKM2 promotes inflammatory response in allergic rhinitis. Inflamm Res. 2025;74:132. doi:10.1007/s00011-025-02097-2
37. Hosoya K, Satoh T, Yamamoto Y, et al. Gene silencing of STAT6 with siRNA ameliorates contact hypersensitivity and allergic rhinitis. Allergy. 2011;66:124–131. doi:10.1111/j.1398-9995.2010.02440.x
38. Gao Z, Lv H, Wang Y, Xie Y, Guan M, Xu Y. TET2 deficiency promotes anxiety and depression-like behaviors by activating NLRP3/IL-1β pathway in microglia of allergic rhinitis mice. Mol Med. 2023;29:160. doi:10.1186/s10020-023-00757-9
39. Quan SH, Zhang YL, Han DH, Iwakura Y, Rhee CS. Contribution of interleukin 17A to the development and regulation of allergic inflammation in a murine allergic rhinitis model. Ann Allergy Asthma Immunol. 2012;108:342–350. doi:10.1016/j.anai.2012.02.014
40. Zhang L, Ma X, Shi R, et al. Allicin ameliorates imiquimod-induced psoriasis-like skin inflammation via disturbing the interaction of keratinocytes with IL-17A. Br J Pharmacol. 2023;180:628–646. doi:10.1111/bph.15983
41. Koscsó B, Csóka B, Kókai E, et al. Adenosine augments IL-10-induced STAT3 signaling in M2c macrophages. J Leukoc Biol. 2013;94:1309–1315. doi:10.1189/jlb.0113043
42. Zhao S, Liang T, Zhang C, et al. IL-27 Rα(+) cells promoted allorejection via enhancing STAT1/3/5 phosphorylation. J Cell Mol Med. 2020;24:10756–10767. doi:10.1111/jcmm.15700
43. Cao G, Memida T, Huang S, et al. Pro-resolving macrophage-induced IL-35(+) but not TGF-β1(+) regulatory B cell activation requires the PD-L1/PD-1 pathway. Int J Mol Sci. 2025;27:26. doi:10.3390/ijms27010026
44. Xu J, Tang Y, Shen C, et al. Melastoma dodecandrum polysaccharide alleviates allergic rhinitis in mice through modulating NLRP3 and IL-17 axis. Int Immunopharmacol. 2025;161:115054. doi:10.1016/j.intimp.2025.115054
45. Jeong HJ, Shin SY, Oh HA, Kim MH, Cho JS, Kim HM. IL-32 up-regulation is associated with inflammatory cytokine production in allergic rhinitis. J Pathol. 2011;224:553–563. doi:10.1002/path.2899
46. Xiaochun L, Cuizhen L, Xiujuan L, et al. Effect of improved Yupingfeng powder prescription on interleukin-33/suppression of tumorigenicity 2 pathway in mice with ovalbumins-induced allergic rhinitis. J Tradit Chin Med. 2025;45:1215–1227. doi:10.19852/j.cnki.jtcm.2025.06.004
47. Liu X, Mao Y, Du R, Liu Z. Experimental study on muscone reducing NF-κB pathway activity by targeting IL-1R1 to attenuate oxidative stress injury in schwann cells. J Vis Exp. 2025;10:e69009.
48. Wang R, Wang Y, Yang Q, et al. Xiaoqinglong decoction improves allergic rhinitis by inhibiting NLRP3-mediated pyroptosis in BALB/C mice. J Ethnopharmacol. 2024;321:117490. doi:10.1016/j.jep.2023.117490
49. Bouffi C, Rochman M, Zust CB, et al. IL-33 markedly activates murine eosinophils by an NF-κB-dependent mechanism differentially dependent upon an IL-4-driven autoinflammatory loop. J Immunol. 2013;191:4317–4325. doi:10.4049/jimmunol.1301465
50. Sanders NL, Mishra A. Role of interleukin-18 in the pathophysiology of allergic diseases. Cytokine Growth Factor Rev. 2016;32:31–39. doi:10.1016/j.cytogfr.2016.07.001
51. Yue L, Yin X, Hao F, et al. Long noncoding RNA Linc00632 inhibits interleukin-13-induced inflammatory cytokine and mucus production in nasal epithelial cells. J Innate Immun. 2020;12:116–128. doi:10.1159/000500420
52. Junttila IS. Tuning the cytokine responses: an update on interleukin (IL)-4 and IL-13 receptor complexes. Front Immunol. 2018;9:888. doi:10.3389/fimmu.2018.00888
53. Pires GV, Souza HS, Elia CC, et al. Small bowel of patients with asthma and allergic rhinitis: absence of inflammation despite the presence of major cellular components of allergic inflammation. Allergy Asthma Proc. 2004;25:253–259.
