Back to Journals » Infection and Drug Resistance » Volume 11
Interaction between rpsL and gyrA mutations affects the fitness and dual resistance of Mycobacterium tuberculosis clinical isolates against streptomycin and fluoroquinolones
Authors Sun H, Zeng J, Li S, Liang P, Zheng C, Liu Y, Luo T, Rastogi N ![]() , Sun Q
, Sun Q
Received 26 September 2017
Accepted for publication 27 December 2017
Published 27 March 2018 Volume 2018:11 Pages 431—440
DOI https://doi.org/10.2147/IDR.S152335
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Eric Nulens
Honghu Sun,1,2,* Jumei Zeng,3,* Song Li,1 Pengkuan Liang,1 Chao Zheng,1 Yong Liu,4 Tao Luo,5 Nalin Rastogi,6 Qun Sun,1
1Key Laboratory of Bio-Resources and Eco-Environment of the Ministry of Education, College of Life Sciences, Sichuan University, 2Chengdu Institutes for Food and Drug Control, Chengdu, Sichuan, People’s Republic of China; 3Department of Infectious Diseases, Boston Children’s Hospital, Harvard Medical School, Boston, MA, USA; 4Public Health Clinical Center of Chengdu, 5West China College of Preclinical Medicine and Forensic Medicine, Sichuan University, Chengdu, Sichuan, People’s Republic of China; 6WHO Supranational TB Reference Laboratory, Institut Pasteur de la Guadeloupe, Abymes, Guadeloupe, France
*These authors contributed equally to this work
Background: The interaction between different drug-resistant mutations is important to the development of drug resistance and its evolution. In this study, we aimed to reveal the potential relationships between mutations conferring resistance to two important antituberculosis drugs streptomycin (STR) and fluoroquinolones (FLQ).
Materials and methods: We used an in vitro competitive fitness assay to reveal the interactions between different mutations of rpsL and gyrA in drug-resistant Mycobacterium smegmatis, followed by the analysis of the frequency of rpsL and gyrA mutation combinations in 213 STR–FLQ dual-resistant clinical Mycobacterium tuberculosis isolates from Sichuan region, which was also investigated by the whole genome data from 3,056 global clinical M. tuberculosis isolates.
Results: The strains with K43R and K88R mutation in rpsL showed no difference in relative fitness compared with their susceptible ancestor, while K43N, K43M, K43T, and K88E exhibited a significantly lower relative fitness (P<0.05). For the FLQ-resistant mutants, all mutation types showed no difference in their relative fitness. Among STR–FLQ dual-resistant M. smegmatis strains, a lower fitness was detected in those with K43N/M/T and K88E instead of K43R and K88R mutations in rpsL. Among M. tuberculosis isolates harboring rpsL and gyrA dual mutations, the most two frequent combinatorial mutation types were K43R/D94G (n=37) and K43R/A90V (n=24), with the former being the most frequent one by both in vitro tests and clinical survey.
Conclusion: Our results suggest that the interaction between rpsL and gyrA mutations affects the fitness cost in STR–FLQ dual-resistant M. smegmatis and also the predilection of mutation combinations in clinical M. tuberculosis isolates.
Keywords: Mycobacterium, fitness, dual resistance, epistasis
Introduction
Tuberculosis (TB) remains one of the deadliest infectious diseases around the world. According to the World Health Organization, an estimated 10.4 million people developed TB, 1.4 million died from the disease in 2015, and an additional 0.4 million deaths resulted from TB disease among people living with HIV.1 The emergence and spread of multidrug-resistant TB (MDR-TB) with 3.3% of newly diagnosed TB cases worldwide, which was associated with poor treatment outcomes, aggravated the burden of TB treatment in many regions of the world.2 When it came to previously treated TB cases, 20% were identified as MDR-TB.1 However, the understanding of the interaction among various drug resistances in TB is quite poor.
Epistasis refers to the phenomenon of phenotypic effect exerted by one gene (locus) over another leading to the fact that epistatic mutations have different effects in combination than individually.3 Acting as the primary factor in molecular evolution, epistasis has been revealed in many processes.4 Recently, epistasis was reported as an important evolutionary force for the emergence of multidrug-resistant bacteria.5,6 In mycobacteria, resistance to rifampicin (RIF) and ofloxacin was reported to be affected by epistasis, in which different combinations of mutations in drug-resistant genes resulted in the varying degree of loss in fitness.7 However, a more generalized epistatic interaction in mutations conferring resistance to other anti-TB drugs remains unknown.
Streptomycin (STR) was first applied in clinical treatment for TB in 1946,8 although its use was reduced gradually in western countries due to its strong side effects and emergence of drug resistance. Nonetheless, in most developing countries, STR has been used as one of the first-line anti-TB drugs up to date, which induced serious STR resistance in these countries.9 Fluoroquinolones (FLQ), as an important second-line anti-TB drug, are critical to the treatment of MDR-TB.10 With the development of FLQ resistance, MDR-TB may progressively lead to the emergence of extensively drug-resistant (XDR), even totally drug-resistant (TDR) TB.2 During the treatment of MDR-TB cases, STR is usually prescribed in combination with FLQ because of the lack of therapeutic activity of isoniazid and RIF against MDR-TB. However, little is known about the interactive effects between STR- and FLQ-resistant mutations in mycobacteria.
In this study, with the aim to explore the interaction between STR- and FLQ-resistant mutations, Mycobacterium smegmatis was used as a model to investigate the epistasis between mutations conferring resistance to STR and FLQ and the data obtained were then compared with the frequency of STR–FLQ dual-resistant gene mutations in clinical Mycobacterium tuberculosis isolates.
Materials and methods
Bacterial strains and culture conditions
The wild-type M. smegmatis strain mc2155 was used as the starting strain for the screening of drug-resistant strains and the reference for the competitive fitness experiments. The mycobacteria were cultured in Middlebrook 7H9 broth supplemented with 0.05% Tween 80 (7H9-Tw). Bacteria were cultured at 37°C with shaking at 180 rpm.
