Back to Journals » Journal of Inflammation Research » Volume 14
Interferon-λ3 Exacerbates the Inflammatory Response to Microbial Ligands: Implications for SARS-CoV-2 Pathogenesis
Authors Read SA ![]() , Gloss BS, Liddle C, George J
, Gloss BS, Liddle C, George J ![]() , Ahlenstiel G
, Ahlenstiel G ![]()
Received 13 January 2021
Accepted for publication 23 February 2021
Published 1 April 2021 Volume 2021:14 Pages 1257—1270
DOI https://doi.org/10.2147/JIR.S301476
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Scott A Read,1– 3 Brian S Gloss,4 Christopher Liddle,3 Jacob George,3 Golo Ahlenstiel1– 3
1Blacktown Clinical School, Western Sydney University, Blacktown, NSW, 2148, Australia; 2Blacktown Hospital, WSLHD, Blacktown, NSW, 2148, Australia; 3Storr Liver Centre, The Westmead Institute for Medical Research, The University of Sydney and Westmead Hospital, Westmead, NSW, 2145, Australia; 4Westmead Research Hub, Westmead Institute for Medical Research, Westmead, NSW, 2145, Australia
Correspondence: Scott A Read Email [email protected]
Introduction: Interferon lambdas (IFN-λs) are antiviral cytokines that restrict pathogen infection and dissemination at barrier surfaces. Controlled expression of IFN-λs efficiently eliminates acute infections by activating a suite of interferon stimulated genes that inhibit viral propagation and activate local immune cells. Excessive or prolonged production of IFN-λs can however mediate tissue inflammation and disrupt epithelial barriers in both viral and non-viral disease. The mechanism by which IFN-λs drive this disease pathogenesis is poorly understood but may be caused by IFN-λ-mediated amplification of other innate immune signaling pathways.
Methods: Monocyte-derived macrophages were differentiated ± IFN-λ 3 and treated with KDO-lipid A, poly I:C or zymosan, representing bacterial, viral or fungal ligands, respectively. Transcriptome and protein expression were quantified by RNA sequencing/PCR and ELISA/bead array, respectively. Bioinformatic analysis was used to define transcription factor profiles and signaling pathways amplified by IFN-λ 3. Finally, the SARS-CoV-2 dataset GSE152075 was queried to compare the effects of IFNL versus IFNA expression in relation to viral load and nasopharyngeal transcriptomes.
Results: IFN-λ 3 exacerbated inflammatory and chemotactic responses unique to each microbial ligand, as measured by RNA sequencing and by ELISA/bead array. Functional annotation identified pathways amplified by IFN-λ 3, including inflammasome activation. Inflammasome amplification was confirmed in vitro, as measured by caspase 1 activity and IL-1β cleavage. Lastly, SARS-CoV-2 infected nasopharyngeal transcriptomes expressing IFN-λs but not IFN-αs were implicated in myeloid cell-driven pathogenesis including neutrophil degranulation, complement and coagulation cascades.
Discussion: These data suggest that IFN-λs contribute to disease pathology by exacerbating innate immune responses during chronic or severe disease states. IFN-λs may contribute to SARS-CoV-2 disease severity, however further study is required to confirm true causation.
Keywords: interferon lambda, inflammation, COVID-19, innate immunity
Introduction
Interferon lambdas (IFN-λs) are a family of innate immune cytokines composed of IFN-λs 1–4 that are critical mediators of barrier immunity.1 IFN-λs signal through interleukin 10 receptor β (IL10Rβ) and interferon lambda receptor 1 (IFNLR1) whose expression is limited to epithelial cells lining the pulmonary, digestive tract and liver, among others.1 Consequently, IFN-λs protect against viral,2 bacterial3 and fungal4 infection and dissemination at barrier sites to facilitate microbial symbiosis and prevent pathogenic infection.
In addition to epithelial cells, dendritic cell (DC) and macrophage populations are highly sensitive to IFN-λs, representing a second line of IFN-λ mediated defense.5 IFN-λs promote numerous macrophage/DC function including direct antiviral activity, antigen presentation, phagocytosis and chemokine production.5,6 In steady-state, most other immune cell populations do not respond to IFN-λs, but can quickly gain sensitivity upon activation/differentiation: neutrophils in response to toll-like receptor 7 (TLR7) activation, B cells in response to B cell receptor (BCR) + CD40 stimulation, T cells in response to CD3 + CD28 stimulation and monocytes in response to granulocyte-macrophage colony-stimulating factor (GM-CSF).5,7 The importance of this gain of sensitivity has not yet been fully understood, but likely strengthens immune responses, particularly following breach of epithelial barriers.
Genome wide association studies (GWAS) have significantly improved our understanding of IFN-λ-mediated immunity. While IFNL polymorphisms are linked to viral clearance (eg, Hepatitis C virus, HCV), they are also associated with inflammation and fibrosis in both viral and non-viral disease. IFNL genotype and IFN-λ1-3 expression are linked to non-viral liver,8,9 pulmonary10 and skin11 inflammation and fibrosis, as well as autoimmune conditions such as psoriasis11 and lupus.12,13 While the etiologies of these conditions differ, they are associated with and/or aggravated by infection with microbial pathogens triggering innate immune pathway activation.13,14 Consequently, we require a better understanding of the mechanisms by which IFN-λs potentiate antimicrobial immune responses, for better or worse. Indeed, sustained IFN-λ production upon chronic HCV infection can aggravate inflammation and tissue damage increasing the likelihood of progressive disease.8,9
We have recently characterized monocyte derived macrophages (MDMs) as an IFN-λ responsive population with the potential to aggravate inflammatory conditions.5 While these studies were performed in vitro, the development of IFN-λ responsiveness likely mirrors monocyte influx and macrophage differentiation in tissues, where IFN-λs can be highly expressed. To evaluate how IFN-λs modulate macrophage differentiation and the anti-microbial response, we differentiated MDMs in the presence of IFN-λ3 and challenged them with different toll-like receptor ligands; Kdo2-Lipid A (KDO) representing the core and lipid component of lipopolysaccharide (LPS), poly I:C representing viral double-stranded RNA (dsRNA) and zymosan, a component of the fungal cell wall. IFN-λ3 amplified PAMP-specific inflammatory responses, demonstrated transcriptionally via RNA sequencing (RNA-Seq), translationally via cytokine secretion, and functionally via an increase in inflammasome activation. Using transcriptomic data from patients infected with the novel coronavirus SARS-CoV-2, we further examined IFNL expression with respect to pathogenic signaling pathways to validate their inflammatory effects in vivo.
Materials and Methods
Sample Collection
Blood was obtained from 12 healthy volunteers at the Westmead Institute of Medical Research in accordance with the Declaration of Helsinki. Participants for individual experiments differed based on availability, ranging from n = 6–10. Ethics approval was obtained from the Sydney West Area Health Service and University of Sydney. Informed consent was obtained for all subjects (HREC2002/12/4.9(1564)).
Immune Cell Isolation and Culture
Peripheral blood mononuclear cells (PBMCs) were isolated using Ficoll Paque Plus (GE Healthcare) density gradient separation. Monocyte isolations were performed using CD14 microbeads (Miltenyi Biotec) and the AutoMACS Pro Separator (Miltenyi Biotec), resulting in monocyte purity >90%. Monocytes were cultured at 37°C and 5% CO2 in RPMI medium containing 10% fetal calf serum (FCS) and differentiated with 50 ng/mL granulocyte macrophage colony-stimulating factor (GM-CSF, PeproTech) ± 50ng/mL IFN-λ3 (R&D Systems) for 7 days. Differentiation media was replaced on day 3 of culture.
TLR Ligand Treatments
On day 6 of macrophage differentiation 50 ng/mL KDO (TLR4 ligand, Sigma-Aldrich), 1 µg/mL high molecular weight poly I:C (TLR3 ligand, Invivogen), or 10 µg/mL Saccharomyces cerevisiae zymosan (TLR2/Dectin-1 ligand, Invivogen) were added to cells in fresh culture media. Poly I:C was delivered by transfection reagent Lipofectamine 2000 according to the manufacturer’s instructions to stimulate endosomal and cytosolic pattern recognition receptors.