54. Teng Y, Zhang R, Liu C, et al. Hong Z: miR-143 inhibits interleukin-13-induced inflammatory cytokine and mucus production in nasal epithelial cells from allergic rhinitis patients by targeting IL13Rα1. Biochem Biophys Res Commun. 2015;457:58–64. doi:10.1016/j.bbrc.2014.12.058
55. Zhou F, Liu P, Lv H, Gao Z, Chang W, Xu Y. miR-31 attenuates murine allergic rhinitis by suppressing interleukin-13-induced nasal epithelial inflammatory responses. Mol Med Rep. 2021;23:42.
56. Yu QN, Guo YB, Li X, et al. ILC2 frequency and activity are inhibited by glucocorticoid treatment via STAT pathway in patients with asthma. Allergy. 2018;73:1860–1870. doi:10.1111/all.13438
57. Rhee CS, Libet L, Chisholm D, et al. Allergen-independent immunostimulatory sequence oligodeoxynucleotide therapy attenuates experimental allergic rhinitis. Immunology. 2004;113:106–113. doi:10.1111/j.1365-2567.2004.01930.x
58. Sehmi R, Wardlaw AJ, Cromwell O, Kurihara K, Waltmann P, Kay AB. Interleukin-5 selectively enhances the chemotactic response of eosinophils obtained from normal but not eosinophilic subjects. Blood. 1992;79:2952–2959. doi:10.1182/blood.V79.11.2952.bloodjournal79112952
59. Saito H, Morikawa H, Howie K, et al. Effects of a cysteinyl leukotriene receptor antagonist on eosinophil recruitment in experimental allergic rhinitis. Immunology. 2004;113:246–252. doi:10.1111/j.1365-2567.2004.01944.x
60. Wang L, Geng Y, Liu L, et al. Synthesis, anti-allergic rhinitis evaluation and mechanism investigation of novel 1,2,4-triazole-enamides as CB1 R antagonist. Eur J Med Chem. 2025;289:117461. doi:10.1016/j.ejmech.2025.117461
61. Fernández-Gallego N, Castillo-González R, Méndez-Barbero N, et al. The impact of type 2 immunity and allergic diseases in atherosclerosis. Allergy. 2022;77:3249–3266. doi:10.1111/all.15426
62. Chen T, Dong L, Wu Y, et al. Bavachinin alleviates allergic rhinitis by modulating gut microbiota and inhibiting NLRP3-mediated epithelial pyroptosis through PI3K/AKT/NF-κB signaling pathway. Cell Signal. 2025;135:112026. doi:10.1016/j.cellsig.2025.112026
63. Nguyen LH, Fakhri S, Frenkiel S, Hamid QA. Molecular immunology and immunotherapy for chronic sinusitis. Curr Allergy Asthma Rep. 2003;3:505–512. doi:10.1007/s11882-003-0062-1
64. Nam SY, Oh HA, Choi Y, Park KY, Kim HM, Jeong HJ. Inhibition of IL-32 signaling by bamboo salt decreases pro-inflammatory responses in cellular models of allergic rhinitis. J Med Food. 2014;17:939–948. doi:10.1089/jmf.2013.2996
65. Kato M, Liu MC, Stealey BA, et al. Production of granulocyte/macrophage colony-stimulating factor in human airways during allergen-induced late-phase reactions in atopic subjects. Lymphokine Cytokine Res. 1992;11:287–292.
66. Guo J, Feng J, Lin L, Zhao X, Wu H. Effect of specific immunotherapy on GM-CSF and IL-5 in the tissues of recurrent nasal polyps. Lin Chuang Er Bi Yan Hou Tou Jing Wai Ke Za Zhi. 2015;29:2023–2025.
67. Liu Y, Liu S, Meng L, et al. The function and mechanism of human nasal mucosa-derived mesenchymal stem cells in allergic rhinitis in mice. Inflamm Res. 2024;73:1819–1832. doi:10.1007/s00011-024-01933-1
68. Peng LY, Li BB, Deng KB, Wang WG. MicroRNA-214-3p facilitates M2 macrophage polarization by targeting GSK3B. Kaohsiung J Med Sci. 2022;38:347–356. doi:10.1002/kjm2.12487
69. Ebrahim Soltani Z, Badripour A, Haddadi NS, et al. Allergic rhinitis in BALB/c mice is associated with behavioral and hippocampus changes and neuroinflammation via the TLR4/ NF-κB signaling pathway. Int Immunopharmacol. 2022;108:108725. doi:10.1016/j.intimp.2022.108725
70. Liu T, Lu BQ, Wang DD, et al. Efficacy and safety of modified yupingfeng nasal spray in controlling the recurrence of persistent and moderate-severe allergic rhinitis: study protocol for a multicenter, open-label, randomized, and parallel-arm trial. Evid Based Complement Alternat Med. 2022;2022:4666332. doi:10.1155/2022/4666332
71. Gopalakrishnan A, Joseph J, Shirey KA, et al. Protection against influenza-induced Acute Lung Injury (ALI) by enhanced induction of M2a macrophages: possible role of PPARγ/RXR ligands in IL-4-induced M2a macrophage differentiation. Front Immunol. 2022;13:968336. doi:10.3389/fimmu.2022.968336
72. McCann MR, Kosloski MP, Xu C, Davis JD, Kamal MA. Dupilumab: mechanism of action, clinical, and translational science. Clin Transl Sci. 2024; 17:e13899.