Microplate-based growth assay for minimal inhibitory concentration (MIC) determination
MIC determination was accomplished as previously described with minor modifications.11 M. smegmatis was shaken at 37°C in 7H9-Tw till reaching optical density (OD600) ~0.8. The cells were spun down, washed twice with 7H9-Tw, and diluted by 100-fold in 7H9-Tw. A total of 200 µL of each culture was placed in a 96-well clear bottom plate. After 4 days of incubation at 37°C, OD was measured from the bottom of the well in a plate reader (Bio-Tek Instruments Inc., Winooski, VT, USA). Growth inhibition was calculated from the following formula:
|
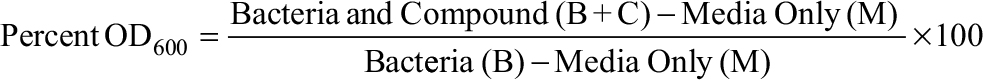
with media only (M) being OD in no growth control wells and bacteria (B) being OD in controls without added antibiotic. The MIC90 was defined as the concentration of antibiotic that caused 90% growth inhibition. Each antibiotic was tested with biological and technical duplicates for every experiment.12
Screening for single- and dual-resistant M. smegmatis mutants
Single STR- and FLQ-resistant M. smegmatis mutants were screened as follows. The wild-type M. smegmatis was cultured till the end of log phase, and 200 µL of bacterial culture was plated onto the minimum of 20–30 plates (or more) of 7H9 agar media containing 16 µg/mL of STR for the isolation of STR-resistant colonies and 10 µg/mL of FLQ (fluoroquinolonic acid) for the isolation of FLQ-resistant colonies. The plates were incubated for 3–5 days at 37°C until colonies became visible. Colonies from these plates were picked and sub-cultured in antibiotic-free Middlebrook 7H9 broth, and the bacteria were cultured to the end of log phase for usage. STR-resistant strains with different rpsL mutants screened above were used subsequently to obtain STR–FLQ dual-resistant strains by culturing in 7H9 agar media containing 10 µg/mL of FLQ.
Mutation type identification
The resistance-related genes, including rpsL for STR and gyrA for FLQ, were amplified by polymerase chain reaction (PCR) using DNA extracted from the single- and dual-resistant mutants. The primers used to amplify the whole gene of rpsL were 5′-GGT CGG CCC TGG TAT TGTG-3′ and 5′-ACG ACG CTG ACG GGA GAAG-3′. Mutations in the quinolone resistance-determining region (QRDR) of gyrA were amplified by primers of 5′-CAT GAG CGT GAT CGT GGG CCG-3′ and 5′-CAG AAC CGT GGG CTC CTG CAC-3′. The 50 μL of PCR mixtures contained 200 μM of dNTP mixture, 0.2 μM of each primer, 50 ng of genomic DNA, and 1.25 U of PrimeSTAR HS DNA Polymerase (Takara Biotechnology, Dalian, China). Amplification consisted of an initial denaturation step at 95°C for 5 min followed by 35 cycles of denaturation at 98°C for 30 s, annealing at 64°C (rpsL)/67°C (gyrA) for 30 s, and extension at 72°C for 1 min, followed by a final extension at 72°C for 10 min. PCR products were purified and sequenced by Thermo Fisher Scientific ( Waltham, MA, USA). The results of sequencing were analyzed by BioEdit using the genome sequence of M. smegmatis strain mc2155 (NC_018289.1) as the control.
Fitness assay and epistasis measurement
The fitness assay was performed as reported previously.7 The mutants were cultured against the wild-type M. smegmatis strain mc2155 in antibiotic-free Middlebrook 7H9 media. For each wild-type-mutant pair, between 1 and 4 replicate competition assays were performed. At the start of the experiment, wild-type and mutant cultures were serially diluted to 10-5–10-6 and plated on nonselective Middlebrook 7H11 media to estimate the baseline colony-forming units (CFU) count in triplicates. About 100 CFU of bacteria/mL from wild-type and mutant cultures were inoculated in 25 mL of Middlebrook 7H9 media in a 1:1 ratio. The competition cultures were incubated at standard conditions on a shaking incubator at 150 rpm. After 72 h, the same competition cultures were serially diluted to 10-5–10-6 and plated on nonselective Middlebrook 7H11 media, as well as diluted to 10-4–10-6 and plated on selective Middlebrook 7H11 media to obtain the endpoint CFU counts. The relative fitness of the drug-resistant strain compared with the wild-type and multiplicative model for pairwise epistasis was determined as previously described.7 Student’s t-test was used to detect differences in the mean fitness, and the limit for statistical significance was set at P=0.05. Test statistics and estimates were based on 1,000 bootstrap replicates. Statistical analysis was performed with STATA SE/10.
Clinical frequency of STR–FLQ-resistant isolates
Isolated from sputum of pulmonary TB patients in the Public Health Clinical Center of Chengdu, China, from 2010 to 2014, all 213 STR–FLQ dual-resistant M. tuberculosis isolates were selected from 2,610 drug-resistant clinical isolates in this study. Genomic DNA was extracted by cetyltrimethyl ammonium bromide (CTAB) and chloroform/isoamyl alcohol extraction method as previously described.13 Primers 5′-GGCCGACAAACAGAACGT-3′ and 5′-GTTCACCAACTGGGTGAC-3′ were used to amplify the full length of STR resistance-related gene rpsL; primers 5′-GGC AAC ACC GAG GTC AAA TC-3′ and 5′-GCC GTC GTA GTT AGG GAT GAA-3′ were used to amplify the QRDR of gyrA gene. PCR mixtures were prepared the same as the detection of resistant M. smegmatis mutants. PCR program was same as the gyrA mutation detection in M. smegmatis mutants. PCR products were purified and sequenced by Thermo Fisher Scientific. The results of sequencing were analyzed by BioEdit with the genome sequence of M. tuberculosis H37Rv (NC_000962.3) as reference. Additionally, a total of 3,056 whole genome data for clinical M. tuberculosis strains were downloaded from The Sequence Read Archive of NCBI (SRA NCBI) database (ERP001072, ERP000192, and SRP018402). The clinical sequence data were aligned with the reference strain H37Rv genome by Bowtie2. The single-nucleotide polymorphisms (SNPs) were identified by Samtools (https://github.com/samtools/samtools). Selection of strains with SNPs in rpsL/gyrA genes and analysis of mutation types and frequency were performed by a Perl script.