Inflammasome Activation and Measurement
Following 7 days of differentiation, cells were treated with pathogen-associated molecular patterns (PAMPs) for 4 h, followed by the addition of 2.5mM adenosine triphosphate (ATP) for 1 h, after which cells and media were harvested. Secreted IL-1β was measured using the IL-1β DuoSet ELISA kit (R&D Systems) and by Western Blot using the polyclonal goat IL-1β antibody (AF-201, R&D Systems). Caspase 1 activity was measured in cell lysates as previously described.15 Briefly, macrophages were lysed in buffer containing 25 mM Hepes pH 7.4, 1 mM dithiothreitol, 2 mM EDTA and 1 mM PMSF. Cell debris was pelleted and 100 µg of protein was incubated in assay buffer containing 25mM Hepes pH 7.4, 100 mM NaCl, 1% CHAPS, 5 mM DTT, 1 mM EDTA and 200 µM of the caspase 1 substrate Ac-YVAD-pNA (Santa Cruz Biotechnology) for 24 h at 37 °C. Absorbance was measured at 405 nm using the SpectraMax plate reader (Molecular Devices).
RNA Sequencing and Bioinformatics
RNA sequencing was performed as described previously.5 Briefly, RNA was extracted using the Favorgen Tissue Total RNA Kit and the sequencing library was prepared using the TruSeq Stranded mRNA Library Prep Kit (Illumina). Single ended RNA sequencing (RNA-seq) was performed at the Australian Genome Research Facility using the Illumina HiSeq 2500 platform (50bp read length; minimum of 107 reads per sample). Sequence alignments and gene counts were performed using STAR RNA-seq aligner version 2.5.1b16 with the GRCh37 reference sequence. Paired comparisons for differential gene expression was accomplished using EdgeR version 3.16.2.17 Gene expression heat maps were generated using Morpheus software from the Broad Institute (https://software.broadinstitute.org/morpheus).
Functional annotation was performed using ConsensusPath-DB.18 Over-representation analysis was used to define biological processes and pathways associated with up-regulated (>1.5x) gene sets. Pscan19 was queried to identify over-represented transcription factor binding sites within gene set promoter regions (from 450 base pairs before transcription start site to 50 base pairs after).
Quantitative PCR
Macrophage RNA was extracted as above, and cDNA was synthesized using MMLV reverse transcriptase (Promega). The Corbett Rotorgene 6000 thermocycler was used to quantify gene expression using primers and Taqman probes as used previously.5 Primer sequences are available in Supplementary Materials. All transcripts were normalized to 18s ribosomal RNA (Applied Biosystems, 4319413E).
ELISAs and Bead Arrays
Protein secretion were measured in macrophage media using the Milliplex Human Cytokine/Chemokine Magnetic Bead Panel, human CXCL10 ELISA MAX set (439904) from Biolegend and the human CCL2 (DY279), CCL3 (DY270) and CCL4 (DY271) DuoSet ELISA kits from R&D Systems. Protein concentrations were determined according to the manufacturer’s instructions.
Western Blotting
Cells were lysed at 4°C using a denaturing buffer containing protease and phosphatase inhibitors. Particulate matter was pelleted and protein was quantified using the BioRad DC protein assay. Protein was subject to sodium dodecyl sulphate poly-acrylamide gel electrophoresis and gels were transferred to nitrocellulose membranes. Blots were blocked with 5% skim milk powder and the following antibodies were used: IL-1β (AF-201-SP, R&D Systems) and Caspase 1 (MAB6215, R&D Systems). Horseradish peroxidase (HRP) conjugated secondary antibodies and the Supersignal West Pico chemiluminescence kit (Pierce Endogen) were used to visualize protein bands with the Bio-Rad ChemiDoc Gel Imaging System.
SARS-CoV-2 Dataset Analysis
Gene-level counts from the GSE152075 dataset20 were analyzed in the R statistical environment.21 Patients were identified as positive where gene counts were above zero in any of IFNL1-3 or IFNA1,2,5,6,7,8,10,13,16,17 or 21. Differential gene expression (DGE) was evaluated using the glmQLF test in EdgeR.17 Briefly, counts data were background filtered (CPM>1 in more than 19 samples) and normalized. DGE in each group (IFNL only, IFNA only and double positive (BH corrected p-value <0.01) were subjected to pathway analysis and visualization as described above. CIBERSORTx was used to estimate immune cell type abundance in bulk RNA sequencing transcriptomes.22 The “Impute cell fractions” function was used with the 22-cell type signature matrix file LM22.
Statistical Analysis
All data was analyzed using GraphPad Prism Version 8. A minimum of two technical replicates were performed for all assays except RNA sequencing and the average replicate value was used for measurement. Statistical tests were performed based on the normality of the data (parametric versus non-parametric) and are indicated in figure legends. In all analyses, a two-tailed p-value of less than 0.05 was considered statistically significant.
Results
Bacterial, Viral and Fungal Ligands Stimulate Unique but Overlapping Macrophage Immune Responses with IFN-λ3
Previous studies by our group have demonstrated that macrophages, but not monocytes are responsive to IFN-λs,5 supporting their contribution to inflammatory disease as a result of excess IFN-λ production.8,9,23 Nonetheless, IFN-λ3 alone does not stimulate a strong inflammatory transcriptional response.5 We confirmed these results by differentiating human monocytes into macrophages with GM-CSF either alone, or in the presence of IFN-λ3 for 7 days (Figure S1). IFN-λ3 stimulated a potent antiviral response, characterized by the induction of a suite of ISGs (eg, IFITM1, RSAD2) but minimal induction of inflammatory genes (eg, IL6, IL12).
To characterize individual PAMP responses in the absence of IFN-λ, monocytes were differentiated into macrophages using GM-CSF for 6 days, then stimulated with bacterial KDO, viral poly I:C (dsRNA), and fungal zymosan for 24 h. Gene expression clustering of RNA-seq (3 samples/treatment) data was performed, followed by functional annotation of differentially regulated gene sets (Figure 1A). Both KDO and zymosan stimulated a potent inflammatory response characterized by inflammatory cytokine and chemokine induction, as well as an inhibition of cellular growth pathways. Poly I:C stimulated gene sets that mediate the antiviral response, and to a lesser extent, inflammation. To determine the transcriptional overlap between individual PAMPs, we next generated lists of significantly up-regulated genes (>1.5x) in response to each treatment (Supplementary Dataset 1). The inflammatory responses mediated by zymosan and KDO significantly overlap, whereas poly I:C stimulates a more unique transcriptional response (Figure 1B). Functional annotation of genes up-regulated by all three stimuli suggests that the induction of type I (α, β) and II (γ) IFN signaling is shared among all three PAMPs.
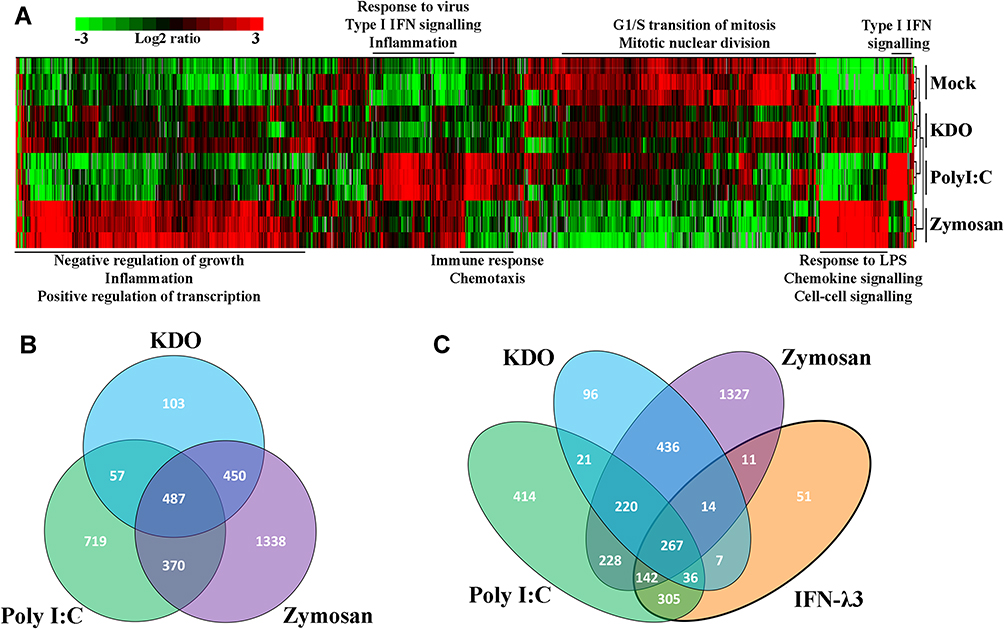 |
Figure 1 Unique transcriptional responses to individual PAMPs overlap with interferon signaling pathways. Macrophage transcriptomes following KDO, poly I:C and zymosan treatment demonstrated unique induction of gene networks (A) (n=3/treatment). Venn diagram demonstrating up-regulated transcripts following PAMP treatments (>1.5 x, p<0.05) (B). Zymosan and KDO demonstrated a significant overlap, dominated by inflammatory and growth regulatory gene expression, while poly I:C gene expression was predominantly antiviral. Poly I:C and IFN-λ3 mediated gene expression demonstrated a significant overlap, supporting their similar antiviral responses (C). |
Since there was an interferon-based gene signature shared by all PAMPs, we compared IFN-λ3 treated macrophages to those treated with KDO, poly I:C and zymosan (Figure 1C). As expected, there was a significant overlap in up-regulated genes between poly I:C and IFN-λ3 treatment, supporting the antiviral nature of IFN-λ3. Minimal overlap was found between IFN-λ3 and inflammatory KDO and zymosan responses.