73. Yang J, Xue J, Mo L, et al. SpaCBA-OVA liposome reprograms immune tolerance to ameliorate allergic rhinitis via DC-T cell crosstalk. Immunology. 2026;177:725–735. doi:10.1111/imm.70078
74. Whiteoak SR, Claridge A, Balendran CA, et al. MicroRNA-31 targets thymic stromal lymphopoietin in mucosal infiltrated CD4+ T cells: a role in achieving mucosal healing in ulcerative colitis? Inflamm Bowel Dis. 2018;24:2377–2385. doi:10.1093/ibd/izy213
75. Jia QN, Zeng YP. Rapamycin blocks the IL-13-induced deficiency of epidermal barrier related proteins via upregulation of miR-143 in HaCaT keratinocytes. Int J Med Sci. 2020;17:2087–2094. doi:10.7150/ijms.45765
76. Wang HR, Wei SZ, Song XY, et al. IL-1β and allergy: focusing on its role in allergic rhinitis. Mediators Inflamm. 2023;2023:1265449. doi:10.1155/2023/1265449
77. Zhang W, Ba G, Tang R, Li M, Lin H. Ameliorative effect of selective NLRP3 inflammasome inhibitor MCC950 in an ovalbumin-induced allergic rhinitis murine model. Int Immunopharmacol. 2020;83:106394. doi:10.1016/j.intimp.2020.106394
78. Zhu Y, Wang Z, Zheng J, et al. RNA-seq revealed the anti-pyroptotic effect of suramin by suppressing NLRP3/caspase-1/GSDMD pathway in LPS-induced MH-S alveolar macrophages. Gene. 2024;893:147888. doi:10.1016/j.gene.2023.147888
79. Jesus AA, Goldbach-Mansky R. IL-1 blockade in autoinflammatory syndromes. Annu Rev Med. 2014;65:223–244. doi:10.1146/annurev-med-061512-150641
80. Bai X, Liu P, Shen H, Zhang Q, Zhang T, Jin X. Water-extracted Lonicera japonica polysaccharide attenuates allergic rhinitis by regulating NLRP3-IL-17 signaling axis. Carbohydr Polym. 2022;297:120053. doi:10.1016/j.carbpol.2022.120053
81. Jeong HJ, Oh HA, Lee BJ, Kim HM. Inhibition of IL-32 and TSLP production through the attenuation of caspase-1 activation in an animal model of allergic rhinitis by Naju Jjok (Polygonum tinctorium). Int J Mol Med. 2014;33:142–150. doi:10.3892/ijmm.2013.1548
82. Lurier EB, Dalton D, Dampier W, et al. Transcriptome analysis of IL-10-stimulated (M2c) macrophages by next-generation sequencing. Immunobiology. 2017;222:847–856. doi:10.1016/j.imbio.2017.02.006
83. Shahzad KA, Wang Z, Cai B, et al. Novel pharmaco-exosomal immunotherapy for united airway diseases: PLGA-encapsulated, mesenchymal stem cell-derived exosomes with PPAR-γ agonist for allergic rhinitis and asthma. Stem Cell Res Ther. 2025;16:488. doi:10.1186/s13287-025-04624-8
84. Kurt S, Kimiz-Gebologlu I, Oncel SS. MSC-exosomes as novel therapeutics in asthma and allergic airway inflammation. Thorac Res Pract. 2025;26:34–36. doi:10.4274/ThoracResPract.2025.s013
85. Gan H, Du J, Ouyang H, Cheng J, Mao H. Interleukin-27 inhibits helper T cell type-2 response in allergic rhinitis. Auris Nasus Larynx. 2020;47:84–89. doi:10.1016/j.anl.2019.05.005
86. Yokota M, Suzuki M, Nakamura Y, Ozaki S, Murakami S. Cytokine modulation by IL-35 in mice with allergic rhinitis. Am J Rhinol Allergy. 2015;29:251–256. doi:10.2500/ajra.2015.29.4188
87. Roca-Ferrer J, Pujols L, Pérez-González M, et al. Superior effect of MP-AzeFlu than azelastine or fluticasone propionate alone on reducing inflammatory markers. Allergy Asthma Clin Immunol. 2018;14:86. doi:10.1186/s13223-018-0311-4
88. Liu Y, Xue M, Han Y, et al. Exosomes from M2c macrophages alleviate intervertebral disc degeneration by promoting synthesis of the extracellular matrix via MiR-124/CILP/TGF-β. Bioeng Transl Med. 2023; 8:e10500.