Results
Selection of STR- and FLQ-single- and STR–FLQ-double-resistant mutants
The MIC of the M. smegmatis on STR and FLQ was 0.5 and 2 mg/L, respectively (determined by microplate-based growth assay), the screening of resistant M. smegmatis mutants was performed by 16 mg/L of STR and 10 mg/L of FLQ. A total of 60 STR- and 89 FLQ-resistant strains were obtained in single-resistant screening, and 90 STR–FLQ dual-resistant mutant strains were obtained by sequential screening on STR-resistant strains. Mutation types of K43R, K43N, K43M, K43T, K88R, and K88E in rpsL and D94A, D94Y, A90V, G88A, D94N, and D94G in gyrA were identified in single-resistant mutants, respectively. The amino acid sequence alignment of rpsL and gyrA showed that those mutation sites were highly conserved among eight Mycobacterium spp. In addition, positions of mutations K43 and K88 of rpsL were marked as STR interaction site (Figure 1), found to be highly conserved between Escherichia coli and M. tuberculosis rpsL (Figure S1A). Accordingly, we further examined the different combination of mutations in rpsL and gyrA presenting in dual-resistant mutants (Table 1). Most mutation sites of gyrA found in STR–FLQ dual-resistant mutants were consistent with those in FLQ single-resistant mutants.
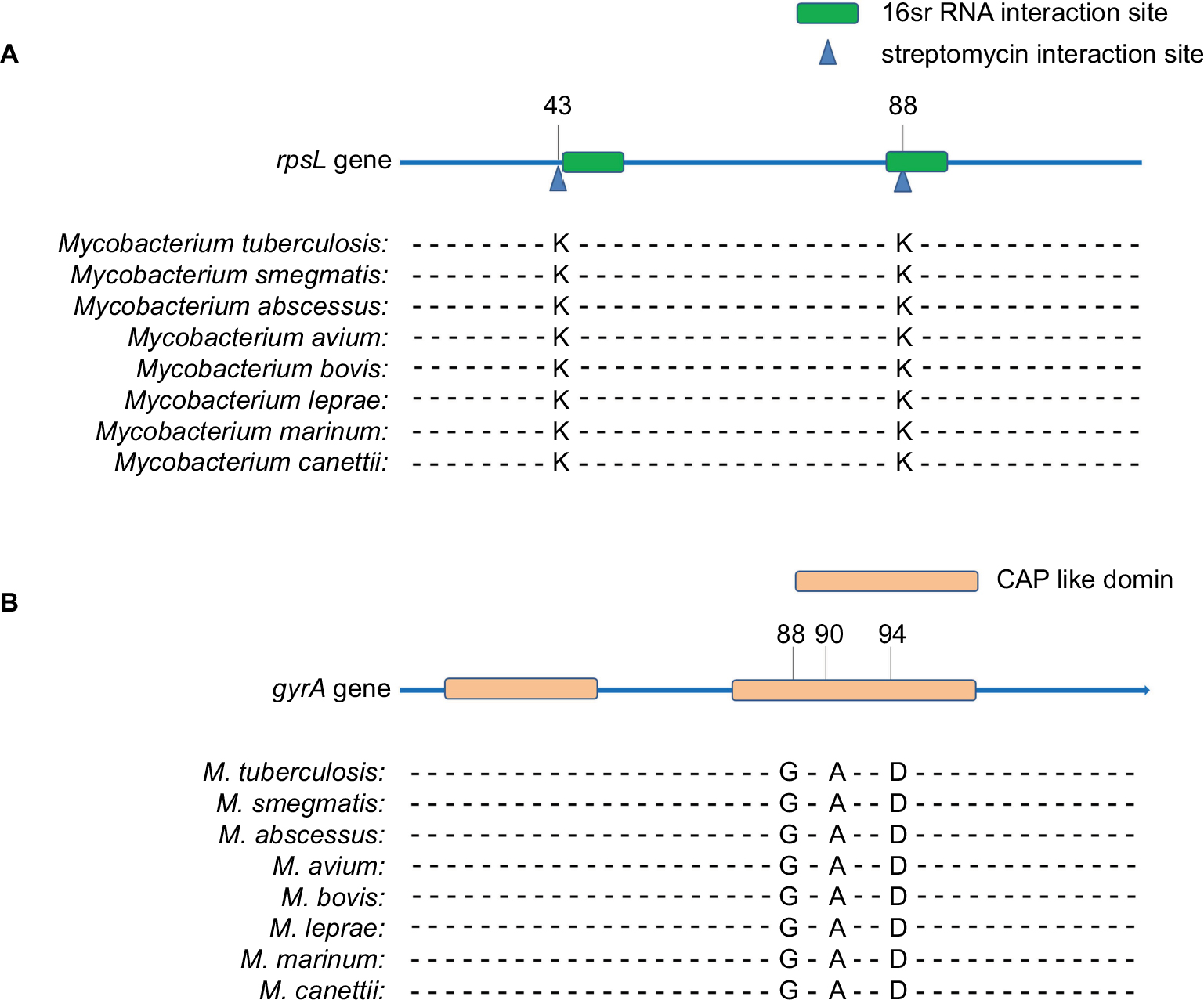 | Figure 1 Functional domain and conservation of rpsL (A) and gyrA (B) mutation sites in Mycobacterium spp. |
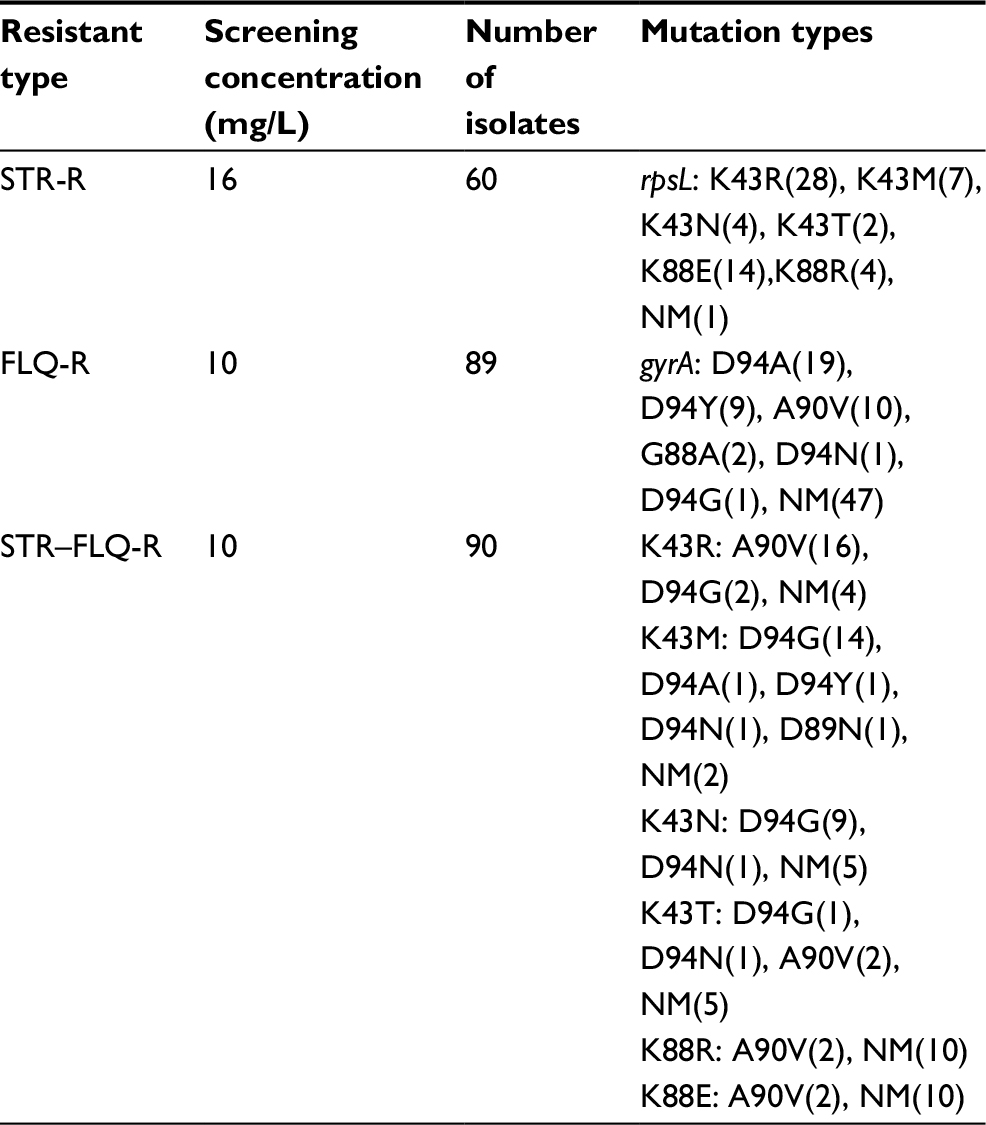 | Table 1 Mutation types of single- and dual-resistant Mycobacterium smegmatis mutants under the selection of antibiotics Abbreviations: FLQ, fluoroquinolones; STR, streptomycin. |
Fitness cost of STR–FLQ-resistant mutants