Interferon Lambda 3 Exacerbates the PAMP-Specific Inflammatory Response
To determine how IFN-λ3 modulates PAMP responses, monocytes were differentiated with GM-CSF either alone, or in the presence of IFN-λ3 for 6 days, then stimulated them for 24 h with PAMPs: bacterial KDO, viral poly I:C (dsRNA), and fungal zymosan. PAMP-specific up/down-regulated gene sets generated as in Figure 2 were first collected, then used to query whether macrophages differentiated in the presence of IFN-λ3 could further increase or decrease their expression (p<0.05, up/down regulation >1.5x) (Figure 2A). For example, of 1097 genes significantly up-regulated in response to KDO, 318 genes were further up-regulated by KDO in macrophages differentiated in the presence of IFN-λ3. Importantly, this method was used to limit the effect of IFN-λ3 stimulated gene expression on subsequent functional annotation studies observed when comparing mock versus PAMP+IFN-λ3 treatment.
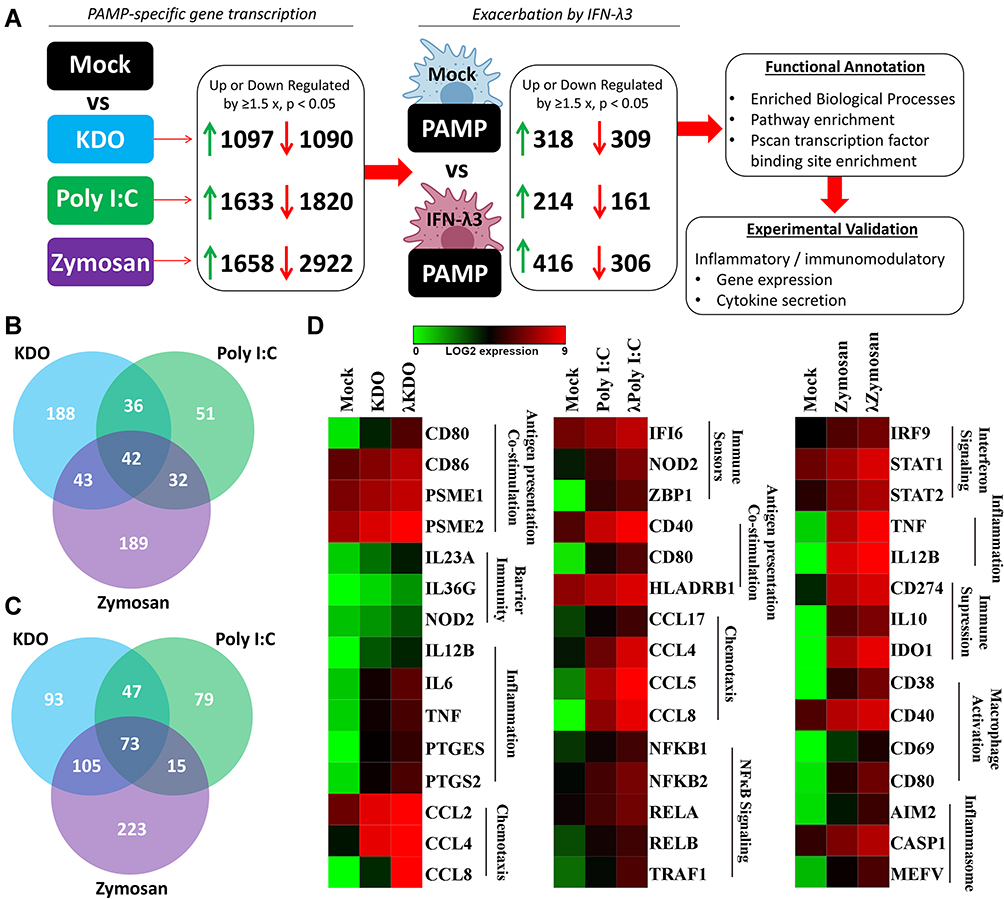 |
Figure 2 IFN-λ3 exacerbates PAMP-specific inflammatory gene expression. To determine if IFN-λ3 can amplify PAMP-specific gene expression, PAMP-specific gene sets (1.5x up or down-regulation, p<0.05) were queried to assess further up/down-regulation by IFN-λ3 (A). IFN-λ3 treatment resulted in down-regulation of 309 (KDO), 161 (poly I:C) and 306 (zymosan) transcripts (B), and up-regulation of 318 (KDO), 214 (poly I:C) and 416 (zymosan) transcripts (C), with minimal overlap particularly among up-regulated genes. Functional annotation revealed IFN-λ3-mediated amplification of unique biological processes among PAMPs (D). Differences are based on 3 biological replicates per treatment. |
IFN-λ3-exacerbated gene sets were functionally annotated to define enriched biological processes, pathways and transcription factor binding to determine their immunological significance. From these PAMP-specific gene lists, Venn diagrams were generated demonstrating shared up- (Figure 2B) and down-regulated (Figure 2C) IFN-λ3 exacerbated gene sets. Of the combined 635 up-regulated genes, 290 are not induced by IFN-λ3 alone, indicating that IFN-λ3 can amplify transcriptional responses to KDO, poly I:C or zymosan that are not IFN-λ3-driven ISGs. Figure 3D is a heat map representation of select genes that are exacerbated by IFN-λ3 during macrophage differentiation based on functional annotation studies outlined in the coming section (full gene lists are available in Supplementary Dataset 1). In response to KDO, IFN-λ3 increased the expression of inflammatory genes such as IL6 and TNF, as well as key regulators of barrier integrity in the gut such as IL23A, IL36G and NOD2. In response to poly I:C, IFN-λ3 increased antigen recognition and chemotactic gene expression, but also substantially increased the expression of numerous NFκB transcription factor genes. In response to zymosan, IFN-λ3 increased both inflammatory cytokine expression, but also genes that drive immune suppression like CD274 (PD-L1) and inflammasome components such as CASP1.
We next performed qPCR and cytokine measurements on antiviral, inflammatory and chemokine targets. In agreement with RNA-seq findings, IFN-λ3 enhanced PAMP-specific antiviral, immunomodulatory and inflammatory gene expression (Figure S2). Bead array and ELISA based measurements demonstrated that Poly I:C stimulated a strong IFN response compared to KDO and zymosan (Figure 3A). IFN-λ3 further increased the secretion of IFN-α2 and IFN-λ1, indicating that an IFN-based positive feedback loop may be in effect. Consistent with a potent IFN response, IFN-λ3 increased the secretion of IFN-responsive IL-15 significantly in all treatments except KDO. In agreement with gene expression studies, IFN-λ3 increased the secretion of inflammatory cytokines IL-6, IL-12p70 and TNF in response to KDO (Figure 3B). Further, IL-6 and TNF secretion were increased in response to poly I:C, reflecting the pro-inflammatory gene signature evident following IFN-λ3 pre-treatment. Lastly, IFN-λ3 increased the secretion of chemokines CCL2 and CXCL10 in response to KDO, CCL3, CCL4 and CXCL10 in response to poly I:C and had little effect on chemokine secretion in response to zymosan, significantly reducing CCL2 secretion (Figure 3C).