89. Dudakov JA, Hanash AM, van den Brink MR. Interleukin-22: immunobiology and pathology. Annu Rev Immunol. 2015;33:747–785. doi:10.1146/annurev-immunol-032414-112123
90. Huang J, Chen X, Xie A. Formononetin ameliorates IL‑13‑induced inflammation and mucus formation in human nasal epithelial cells by activating the SIRT1/Nrf2 signaling pathway. Mol Med Rep. 2021;24:1–9.
91. Sun L, Ma W, Gao W, et al. Propofol directly induces caspase-1-dependent macrophage pyroptosis through the NLRP3-ASC inflammasome. Cell Death Dis. 2019;10:542. doi:10.1038/s41419-019-1761-4
92. Guo J, Xu S. Astragaloside IV suppresses histamine-induced inflammatory factors and mucin 5 subtype AC overproduction in nasal epithelial cells via regulation of inflammation-related genes. Bioengineered. 2021;12:6045–6056. doi:10.1080/21655979.2021.1965813
93. Wang J, Wang D, Feng L, et al. Eremophilane-type and xanthanolide-type sesquiterpenes from the aerial parts of Xanthium sibiricum and their anti-inflammatory activities. Phytochemistry. 2023;208:113603. doi:10.1016/j.phytochem.2023.113603
94. Gu Z, Wei P, Kou W, Tang XY, Yao HB, Liu EM. Analysis of multimorbidity of moderate to severe allergic rhinitis in children: a real-world study. Int Arch Allergy Immunol. 2023;184:882–892. doi:10.1159/000530842
95. Tantilipikorn P, Julian YKC, Yeh TH, et al. An observational study to determine the real-life effectiveness of MP-AzeFlu in Asian patients with allergic rhinitis. Drugs Real World Outcomes. 2026;13:171–182. doi:10.1007/s40801-026-00546-w
96. van Breugel M, Qi C, Xu Z, et al. Nasal DNA methylation at three CpG sites predicts childhood allergic disease. Nat Commun. 2022;13:7415. doi:10.1038/s41467-022-35088-6
97. Wong CK, Zhang J, Ip WK, Lam CW. Intracellular signal transduction in eosinophils and its clinical significance. Immunopharmacol Immunotoxicol. 2002;24:165–186. doi:10.1081/IPH-120003748
98. Lee SB, Song JA, Choi GE, Kim HS, Jang YJ. Rhinovirus infection in murine chronic allergic rhinosinusitis model. Int Forum Allergy Rhinol. 2016;6:1131–1138. doi:10.1002/alr.21805
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Shensong Yangxin Capsule Reduces the Susceptibility of Arrhythmia in db/db Mice via Inhibiting the Inflammatory Response Induced by Endothelium Dysfunction
Zhang J, Li H, Wang D, Gu J, Hou Y, Wu Y
Drug Design, Development and Therapy 2023, 17:313-330
Published Date: 5 February 2023
Common Allergens and Immune Responses Associated with Allergic Rhinitis in China
Li Q, Zhang X, Feng Q, Zhou H, Ma C, Lin C, Wang D, Yin J
Journal of Asthma and Allergy 2023, 16:851-861
Published Date: 17 August 2023
The Mechanism of Pyroptosis and Its Application Prospect in Diabetic Wound Healing
Al Mamun A, Shao C, Geng P, Wang S, Xiao J
Journal of Inflammation Research 2024, 17:1481-1501
Published Date: 6 March 2024
Thymosin β4 Regulates Tissue Inflammatory Response in Mouse Nonalcoholic Fatty Liver Disease by Promoting Macrophage M2-Type Polarization
Zhu Z, Liao Y, Mou Q, Liu H, Shen Y, Zhu L, Cong S
Journal of Inflammation Research 2025, 18:5791-5809
Published Date: 29 April 2025
NLRP3 Inflammasome-Mediated Pyroptosis in Diabetic Nephropathy: Pathogenic Mechanisms and Therapeutic Targets
Chen Y, Chen R, Ji X, Zeng Z, Guan C
Journal of Inflammation Research 2025, 18:8399-8418
Published Date: 25 June 2025