Relative fitness of M. smegmatis mutants were obtained by co-cultivation in vitro against their susceptible ancestor. For the STR-resistant mutants, we found that the strains with K43R showed no difference in relative fitness compared with their susceptible ancestor, while K43N, K43M, K43T, and K88E showed a significantly lower relative fitness (P<0.05). Additionally, with the highest relative fitness, K43R mutants presented at the highest frequency (7/15) in STR-screened mutants (Table 1). Interestingly, the strains with K88R mutation showed considerable differences in fitness cost (Figure 2A). For the FLQ-resistant mutants, all mutation types were detected with no difference in relative fitness compared with their susceptible ancestor (Figure 2B). No fitness cost was caused by FLQ-resistant mutations in the resistant strains screened. Except strains harboring K43R and K88R mutations, other STR–FLQ dual-resistant strains showed a significant decrease in relative fitness compared with wild strains (Figure 2C). Significantly negative epistasis was found in the strain with K43M/D94Y, K43M/D94G, and K43T/D94G mutations (P<0.05) (Table S1).
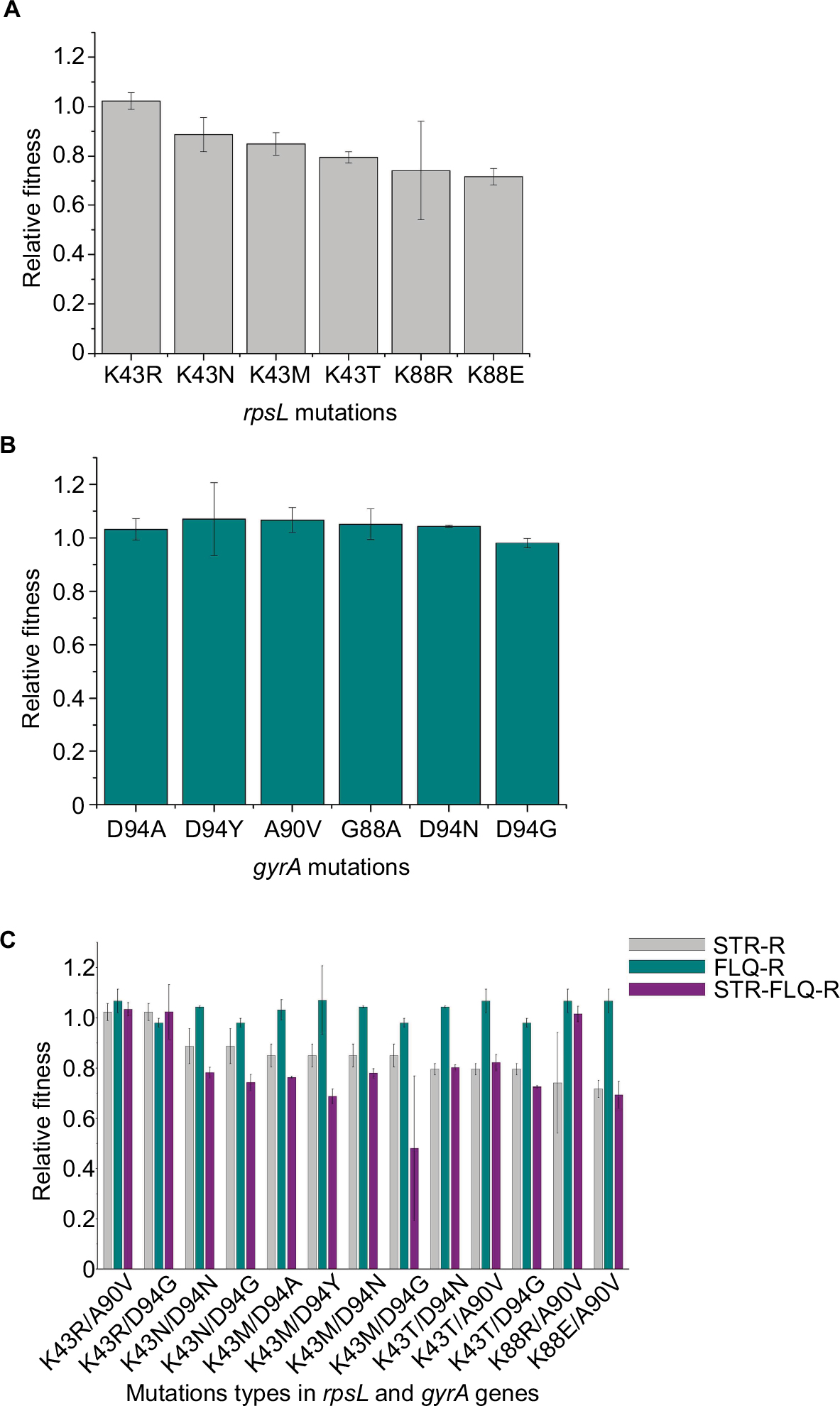 | Figure 2 Relative fitness of Mycobacterium smegmatis mutants. Notes: (A) Relative fitness of STR-resistant M. smegmatis. (B) Relative fitness of FLQ-resistant M. smegmatis. (C) Relative fitness of STR and FLQ dual-resistant M. smegmatis. Gray bar is for the STR-resistant M. smegmatis, turquoise is for FLQ-resistant, and purple is for the STR–FLQ dual-resistant M. smegmatis. Abbreviations: FLQ, fluoroquinolones; STR, streptomycin. |
Remarkably, the model of mutations appearing in rpsL and gyrA in this study (Figure S1) allows to have an insight on the structural evidence of dispensable feature for these mutations. There are 69% identities between M. tuberculosis and E. coli rpsL (Figure S2). Mutation sites of M. tuberculosis rpsL could be seen through the structure of E. coli rpsL (Protein Data Bank accession number 5U4I). Indeed, K43 and K88 of M. tuberculosis rpsL are located