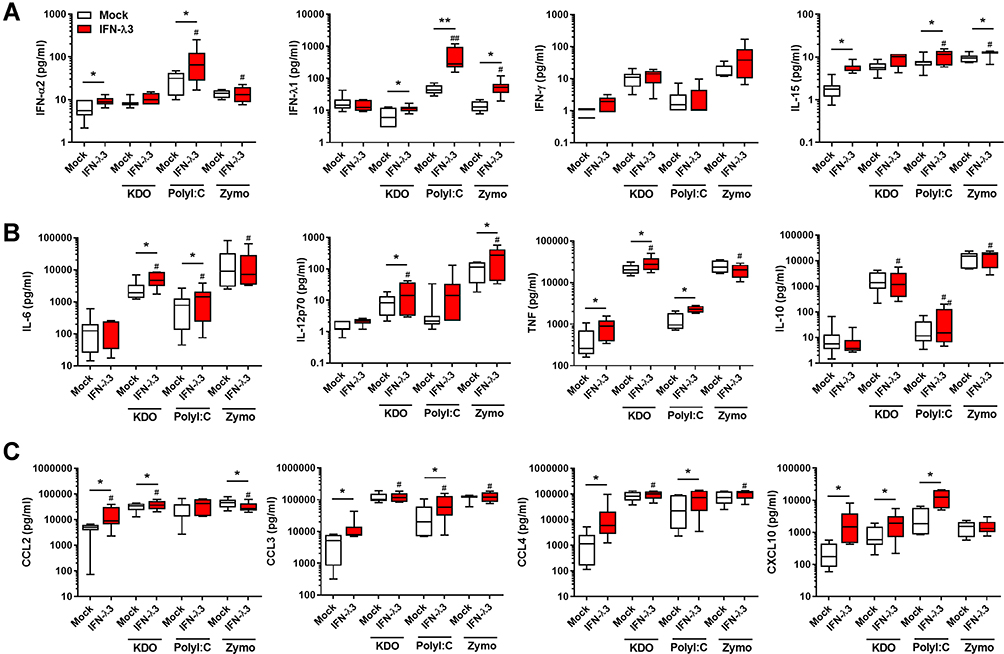 |
Figure 3 Antiviral, inflammatory cytokine and chemokine secretion is exacerbated by IFN-λ3. Macrophages differentiated with IFN-λ3 display enhanced cytokine secretion in response to KDO, poly I:C and zymosan (n=6–8/treatment). IFNs and IFN-stimulated cytokine IL-15 (A), pro- and anti-inflammatory cytokines (B) and chemokines (C). Wilcoxon matched pairs signed rank test, */#p < 0.05, **/##p < 0.01, (median and interquartile range). Logarithmically displayed data was analyzed using linear values and significance tests. *Mock versus IFN-λ3, #IFN-λ3 versus PAMP+IFN-λ3. |
Functional Annotation to Assess the Effects of IFN-λ3 on PAMP-Specific Responses
To determine the functional significance of the IFN-λ3 exacerbated gene sets, ConsensusPathDB was queried to define over-represented biological processes and signaling pathways.18 Among the three PAMP treatments, IFN-λ3 stimulated a significant enrichment in both type I and II IFN and inflammatory responses (Table 1). Moreover, IFN-λ3 enriched biological processes unique to each of the PAMPs studied: IL-23 and IL-12 signaling in response to KDO, death receptor activity and IL-27 binding in response to poly I:C, and myeloid cell differentiation and IL-15 signaling in response to zymosan, among others (full list in Supplementary Dataset 2). ISGs in Table 1 are listed in bold.
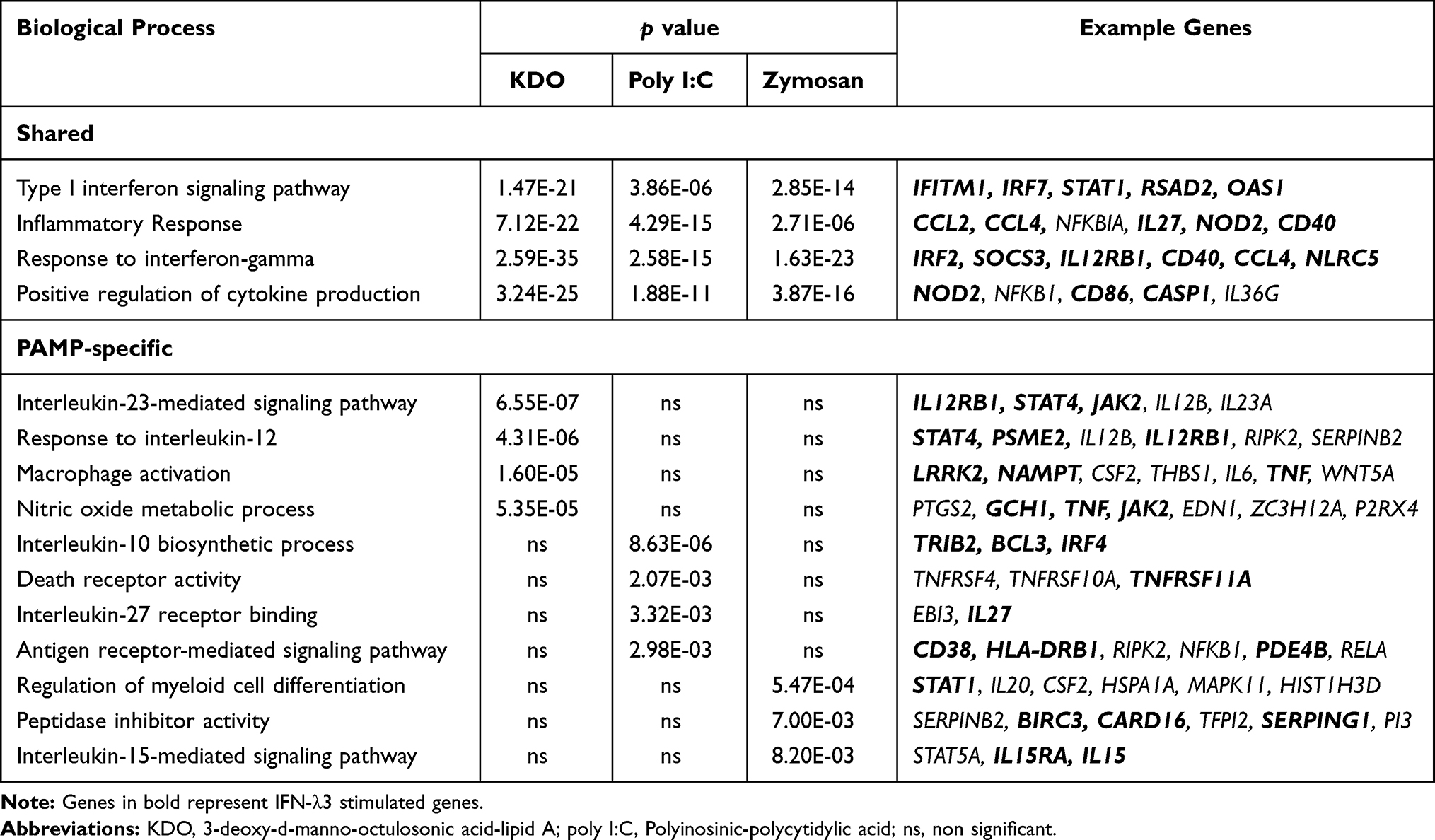 |
Table 1 PAMP-Specific Biological Processes Enriched by IFN-λ3 |
To identify transcription factors that are driving the expression of PAMP-specific and IFN-λ3 treated gene sets, we queried the Pscan web interface.19 Pscan identifies conserved transcription factor binding sites within the promoter regions of gene sets to quantify transcription factor enrichment. Significantly up-regulated (p<0.05, >1.5x increase) genes from KDO, poly I:C and zymosan treatment were first queried, to provide a baseline transcription factor profile for each PAMP. Both KDO and zymosan treatments stimulated an inflammatory transcription factor profile dominated by NFκB binding (Table 2), whereas poly I:C stimulated an IFN-based dominated by IFN regulatory factors (IRFs), STAT1 and STAT1:2 heterodimer binding. Gene sets belonging to macrophages differentiated in the presence of IFN-λ3 demonstrated a potent enrichment in IRF/STAT1:2 and NFκB binding sites following KDO and zymosan treatment. IFN-λ3 pre-treatment decreased IRF/STAT1:2 enrichment following poly I:C, but drastically increased NFκB transcription factor binding. These data are consistent RNA-seq data demonstrating an increase in NFκB expression following poly I:C in macrophages differentiated with IFN-λ3. Full transcription factor profiles are available in Table S1.