closely in the conformation of E. coli 30S ribosomal protein S12 (5U4I as the structure of E. coli entire ribosomal complex).14 Similarly, the G88, A90, and D94 of M. tuberculosis gyrA, crucial for its proper function, sit at the interacting area of chain A and chain B of 3IFZ, which is the crystal structure of M. tuberculosis DNA gyrase.15
Clinical epistasis in STR–FLQ-resistant M. tuberculosis
Among the 213 STR–FLQ dual-resistant clinical isolates collected, we identified 122 isolates with mutations in both rpsL and gyrA genes and 25 isolates with no mutations in them. The other 17 and 49 isolates were detected with mutations in rpsL and gyrA, respectively. Four mutation types, such as K43R (n=107), K88R (n=30), K88T (n=1), and K88M (n=1), were seen in rpsL, while eight mutation types, such as D94G (n=72), A90V (n=41), D94A (n=17), S91P (n=12), D94Y (n=15), D94N (n=12), D94H (n=2), and G88C (n=1), were seen in gyrA (Table 2). Several mutation types in rpsL (K43N, K43M, K43T, and K88E), present in STR-resistant M. smegmatis mutants, were not identified in clinical M. tuberculosis isolates. Similarly, the gyrA mutation present in FLQ-resistant M. smegmatis strains (G88A) was not identified in M. tuberculosis strains. Among the M. tuberculosis strains harboring rpsL and gyrA dual mutations, the most frequent combinatorial mutation types were K43R/D94G (n=37) and K43R/A90V (n=24) (Figure 3A). By searching 3,056 M. tuberculosis genome sequence in global SRA NCBI database, we found 148 strains with rpsL and gyrA combinatorial mutations (Figure 3B). Similar to our local clinical data, only K43R and K88R mutation types were found in rpsL in M. tuberculosis, with D94G, A90V, D94A, S91P, D94Y, and D94N being the predominant ones (79.7%) in gyrA. Based on the information available, K43R and D94G mutations exhibited the highest frequency in rpsL and gyrA, respectively.
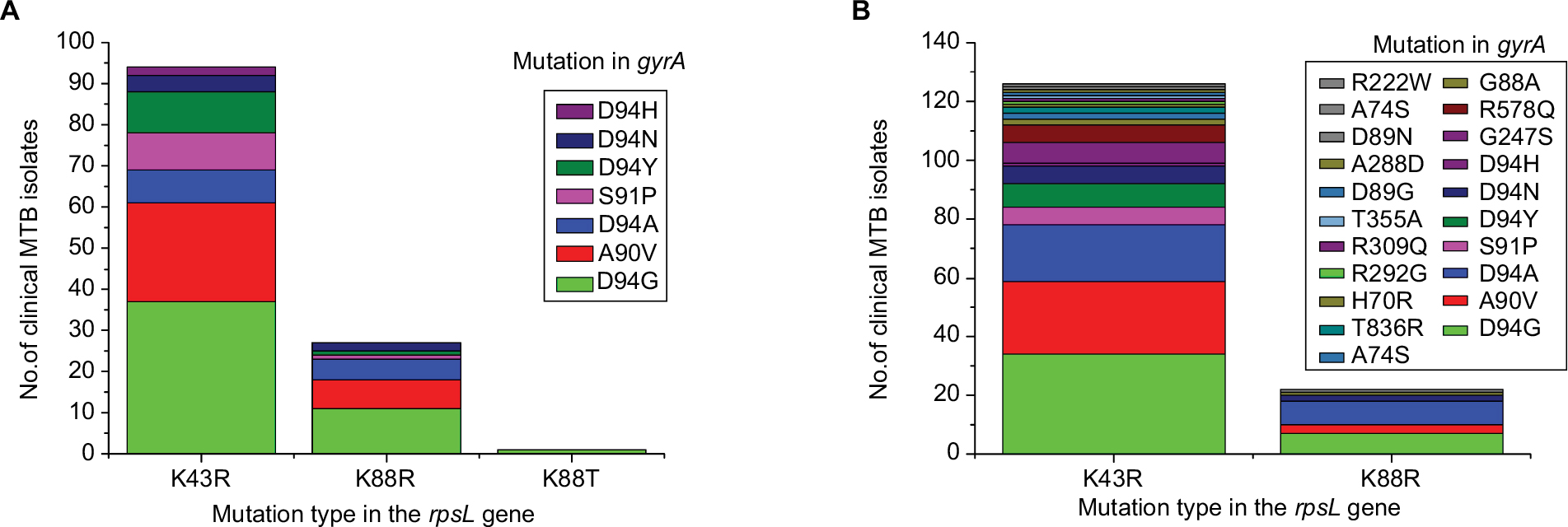 | Figure 3 Frequency of dual-mutation of rpsL and gyrA found in clinical Mycobacterium tuberculosis isolates. Notes: (A) Mutation combinations of clinical M. tuberculosis isolates from Sichuan. (B) Mutation combinations of 3,056 M. tuberculosis genome sequence in database. Abbreviation: MTB, Mycobacterium tuberculosis. |
 | Table 2 Gene mutations in clinical STR- and FLQ-resistant Mycobacterium tuberculosis isolates Note: NM, no mutation detected. |
Discussion
The development of drug resistance in M. tuberculosis is commonly influenced by different factors, such as the lineages preferentially associated with resistance to multiple drugs,16 enhanced heterogeneity at low pH,17 and sub-lethal antibiotic treatment.18 Epistatic interactions in strains of different genetic background, drug-resistant mutations, and compensatory mutations could play a role in the emergence of drug-resistant M. tuberculosis.19 In our study, significant epistasis was found in mutations occurring in rpsL and gyrA of mycobacteria.
The genes of rpsL and gyrA have been widely reported to be related to STR and FLQ resistance in M. tuberculosis, respectively.20 Mutations in the DNA gyrase genes (gyrA and gyrB) may lead to the resistance to FLQ. However, most of the clinical isolates showed mutations mainly on codons 90, 91, and 94, which are located in the QRDR of gyrA. Thus, the FLQ-resistant mutants were analyzed by sequencing the QRDR of gyrA in the present study. In our study, 59/60 STR-resistant strains selected from susceptible M. smegmatis were identified to harbor the mutations in codon 43 and 88 of rpsL, suggesting that the nucleotide change in rpsL was the main cause of STR resistance in spontaneous mutation in vitro, which was similar for STR-resistant M. smegmatis strains screened in the previous study.21 However, only 42/89 FLQ-resistant M. smegmatis strains were detected with mutations in gyrA and this implied that other important mechanisms might exist in the development of resistance to FLQ, for example, the phenotypic resistance to FLQ by mycobacterial mistranslation.22 In previous report by Borrell et al,7 only four mutation types were found in gyrA, whereas more gyrA mutations were detected in our screening, providing a more comprehensive information on their functions. Various combinations of rpsL and gyrA mutations were detected in dual-resistant M. smegmatis screening, suggesting that some kinds of preferences might exist favoring the development of dual-mutations.
It has been generally accepted that the acquisition of drug resistance typically confers a reduction in fitness when the antibiotic is absent.23 However, drug-resistant-related mutations with low or no fitness cost were reported recently in M. tuberculosis and Salmonella typhi,24–26 and the evolution of compensatory mutations may further contribute to the spreading of drug-resistant TB disease.27 In our study, significant fitness differences were detected among M. smegmatis strains of different STR-resistant mutations. K43R in rpsL, the most important mutation causing STR-R, showed a significant higher fitness than other mutations in rpsL. The absence of fitness cost in K43R mutants presumably lead to the increased frequency of occurrence observed for resistant strains harboring this mutation. Therefore, we could observe a dominant (107/139) K43R mutation in clinical STR-resistant M. tuberculosis isolates. The strains with other mutations, such as K43M/T/N and K88E, presented a lower relative fitness when compared with the K43R mutant in competition assays with the wild-type strain of M. smegmatis. This property could explain the absence of these mutations in the clinical M. tuberculosis isolates analyzed. Notably, varied fitness was found in strains harboring K88R mutation, which might result in the changed frequency of K88R, the second highest in clinical STR-resistant mutations. Although the G88C mutation showed a significant decrease in relative fitness reported by Borrell et al,7 gyrA mutations in M. smegmatis showed no fitness cost in all six mutation types, including G88A, in our study. Surprisingly, in clinical gyrA mutations, D94G/A and A90V contributed >76% (130/171) in 213 STR–FLQ dual-resistant M. tuberculosis isolates, which suggested that the preference of gyrA mutations for FLQ-R exists in clinical isolates and the fitness of gyrA in M. smegmatis might not sufficiently reflect the situation of clinical model. The frequency of mutation types in clinical M. tuberculosis genome database showed similar trend to our local clinical detection, supporting this preference existing wildly in clinical cases.
Epistasis between different antibiotic resistance mutations can affect the fitness-related traits and has an important impact on the evolutionary dynamics of a population.28–30 In our study, the relative fitness of STR–FLQ dual-resistant M. smegmatis strains showed a low fitness except those with K43R/D94G, K43R/A90V, and K88R/A90V mutations, indicating that the fitness of dual-resistant strains was mainly determined by the mutation type of rpsL. High fitness of K43R and K88R always exhibited high relative fitness in STR–FLQ dual-resistant mutations; low fitness K43N/M/T and K88E came with low relative fitness in STR–FLQ dual-resistant mutants with these mutations. The rpsL/gyrA dual-mutation types, such as K43R/D94G, K43R/A90V, K88R/D94G, and K88R/A90V, also presented with high frequency in clinical M. tuberculosis isolates (79/122). Negative epistasis in K43M/D94Y, K43M/D94G, and K43T/D94G of significant lower fitness was found in our study, and this adverse dual mutation was also absent in clinical M. tuberculosis isolates. It suggested that epistatic effect between rpsL and gyrA may affect the preference of mutation type in clinical isolates. Similar to our study, in a household-based case–control study by Salvatore et al,31 K43R mutation in rpsL was less likely to be found in multiple-case households, suggesting a negative epistatic interaction. In the work by Borrell et al,7 sign-epistasis (when one mutation has the opposite effect in the presence of another mutation) occurred in six rpoB/gyrA dual-mutation strains with fitness of the double-resistant mutant being higher than at least one of the corresponding single-resistant mutants. It is possible that various epistatic effects exist in different genes, and future research should focus more on the mechanism of negative epistasis between specific mutation types of rpsL and gyrA.