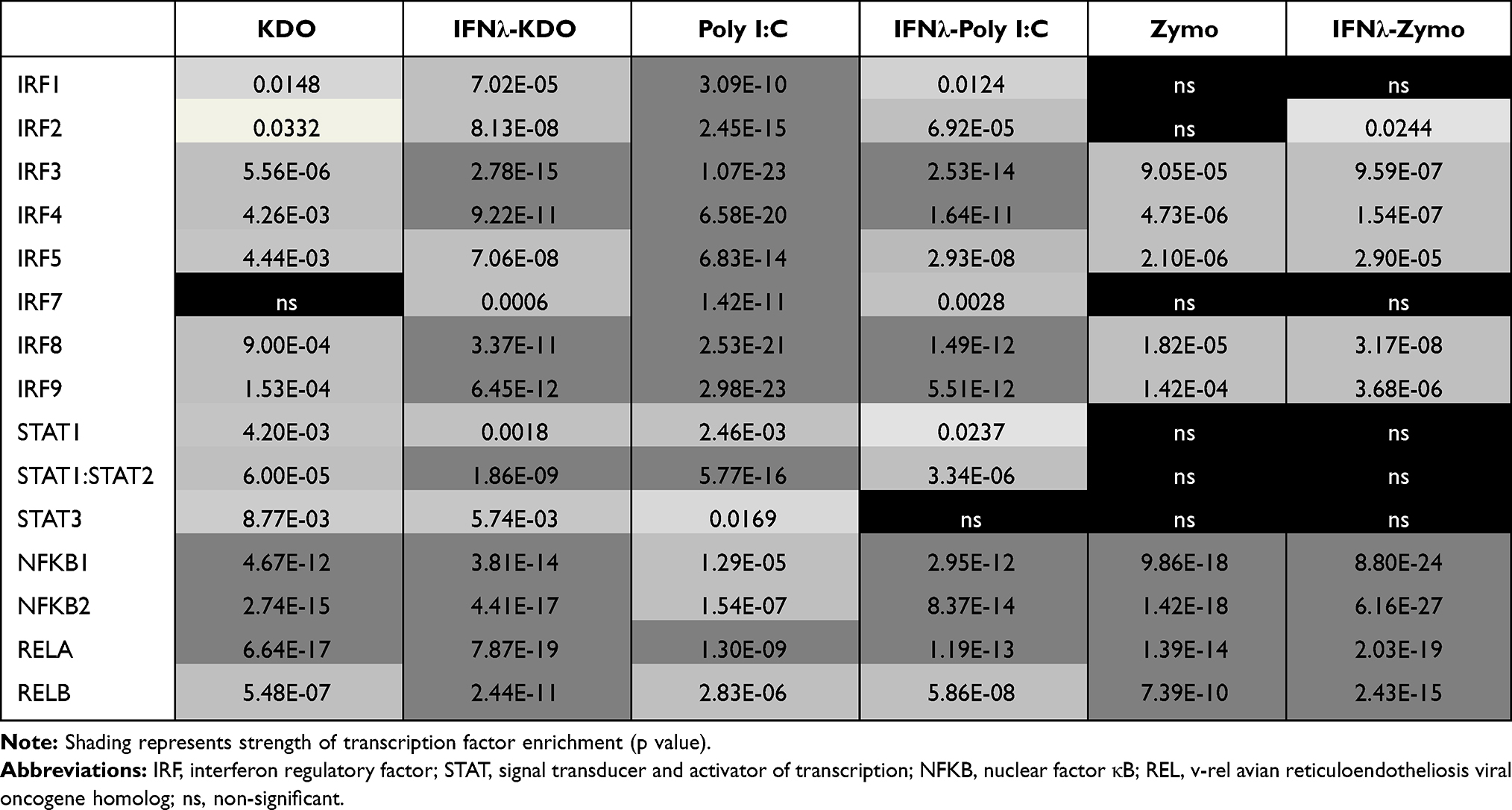 |
Table 2 Transcription Factor Enrichment Following PAMP Stimulation |
IFN-λ3 Exacerbates Inflammasome Activation
We next sought to experimentally validate inflammasome pathway enrichment that was defined by functional annotation in KDO and zymosan treated macrophages. RNA-seq demonstrated an up-regulation of many inflammasome sensors, intermediates and inflammatory cytokines in response to KDO, poly I:C and zymosan, and was confirmed by qPCR (Figure 4A and B). Importantly, while IFN-λ3 alone could significantly increase inflammasome RNA transcripts, it was unable to stimulate inflammasome activation (Figure S3). Stimulation with KDO or zymosan was necessary for the production of pro-inflammatory cytokines such as IL1B and IL18.
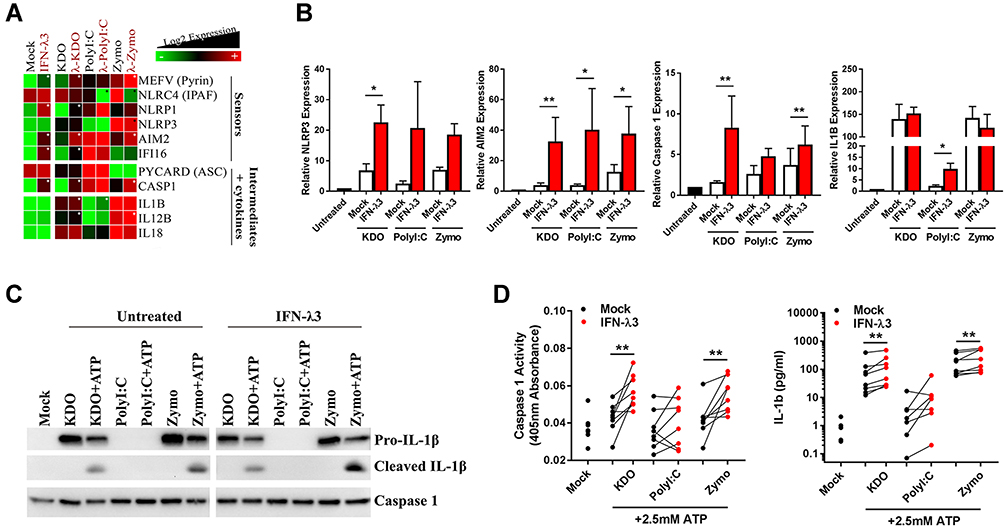 |
Figure 4 IFN-λ3 exacerbates inflammasome activation in response to KDO and zymosan. Transcription of inflammasome components was measured by RNA-seq (n=3/treatment) (A) and qPCR (n=8/treatment) (B) to assess their induction in response to KDO, poly I:C or zymosan in macrophages differentiated ± IFN-λ3. Heat map of RNA-seq data demonstrating IFN-λ3 mediated increase (white star) or decrease (black star) in inflammasome component expression. Inflammasome activation was next measured by treating macrophages with ATP in combination with KDO, poly I:C or zymosan. Caspase 1 expression and IL-1β cleavage was measured by Western blot (n=2 replicates) (C) demonstrating an increase in inflammasome activation that was confirmed by caspase activation assay and IL-1β ELISA (n=7–8/treatment) (D). Mann–Whitney test, *p<0.05, **p<0.01, (mean ± SE). |
Inflammasome activation is the result post-transcriptional protein interaction and cleavage and requires validation at the protein level. Consequently, we utilized “two hits” required to activate the inflammasome in macrophages: 4 h PAMP (KDO, poly I:C or zymosan) + 1 h ATP stimulus. Importantly, IFN-λ3 alone is not sufficient to activate the inflammasome. Caspase expression and functional cleavage was measured by Western blot and by colorimetric assay using the caspase 1 substrate Ac-YVAD-pNA, respectively and cleaved IL-1β was measured by Western blot and ELISA. Both KDO and zymosan were able to activate the inflammasome as demonstrated by IL-1β cleavage, albeit only in the presence of 2.5 mM ATP (Figure 4C). IFN-λ3 treatment was able to further increase inflammasome activation, as demonstrated by increased caspase activation and IL-1β production following KDO and zymosan treatments (Figure 4D).
IFNLs and the SARS-CoV-2 Inflammatory Transcriptome
We next utilized the RNA-Seq dataset GSE152075 (NCBI GeoDatasets) to examine IFNA and IFNL expression in nasopharyngeal swabs from 430 SARS-CoV-2 infected individuals and 54 healthy controls.20 Patient sex, age and viral load (high [N1 ct <19], medium [N1 ct 19–24], low [N1 ct>24]) were available for query. In agreement with previous studies,24 the majority of SARS-CoV-2 infected patients displayed ISG rich profiles with minimal expression of type I and III IFNs. Indeed, only 57/430 patients expressed any of the IFNL genes IFNL1/2/3, and 92/430 expressed any IFNA genes. Most individuals expressing IFNLs had high and medium viral loads, (Figure 5A), whereas IFNA expression was more widely distributed among patients, even appearing in uninfected controls (Figure 5B). Although IFNA and IFNL genes demonstrated minor correlation amongst each other (Figure S4), there was minimal overlap between IFNA and IFNL gene expression in SARS-CoV-2 infected patients (Figure 5B).
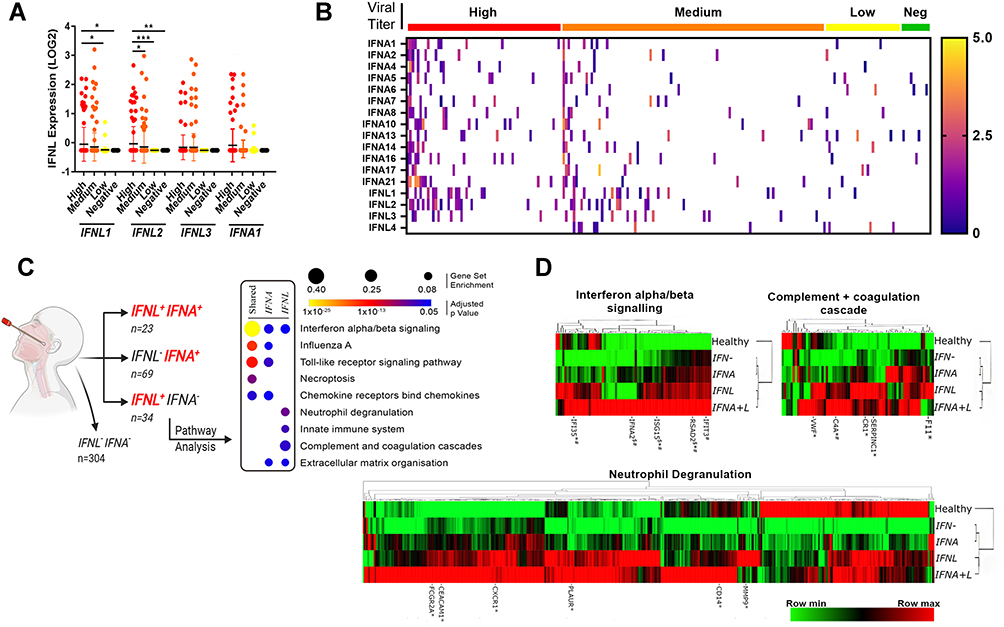 |
Figure 5 IFNL expression upon SARS-CoV-2 infection stimulates antiviral and inflammatory responses. Nasopharyngeal swab transcriptomes from dataset GSE152075 were queried for IFNA and IFNL gene expression. IFNL expression was concentrated within patients with high and medium viral loads (A) and displayed minor overlap with IFNA gene expression in CoV patients and healthy controls (Neg) (B). Differentially expressed genes among subgroups of CoV patients expressing IFNLs (n=34), IFNAs (n=69) or both (n=23), demonstrated that shared and IFNA subgroups occupy primarily an antiviral role (C). IFNL expressing patients display an amplified neutrophil degranulation, complement and coagulation cascade response, supporting their role in disease pathogenesis. This was confirmed by comparing IFNA+, IFNL+ and IFNA+L SARS-CoV-2 swab transcriptomes to patients lacking IFN expression (IFN-) and healthy controls (D). Kruskal–Wallis analysis of variance and Dunn’s multiple comparisons test, *p<0.05, **p<0.01, ***p<0.001 (A). EdgeR glmQLF test, $Significantly up-regulated in IFNA subgroup vs other SARS-CoV-2 transcriptomes, *Significantly up-regulated in IFNL subgroup vs other SARS-CoV-2 transcriptomes, #Significantly up-regulated in IFNA+L subgroup vs other SARS-CoV-2 transcriptomes (D). |
To separate the IFNA from the IFNL gene signature, patient transcriptomes were grouped based on their IFN expression: IFNL expression alone (n=34), IFNA expression alone (n=69) or co-expression of both IFNL and IFNA genes (n=23) (Figure 5C). Subgroup transcriptomes were compared to the remainder of the SARS-CoV-2+ dataset, and differential gene expression was determined (p<0.05, LogFC ≥1, Supplementary Dataset 3). Gene sets were entered into ConsensusPath-DB to identify enriched pathways by which IFNs may be influencing the course of CoV-2 infection. As expected, all three subgroups demonstrated enrichment for type I signaling pathways shared by the IFN-λs. Moreover, the IFN-shared and IFNA subgroups were enriched for TLR response and chemokine signaling pathways supporting their role in antiviral immunity. Unlike IFNA expressing subgroups, the IFNL gene signature was enriched for neutrophil degranulation, complement and coagulation cascade pathways, suggesting that IFNL expression is associated with SARS-CoV-2 pathogenesis (Figure 5C). This is consistent with the transcriptional signature of IFN-λ3 differentiated MDMs. Indeed, complement and coagulation cascades are significantly enriched in IFN-λ3 differentiated MDMs (p=2.86 x 10−6), stimulating induction of numerous complement proteins including C1R, C1S, C1QA/B/C, C2, C5 and CFB (complement factor B).
Using gene sets identified by ConsensusPath-DB from the KEGG and Reactome databases, heat maps were generated to compare healthy, IFN- (SARS-CoV-2+ individuals with no IFN expression), IFNA, IFNL, and IFN-shared expressing individuals. All IFN-expressing subgroups shared activation of IFN signaling pathways (Figure 5D) upon CoV-2 infection, however neutrophil degranulation, complement and coagulation cascade gene expression was significantly amplified in IFNL+ patients. CIBERSORTx was queried using bulk gene expression data and immune cell reference matrices to estimate the abundance of myeloid immune cell types in nasal swab transcriptomes (Figure S5). Importantly, nasopharyngeal swabs can collect immune cells as well as epithelial cells that are in greater abundance during symptomatic respiratory infections.25–27 SARS-CoV-2 transcriptomes, irrespective of their IFN expression, were significantly enriched for M1 (inflammatory), but not M2 (anti-inflammatory) macrophages. The ratio of M1/M2 macrophages was significantly increased in all SARS-CoV-2 transcriptomes, with the strongest enrichment in IFNL expressing patients. There were, however, no significant differences among the IFNL expressing subgroup and the other SARS-CoV-2 infected groups. IFNL expressing transcriptomes also possessed the largest abundance of neutrophils, as calculated by CIBERSORTx, however no significant differences were calculated. Lastly, no significant differences were calculated for resting or activated DCs among subgroups. Nonetheless, the ratio of activated/resting DCs was significantly increased in IFNL and IFNA+L compared to IFN- transcriptomes, suggesting that activated DCs may be a source of IFN-λs in the nasal mucosa.
Discussion
Defining the inflammatory role of IFN-λs, if any, has proven difficult due to their potent antiviral role at barrier surfaces. Indeed, numerous studies have demonstrated that IFNLR1-/- mice cannot adequately control viral dissemination, and consequently develop more severe pathologies.28 It can therefore be suggested that IFN-λs possess an anti-inflammatory role by restricting virus-mediated inflammation. While this is the case for many acute infections,29 IFN-λs are immuno-stimulatory cytokines that can amplify the inflammatory responses to microbial stimuli, as we have shown herein. Excessive or chronic production of IFN-λs therefore have the potential to drive chronic inflammation, tissue damage and fibrosis.8,13 Our study confirms that IFN-λs can amplify inflammatory signaling in a PAMP-specific manner.
Tissue macrophages represent a second line of defense against pathogens that breach the epithelial barriers of the airways, lung, gut and liver. In the absence of infection, tissue macrophages exert a homeostatic role, but can quickly become key mediators of chronic inflammation and tissue damage in the context of autoimmunity and infection.30,31 This is particularly relevant for monocytes, as their differentiation status is dependent on the cytokine milieu present in tissue. We previously demonstrated that IFN-λ3 stimulates an M1 phenotype in MDMs,32 suggesting that they are geared towards an inflammatory phenotype, but are nonetheless not inherently inflammatory as we have shown. This study aims to understand how IFN-λs modulate monocyte differentiation and function, as can occur following monocyte migration in tissue in response to infection in vivo.
To understand the role of IFN-λs in the context of TLR stimulation, we differentiated blood monocytes with IFN-λ3 for 6 days to stimulate maximum effect prior to PAMP treatment. While IFN-λs are likely not a primary cause most inflammatory diseases, they are expressed in response to a variety of microbial ligands and can exacerbate antimicrobial responses as we have shown. Chemokine receptors CCR2, CCR5 and CXCR3 for example, are therapeutic targets for a multitude of inflammatory diseases including NASH (Cenicriviroc33), airway inflammation34 and colitis.35 Their chemokine ligands including CCL2, 4, 8 and CXCL10 are significantly up-regulated in PAMP treated macrophages pre-treated with IFN-λ3. Moreover, IFN-λ3 up-regulated PAMP-mediated production of pro-inflammatory mediators such as IL-6, IL-12 and TNF that are expressed from infiltrating monocytes and resident macrophages in barrier surface disease, resulting in further exacerbation of tissue damage.36 Importantly, our study was limited to testing three microbial ligands representing only a small fraction of inflammatory PAMPs. Indeed, many co-infections in people infected with SARS-CoV-2 are not gram negative species (eg Mycoplasma pneumoniae), and thus do not contain LPS.37 Further study is required using broad PRR agonists such as peptidoglycan (TLR2) or unmethylated CpG DNA (TLR9) to ascertain how IFN-λs can influence disease progression and mortality during co- or super-infection at IFN-λ sensitive barrier locations.