Conclusion
The fitness of dual-resistant strains was primarily determined by the mutation type of rpsL, which was verified by clinical frequency distortion of drug-resistant mutations. Although only one negatively epistatic mutation combination of K43M/D94Y was detected in our study and significant absence of this combination was found in clinical investigation, the further work should be performed to explore the negatively epistatic mutation combination in other anti-TB drugs. Understanding how the resistance conferring mutations interact with negative epistasis could help us optimize the treatment of combined use of drugs and thus reduce the possibility of drug resistance emergence.
Acknowledgments
We sincerely thank our colleagues from Public Health Clinical Center of Chengdu for the collection of samples and the performance of drug susceptibility tests. This work was supported by the Science and Technology Department of Sichuan Province (2017TJPT0014 and 2016SZ0068), and the Ministry of Science & Technology of China (2015DFR31060).
Disclosure
The authors report no conflicts of interest in this work.
References
WHO. Global Tuberculosis Report. 2016. Geneva: World Health Organization; 2016. | ||
Dheda K, Gumbo T, Gandhi NR, et al. Global control of tuberculosis: from extensively drug-resistant to untreatable tuberculosis. Lancet Respir Med. 2014;2(4):321–338. | ||
Lehner B. Molecular mechanisms of epistasis within and between genes. Trends Genet. 2011;27(8):323–331. | ||
Breen MS, Kemena C, Vlasov PK, Notredame C, Kondrashov FA. Epistasis as the primary factor in molecular evolution. Nature. 2012;490(7421):535–538. | ||
Trindade S, Sousa A, Xavier KB, Dionisio F, Ferreira MG, Gordo I. Positive epistasis drives the acquisition of multidrug resistance. PLoS Genet. 2009;5(7):e1000578. | ||
Müller B, Borrell S, Rose G, Gagneux S. The heterogeneous evolution of multidrug-resistant Mycobacterium tuberculosis. Trends Genet. 2013;29(3):160–169. | ||
Borrell S, Teo Y, Giardina F, et al. Epistasis between antibiotic resistance mutations drives the evolution of extensively drug-resistant tuberculosis. Evol Med Public Health. 2013;2013(1):65–74. | ||
Committee TT. Streptomycin treatment of pulmonary tuberculosis: a medical research council investigation. Br Med J. 1948;2(4582):769–782. | ||
Pourakbari B, Mamishi S, Mohammadzadeh M, Mahmoudi S. First-line anti-tubercular drug resistance of Mycobacterium tuberculosis in Iran: a systematic review. Front Microbiol. 2016;7:1139. | ||
Caminero JA, Sotgiu G, Zumla A, Migliori GB. Best drug treatment for multidrug-resistant and extensively drug-resistant tuberculosis. Lancet Infect Dis. 2010;10(9):621–629. | ||
Mir M, Prisic S, Kang CM, et al. Mycobacterial gene cuvA is required for optimal nutrient utilization and virulence. Infect Immun. 2014;82(10):4104–4117. | ||
Singh AK, Carette X, Potluri LP, et al. Investigating essential gene function in Mycobacterium tuberculosis using an efficient CRISPR interference system. Nucleic Acids Res. 2016;44(18):e143. | ||
Guo JH, Xiang WL, Zhao QR, et al. Molecular characterization of drug-resistant Mycobacterium tuberculosis isolates from Sichuan Province in China. Jpn J Infect Dis. 2008;61(4):264–268. | ||
Zeng F, Chen Y, Remis J, et al. Structural basis of co-translational quality control by ArfA and RF2 bound to ribosome. Nature. 2017;541(7638):554–557. | ||
Piton J, Petrella S, Delarue M, et al. Structural insights into the quinolone resistance mechanism of Mycobacterium tuberculosis DNA gyrase. PLoS One. 2010;5(8):e12245. | ||
Ford CB, Shah RR, Maeda MK, et al. Mycobacterium tuberculosis mutation rate estimates from different lineages predict substantial differences in the emergence of drug-resistant tuberculosis. Nat Genet. 2013;45(7):784–790. | ||