Similar to type I IFNs, IFN-λs signal primarily through the JAK-STAT pathway to drive ISG expression.1 IFN-λs can however exclusively activate signaling via JAK2 and MAPKs, whose signaling drives the expression of at least a subset of ISGs.38,39 While NF-κB activation has not been well characterized in response to IFN-λs, it is potently activated in response to type I IFNs.40 Consequently, IFN-λ mediated activation of STAT,41 MAPK and NF-κB transcription factors may amplify PAMP-mediated gene expression through shared signaling pathways. Indeed, IFN-λ amplified the expression of transcripts possessing NF-κB subunit promoter binding following treatment with all PAMPs (Table 2). This was particularly evident following poly I:C stimulation, where IFN-λ3 amplified inflammatory gene expression and transcription factor binding that was largely absent in response to poly I:C alone.
IFN-λ3 amplified inflammasome transcript expression both alone and in combination with PAMPs. Like type I IFNs,42 IFN-λ3 amplified the expression ISGs NLRP1, AIM2, IFI16 and CASP1. While IFN-λ3 was unable to stimulate the inflammasome alone, it amplified inflammasome activation (Caspase 1 activity and IL-1β cleavage) in response to KDO and zymosan (Figure S3, Figure 5). These data are consistent with mouse studies of respiratory tract infection with Staphylococcus aureus and Pseudomonas aeruginosa, where IL-1β production was significantly dampened in mice lacking IFNLR1.43 IFNs have nonetheless been shown to amplify and dampen inflammasome activation (reviewed in44), indicating that their effects are cell type and context dependent. Poly I:C has been shown to stimulate inflammasome activation in mice,45 but we could not observe any response in human MDMs.
SARS-CoV-2 infection has been shown to evoke an ISG signature both in vitro46 and in vivo,47 however type I and III IFNs appear to be only modestly produced. Impaired IFN production by infected pneumocytes may therefore be the result of subversion of the antiviral response by CoV proteins (reviewed in48). SARS-CoV-2 can infect but not replicate within monocyte-derived macrophages and dendritic cells,49 suggesting that the CoV-2 infection of lung macrophages observed in vivo24 does not interfere with their antiviral and inflammatory responses. Instead, SARS-CoV-2 appears to exacerbate monocyte/macrophage mediated inflammation. Indeed, the myeloid IFN response has been linked to aggravated inflammation, and “cytokine storm” mediators such as IL-1β and TNF in severe SARS-CoV-2 patients.47 Moreover, while resident pulmonary macrophages are reduced in numbers and function,50,51 there is an increase in monocyte-derived macrophages that secrete large amounts of inflammatory cytokines including IL-1β, IL-18, IL-6, and TNF.51 This monocyte infiltration is likely due, in part, to increased CCL2 and CXCL10 chemokine production following SARS-CoV-2 infection.24 These chemokines in particular are linked to disease severity in the form of hospitalization52 and duration of mechanical ventilation.53 These data suggest that infiltrating inflammatory monocytes are significant drivers of SARS-CoV-2 pathologies and support our hypothesis that elevated tissue IFN-λs can modulate macrophage differentiation to drive tissue damage. These data are supported by immune cell abundance estimations using CIBERSORTx. IFNL expressing subgroups demonstrated increased M1/M2 ratios (p>0.05) when compared to IFN- and IFNA subgroups, as well as increased activated/resting DC ratios (p<0.001 versus IFN-, Figure S4). These data suggest that DC populations may represent a significant source of IFN-λs that help drive pro-inflammatory differentiation of infiltrating monocytes in response to infection. Of course, the association between IFNL expression and increased M1/M2 ratio is simply a trend, and therefore requires further study to determine whether the inflammatory effect of IFN-λs is simply correlative or truly causative.
Our data suggests that IFN-λs may play a role in COVID-19 disease severity due to their strong association with neutrophil chemotaxis and the complement/coagulation cascade. Excessive pulmonary neutrophil activation is a hallmark of SARS-CoV-2 disease,54,55 which can generate a coagulopathic milieu when combined with complement activation.56 While macrophage populations during SARS-CoV-2 infection are significant producers of chemokines that drive neutrophil migration including CXCL1,2 and 8,51 we found no significant induction of neutrophil chemoattractants following IFN-λ3 treated differentiation of MDMs. Whether their expression is amplified by IFN-λs during SARS-CoV-2 infection remains to be determined. Nonetheless, key genes within the complement and coagulation cascades were up-regulated by IFN-λ differentiated MDMs, as well as within the IFNL expressing SARS-CoV-2 subgroup.
Importantly, limitations to this analysis support further, direct examination of the role of IFN-λs in SARS-CoV-2 infection. Firstly, nasopharyngeal swabs do not fully recapitulate lower respiratory tract SARS-CoV-2 infection that often dictates disease progression and mortality. In addition, our analysis demonstrated an association between IFN-λs and inflammatory signaling pathways, but no clear causation. Further direct examination of the IFN-λs on SARS-CoV-2 inflammatory responses are required to confirm these hypotheses via IFN-λ treatment or antagonistic studies.
In summary, we have demonstrated that IFN-λs amplify responses to bacterial, viral and fungal PAMPs, likely via synergistic activation of immune pathways. Because tissue macrophages are IFN-λ-responsive sentinel populations at barrier surfaces, they represent a prospective population able to exacerbate inflammatory responses to pathogens. We confirmed these findings using SARS-CoV-2 patient transcriptomes, demonstrating that IFNL gene expression can potentially stimulate signaling pathways that drive tissue pathologies and influence mortality. Together, these data suggest that IFN-λs may represent novel therapeutic targets for acute or chronic barrier inflammation.
Acknowledgments
RNA sequencing was performed in the Genomics Core Facility that is supported by the Westmead Research Hub, Cancer Institute New South Wales and National Health and Medical Research Council. In particular, we would like to Joey Lai for his help preparing mRNA libraries for RNA sequencing. This project was supported by the Ainsworth Bequest to the School of Medicine of Western Sydney University and the Robert W. Storr Bequest to the Sydney Medical Foundation of the University of Sydney.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Lazear HM, Nice TJ, Diamond MS. Interferon-lambda: immune functions at barrier surfaces and beyond. Immunity. 2015;43(1):15–28. doi:10.1016/j.immuni.2015.07.001
2. Baldridge MT, Lee S, Brown JJ, et al. Expression of Ifnlr1 on intestinal epithelial cells is critical to the antiviral effects of IFN-lambda against norovirus and reovirus. J Virol. 2017;91(7). doi:10.1128/JVI.02079-16
3. Odendall C, Voak AA, Kagan JC. Type III IFNs are commonly induced by bacteria-sensing TLRs and reinforce epithelial barriers during infection. J Immunol. 2017;199(9):3270–3279. doi:10.4049/jimmunol.1700250
4. Espinosa V, Dutta O, McElrath C, et al. Type III interferon is a critical regulator of innate antifungal immunity. Sci Immunol. 2017;2(16).
5. Read SA, Wijaya R, Ramezani-Moghadam M, et al. Macrophage coordination of the interferon lambda immune response. Front Immunol. 2019;10:2674.
6. Finotti G, Tamassia N, Cassatella MA. Interferon-lambda s and plasmacytoid dendritic cells: a close relationship. Front Immunol. 2017;8.
7. Santer DM, Minty GES, Golec DP, et al. Differential expression of interferon-lambda receptor 1 splice variants determines the magnitude of the antiviral response induced by interferon-lambda 3 in human immune cells. PLoS Pathog. 2020;16(4).
8. Eslam M, McLeod D, Kelaeng KS, et al. IFN-lambda3, not IFN-lambda4, likely mediates IFNL3-IFNL4 haplotype-dependent hepatic inflammation and fibrosis. Nat Genet. 2017;49(5):795–800.
9. Eslam M, Hashem AM, Leung R, et al. Interferon-lambda rs12979860 genotype and liver fibrosis in viral and non-viral chronic liver disease. Nat Commun. 2015;6:6422.
10. Metwally M, Thabet K, Bayoumi A, et al. IFNL3 genotype is associated with pulmonary fibrosis in patients with systemic sclerosis. Sci Rep. 2019;9.
11. Witte E, Kokolakis G, Witte K, et al. Interleukin-29 induces epithelial production of CXCR3A ligands and T-cell infiltration. J Mol Med. 2016;94(4):391–400. doi:10.1007/s00109-015-1367-y
12. Zahn S, Rehkamper C, Kummerer BM, et al. Evidence for a pathophysiological role of keratinocyte-derived type III interferon (IFN lambda) in cutaneous lupus erythematosus. J Invest Dermatol. 2011;131(1):133–140. doi:10.1038/jid.2010.244
13. Goel RR, Wang X, O’Neil LJ, et al. Interferon lambda promotes immune dysregulation and tissue inflammation in TLR7-induced lupus. Proc Natl Acad Sci U S A. 2020;117(10):5409–5419. doi:10.1073/pnas.1916897117
14. Getts DR, Chastain EML, Terry RL, Miller SD. Virus infection, antiviral immunity, and autoimmunity. Immunol Rev. 2013;255(1):197–209. doi:10.1111/imr.12091
15. Chopra P, Gupta S, Dastidar SG, Ray A. Development of cell death-based method for the selectivity screening of caspase-1 inhibitors. Cytotechnology. 2009;60(1–3):77–83. doi:10.1007/s10616-009-9217-9
16. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi:10.1093/bioinformatics/bts635
17. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi:10.1093/bioinformatics/btp616
18. Kamburov A, Wierling C, Lehrach H, Herwig R. ConsensusPathDB-a database for integrating human functional interaction networks. Nucleic Acids Res. 2009;37(suppl_1):D623–D628. doi:10.1093/nar/gkn698
19. Zambelli F, Pesole G, Pavesi G. Pscan: finding over-represented transcription factor binding site motifs in sequences from co-regulated or co-expressed genes. Nucleic Acids Res. 2009;37(WebServer issue):W247–252. doi:10.1093/nar/gkp464
20. Lieberman NAP, Peddu V, Xie H, et al. In vivo antiviral host response to SARS-CoV-2 by viral load, sex, and age. bioRxiv. 2020. doi:10.1101/2020.06.22.165225
21. R: a language and environment for statistical computing [computer program]. Vienna, Austria: Foundation for Statistical Computing; 2013.
22. Newman AM, Steen CB, Liu CL, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol. 2019;37(7):773–782. doi:10.1038/s41587-019-0114-2
23. Chiriac MT, Buchen B, Wandersee A, et al. Activation of epithelial signal transducer and activator of transcription 1 by interleukin 28 controls mucosal healing in mice with colitis and is increased in mucosa of patients with inflammatory bowel disease. Gastroenterology. 2017;153(1):123–138e128. doi:10.1053/j.gastro.2017.03.015
24. Chu H, Chan JF, Wang Y, et al. Comparative replication and immune activation profiles of SARS-CoV-2 and SARS-CoV in human lungs: an ex vivo study with implications for the pathogenesis of COVID-19. Clin Infect Dis. 2020;71(6):1400–1409. doi:10.1093/cid/ciaa410
25. Alho OP, Karttunen TJ, Karttunen R, Tuokko H, Koskela M, Uhari M. Lymphocyte and mast cell counts are increased in the nasal mucosa in symptomatic natural colds. Clin Exp Immunol. 2003;131(1):138–142. doi:10.1046/j.1365-2249.2003.02037.x
26. Vanders RL, Hsu A, Gibson PG, Murphy VE, Wark PAB. Nasal epithelial cells to assess in vitro immune responses to respiratory virus infection in pregnant women with asthma. Respir Res. 2019;20(1):259. doi:10.1186/s12931-019-1225-5
27. Sungnak W, Huang N, Becavin C, et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat Med. 2020;26(5):681–687. doi:10.1038/s41591-020-0868-6
28. Pott J, Mahlakoiv T, Mordstein M, et al. IFN-lambda determines the intestinal epithelial antiviral host defense. Proc Natl Acad Sci U S A. 2011;108(19):7944–7949. doi:10.1073/pnas.1100552108
29. Galani IE, Triantafyllia V, Eleminiadou EE, et al. Interferon-lambda mediates non-redundant front-line antiviral protection against influenza virus infection without compromising host fitness. Immunity. 2017;46(5):875. doi:10.1016/j.immuni.2017.04.025
30. Gadd VL, Skoien R, Powell EE, et al. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology. 2014;59(4):1393–1405. doi:10.1002/hep.26937
31. Jones GR, Bain CC, Fenton TM, et al. Dynamics of colon monocyte and macrophage activation during colitis. Front Immunol. 2018;9:9. doi:10.3389/fimmu.2018.02764
32. Read SA, Wijaya R, Ramezani-Moghadam M, et al. Macrophage coordination of the interferon lambda immune response. Front Immunol. 2019;10:10. doi:10.3389/fimmu.2019.02674
33. Friedman SL, Ratziu V, Harrison SA, et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology. 2017.
34. Suzaki Y, Hamada K, Nomi T, et al. A small-molecule compound targeting CCR5 and CXCR3 prevents airway hyperresponsiveness and inflammation. Eur Respir J. 2008;31(4):783–789. doi:10.1183/09031936.00111507
35. Mencarelli A, Cipriani S, Francisci D, et al. Highly specific blockade of CCR5 inhibits leukocyte trafficking and reduces mucosal inflammation in murine colitis. Sci Rep. 2016;6.
36. Na YR, Stakenborg M, Seok SH, Matteoli G. Macrophages in intestinal inflammation and resolution: a potential therapeutic target in IBD. Nat Rev Gastroenterol Hepatol. 2019;16(9):531–543. doi:10.1038/s41575-019-0172-4
37. Lansbury L, Lim B, Baskaran V, Lim WS. Co-infections in people with COVID-19: a systematic review and meta-analysis. J Infect. 2020;81(2):266–275. doi:10.1016/j.jinf.2020.05.046
38. Odendall C, Dixit E, Stavru F, et al. Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat Immunol. 2014;15(8):717–726. doi:10.1038/ni.2915
39. Pervolaraki K, Stanifer ML, Munchau S, et al. Type I and type III interferons display different dependency on mitogen-activated protein kinases to mount an antiviral state in the human gut. Front Immunol. 2017;8:459. doi:10.3389/fimmu.2017.00459
40. Yang CH, Murti A, Pfeffer SR, Basu L, Kim JG, Pfeffer LM. IFNalpha/beta promotes cell survival by activating NF-kappa B. Proc Natl Acad Sci U S A. 2000;97(25):13631–13636.
41. Maher SG, Sheikh F, Scarzello AJ, et al. IFNalpha and IFNlambda differ in their antiproliferative effects and duration of JAK/STAT signaling activity. Cancer Biol Ther. 2008;7(7):1109–1115.
42. Veeranki S, Duan X, Panchanathan R, Liu H, Choubey D. IFI16 protein mediates the anti-inflammatory actions of the type-I interferons through suppression of activation of caspase-1 by inflammasomes. PLoS One. 2011;6(10):e27040.
43. Cohen TS, Prince AS. Bacterial pathogens activate a common inflammatory pathway through IFNlambda regulation of PDCD4. PLoS Pathog. 2013;9(10):e1003682.
44. Kopitar-Jerala N. The role of interferons in inflammation and inflammasome activation. Front Immunol. 2017;8:873.
45. Rajan JV, Warren SE, Miao EA, Aderem A. Activation of the NLRP3 inflammasome by intracellular poly I:C. FEBS Lett. 2010;584(22):4627–4632.
46. Vishnubalaji R, Shaath H, Alajez NM. Protein coding and long noncoding RNA (lncRNA) transcriptional landscape in SARS-CoV-2 infected bronchial epithelial cells highlight a role for interferon and inflammatory response. Genes (Basel). 2020;11(7).
47. Lee JS, Park S, Jeong HW, et al. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci Immunol. 2020;5(49).
48. Sa Ribero M, Jouvenet N, Dreux M, Nisole S. Interplay between SARS-CoV-2 and the type I interferon response. PLoS Pathog. 2020;16(7):e1008737.
49. Yang D, Hou Y, Chai Y, et al. Attenuated interferon and proinflammatory response in SARS-CoV-2–infected human dendritic cells is associated with viral antagonism of STAT1 phosphorylation. J Infect Dis. 2020;222(5).
50. Bost P, Giladi A, Liu Y, et al. Host-viral infection maps reveal signatures of severe COVID-19 patients. Cell. 2020;181(7):1475–1488e1412.
51. Xu G, Qi F, Li H, et al. The differential immune responses to COVID-19 in peripheral and lung revealed by single-cell RNA sequencing. Cell Discov. 2020;6:73.
52. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506.
53. Blot M, Jacquier M, Glele LSA, et al. CXCL10 could drive longer duration of mechanical ventilation during COVID-19 ARDS. Crit Care. 2020;24(1).
54. Zuo Y, Yalavarthi S, Shi H, et al. Neutrophil extracellular traps in COVID-19. JCI Insight. 2020;5(11).
55. Liu J, Liu Y, Xiang P, et al. Neutrophil-to-lymphocyte ratio predicts critical illness patients with 2019 coronavirus disease in the early stage. J Transl Med. 2020;18(1):206.
56. Java A, Apicelli AJ, Liszewski MK, et al. The complement system in COVID-19: friend and foe? JCI Insight. 2020.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.