Jenkins C, Bacon J, Allnutt J, et al. Enhanced heterogeneity of rpoB in Mycobacterium tuberculosis found at low pH. J Antimicrob Chemother. 2009;63(6):1118–1120. | ||
Kohanski MA, DePristo MA, Collins JJ. Sublethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis. Mol Cell. 2010;37(3):311–320. | ||
Borrell S, Gagneux S. Strain diversity, epistasis and the evolution of drug resistance in Mycobacterium tuberculosis. Clin Microbiol Infect. 2011;17(6):815–820. | ||
Walker TM, Kohl TA, Omar SV, et al. Whole-genome sequencing for prediction of Mycobacterium tuberculosis drug susceptibility and resistance: a retrospective cohort study. Lancet Infect Dis. 2015;15(10):1193–1202. | ||
Pelchovich G, Zhuravlev A, Gophna U. Effect of ribosome-targeting antibiotics on streptomycin-resistant Mycobacterium mutants in the rpsL gene. Int J Antimicrob Agents. 2013;42(2):129–132. | ||
Javid B, Sorrentino F, Toosky M, et al. Mycobacterial mistranslation is necessary and sufficient for rifampicin phenotypic resistance. Proc Natl Acad Sci U S A. 2014;111(3):1132–1137. | ||
Andersson DI. Persistence of antibiotic resistant bacteria. Curr Opin Microbiol. 2003;6(5):452–456. | ||
Lee JH, Ammerman NC, Nolan S, et al. Isoniazid resistance without a loss of fitness in Mycobacterium tuberculosis. Nat Commun. 2012;3(2):753. | ||
Spies FS, von Groll A, Ribeiro AW, et al. Biological cost in Mycobacterium tuberculosis with mutations in the rpsL, rrs, rpoB, and katG genes. Tuberculosis. 2013;93(2):150–154. | ||
Baker S, Duy PT, Nga TV, et al. Fitness benefits in fluoroquinolone-resistant Salmonella Typhi in the absence of antimicrobial pressure. Elife. 2013;2(49):e01229. | ||
Shcherbakov D, Akbergenov R, Matt T, Sander P, Andersson DI, Böttger EC. Directed mutagenesis of Mycobacterium smegmatis 16S rRNA to reconstruct the in vivo evolution of aminoglycoside resistance in Mycobacterium tuberculosis. Mol Microbiol. 2010;77(4):830–840. | ||
Vogwill T, Kojadinovic M, MacLean RC. Epistasis between antibiotic resistance mutations and genetic background shape the fitness effect of resistance across species of Pseudomonas. Proc Biol Sci. 2016;283(1830):20160151. | ||
Durão P, Trindade S, Sousa A, Gordo I. Multiple resistance at no cost: rifampicin and streptomycin a dangerous liaison in the spread of antibiotic resistance. Mol Biol Evol. 2015;32(10):2675–2680. | ||
Stower H. Molecular evolution: epistasis prevails. Nat Rev Genet. 2012;13(12):828. | ||
Salvatore PP, Becerra MC, Abel zur Wiesch P, et al. Fitness costs of drug resistance mutations in multidrug-resistant Mycobacterium tuberculosis: a household-based case-control study. J Infect Dis. 2016;213(1):149–155. |
Supplementary materials
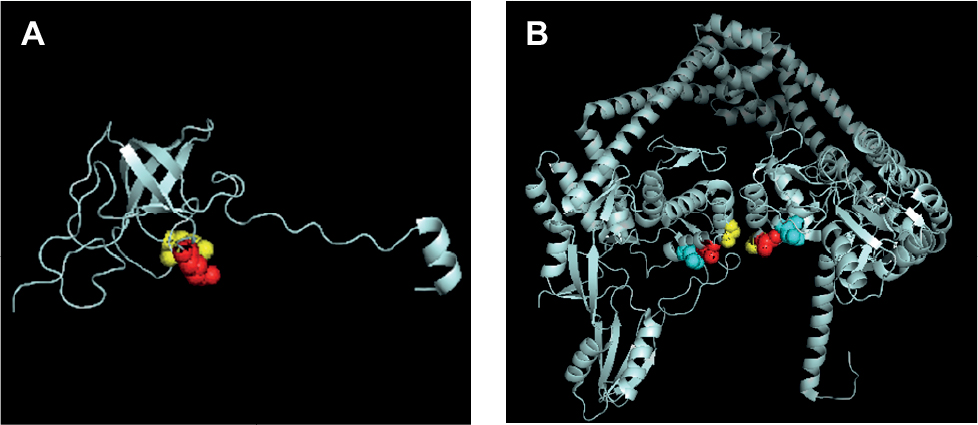 | Figure S1 Model of mutations appearing in rpsL and gyrA in this study. Notes: (A) Structure of Escherichia coli rpsL (Protein Data Bank accession number 5U4I). The position of K43 residue (yellow) and K88 residue (red) screened in this study was highly conserved between E. coli and Mycobacterium tuberculosis. (B) M. tuberculosis gyrA (Protein Data Bank accession number 3IFZ). The position of G88 residue (yellow), A90 residue (red), and D94 residue (cyan) screened in present study is shown. Amino acids were placed in the cartoon structure in dots format using the PyMOL Viewer. |
 | Figure S2 The alignment of rpsL in M. tuberculosis and E. coli. |
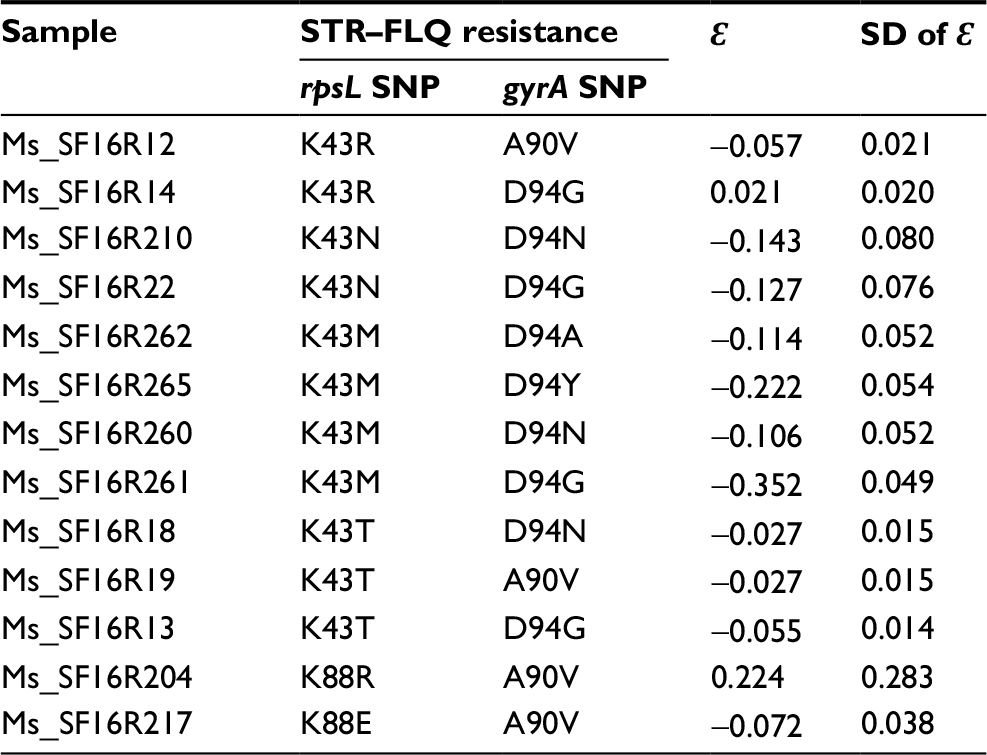 | Table S1 Epistasis (ε) in mutants resistant to STR and FLQ Abbreviations: FLQ, fluoroquinolones; STR, streptomycin. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.